
Abstract
Resistance of important bacterial pathogens to common antimicrobial therapies and the emergence of multidrug-resistant bacteria are increasing at an alarming rate and constitute one of our greatest challenges in the combat of bacterial infection and accompanied diseases. The current shortage of effective drugs, lack of successful prevention measures and only a few new antibiotics in the clinical pipeline demand the development of novel treatment options and alternative antimicrobial therapies. Our increasing understanding of bacterial virulence strategies and the induced molecular pathways of the infectious disease provides novel opportunities to target and interfere with crucial pathogenicity factors or virulence-associated traits of the bacteria while bypassing the evolutionary pressure on the bacterium to develop resistance. In the past decade, numerous new bacterial targets for anti-virulence therapies have been identified, and structure-based tailoring of intervention strategies and screening assays for small-molecule inhibitors of such pathways were successfully established. In this chapter, we will take a closer look at the bacterial virulence-related factors and processes that present promising targets for anti-virulence therapies, recently discovered inhibitory substances and their promises and discuss the challenges, and problems that have to be faced.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
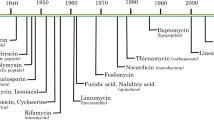
Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction—Challenges in Fighting Bacterial Infections
Although prevention and treatment of infectious diseases have improved over the last decades due to the widespread use of vaccines and anti-infectives and the development of infection control measurements, bacterial infections are still a major cause of morbidity and mortality worldwide. In particular, global spreading of antibiotic resistance genes and their acquisition by clinically relevant bacterial pathogens constitute a serious public health problem. In the European Union alone, about 3 million healthcare-associated infections were reported in 2004 leading to an approximate 50,000 deaths (McHugh et al. 2010). A report published by the Centre for Disease Control and Prevention (CDC) in 2013 estimates that more than 2 million infections and 23,000 deaths annually are caused by antibiotic-resistant bacteria in the USA alone and lists the top 18 drug-resistant pathogens considered a threat in the USA (Control 2013). These pathogens are categorized into three threat classes, urgent, serious and concerning, and include the clinically most relevant pathogens Enterococcus faecium, Staphylococcus aureus, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species summarized as ESKAPE pathogens (Rice 2008) to emphasize that they efficiently “escape” the effects of common antibacterial drugs. This list also includes Clostridium difficile, Neisseria gonorrhoeae, carbapenem- and Extended Spectrum β-Lactam-resistant Enterobacteriaceae (ESBL), drug-resistant Campylobacter, Salmonella and Shigella, and Streptococcus pneumoniae (Control 2013).
At a time, when antibiotic and multidrug resistance is becoming increasingly common in the clinic, new treatments are urgently needed to circumvent high mortality rates due to untreatable infections. Increasing effort is currently being put into the discovery and development of new resistance-breaking antibiotics, and some novel antimicrobial agents are currently in the preclinical and clinical pipeline (Hesterkamp 2015). However, we do not yet know whether this classical approach will succeed to identify new agents with activities against these pan-resistant pathogens in the foreseeable future. Recent work on the occurrence of antibiotic resistance determinants in bacterial populations further demonstrated that non-pathogenic bacteria can be identified in the environment that are already resistant to recently developed antimicrobial drugs (D’Costa et al. 2006; Martinez et al. 2009). For instance, many antibiotic-producing microorganisms encode resistance genes to the antibiotics they synthesize for self-protection (Hopwood 2007). In addition, many of the acquired antibiotic resistance genes are carried on mobile genetic elements such as transposons, integrons, and plasmids, which are frequently transferred to other bacteria of the same or related species by horizontal gene transfer. Evidence exists that the transfer of resistance determinants between commensal bacteria and pathogens, in particular in the intestinal tract, is extensive, leading to the rapid spread of resistance in bacterial populations and communities (Aarestrup 2005; Allen et al. 2010; Salyers et al. 2004). Moreover, an alarmingly high prevalence of antibiotic-resistant bacterial strains has been reported both in domestic and wild animals and in the environment. Prophylactic use of antibiotics in agricultural settings and in feed/water in the animal food production has significantly enhanced the evolution and global spread of antibiotic resistances (Allen et al. 2010; Berendonk et al. 2015; Canton 2009). These findings are daunting as bacteria can rapidly catch up or are even on par with the development of new chemical entities and the danger that we re-enter an apocalyptic preantibiotic era is looming on the horizon.
Consequently, novel intervention strategies are required to respond to current antimicrobial resistance and anticipate evolving resistance mechanisms. One compelling approach to antibiotic therapy is the development of anti-virulence strategies, by which only virulence-associated, but not survival/fitness-relevant traits are targeted. In other words, the anti-infective drug interferes with pathogenicity mechanisms, in particular properties of the bacteria that cause disease. This targeted intervention effectively disarms the pathogen and enables its clearance by the host immune system, but contrary to common antibiotic therapies, it is not bacteriostatic (inhibiting bacterial growth) or bacteriocidal (killing bacteria). Active agents acting in this fashion, the so-called patho- or virulence blockers, alleviate the pressure on the pathogen to develop resistance by solely affecting pathogens expressing the targeted pathogenicity factor (Baron 2010; Beckham and Roe 2014; Cegelski et al. 2008; Clatworthy et al. 2007; Escaich 2008; Lee and Boucher 2015; Lynch and Wiener-Kronish 2008; O’Connell et al. 2013; Rasko and Sperandio 2010; Zambelloni et al. 2015). Consequently, they neither damage nor modify the composition of the natural host microbiota, a process, which is increasingly recognized to facilitate the development, progress, and persistence of chronic inflammatory diseases and other morbidities such as diabetes.
2 Potential Virulence Targets and Strategies
Recent efforts in the development of anti-virulence therapies are directed to target various factors or mechanisms of pathogens that are crucial to initiate an infection and cause disease. This includes interference with various pathogenicity factors promoting cell adhesion, cell invasion, intracellular replication and damage of host tissues, biofilm formation and maintenance (Fig. 1), stress adaptation and metabolic functions important to adapt to the different host environments, mechanisms to evade or overcome the host immune defense, and control systems regulating the expression of virulence-relevant genes.

Schematic representation of the infection processes that can be targeted by anti-virulence drugs. Bacterial pathogens coordinate highly complex and organized temporal and spatial events to colonize and to disseminate to different sites within the host. They produce special pathogenicity factors to adhere to, invade, persist, and in some cases replicate within host cells. Many pathogens are also able to form and replicate with a biofilm-like bacterial community on and/or within cells. Knowledge about the molecular mechanisms will allow the interference with colonization by the (i) inhibition of adhesin biosynthesis and function and (ii) inhibition of invasion, persistence, and proliferation in cells or outside cells in distinct host tissues
The development of novel technologies, such as in vivo transcriptome analysis by RNA-Seq (dual RNA-Seq) and transposon-directed insertion site sequencing (TraDIS), now enables us to identify in vivo active genes and gene functions that are crucial for the survival of pathogens in certain tissues (Chaudhuri et al. 2013; Langridge et al. 2009; Westermann et al. 2012). The discovery of the bottlenecks of an infection will allow us to develop anti-infectives that particularly target fundamental virulence mechanisms (the Achilles heel) of a pathogen. Particularly, approaches that (i) target classical virulence factors, such as adhesins/invasins, (ii) inhibit pathogen-induced host signaling disruption by toxins, effectors, and immune modulators, (iii) manipulate microbial signal transduction and regulation, or (iv) interfere with functions required for bacterial survival and/or persistence during the infection appear promising for the development of new therapeutics against infections (Fig. 2).

Schematic overview of the targets of current anti-virulence strategies designed for Gram-negative bacterial pathogens. Pathoblockers can target toxin function, quorum and environmental parameter sensing, gene regulation by global or specific regulators, sensory or regulatory RNAs, cell adhesion and invasion-promoting surface structures, and bacterial secretion systems (i.e., T3SS, T4SS)
2.1 Denying Access: Adherence and Invasion Inhibitors
Upon entering the host, bacterial pathogens must travel to their respective site of infection in order to initialize the disease process. Once the bacteria reach the site of infection, bacterial cell surface structures and appendages such as pili/fimbriae and afimbrial adhesins detect and interact tightly with specific host cell receptors to adhere to the host cell (Thanassi et al. 2012). Cell attachment enables bacteria to withstand host mechanical and immunological clearance and is crucial for the initiation of an infection. Furthermore, adhesion is essential for the pathogen to get into close proximity with the host cell surface, a process required for the formation and activation of secretion systems (Thanassi et al. 2012). Agents targeting bacterial adherence, e.g., by the inhibition of pili/fimbriae or adhesin formation would not only deny access to host tissues, but would also promote rapid clearance of the bacteria and avoid the release and translocation of tissue-damaging factors (e.g., toxins) (Krachler and Orth 2013). As the adhesion structures are further specific to the pathogen and will mediate only attachment to host cells that express the corresponding receptor, they are potential targets for new anti-infectives.
Pili or fimbriae are hairlike, multi-subunit bacterial cell surface protrusions that facilitate bacterial adhesion and aid colonization. They are important virulence factors in a wide range of bacterial pathogens, including E. coli, Salmonella, Yersinia, Haemophilus, Pseudomonas, and Klebsiella (Krachler and Orth 2013). Many pili structures are assembled in the bacterial cell via the chaperone–usher pathway. In this pathway, the pilin subunits reach the periplasm via the Sec pathway, where they interact with the chaperone, which aids the folding of the pilin subunit’s immunoglobulin fold (Li et al. 2004). The chaperone–pilin complex traffics to bacterial outer membrane usher proteins, where the pilus is formed by the incorporation of the pilin subunit into the growing pilin fiber resulting in the liberation of the chaperone (Barnhart et al. 2003).
Bicyclic 2-pyridones and N-substituted amino acid derivatives were discovered as potential pilicides (inhibitors of pili formation/biogenesis) that target conserved regions on the chaperone and competitively inhibit the binding of the chaperone to the pilin subunits. They were shown to inhibit biogenesis of type 1 and P pili of uropathogenic E. coli (UPEC), the major cause of urinary tract infections, which decrease UPEC binding to bladder cells by 90 % (Pinkner et al. 2006). Furthermore, they also inhibit hemagglutination and biofilm formation in E. coli strains (Pinkner et al. 2006). While none of the substances affect bacterial growth, pilicides may have broad-spectrum activity as both the chaperone structure and the chaperone–usher pathway are highly conserved among bacteria.
The interaction of pili or fimbriae with host cell surface receptors is often mediated through binding of the terminal pilin subunits and usually involves specific sugar or peptide moieties at the receptor (Ofek et al. 2003). Therefore, competitive inhibition of bacterial binding by tailoring anti-adhesive compounds to the specific receptor can be a promising strategy for anti-adhesion therapies. For instance, FimH or PapG are bacterial lectins that are located at the tip of type 1 or P pili of UPEC and they are one of the bacterium’s main virulence factors, responsible for colonization, invasion of host bladder epithelial cells, and biofilm formation (Wright and Hultgren 2006). FimH recognizes mannosylated receptors on the host cell surface. Initial studies showed that monovalent mannose derivatives display rather weak inhibitory effects (Firon et al. 1987), but recent use of multivalent compounds with increased binding activity is promising success (Han et al. 2010). Biphenyl mannosides were shown to be 200,000-fold more potent than the originally tested monovalent mannose. The substances are orally available and demonstrate an overall low toxicity (Han et al. 2012; Hartmann et al. 2012). Additionally, mouse models indicate that biphenyl mannosides decrease colonization levels significantly (Klein et al. 2010). Alternatively, adhesin binding to glycosylated receptor proteins can be inhibited by mucins. Mucins are glycoproteins of the mucus that mimic the glycosylation pattern found on host cell receptors. Purified Muc1, a mucin derived from cow milk that is highly glycosylated, can selectively inhibit binding of Gram-negative pathogens such as E. coli and Salmonella (Parker et al. 2010). It can also limit infection with Helicobacter pylori (Linden et al. 2009). Muc1 does, however, have a less pronounced effect on Gram-positive bacteria (Parker et al. 2010). Another very effective option to block cell adhesion by a particular bacterial adhesin is the application of synthetic peptides that mimic the epitope or use of monoclonal antibodies raised against the binding epitope that interacts with the host cell receptor. This strategy has been successfully applied to the streptococcal antigen (SA) I/II, an adhesin of Streptococcus mutants responsible for bacterial binding to host salivary receptors. A synthetic peptide and an antibody directed against a binding epitope of SA I/II have been shown to prevent colonization of the oral cavity by S. mutans (Lehner et al. 1985; Ma et al. 1989).
Another option to inhibit bacterial adhesion is interference with the biogenesis and presentation of the host receptor. Many bacterial adhesins and toxins bind to host glycosphingolipids. Blocking of the ceramide-specific glycosyltransferase that catalyzes the formation of a glycosphingolipid precursor has been used to successfully diminish the amount of colonization of UPEC in cell culture and mouse studies (Svensson et al. 2003). Also, depletion of glycosphingolipids using enzyme replacement therapy was successfully used to treat systemic salmonellosis (Margalit et al. 2002).
2.2 Fighting Biofilms and Chronic Infections
More than 80 % of all microbial infectious diseases, which are difficult to treat, involve the formation of biofilms (Romling and Balsalobre 2012). Therefore, biofilms and quorum sensing implicated in the control of biofilm formation constitute other important treatment targets.
2.2.1 Interference with Cell–Cell Communication—Quorum Sensing
Quorum sensing was discovered in the early 1970s as a means of cell–cell communication between bacteria of a species. It allows the cells to react to environmental changes/stresses as a group by coordinating gene expression according to the local cell population density (LaSarre and Federle 2013). Quorum sensing is a three-step process, in which the bacteria produce small signal molecules, so-called autoinducers that are released from the cell and detected by membrane-bound or cytoplasmic receptors (Ng and Bassler 2009). When bacterial numbers are low, the concentration of autoinducers is also low and the molecules dissipate in the environment. If, however, the concentration of bacteria reaches a threshold level, they come into contact with the released autoinducer molecules, inferring that they have neighbors. This, in turn, induces the formation of biofilms, antibiotic resistance, and expression of multiple pathogenicity factors (Deep et al. 2011). By regulating genes that are mainly associated with virulence and persistence, quorum sensing poses an important target for anti-virulence therapies. Different strategies identified for its inhibition include the blockage of signal production, interference with or degradation of signal molecules (signal dissemination), and disruption of the signal reception and response (Hentzer and Givskov 2003; LaSarre and Federle 2013; Lu et al. 2014a, b).
One important strategy to interfere with quorum sensing is to prevent the production of autoinducers. Gram-negative bacteria generate autoinducer molecules such as acyl-homoserine lactone (AHL) from S-adenosyl methionine (SAM) by proteins homologous to LuxI of Vibrio fischeri. This knowledge was exploited for the generation of quorum sensing inhibitors. For instance, analogues of SAM (e.g., S-adenosyl-homocysteine) were shown to inhibit AHL synthesis in P. aeruginosa (Parsek et al. 1999), and analogues of AHLs resulted in the inhibition of the expression of quorum sensing-regulated genes (Smith et al. 2003b) and, subsequently, biofilm formation (Smith et al. 2003a). Furthermore, substances blocking regulators of AHL synthesis were identified (Park et al. 2015; Soheili et al. 2015; Lu et al. 2014a, b). Gram-positive bacteria such as S. aureus produce autoinducing peptides that require cleaving for activation and recognition. Administration of inhibitory autoinducer peptides to mice during the initial stages of S. aureus infection was shown to inhibit S. aureus-induced abscess formation (Wright et al. 2005). In addition, diverse small-molecule inhibitors of biofilm formation have been identified. These include hydnocarpin-type flavonolignans and streptorubin B for S. aureus and cyclosporine and valspodar for Streptococcus biofilm inhibition (Aggarwal et al. 2015; Suzuki et al. 2015; Vimberg et al. 2015).
Several species of bacteria also produce enzymes that are capable of inactivating AHL molecules. Bacillus subtilis produces an acyl-homoserine lactonase, which hydrolyzes the lactone ring of AHL molecules, resulting in the ablation of AHL function (Dong et al. 2000). Interestingly, tobacco plants expressing this AHL-lactonase show an enhanced resistance to Erwinia infection suggesting at a broad spectrum function of these enzymes (Dong et al. 2001, 2002). Moreover, AHL-acylases, which cleave AHL after the N-terminal acyl, inactivating AHL molecules in a substrate-specific manner have been identified in different strains of Pseudomonas and Ralstonia (LaSarre and Federle 2013) and are also of interest as anti-quorum sensing agents.
Another possibility is the blockage of AHL signaling by competitive inhibitors that prevent AHL binding to its LuxR-type receptor (Hentzer and Givskov 2003). Several AHL analogues with modified acyl side chains were identified with agonistic activities (Chhabra et al. 1993; Kline et al. 1999; Smith et al. 2003b), and halogenated furanones of marine algae were shown to modulate quorum sensing regulators at a post-transcriptional level (Hentzer and Givskov 2003; Hentzer et al. 2002; Manefield et al. 1999).
Cyclic-di-GMP (c-di-GMP) is a widely used second messenger that plays a crucial role in bacterial biofilm formation as it stimulates the biosynthesis of adhesins and exopolysaccharide matrix components and prevents bacterial motility. C-di-GMP is synthesized by diguanylate cyclase (DGC), which is considered to be an attractive drug target. Recent research led to the identification of a catechol-containing sulfonohydrazide compound that inhibits the DGC PleD (Fernicola et al. 2015).
2.2.2 Prevention and Resolution of Biofilms
Biofilms form once bacteria sense a sufficiently high population density in their vicinity. This alters their gene expression and induces the secretion of a mix of polysaccharides, proteins, and extracellular DNA. These secreted substances interact to form an extracellular matrix for the pathogens. Once the biofilm has been formed and bacteria are embedded in the matrix, the targeting of these bacteria becomes difficult. The formation of a biofilm increases bacterial resistance to exogenous stresses such as antibiotics, UV damage, acidity, metal toxicity, and to host immune clearance and phagocytosis (Costerton et al. 1999; Hall-Stoodley et al. 2004; Romling and Balsalobre 2012). An additional disadvantage of biofilm formation in terms of treatment is that growth in biofilms aids the occurrence of processes that lead to the acquisition of inheritable resistance traits, such as horizontal gene transfer and adaptive mutations (Madsen et al. 2012). Growth within biofilms also raises the probability of bacterial persistence and antibiotic tolerance as bacteria within the biofilm alter their metabolism (Lewis 2005, 2008). This may lead to resistance to bacterial clearance, enabling the bacteria to regrowth within the host when the conditions become more hospitable. To target biofilm formation to inhibit persistence and recurrent infection, substances are being investigated that can inhibit secretion of biofilm components and biofilm matrix formation or destroy or resolve existing biofilm matrices.
Strategies to target bacteria within the context of a mature biofilm or to prevent biofilms from forming include the induction of bacterial motility, inhibition of host cell adhesion (see also 2.1), and/or the initiation of cell dispersal. Under natural circumstances, molecules inducing cell dispersal are produced by bacteria within the biofilm, allowing some cells to detach and infect new cells or surfaces. B. subtilis produces D-amino acids that result in the disruption of amyloid fibers, which link bacteria within the biofilm (Kolodkin-Gal et al. 2010). Exogenous treatment of biofilms with low levels of D-amino acids results in the disruption of mature biofilms and inhibition of biofilm formation by B. subtilis (Kolodkin-Gal et al. 2010). Norspermidine is another molecule produced by bacteria and plants that targets polysaccharides present in the matrix. It disperses biofilms formed by B. subtilis and was further shown to inhibit the formation of biofilms by E. coli and S. aureus. Administration of a combination of D-amino acids and norspermidine enhances their activity against mature biofilms (Kolodkin-Gal et al. 2012). Moreover, nitric oxide or the addition of the nitric oxide donor sodium nitroprusside (SNP) induces the dispersion of P. aeruginosa biofilms, and cotreatment of biofilms with SNP and antibiotics increases the efficacy of the antibiotics (Barraud et al. 2006). Nitric oxide was suggested to reduce biofilms by stimulating c-di-GMP-degrading phosphodiesterases, decreasing local c-di-GMP levels (Barraud et al. 2009). Furthermore, DNase I can degrade the extracellular DNA present in the biofilm matrix (Okshevsky et al. 2015; Qin et al. 2007), interfering with its formation and stability as shown for Bordetella pertussis (Conover et al. 2011), Listeria monocytogenes (Harmsen et al. 2010), and Campylobacter jejuni (Brown et al. 2015). A bacterial glycoside hydrolase, named Dispersin B, has been isolated from Actinobacillus actinomycetemcomitans, which was shown to disrupt mature Actinobacillus biofilms (Kaplan et al. 2004) and to inhibit S. aureus biofilms when added exogenously to the cells (Izano et al. 2008). Furthermore, the use of lytic bacteriophages to treat bacteria within a biofilm has been investigated, and it was shown that using a phage engineered to express Dispersin B could lead to complete dispersal of E. coli biofilms (Lu and Collins 2007) targeting even potential persister cells.
2.3 Interference with Global Virulence Control (Regulation of Virulence Gene Expression)
Bacteria tightly control the production of energy-consuming pathogenicity factors and virulence-associated traits to avoid unnecessary energy expenses and optimize their biological fitness. Recent advances in our understanding of virulence regulation have identified many control circuits and networks implicating many different often conserved sensory and regulatory components acting at the transcriptional or post-transcriptional level that could be targeted. This includes bacterial sensory and signal transduction molecules, global and specific transcriptional regulators, and RNA-based regulatory mechanisms. A major advantage of targeting signal transduction and regulatory mechanism is that these control systems are specific for bacteria and not present in eukaryotic host cells.
2.3.1 Two-Component Systems Involved in Virulence
Environmental cues, which are important for the pathogenesis and the biological fitness of bacterial pathogens during infection, are sensed by ubiquitous, highly conserved two-component systems. They constitute a membrane-bound histidine sensor kinase, which activates a corresponding cytoplasmic response regulator by phosphorylation. In particular, the two-component systems EnvZ/OmpR, RcsB/RcsC, PhoP/PhoQ, BarA/SirA, CpxR/CpxS, AgrC/AgrA, and QseC/QseB of bacterial pathogens have been well characterized and shown to control complex gene networks important for virulence in response to temperature, osmolarity, nutrients, secondary metabolites, and ions (Altier et al. 2000; Arya and Princy 2013; Clarke 2010; Forst and Roberts 1994; Groisman and Mouslim 2006; Vogt and Raivio 2012; Weigel and Demuth 2015). Moreover, studies exist that inhibitors of bacterial two-component systems worked in animal models and blocked pathogenesis of important pathogens (Rasko et al. 2008; Stephenson et al. 2000; Wilke et al. 2015; Worthington et al. 2013). One prominent example is the small-molecule inhibitor savirin, which reduces the expression of AgrCA-regulated genes in S. aureus, affecting its virulence, but not its survival and has no impact on the commensal Staphylococcus epidermidis (Sully et al. 2014). Another promising approach has identified a small-molecule LED209, which prevents the autophosphorylation of the sensor kinase QseC without influencing bacterial growth or inducing cell cytotoxicity. The QseC kinase contributes to virulence in a number of Gram-negative pathogens and has been studied extensively in enterohemorrhagic E. coli. Here, QseC phosphorylates three transcription factors, QseB (regulates flagella and motility genes by binding the master regulator flhDC), QseF (activates Stx production), and KdpE (binds to ler, the regulator of the main EHEC pathogenicity island LEE). Furthermore, QseC knockout strains of Salmonella and Francisella were shown to be attenuated in animal models. Strikingly, the inhibitor LED209 prevents the expression of the EHEC pathogenicity island LEE and Shiga toxin 2 without triggering an SOS response in EHEC, a reaction, which has been shown to result in the activation of Shiga toxin expression. On the contrary, a decrease in Stx2 expression could be observed (Curtis et al. 2014; Rasko et al. 2008).
2.3.2 Global Transcriptional Regulators of Virulence
A growing number and range of global transcriptional regulators of virulence have been identified which are highly conserved among bacteria, but specific for prokaryotic gene expression control, and which adjust coexpression of host-adapted metabolic processes, adaptation to host stresses, and virulence factors. Targeting of these regulators is an attractive concept as rapid adjustment to continuously changing environments during the course of an infection is a prerequisite for successful persistence in the host and the development of the infectious disease. Among the most promising drug targets are transcriptional regulators, such as the cAMP repressor protein (Crp) of Gram-negative bacteria, the equivalent catabolite control protein (CcpA) of Gram-positive bacteria, the carbon storage regulator (CsrA/RsmA, see also below), and AraC-type activators (e.g., RhaR). They are produced by both Gram-negative and Gram-positive bacteria which all adjust expression of important virulence-relevant processes (e.g., toxin production) in response to available nutrients in the infected tissues (Brautaset et al. 2009; Bruckner and Titgemeyer 2002; Deutscher et al. 2005; Heroven et al. 2012; Romeo et al. 2013; Skredenske et al. 2013; Vakulskas et al. 2015). The strength of this approach is supported by a recent study demonstrating that the phenylpropanoid anethole, which influences the virulence regulatory cascade by overproduction of Crp suppressed toxigenic V. cholerae-mediated fluid accumulation in ligate ileum of rabbits (Zahid et al. 2015).
2.3.3 Regulatory and Sensory RNAs
Novel deep-sequencing-based strategies have discovered an unprecedented level of complexity of transcriptional networks by the identification of small trans -acting regulatory RNAs (sRNAs), antisense RNAs, and sensory RNAs, such as RNA thermometers and riboswitches. Many of these RNA-based control elements were shown to influence the expression of virulence-relevant processes (Oliva et al. 2015; Papenfort and Vogel 2014). Several regulatory RNAs (e.g., Qrr RNAs, CsrB/C, and RsmZ/Y RNAs) have redundant functions and are used to fine-tune the regulation of pathogenicity, stress adaptation, and/or metabolic genes by sequestration of regulatory proteins, hindrance of translation, and/or control of RNA degradation (Feng et al. 2015; Heroven et al. 2012; Vakulskas et al. 2015). Redundancy and distinct regulatory mechanisms hamper the development of anti-regulatory RNA inhibitors. However, mechanistic insights further revealed that the function of many regulatory RNAs is governed by highly conserved, global RNA-binding proteins, such as the RNA chaperone Hfq and CsrA/RsmA (Heroven et al. 2012; Lucchetti-Miganeh et al. 2008; Oliva et al. 2015; Papenfort and Vogel 2014; Vakulskas et al. 2015). Hfq is known as the central mediator of sRNA-based gene regulation in bacteria as it establishes dynamic interactions of a wide range of RNA molecules and manipulates translation and degradation of many mRNAs important for pathogenesis (Chao and Vogel 2010; Vogel and Luisi 2011). In fact, hfq knockout derivatives of many important pathogens (e.g., Francisella, Neisseria, Legionella, Salmonella, Yersinia, and Listeria) showed a severe growth defect and were drastically attenuated in animal infection models (Oliva et al. 2015). Another well-characterized RNA-binding protein implicated in the control of multiple regulatory RNAs and a large set of virulence-linked traits is the CsrA/RsmA protein. This RNA-binding regulator is highly conserved among bacteria, but it is not produced in archaea and eukaryotes. It predominantly controls RNA translation and degradation by binding to A(N)GGA motifs within the 5′-untranslated region of the mRNA targets (Duss et al. 2014). The CsrA/RsmA-controlled network is very versatile and includes target mRNAs implicated in the control of cell morphology, motility, biofilm formation, multiple stress responses, and crucial virulence factors/regulators (i.e., secretion systems and secreted effectors, adhesins, and invasins). Accordingly, csrA/rsmA mutants of many pathogens are avirulent or strongly attenuated (Heroven et al. 2012; Lucchetti-Miganeh et al. 2008; Vakulskas et al. 2015). Furthermore, multiple regulatory RNAs of pathogenic Enterobacteriaceae implicated in the control of virulence functions are controlled by the global transcriptional regulator Crp (see above) in response to the available nutrients in the medium. Strikingly, transcriptional profiling using RNA-Seq recently revealed Crp as a master regulator of 50 % of all identified small RNAs in Y. pseudotuberculosis which are reprogrammed by Crp in response to temperature (Nuss et al. 2015). The major impact of Hfq, CsrA/RsmA, and Crp on the expression of the virulence phenotype of many pathogens makes these global regulators also to promising targets for anti-virulence strategies.
RNA riboswitches and thermometers represent another type of regulatory RNA elements. They are predominantly located in the 5′-untranslated regions of target mRNAs and comprise complex RNA structures (e.g., stem loops, which include the ribosome-binding site and/or the start codon in base-pairing of the stem structure) that sense and react to thermal or biochemical signals by conformational changes, which mainly affect translation of the downstream gene(s) (Kortmann and Narberhaus 2012). Prominent examples are the RNA thermometers controlling the expression of major virulence regulators, such as PrfA of Listeria monocytogenes (Johansson et al. 2002), LcrF/VirF of Yersinia (Böhme et al. 2012), and the cholera toxin regulator ToxT of Vibrio cholerae (Weber et al. 2014). Additionally, three RNA thermosensors were described to be essential for Neisseria meningitidis resistance against immune killing (Loh et al. 2013).
Several virulence-related metabolic functions are also controlled by riboswitches—metabolite-binding mRNA or part of regulatory RNA structures (Mellin et al. 2014; Peselis and Serganov 2014; Serganov and Nudler 2013). Utilization of ethanolamine and propanediol, by-products of rhamnose and fucose fermentation by the intestinal microbiota, is controlled by riboswitches in response to vitamin B12 binding. This leads to the synthesis of short or longer regulatory RNAs, which differentially modulate transcription of the pdu and eut genes (Mellin et al. 2013, 2014; Toledo-Arana et al. 2009). Moreover, trans-regulatory riboswitches can function as small regulatory RNAs to link metabolic and virulence control. Several potential S-adenosylmethionine (SAM)-binding riboswitches were identified in enteric pathogens and two of them—SreA and SreB—were shown to bind the 5′-untranslated region of the prfA transcript encoding the master virulence regulator of L. monocytogenes (Toledo-Arana et al. 2009). Rapidly increasing numbers of crucial RNA thermometers and riboswitches involved in virulence control may warrant the design of potential RNA-based inhibitors, e.g., RNA fragments that interfere with these crucial RNA elements or show perfect complementarities to the ribosomal binding site and the start codon of crucial virulence regulators.
2.4 Preventing Host Damage and Development of the Disease
Toxin-producing pathogens exhibit the severest effect on their host. The most serious clinical symptoms associated with infectious diseases are the results of severe tissue damage, cellular malfunction, or destruction caused by bacterial exotoxins such as botulinum, cholera, diphtheria, anthrax, tetanus, and Shiga toxins (Henkel et al. 2010; Schmitt et al. 1999). A deletion of the toxin gene(s) generally disarms the bacteria and results in avirulence without harming their overall biological fitness. This makes these virulence factors ideal targets for new inhibitors, and multiple approaches are currently being followed to prevent toxin-mediated damages of the host.
2.4.1 Targeting Exotoxin Trafficking or Function
Exotoxins are bacterial virulence factors that are actively released into the surrounding environment from the bacterium during its growth, commonly via bacterial type II secretion systems (Henkel et al. 2010) and/or outer membrane vesicles (Kulp and Kuehn 2010; Kunsmann et al. 2015). As toxins remain outside the cell for a period of time prior to binding of their specific target cells, the toxins themselves make for good therapeutic targets, for example for competitive inhibitors or neutralizing antibodies. Once released into the environment, the toxins traffic through the host until they recognize and bind to specific host cell receptors. Interaction of the toxin with its receptor induces the uptake of the receptor from the cell surface by endocytosis. Once inside the host cell cytosol, the trafficking and destination of toxins varies according to their specificity. Some toxins are activated at neutral pH, while others require the acidification of the endosome for activation or trafficking to a particular compartment (Henkel et al. 2010; Schmitt et al. 1999). Knowledge of the route of action of a specific toxin now enables us to develop new toxin-trafficking inhibitors.
For many exotoxins, screening assays have been designed to identify small-molecule compounds that inhibit the action of the particular exotoxin. For instance, several potent inhibitors have been identified for the Bacillus anthracis-encoded anthrax toxin, a multimeric toxin consisting of three proteins, the protective antigen (PA), the edema factor (EF), and the lethal factor (LF) (Montecucco et al. 2004; Nestorovich and Bezrukov 2014; Tonello et al. 2002). The PA binds to the host cell surface, where it gets cleaved and forms multimeric channels in the host cell membrane through which the EF and the LF can enter the cell. The EF, an adenylate cyclase, raises the level of cAMP within the host cell, leading to edema (Baldari et al. 2006; Dell’Aica et al. 2004). The LF, a zinc metalloprotease, which has been shown to be critical for infection, disrupts host MAP kinase signaling pathways. Initial screening of a library of known zinc metalloprotease inhibitors identified a potent sulfonamide derivative, anthrax LF inhibitor 40 ((2R)-2-[(4-fluoro-3-methylphenyl) sulfonylamino]-N-hydroxy-2-(tetrahydro-2H-pyran-4-yl)acetamide), which was further shown to be effective in several animal models (Xiong et al. 2006). The compound binds competitively within the active site of LF and was found to be efficient in prophylactic therapy as well as for therapeutic treatment when used in combination with classic antibiotics (Shoop et al. 2005). Other screening assays using small-molecule libraries and application of a mixture-based peptide library approach identified additional small-molecule inhibitors and peptide analogues, which show competitive inhibition of anthrax LF (Bannwarth et al. 2012; Goldman et al. 2006; Kim et al. 2011; Panchal et al. 2004; Shoop et al. 2005; Turk et al. 2004). Furthermore, some polyphenols such as catechin gallate (CG) and epigallocatechin-3-gallate (EGCG) abundantly identified in green tea were found to exert a strong inhibition of the LF proteolytic activity (Dell’Aica et al. 2004). Several non-competitive/exosite-targeting inhibitors that prevent LF function were also identified (Bannwarth et al. 2012; Kuzmic et al. 2006). Similarly, antitoxins targeting the EF enzymatic activity have been identified of which the most potent inhibitors interact with the catalytic site of the protein (Nestorovich and Bezrukov 2014).
As the PA subunit of the anthrax toxin is responsible for LF and EF delivery and represents the major antigen for toxin-neutralizing antibodies, it has been the most important target for preventive and therapeutic measures. Several attempts have been made to (i) prevent PA binding to its host cell receptors (ATR/TEM8, CMG2) by blocking the receptor-binding domain of PA or the cell receptors or by the design of soluble polyvalent peptide analogues which compete with the natural receptors for PA binding (Cryan et al. 2013; Cryan and Rogers 2011; Rogers et al. 2012; Scobie et al. 2005), (ii) to prevent endocytosis of the toxin by blocking pH-dependent cell entry and endosomal trafficking, or (iii) to prevent PA-promoted translocation of the LF and the EF. The most potent inhibitors include liposome-functionalized multiple copies of the AWPLSQLDHSYN peptide that binds and neutralizes the LF (Basha et al. 2006), dominant negative derivatives of the PA that coassemble with wild-type PA, and the small-molecule inhibitor (3-aminopropylthio)-β-cyclodextrin, which disrupts proper PA channel formation and/or blocks its activity to translocate the LF and the EF (Karginov et al. 2006; Nestorovich and Bezrukov 2014).
Shiga toxin and Shiga-like toxin are found in Shigella and Shiga toxin-producing E. coli strains (EHEC). In EHEC pathogenesis, Shiga toxin has been identified as the main virulence factor responsible for bloody diarrhea, destruction of red blood cells and platelets, and the development of hemolytic uremic syndrome (HUS) resulting in severe kidney and neurological damage (Greener 2000; Kaplan et al. 1998; Tarr et al. 2005). Shiga toxin is an AB5 toxin, of which the B subunit promotes binding to Gb3 glycolipid receptors most commonly found on kidney cells, but also on thrombocytes and neuronal cells (Boyd and Lingwood 1989; Kaplan et al. 1998). Several groups have designed strategies to interfere with receptor binding of the Shiga toxin. Sugars mimicking the Gb3 receptor have been designed in previous studies with varying success at neutralizing the free toxin (Kitov et al. 2000; Nishikawa et al. 2005; Trachtman et al. 2003). Despite toxin binding and removal, clinical symptoms were not significantly reduced. In another approach, glycan-encapsulated gold nanoparticles have been used to display ligands for Shiga toxin in an attempt to bind the free toxin. The nanoparticles were able to limit Vero cell cytotoxicity in response to Shiga toxins 1 and 2, but were unable to neutralize certain Shiga toxin 2 variants (Kulkarni et al. 2010). Furthermore, C-9, an inhibitor of glucosylceramide synthase, was applied to downregulate Gb3 expression and prevented the cytotoxic effect of Stx2 on Vero cells (Silberstein et al. 2011). Using a rat model of infection, C-9 protected animals against Shiga toxin 2-associated disease and both prophylactic and therapeutic treatment of rats with C-9 decreased expression of Gb3 receptors and the development of a disease phenotype (Kulkarni et al. 2010).
2.4.2 Inhibition of Exotoxin Synthesis
The two main virulence factors of V. cholerae are cholera toxin (CT) and toxin coregulated pilus (TCP). CT is another AB5 toxin of which the catalytically active A subunit activates host cell G proteins upon uptake into the host cell. This in turn leads to the activation of adenylate cyclase, increasing the concentration of cAMP in intestinal epithelial cells, inducing strong secretory diarrhea (Field 2003). The specific transcription factor ToxT is known to directly activate the expression of both cholera toxin (ctxAB) and tcp genes. The small-molecule virstatin was found to interfere with the homodimerization of the ToxT N-termini and thus blocks CT and TCP production resulting in reduced colonization of V. cholerae in mice (Hung et al. 2005; Shakhnovich et al. 2007). Unfortunately, virstatin-resistant toxT mutants containing a single amino acid substitution in the N-terminus have already been isolated (Hung et al. 2005).
2.4.3 Antibody-Mediated Exotoxin Neutralization
A very effective strategy to block toxin function and prevent the deleterious effect of very aggressive exotoxins is the treatment with antibodies that specifically bind and neutralize the toxin. One example for a highly potent neurotoxin that can be efficiently treated with neutralizing antibodies is the Clostridium botulinum-produced botulinum toxin. In the case of infections of adults, the US Centers for Disease Control supply an antitoxin that contains horse antibodies raised against type A, B, and/or E strains of the neurotoxin. For the treatment of children, the FDA approved a drug containing anti-botulinum toxin antibodies produced from human [marketed as human botulism immune globulin (BabyBIG)] (Arnon et al. 2006). Due to the high cost of the antibodies, a new equine alternative of the antibody is being tested (Vanella de Cuetos et al. 2011).
In addition, monoclonal antibodies binding directly to the PA subunit of the anthrax toxin and thus preventing its interaction with host cells were developed, which were shown to protect rats and chimpanzees against B. anthracis infections (Chen et al. 2011).
Furthermore, antibodies targeting Shiga toxin have been isolated from rabbits immunized with a fusion protein consisting of an epitope of the B subunit of heat-labile toxin (LT) of enterotoxic E. coli (ETEC) and the A subunit of Shiga toxin (Stx) fused to the fimbrial protein FaeG. The produced antibodies were able to inhibit the adhesion of E. coli to enterocytes and neutralize Shiga toxin (Zhang and Zhang 2010). Similarly, a fusion protein of two different Shiga toxin antigens as well as the bacterial cell surface protein intimin was used to immunize mice and shown to result in the production of anti-Stx and anti-intimin antibodies. Mice immunized with this fusion protein were immune to lethal doses of EHEC (Gu et al. 2011).
2.4.4 Targeting of Secretion Systems
Secretion systems are used by the bacterium to translocate virulence factors (effectors) directly into the host cytosol. Several types of secretion systems have a strong association with disease. The secretion systems most innately connected to pathogenesis are the type III (T3SS) and type IV (T4SS) secretion systems. T3SSs are needle-like structures with a high similarity to flagella, and T4SSs are evolutionary related to bacterial conjugation systems. Both secretion systems are highly conserved in structure between the pathogens that employ them. They span the bacterial inner and outer membrane and connect to the host cell. Here, they form a pore in the host cell membrane enabling the pathogen to translocate virulence proteins directly into the host cell cytosol in which they can interfere with cell signaling pathways in favor of bacterial persistence (Chandran 2013; Galan and Wolf-Watz 2006; Hueck 1998; Trokter et al. 2014; Waksman and Orlova 2014).
The genes for T3SSs and T4SSs are only found in pathogenic bacteria and are usually encoded within mobile genetic regions that are associated with virulence (pathogenicity islands). T3SSs are found in more than 25 species of Gram-negative pathogens including pathogenic Chlamydia, E. coli, Salmonella, Shigella, Yersinia, and Pseudomonas (Coburn et al. 2007; Galan and Wolf-Watz 2006; Schroeder and Hilbi 2008). T4SSs are associated with virulence in pathogens such as Legionella, Bartonella, Helicobacter, Coxiella, and Brucella (Nagai and Kubori 2011; Voth et al. 2012). As the structures and functions of the secretion systems themselves show a high conservation between the different strains of bacteria, so does their synthesis and assembly. Furthermore, the proteins that make up the secretion systems are exposed on the bacterial cell surface, making them accessible. Taken everything into consideration, a number of different potential targets present themselves: synthesis of needle components, assembly of the secretion system, interaction with the host cell, and secretion/translocation of the substrates. Due to the high similarity of the secretion systems, it is likely that inhibitors can be found which target not only one but also several different pathogens at once. Furthermore, as only pathogenic bacteria express these types of secretion systems, non-pathogenic bacteria will not be targeted. Additionally, as secretion system inhibition does not influence the overall survival of the bacterium, the selective pressure to develop resistance is low.
The design and use of special secretion test assays, i.e., fusion of the β-lactamase gene to the translocation signal of effectors secreted by T3SSs, GFP-labeled chaperone, and tagged effector labeling, allowed the identification of different natural compounds and chemical inhibitors that block the function of T3SSs of different important pathogens (Baron 2010; Izore et al. 2011; Keyser et al. 2008; Marshall and Finlay 2014; McShan and De Guzman 2015; Pan et al. 2009; Tsou et al. 2013). The glycolipids caminoside A, B, and C isolated from the marine sponge Caminus sphaeroconia were the first T3SS inhibitors. They block the secretion of the effector EspB, a protein that makes up part of the translocon of the E. coli T3SS, by varying degrees. Caminoside B appears to be the most potent inhibitor of this class; however, the cellular target remains unknown (Linington et al. 2002). The kirromycin derivative aurodox, isolated from Streptomyces, inhibited T3SS-mediated hemolysis of red blood cells in response to incubation with enteropathogenic E. coli (EPEC). It was further shown to decrease the secretion of the effectors EspB, EspF, and Map without affecting bacterial growth. Furthermore, aurodox protected mice against infection with a lethal dose of Citrobacter rodentium, the mouse homologue of EPEC. The mode of action of aurodox is still unknown; however, it was suggested that it might interact with transcriptional regulators (Kimura et al. 2011). Guadinomines A and B are Streptomyces-produced natural compounds with a strong activity against the T3SS of EPEC as shown by the inhibition of T3S-induced hemolysis. The mode of action of these compounds is also unknown (Duncan et al. 2014; Iwatsuki et al. 2008). The insecticidal, actinomycete-derived respiratory chain inhibitor piericidin A1 and its closely related derivative Mer-A2026B were also shown to act against T3SS of Y. pseudotuberculosis, inhibiting the secretion and translocation of the Yersinia virulence proteins (Yops) into host cells (Duncan et al. 2014). A series of thiazolidinone inhibitors were discovered to inhibit both the Salmonella and the Yersinia T3SSs. However, they seem to also block T2SS and type IV pili, most likely due to an inhibition of the common outer membrane protein secretin (Felise et al. 2008). The polyphenol (-)-hopeaphenol is a natural product that also decreases Yop secretion in Y. pseudotuberculosis without affecting Yop expression. The compound is also active against Pseudomonas, but the exact target of (-)-hopeaphenol is still unknown (Zetterstrom et al. 2013). The salicylidene acylhydrazides (SAHs) are by far the best-studied chemical substances targeting T3SSs. They are effective against the T3SS of a range of pathogens including Salmonella Typhimurium (Hudson et al. 2007; Negrea et al. 2007), Y. pseudotuberculosis (Nordfelth et al. 2005), Chlamydia (Bailey et al. 2007; Muschiol et al. 2006; Slepenkin et al. 2007; Wolf et al. 2006), E. coli (Tree et al. 2009), and Shigella (Veenendaal et al. 2009). The mode of action seems to be via the inhibition of needle subunit secretion or assembly, but recent analysis further demonstrated that SAHs significantly repressed the expression of main regulators of the T3SS machinery and modulate the function or activity of several protein targets (Layton et al. 2010; Tree et al. 2009; Wang et al. 2011). An involvement in iron chelation (Layton et al. 2010; Slepenkin et al. 2007) and a role for the metabolism (Wang et al. 2011) are also debated. In addition to these inhibitors, several other virulence blockers have been identified that inhibit transcription factors important for the expression of the T3SS in different pathogens. For instance, N-hydroxybenzimidazole derivatives block T3SS expression in Pseudomonas and were found to prevent T3SS expression of Yersinia by the inhibition of the MarA-type transcriptional activator LcrF/VirF of Yersinia and ExsA of Pseudomonas (Bowser et al. 2007; Garrity-Ryan et al. 2010; Harmon et al. 2010; Kim et al. 2009). Furthermore, salicylanilides were identified as inhibitors of LcrF expression (Kauppi et al. 2003), and the small-molecule inhibitor SE-1 inhibits the master T3SS regulator VirF of Shigella (Koppolu et al. 2013).
Another very promising approach includes the use of antibodies directed against the tip complex of the secretion systems, e.g., PcrV for the Pseudomonas T3SS. A PcrV-specific antibody of KaloBios Pharmaceuticals is in clinical phase II and already showed that it is non-immunogenic and safe in pharmacokinetic studies (Francois et al. 2012).
The Brucella abortus protein VirB8 is an essential component of the T4SS and indispensable for its assembly. B8I-2 was identified to inhibit the dimerization of VirB8 as well as the interaction with other VirB proteins (Paschos et al. 2011). The compound markedly reduced virB transcription and subsequently VirB protein levels, but it also strongly reduced the intracellular survival of Brucella in macrophages (Paschos et al. 2011). Another study identified three substances to inhibit VirB11, the crucial ATPase of the T4SS of H. pylori, which were named CHIR-1, CHIR-2, and CHIR-3. All were shown to inhibit the secretion of CagA into cells in vitro which resulted in a strong reduction of H. pylori colonization in mice (Hilleringmann et al. 2006).
2.4.5 Sortases
Gram-positive bacteria use enzymes named sortases to present proteins such as pilins or glycoproteins on their surface. In addition, pathogens such as S. aureus use these proteins to display their virulence factors (Cascioferro et al. 2014). This presentation is essential, as sortase A (srtA) mutant strains are impaired in their ability to cause infection in the mouse model (Jonsson et al. 2002). Sortase A mutants in Streptococcus suis and L. monocytogenes also show a marked reduction in pathogenicity (Bierne et al. 2002; Vanier et al. 2008). The conservation, essentiality, and widespread use of sortase by pathogens suggest that compounds, which inhibit their activity, will function as potent anti-infective agents (Cascioferro et al. 2015). The surface proteins anchored to the cell wall by S. aureus SrtA include virulence factors that play key roles in the infection process by promoting nutrient acquisition from the host, bacterial adhesion, and immune evasion. Among the first sortase inhibitors were peptidomimetic molecules, the small-molecule inhibitor diarylacrylonitriles, and flavonols such as morin (Cascioferro et al. 2015). More recently, the compound (2-(2,3-dihydro-1H-perimidin-2-yl)-phenoxy)-acetic acid was found to inhibit SrtA without affecting bacterial growth (Chan et al. 2013; Zhang et al. 2014). It also protects mice from bacteremia (Zhang et al. 2014). Furthermore, aryl beta-amino(ethyl) ketone (AAEK) was identified as a sortase inhibitor in a high-throughput screen and selected for further investigation on the basis of its marked effect on sortases of staphylococci and bacilli (Maresso et al. 2007).
2.5 Anti-resistance Drugs
The development and use of anti-resistance drugs that are administered together with known antibiotics, to circumvent acquired resistance mechanisms of bacterial pathogens, opens a door to extending the life span of known antibiotics.
2.5.1 Resistance Against Antibiotics
β-lactamase Inhibitors
β-lactam antibiotics have been widely used for almost 80 years. They are bactericidal compounds that act by inhibiting cell wall-synthesizing enzymes found only in bacteria (Kong et al. 2010). The continuous use of this family of antibiotics has given rise to the extensive proliferation of bacterial β-lactamases, which hydrolyze the β-lactam ring of the antibiotic, rendering the bacteria resistant to antibiotics of the carbapenem family (Palzkill 2013). A combination therapy of β-lactamase inhibitors as an adjuvant to suppress resistance with a β-lactam antibiotic increases the efficacy and the spectrum of the antibiotic. Successful combinations of antibiotics and β-lactamase inhibitors include clavulanic acid, sulbactam or tazobactam, and penicillins (Carlier et al. 2014; Totir et al. 2007). Avibactam, a non-beta-lactam bicyclic diazabicyclooctane with no antibiotic activity, forms reversible bonds with several beta-lactamases including K. pneumoniae carbapenemase and ESBL- or AmpC-overexpressing strains (Castanheira et al. 2014; Coleman et al. 2014). Similar modes of action have been described for Merck MK-7655, a piperidine analogue used with imipenem (Blizzard et al. 2014).
Efflux Pump Inhibitors
Overexpression of efflux pumps to expel toxic compounds such as antibiotics from the cell is a common resistance strategy found in bacteria. With the added advantage that uptake of antibiotics into Gram-negative bacteria is slowed down by decreased permeability of the bacterial outer membrane, the bacteria are less susceptible to efflux pump substrates even those with poor affinity, as the rate of efflux usually exceeds that of influx (Nikaido and Pages 2012). Discovering compounds that selectively inhibit the bacterial efflux pumps may lead to the development of therapies that could restore sensitivity to antibiotics. Several bacterial efflux pumps have been studied in detail and would make for promising therapeutic targets.
Resistance–nodulation–division (RND) efflux pumps are encoded by Gram-negative Enterobacteriaceae and Pseudomonas. They transport a variety of toxic substances from the bacterial cell, including antibiotics such as fluoroquinolones, β-lactams, tetracyclines, and oxazolidines, mediating intrinsic resistance of the bacteria to these substances (Nikaido and Pages 2012). Phenylalanyl arginyl β-naphthylamide (PAβN) was discovered in a high-throughput screen for molecules that sensitize efflux pump-overexpressing P. aeruginosa to levofloxacin (Renau et al. 1999). PAβN inhibits the efflux pumps of several bacterial pathogens such as E. coli, S. Typhimurium, K. pneumoniae, and Campylobacter sp. with divergent efficacy. It was shown that while PAβN sensitizes Pseudomonas to antibiotics such as levofloxacin, erythromycin, and chloramphenicol, it showed little effect for enhancing the susceptibility to carbenicillin, suggesting that its activity is strongly dependent on the antibiotic (Lomovskaya et al. 2001). It was therefore suggested that PAβN is a competitive inhibitor that binds within the substrate-binding pocket used by a specific set of antibiotics. PAβN was found to increase the permeability of the cellular membrane (Lomovskaya et al. 2001) in addition to its function as an efflux pump. Increased uptake into the bacterial cell increases its potency. Naphtylpiperazines (NMPs) were identified as potentiators of levofloxacin in RND efflux pump-overexpressing E. coli (Bohnert and Kern 2005). Addition of NMP to cells increases the intracellular concentration of levofloxacin. It further affects the susceptibility of the bacteria to other antibiotic substances such as rifampicin and chloramphenicol. NMP is able to reverse multidrug resistance in clinical E. coli isolates (Kern et al. 2006) and shows some effects against multidrug-resistant K. pneumoniae (Schumacher et al. 2006) and A. baumannii (Pannek et al. 2006). However, NMP is ineffective in increasing antibiotic susceptibility of P. aeruginosa (Coban et al. 2009). Notably, antisense peptide nucleic acids (PNAs) are synthetic homologues of nucleic acids that bind complementary DNA and RNA sequences with very high specificity (Paulasova and Pellestor 2004). They can be used as antisense peptides to specifically inhibit the gene expression of efflux pump genes. This approach was employed to decrease the expression of the C. jejuni RND efflux pump CmeABC, resulting in a significant increase of susceptibility to antibiotic treatment (Jeon and Zhang 2009; Mu et al. 2013). NorA efflux pumps are found in Gram-positive pathogens such as S. aureus. They confer resistance to antibiotics including those of the family of fluoroquinolones. Capsaicin, an alkaloid, has recently been shown to potently reduce the resistance of S. aureus to ciprofloxacin and inhibit the efflux of ethidium bromide in vitro (Kalia et al. 2012).
2.5.2 Environmental Stress Resistance
Depletion of Iron
Pathogenic bacteria are exposed to multiple host-associated stresses directed to prevent proliferation and eliminate the invaders. Among the most important stresses is the depletion of iron from the blood and lymph systems by specific iron-complexing molecules. To overcome this problem, bacteria synthesize a plethora of iron-chelating siderophores for iron uptake, which are essential for virulence (Miethke and Marahiel 2007). Several attempts were made to identify compounds that prevent siderophore synthesis and transport. A salicylsulfamoyl adenosine and other nucleoside bisubstrate analogues were found to prevent the early step of siderophore synthesis of Yersinia and Mycobacterium (Ferreras et al. 2005; Miethke and Marahiel 2007; Neres et al. 2008).
2.5.3 Resistance Against Reactive Oxygen and Nitrogen Species (ROS, NOS)
In a similar manner, virulence factors can be targeted which are essential to promote resistance against reactive oxygen or nitrogen species used by innate immune cells to destroy bacteria. Enzymes that have been successfully targeted by inhibitors are staphyloxanthin, a S. aureus pigment with antioxidant activity, which is blocked by phosphosulfonate, and the Mycobacterium tuberculosis factor DlaT essential to resist NOS intermediates which are targeted by rhodanine analogues (Escaich 2010).
2.5.4 Interference with Bacterial Host Resistance Mechanisms
Components of the innate immunity, such as complement factors, antimicrobial peptides (e.g., defensins), and professional phagocytes (e.g., neutrophils, macrophages) circulating in the blood, lymph system, and tissues, are able to efficiently eliminate invading pathogens. Lipopolysaccharides (LPSs) of the outer membrane of Gram-negative bacteria are required for the resistance to complement and cationic antimicrobial peptides (CAMPs). Modification of LPS to decrease surface charges (e.g., by the addition of aminoarabinose or heptoses) and inhibition of several enzymes of LPS biosynthesis have been identified as treatment strategies (Desroy et al. 2013; Escaich 2010). Bacterial resistance to CAMPs is linked to an increase in D-alanylation of the lipoteichoic acids in the cell wall of staphylococci and streptococci (Peschel et al. 1999; Saar-Dover et al. 2012). Furthermore, inhibitors of D-alanylation enzymes have been identified, some of which were also shown to reduce bacteremia (Escaich 2010; Santa Maria et al. 2014).
3 Challenges and Problems Associated with Virulence Blockers
The large variety of newly identified natural compounds, structural analogues, mimetic peptides, and antibodies that act as inhibitors of crucial virulence traits over the last ten years has demonstrated that anti-virulence strategies can be successfully applied to combat bacterial infections. However, there are still many challenges and problems for the development of anti-virulence drugs, which will have to be addressed in the near future.
Recent development of target-based high-throughput screening or rational drug design with chemical and natural compound libraries or structural analogues has furthered the field and facilitated the identification of inhibitors. However, for many inhibitors, the demonstration that inhibition of a particular virulence strategy leads to an inhibition of the bacterial infection in vitro and the validation of inhibitor activities in animal models is still missing.
Another important issue concerns the mode of action of many compounds. Although several effective inhibitors of certain virulence traits (e.g., T3SS-mediated translocation of bacterial effectors) have been identified, the precise molecular mechanism of the inhibition and the exact targets are often unclear. Moreover, some of the identified inhibitor classes (i.e., SAHs) seem to have multiple molecular targets, which contribute to the inhibitory effect of the compounds, and other inhibitors are active on more than one species.
One major challenge in the development of successful anti-virulence strategies is the general redundancy of many crucial virulence mechanisms of pathogens. Bacterial pathogens usually expose multiple adhesive surface structures with distinct cell receptor specificities, which allow binding and colonization of different tissues during the infection cycle. Furthermore, alternative infection routes are used by several pathogens to disseminate into deeper tissues. Hence, the development of a successful universal adherence inhibitor is rather difficult. Nonetheless, certain adhesion structures have been identified as hallmark requirements for specific pathogens, which are promising targets for drug discovery and development. Other examples are bacterial effector proteins, which are injected into host cells by T3SS or T4SSs to manipulate host cells and circumvent immune responses. Many bacteria that cannot evade detection by the receptors encode multiple effector proteins, which are able to specifically modify the host innate immune response in their favor. Pathogenic Yersinia species encode 5 effector proteins YopH, YopE, YopJ/YopP, YopT, and YopM (Bliska et al. 2013), and in EPEC and enterohemorrhagic E. coli (EHEC), at least seven translocated effector proteins (NleB1, NleC, NleD, NleE, NleF, NleH2, and Tir) are known to modulate different aspects of the proinflammatory and apoptotic cell signaling pathways (Wong et al. 2011). In Legionalla pneumophila, which encodes a staggering amount of at least 300 effector proteins, the redundancy of effector function is consequently much higher (Pearson et al. 2015). In addition, many host-adapted metabolic processes, ion/nutrient uptake systems, and regulatory circuits are redundant, and elimination of one is almost fully compensated by the upregulation or activation of (an) alternative pathway(s).
Crucial pathogenicity traits are often only expressed at distinct sites and at certain time points when they are required during the infection to prevent immune responses and balance their energy budget to optimize their fitness. Hence, detailed knowledge about the tempo-spatial expression pattern and the role of the potential targets is required for the design/development of effective anti-virulence drugs.
Over the past years, an increasing number of reports demonstrated that certain crucial virulence traits are only expressed by a certain ratio of the bacterial population at any time during the infection (Burton et al. 2014; Claudi et al. 2014; Diard et al. 2013; Helaine et al. 2014; Manina et al. 2015; Putrins et al. 2015; Sturm et al. 2011). The presence of genetically identical but phenotypically heterogeneous subpopulations is advantageous for a pathogen to survive within a fluctuating environment with varying nutrient, ion, and stress conditions. It allows the bacteria to prepare themselves for uncertainties (bet-hedging) and to divide a biological task into different subtasks executed by the different subpopulations (division of labor) (Ackermann 2015; Avery 2006; Smits et al. 2006). Furthermore, certain pathogen subsets reside in diverse tissue microenvironments and biofilms as a response to local conditions and molecular interactions, and this has disparate consequences on the expression of virulence-relevant traits. Differential expression of certain virulence traits in individual subpopulations can result in a failure to control the infection foci by identified inhibitors or in selective targeting of pathogen subsets.
Even if the selective pressure of virulence blockers is less than that induced by antibiotics, the appearance of resistance against anti-virulence drugs may result in pathogens that have an advantage over the remaining population. This allows for (involuntary) selection for these better-adapted pathogens over time. Many crucial virulence factors are located on mobilizable DNA elements, such as plasmids and phages. Rapid exchange of mutated gene variants of a targeted virulence trait by horizontal DNA transfer could result in the rapid evolution of drug-resistant bacterial populations. Application of combinational therapies may be a good option to suppress these better-adapted pathogens at the moment they appear.
4 Conclusions and Outlook
The present review takes a close look at current anti-virulence strategies and identified classes of novel virulence blockers and illustrates the promising advances made in our attempts to develop alternatives to antibiotic therapies. To fully exploit this strategy, it is imperative that we improve our understanding of the molecular mechanisms and the consequences of host–pathogen interactions, as many crucial virulence-associated processes remain unclear. Future research in this field will also be aided by the increasing number of crystal structures of crucial pathogenicity factors allowing directed drug design for high-throughput screening assays. This will not only facilitate structure–function analyses and optimization of identified inhibitors, but also allow an exploitation of other less well-characterized adhesion structures and secretion systems (T6SS, T7SS).
The loss of interest in antimicrobial drug development by large pharmaceutical companies and the urgent need for alternatives have triggered science funding agencies around the world to support projects that advance the development of alternatives to antibiotics. This generated a boost for academic researchers and small biotechnology enterprises to pursue different strategies with the goal to find alternative solutions for future applications. However, the following pipeline for the development of novel antibiotics is often not available. There is an urgent task to provide test systems to study pharmacokinetics/dynamics parameters and evaluate the efficacy in animal models. In particular, the specificity, safety, and tolerability of the compounds need to be assessed, an appropriate formulation for delivery must be developed, and setups must be installed that help transfer successful virulence blockers into broad-scale clinical trials. A major task will be to convince the pharmaceutical industry to participate and support this mission early on.
Another important aspect is that an effective application of virulence blockers requires a rapid and precise diagnosis of infectious agents in the clinics. This will include a more detailed profiling of the pathogen and its virulence traits and demands a more patient-specific, personalized analysis of the responsible disease agent. Such advancements are not only attractive due to their potential to combat resistant pathogens, but also help to improve the use of standard antibiotics and offer the possibility to use both antibiotics and virulence blockers in a synergistic therapy approach to minimize the selection of resistant variants.
References
Aarestrup FM (2005) Veterinary drug usage and antimicrobial resistance in bacteria of animal origin. Basic Clin Pharmacol Toxicol 96:271–281
Ackermann M (2015) A functional perspective on phenotypic heterogeneity in microorganisms. Nat Rev Microbiol 13:497–508
Aggarwal C, Jimenez JC, Lee H, Chlipala GE, Ratia K, Federle MJ (2015) Identification of quorum-sensing inhibitors disrupting signaling between Rgg and short hydrophobic peptides in Streptococci. MBio 6:e00393–e00315
Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J (2010) Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 8:251–259
Altier C, Suyemoto M, Ruiz AI, Burnham KD, Maurer R (2000) Characterization of two novel regulatory genes affecting Salmonella invasion gene expression. Mol Microbiol 35:635–646
Arnon SS, Schechter R, Maslanka SE, Jewell NP, Hatheway CL (2006) Human botulism immune globulin for the treatment of infant botulism. N Engl J Med 354:462–471
Arya R, Princy SA (2013) An insight into pleiotropic regulators Agr and Sar: molecular probes paving the new way for antivirulent therapy. Future Microbiol 8:1339–1353
Avery SV (2006) Microbial cell individuality and the underlying sources of heterogeneity. Nat Rev Microbiol 4:577–587
Bailey L, Gylfe A, Sundin C, Muschiol S, Elofsson M, Nordstrom P, Henriques-Normark B, Lugert R, Waldenstrom A, Wolf-Watz H et al (2007) Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett 581:587–595
Baldari CT, Tonello F, Paccani SR, Montecucco C (2006) Anthrax toxins: a paradigm of bacterial immune suppression. Trends Immunol 27:434–440
Bannwarth L, Goldberg AB, Chen C, Turk BE (2012) Identification of exosite-targeting inhibitors of anthrax lethal factor by high-throughput screening. Chem Biol 19:875–882
Barnhart MM, Sauer FG, Pinkner JS, Hultgren SJ (2003) Chaperone-subunit-usher interactions required for donor strand exchange during bacterial pilus assembly. J Bacteriol 185:2723–2730
Baron C (2010) Antivirulence drugs to target bacterial secretion systems. Curr Opin Microbiol 13:100–105
Barraud N, Hassett DJ, Hwang SH, Rice SA, Kjelleberg S, Webb JS (2006) Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J Bacteriol 188:7344–7353
Barraud N, Schleheck D, Klebensberger J, Webb JS, Hassett DJ, Rice SA, Kjelleberg S (2009) Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels, and enhanced dispersal. J Bacteriol 191:7333–7342
Basha S, Rai P, Poon V, Saraph A, Gujraty K, Go MY, Sadacharan S, Frost M, Mogridge J, Kane RS (2006) Polyvalent inhibitors of anthrax toxin that target host receptors. Proc Natl Acad Sci USA 103:13509–13513
Beckham KS, Roe AJ (2014) From screen to target: insights and approaches for the development of anti-virulence compounds. Front Cell Infect Microbiol 4:139
Berendonk TU, Manaia CM, Merlin C, Fatta-Kassinos D, Cytryn E, Walsh F, Burgmann H, Sorum H, Norstrom M, Pons MN et al (2015) Tackling antibiotic resistance: the environmental framework. Nat Rev Microbiol 13:310–317
Bierne H, Mazmanian SK, Trost M, Pucciarelli MG, Liu G, Dehoux P, Jansch L, Garcia-del Portillo F, Schneewind O, Cossart P et al (2002) Inactivation of the srtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol Microbiol 43:869–881
Bliska JB, Wang X, Viboud GI, Brodsky IE (2013) Modulation of innate immune responses by Yersinia type III secretion system translocators and effectors. Cell Microbiol 15:1622–1631
Blizzard TA, Chen H, Kim S, Wu J, Bodner R, Gude C, Imbriglio J, Young K, Park YW, Ogawa A et al (2014) Discovery of MK-7655, a beta-lactamase inhibitor for combination with Primaxin(R). Bioorg Med Chem Lett 24:780–785
Böhme K, Steinmann R, Kortmann J, Seekircher S, Heroven AK, Berger E, Pisano F, Thiermann T, Wolf-Watz H, Narberhaus F et al (2012) Concerted actions of a thermo-labile regulator and a unique intergenic RNA thermosensor control Yersinia virulence. PLoS Pathog 8:e1002518
Bohnert JA, Kern WV (2005) Selected arylpiperazines are capable of reversing multidrug resistance in Escherichia coli overexpressing RND efflux pumps. Antimicrob Agents Chemother 49:849–852
Bowser TE, Bartlett VJ, Grier MC, Verma AK, Warchol T, Levy SB, Alekshun MN (2007) Novel anti-infection agents: small-molecule inhibitors of bacterial transcription factors. Bioorg Med Chem Lett 17:5652–5655
Boyd B, Lingwood C (1989) Verotoxin receptor glycolipid in human renal tissue. Nephron 51:207–210
Brautaset T, Lale R, Valla S (2009) Positively regulated bacterial expression systems. Microb Biotechnol 2:15–30
Brown HL, Hanman K, Reuter M, Betts RP, van Vliet AH (2015) Campylobacter jejuni biofilms contain extracellular DNA and are sensitive to DNase I treatment. Front Microbiol 6:699
Bruckner R, Titgemeyer F (2002) Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett 209:141–148
Burton NA, Schurmann N, Casse O, Steeb AK, Claudi B, Zankl J, Schmidt A, Bumann D (2014) Disparate impact of oxidative host defenses determines the fate of Salmonella during systemic infection in mice. Cell Host Microbe 15:72–83
Canton R (2009) Antibiotic resistance genes from the environment: a perspective through newly identified antibiotic resistance mechanisms in the clinical setting. Clin Microbiol Infect 15(Suppl 1):20–25
Carlier M, Carrette S, Stove V, Verstraete AG, De Waele JJ (2014) Does consistent piperacillin dosing result in consistent therapeutic concentrations in critically ill patients? A longitudinal study over an entire antibiotic course. Int J Antimicrob Agents 43:470–473
Cascioferro S, Totsika M, Schillaci D (2014) Sortase A: an ideal target for anti-virulence drug development. Microb Pathog 77:105–112
Cascioferro S, Raffa D, Maggio B, Raimondi MV, Schillaci D, Daidone G (2015) Sortase A inhibitors: recent advances and future perspectives. J Med Chem
Castanheira M, Williams G, Jones RN, Sader HS (2014) Activity of ceftaroline-avibactam tested against contemporary Enterobacteriaceae isolates carrying beta-lactamases prevalent in the United States. Microb Drug Resist 20:436–440
Cegelski L, Marshall GR, Eldridge GR, Hultgren SJ (2008) The biology and future prospects of antivirulence therapies. Nat Rev Microbiol 6:17–27
Chan AH, Wereszczynski J, Amer BR, Yi SW, Jung ME, McCammon JA, Clubb RT (2013) Discovery of Staphylococcus aureus sortase A inhibitors using virtual screening and the relaxed complex scheme. Chem Biol Drug Des 82:418–428
Chandran V (2013) Type IV secretion machinery: molecular architecture and function. Biochem Soc Trans 41:17–28
Chao Y, Vogel J (2010) The role of Hfq in bacterial pathogens. Curr Opin Microbiol 13:24–33
Chaudhuri RR, Morgan E, Peters SE, Pleasance SJ, Hudson DL, Davies HM, Wang J, van Diemen PM, Buckley AM, Bowen AJ et al (2013) Comprehensive assignment of roles for Salmonella typhimurium genes in intestinal colonization of food-producing animals. PLoS Genet 9:e1003456
Chen Z, Moayeri M, Purcell R (2011) Monoclonal antibody therapies against anthrax. Toxins (Basel) 3:1004–1019
Chhabra SR, Stead P, Bainton NJ, Salmond GP, Stewart GS, Williams P, Bycroft BW (1993) Autoregulation of carbapenem biosynthesis in Erwinia carotovora by analogues of N-(3-oxohexanoyl)-L-homoserine lactone. J Antibiot (Tokyo) 46:441–454
Clarke DJ (2010) The Rcs phosphorelay: more than just a two-component pathway. Future Microbiol 5:1173–1184
Clatworthy AE, Pierson E, Hung DT (2007) Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 3:541–548
Claudi B, Sprote P, Chirkova A, Personnic N, Zankl J, Schurmann N, Schmidt A, Bumann D (2014) Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158:722–733
Coban AY, Tanriverdi Cayci Y, Erturan Z, Durupinar B (2009) Effects of efflux pump inhibitors phenyl-arginine-beta-naphthylamide and 1-(1-naphthylmethyl)-piperazine on the antimicrobial susceptibility of Pseudomonas aeruginosa isolates from cystic fibrosis patients. J Chemother 21:592–594
Coburn B, Sekirov I, Finlay BB (2007) Type III secretion systems and disease. Clin Microbiol Rev 20:535–549
Coleman K, Levasseur P, Girard AM, Borgonovi M, Miossec C, Merdjan H, Drusano G, Shlaes D, Nichols WW (2014) Activities of ceftazidime and avibactam against beta-lactamase-producing Enterobacteriaceae in a hollow-fiber pharmacodynamic model. Antimicrob Agents Chemother 58:3366–3372
Conover MS, Mishra M, Deora R (2011) Extracellular DNA is essential for maintaining Bordetella biofilm integrity on abiotic surfaces and in the upper respiratory tract of mice. PLoS ONE 6:e16861
Control CfD (2013) Antibiotic resistance threats in the United States
Costerton JW, Stewart PS, Greenberg EP (1999) Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322
Cryan LM, Rogers MS (2011) Targeting the anthrax receptors, TEM-8 and CMG-2, for anti-angiogenic therapy. Front Biosci (Landmark Ed) 16:1574–1588
Cryan LM, Habeshian KA, Caldwell TP, Morris MT, Ackroyd PC, Christensen KA, Rogers MS (2013) Identification of small molecules that inhibit the interaction of TEM8 with anthrax protective antigen using a FRET assay. J Biomol Screen 18:714–725
Curtis MM, Russell R, Moreira CG, Adebesin AM, Wang C, Williams NS, Taussig R, Stewart D, Zimmern P, Lu B et al (2014) QseC inhibitors as an antivirulence approach for Gram-negative pathogens. MBio 5:e02165
D’Costa VM, McGrann KM, Hughes DW, Wright GD (2006) Sampling the antibiotic resistome. Science 311:374–377
Deep A, Chaudhary U, Gupta V (2011) Quorum sensing and bacterial pathogenicity: from molecules to disease. J Lab Phys 3:4–11
Dell’Aica I, Dona M, Tonello F, Piris A, Mock M, Montecucco C, Garbisa S (2004) Potent inhibitors of anthrax lethal factor from green tea. EMBO Rep 5:418–422
Desroy N, Denis A, Oliveira C, Atamanyuk D, Briet S, Faivre F, LeFralliec G, Bonvin Y, Oxoby M, Escaich S et al (2013) Novel HldE-K inhibitors leading to attenuated Gram negative bacterial virulence. J Med Chem 56:1418–1430
Deutscher J, Herro R, Bourand A, Mijakovic I, Poncet S (2005) P-Ser-HPr—a link between carbon metabolism and the virulence of some pathogenic bacteria. Biochim Biophys Acta 1754:118–125
Diard M, Garcia V, Maier L, Remus-Emsermann MN, Regoes RR, Ackermann M, Hardt WD (2013) Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature 494:353–356
Dong YH, Xu JL, Li XZ, Zhang LH (2000) AiiA, an enzyme that inactivates the acylhomoserine lactone quorum-sensing signal and attenuates the virulence of Erwinia carotovora. Proc Natl Acad Sci USA 97:3526–3531
Dong YH, Wang LH, Xu JL, Zhang HB, Zhang XF, Zhang LH (2001) Quenching quorum-sensing-dependent bacterial infection by an N-acyl homoserine lactonase. Nature 411:813–817
Dong YH, Gusti AR, Zhang Q, Xu JL, Zhang LH (2002) Identification of quorum-quenching N-acyl homoserine lactonases from Bacillus species. Appl Environ Microbiol 68:1754–1759
Duncan MC, Wong WR, Dupzyk AJ, Bray WM, Linington RG, Auerbuch V (2014) An NF-kappaB-based high-throughput screen identifies piericidins as inhibitors of the Yersinia pseudotuberculosis type III secretion system. Antimicrob Agents Chemother 58:1118–1126
Duss O, Michel E, Yulikov M, Schubert M, Jeschke G, Allain FH (2014) Structural basis of the non-coding RNA RsmZ acting as a protein sponge. Nature 509:588–592
Escaich S (2008) Antivirulence as a new antibacterial approach for chemotherapy. Curr Opin Chem Biol 12:400–408
Escaich S (2010) Novel agents to inhibit microbial virulence and pathogenicity. Expert Opin Ther Pat 20:1401–1418
Felise HB, Nguyen HV, Pfuetzner RA, Barry KC, Jackson SR, Blanc MP, Bronstein PA, Kline T, Miller SI (2008) An inhibitor of gram-negative bacterial virulence protein secretion. Cell Host Microbe 4:325–336
Feng L, Rutherford ST, Papenfort K, Bagert JD, van Kessel JC, Tirrell DA, Wingreen NS, Bassler BL (2015) A qrr noncoding RNA deploys four different regulatory mechanisms to optimize quorum-sensing dynamics. Cell 160:228–240
Fernicola S, Paiardini A, Giardina G, Rampioni G, Leoni L, Cutruzzola F, Rinaldo S (2015) In silico discovery and in vitro validation of catechol-containing sulfonohydrazide compounds as potent inhibitors of the diguanylate cyclase PleD. J Bacteriol
Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE (2005) Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol 1:29–32
Field M (2003) Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest 111:931–943
Firon N, Ashkenazi S, Mirelman D, Ofek I, Sharon N (1987) Aromatic alpha-glycosides of mannose are powerful inhibitors of the adherence of type 1 fimbriated Escherichia coli to yeast and intestinal epithelial cells. Infect Immun 55:472–476
Forst SA, Roberts DL (1994) Signal transduction by the EnvZ-OmpR phosphotransfer system in bacteria. Res Microbiol 145:363–373
Francois B, Luyt CE, Dugard A, Wolff M, Diehl JL, Jaber S, Forel JM, Garot D, Kipnis E, Mebazaa A et al (2012) Safety and pharmacokinetics of an anti-PcrV PEGylated monoclonal antibody fragment in mechanically ventilated patients colonized with Pseudomonas aeruginosa: a randomized, double-blind, placebo-controlled trial. Crit Care Med 40:2320–2326
Galan JE, Wolf-Watz H (2006) Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573
Garrity-Ryan LK, Kim OK, Balada-Llasat JM, Bartlett VJ, Verma AK, Fisher ML, Castillo C, Songsungthong W, Tanaka SK, Levy SB et al (2010) Small molecule inhibitors of LcrF, a Yersinia pseudotuberculosis transcription factor, attenuate virulence and limit infection in a murine pneumonia model. Infect Immun 78:4683–4690
Goldman ME, Cregar L, Nguyen D, Simo O, O’Malley S, Humphreys T (2006) Cationic polyamines inhibit anthrax lethal factor protease. BMC Pharmacol 6:8
Greener M (2000) How Escherichia coli kills. Mol Med Today 6:411
Groisman EA, Mouslim C (2006) Sensing by bacterial regulatory systems in host and non-host environments. Nat Rev Microbiol 4:705–709
Gu J, Ning Y, Wang H, Xiao D, Tang B, Luo P, Cheng Y, Jiang M, Li N, Zou Q et al (2011) Vaccination of attenuated EIS-producing Salmonella induces protective immunity against enterohemorrhagic Escherichia coli in mice. Vaccine 29:7395–7403
Hall-Stoodley L, Costerton JW, Stoodley P (2004) Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108
Han Z, Pinkner JS, Ford B, Obermann R, Nolan W, Wildman SA, Hobbs D, Ellenberger T, Cusumano CK, Hultgren SJ et al (2010) Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J Med Chem 53:4779–4792
Han Z, Pinkner JS, Ford B, Chorell E, Crowley JM, Cusumano CK, Campbell S, Henderson JP, Hultgren SJ, Janetka JW (2012) Lead optimization studies on FimH antagonists: discovery of potent and orally bio-available ortho-substituted biphenyl mannosides. J Med Chem 55:3945–3959
Harmon DE, Davis AJ, Castillo C, Mecsas J (2010) Identification and characterization of small-molecule inhibitors of Yop translocation in Yersinia pseudotuberculosis. Antimicrob Agents Chemother 54:3241–3254
Harmsen M, Lappann M, Knochel S, Molin S (2010) Role of extracellular DNA during biofilm formation by Listeria monocytogenes. Appl Environ Microbiol 76:2271–2279
Hartmann M, Papavlassopoulos H, Chandrasekaran V, Grabosch C, Beiroth F, Lindhorst TK, Rohl C (2012) Inhibition of bacterial adhesion to live human cells: activity and cytotoxicity of synthetic mannosides. FEBS Lett 586:1459–1465
Helaine S, Cheverton AM, Watson KG, Faure LM, Matthews SA, Holden DW (2014) Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science 343:204–208
Henkel JS, Baldwin MR, Barbieri JT (2010) Toxins from bacteria. EXS 100:1–29
Hentzer M, Givskov M (2003) Pharmacological inhibition of quorum sensing for the treatment of chronic bacterial infections. J Clin Invest 112:1300–1307
Hentzer M, Riedel K, Rasmussen TB, Heydorn A, Andersen JB, Parsek MR, Rice SA, Eberl L, Molin S, Hoiby N et al (2002) Inhibition of quorum sensing in Pseudomonas aeruginosa biofilm bacteria by a halogenated furanone compound. Microbiology 148:87–102
Heroven AK, Böhme K, Dersch P (2012) The Csr/Rsm system of Yersinia and related pathogens: a post-transcriptional strategy for managing virulence. RNA Biol 9:379–391
Hesterkamp T (2015) Antibiotics clinical development and pipeline. Curr Top Microbiol Immunol
Hilleringmann M, Pansegrau W, Doyle M, Kaufman S, MacKichan ML, Gianfaldoni C, Ruggiero P, Covacci A (2006) Inhibitors of Helicobacter pylori ATPase Cagalpha block CagA transport and cag virulence. Microbiology 152:2919–2930
Hopwood DA (2007) How do antibiotic-producing bacteria ensure their self-resistance before antibiotic biosynthesis incapacitates them? Mol Microbiol 63:937–940
Hudson DL, Layton AN, Field TR, Bowen AJ, Wolf-Watz H, Elofsson M, Stevens MP, Galyov EE (2007) Inhibition of type III secretion in Salmonella enterica serovar Typhimurium by small-molecule inhibitors. Antimicrob Agents Chemother 51:2631–2635
Hueck CJ (1998) Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev 62:379–433
Hung DT, Shakhnovich EA, Pierson E, Mekalanos JJ (2005) Small-molecule inhibitor of Vibrio cholerae virulence and intestinal colonization. Science 310:670–674
Iwatsuki M, Uchida R, Yoshijima H, Ui H, Shiomi K, Kim YP, Hirose T, Sunazuka T, Abe A, Tomoda H et al (2008) Guadinomines, type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. II: physico-chemical properties and structure elucidation. J Antibiot (Tokyo) 61:230–236
Izano EA, Amarante MA, Kher WB, Kaplan JB (2008) Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl Environ Microbiol 74:470–476
Izore T, Job V, Dessen A (2011) Biogenesis, regulation, and targeting of the type III secretion system. Structure 19:603–612
Jeon B, Zhang Q (2009) Sensitization of Campylobacter jejuni to fluoroquinolone and macrolide antibiotics by antisense inhibition of the CmeABC multidrug efflux transporter. J Antimicrob Chemother 63:946–948
Johansson J, Mandin P, Renzoni A, Chiaruttini C, Springer M, Cossart P (2002) An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110:551–561
Jonsson IM, Mazmanian SK, Schneewind O, Verdrengh M, Bremell T, Tarkowski A (2002) On the role of Staphylococcus aureus sortase and sortase-catalyzed surface protein anchoring in murine septic arthritis. J Infect Dis 185:1417–1424
Kalia NP, Mahajan P, Mehra R, Nargotra A, Sharma JP, Koul S, Khan IA (2012) Capsaicin, a novel inhibitor of the NorA efflux pump, reduces the intracellular invasion of Staphylococcus aureus. J Antimicrob Chemother 67:2401–2408
Kaplan BS, Meyers KE, Schulman SL (1998) The pathogenesis and treatment of hemolytic uremic syndrome. J Am Soc Nephrol 9:1126–1133
Kaplan JB, Velliyagounder K, Ragunath C, Rohde H, Mack D, Knobloch JK, Ramasubbu N (2004) Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J Bacteriol 186:8213–8220
Karginov VA, Yohannes A, Robinson TM, Fahmi NE, Alibek K, Hecht SM (2006) Beta-cyclodextrin derivatives that inhibit anthrax lethal toxin. Bioorg Med Chem 14:33–40
Kauppi AM, Nordfelth R, Uvell H, Wolf-Watz H, Elofsson M (2003) Targeting bacterial virulence: inhibitors of type III secretion in Yersinia. Chem Biol 10:241–249
Kern WV, Steinke P, Schumacher A, Schuster S, von Baum H, Bohnert JA (2006) Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J Antimicrob Chemother 57:339–343
Keyser P, Elofsson M, Rosell S, Wolf-Watz H (2008) Virulence blockers as alternatives to antibiotics: type III secretion inhibitors against Gram-negative bacteria. J Intern Med 264:17–29
Kim OK, Garrity-Ryan LK, Bartlett VJ, Grier MC, Verma AK, Medjanis G, Donatelli JE, Macone AB, Tanaka SK, Levy SB et al (2009) N-hydroxybenzimidazole inhibitors of the transcription factor LcrF in Yersinia: novel antivirulence agents. J Med Chem 52:5626–5634
Kim S, Jiao GS, Moayeri M, Crown D, Cregar-Hernandez L, McKasson L, Margosiak SA, Leppla SH, Johnson AT (2011) Antidotes to anthrax lethal factor intoxication. Part 2: structural modifications leading to improved in vivo efficacy. Bioorg Med Chem Lett 21:2030–2033
Kimura K, Iwatsuki M, Nagai T, Matsumoto A, Takahashi Y, Shiomi K, Omura S, Abe A (2011) A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J Antibiot (Tokyo) 64:197–203
Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR (2000) Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 403:669–672
Klein T, Abgottspon D, Wittwer M, Rabbani S, Herold J, Jiang X, Kleeb S, Luthi C, Scharenberg M, Bezencon J et al (2010) FimH antagonists for the oral treatment of urinary tract infections: from design and synthesis to in vitro and in vivo evaluation. J Med Chem 53:8627–8641
Kline T, Bowman J, Iglewski BH, de Kievit T, Kakai Y, Passador L (1999) Novel synthetic analogs of the Pseudomonas autoinducer. Bioorg Med Chem Lett 9:3447–3452
Kolodkin-Gal I, Romero D, Cao S, Clardy J, Kolter R, Losick R (2010) D-amino acids trigger biofilm disassembly. Science 328:627–629
Kolodkin-Gal I, Cao S, Chai L, Bottcher T, Kolter R, Clardy J, Losick R (2012) A self-produced trigger for biofilm disassembly that targets exopolysaccharide. Cell 149:684–692
Kong KF, Schneper L, Mathee K (2010) Beta-lactam antibiotics: from antibiosis to resistance and bacteriology. APMIS 118:1–36
Koppolu V, Osaka I, Skredenske JM, Kettle B, Hefty PS, Li J, Egan SM (2013) Small-molecule inhibitor of the Shigella flexneri master virulence regulator VirF. Infect Immun 81:4220–4231
Kortmann J, Narberhaus F (2012) Bacterial RNA thermometers: molecular zippers and switches. Nat Rev Microbiol 10:255–265
Krachler AM, Orth K (2013) Targeting the bacteria-host interface: strategies in anti-adhesion therapy. Virulence 4:284–294
Kulkarni AA, Fuller C, Korman H, Weiss AA, Iyer SS (2010) Glycan encapsulated gold nanoparticles selectively inhibit shiga toxins 1 and 2. Bioconjug Chem 21:1486–1493
Kulp A, Kuehn MJ (2010) Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol 64:163–184
Kunsmann L, Ruter C, Bauwens A, Greune L, Gluder M, Kemper B, Fruth A, Wai SN, He X, Lloubes R et al (2015) Virulence from vesicles: novel mechanisms of host cell injury by Escherichia coli O104: H4 outbreak strain. Sci Rep 5:13252
Kuzmic P, Cregar L, Millis SZ, Goldman M (2006) Mixed-type noncompetitive inhibition of anthrax lethal factor protease by aminoglycosides. FEBS J 273:3054–3062
Langridge GC, Phan MD, Turner DJ, Perkins TT, Parts L, Haase J, Charles I, Maskell DJ, Peters SE, Dougan G et al (2009) Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res 19:2308–2316
LaSarre B, Federle MJ (2013) Exploiting quorum sensing to confuse bacterial pathogens. Microbiol Mol Biol Rev 77:73–111
Layton AN, Hudson DL, Thompson A, Hinton JC, Stevens JM, Galyov EE, Stevens MP (2010) Salicylidene acylhydrazide-mediated inhibition of type III secretion system-1 in Salmonella enterica serovar Typhimurium is associated with iron restriction and can be reversed by free iron. FEMS Microbiol Lett 302:114–122
Lee B, Boucher HW (2015) Targeting antimicrobial-resistant bacterial respiratory tract pathogens: it is time to ‘get smart’. Curr Opin Pulm Med 21:293–303
Lehner T, Caldwell J, Smith R (1985) Local passive immunization by monoclonal antibodies against streptococcal antigen I/II in the prevention of dental caries. Infect Immun 50:796–799
Lewis K (2005) Persister cells and the riddle of biofilm survival. Biochemistry (Mosc) 70:267–274
Lewis K (2008) Multidrug tolerance of biofilms and persister cells. Curr Top Microbiol Immunol 322:107–131
Li H, Qian L, Chen Z, Thibault D, Liu G, Liu T, Thanassi DG (2004) The outer membrane usher forms a twin-pore secretion complex. J Mol Biol 344:1397–1407
Linden SK, Sheng YH, Every AL, Miles KM, Skoog EC, Florin TH, Sutton P, McGuckin MA (2009) MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog 5:e1000617
Linington RG, Robertson M, Gauthier A, Finlay BB, van Soest R, Andersen RJ (2002) Caminoside A, an antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Org Lett 4:4089–4092
Loh E, Kugelberg E, Tracy A, Zhang Q, Gollan B, Ewles H, Chalmers R, Pelicic V, Tang CM (2013) Temperature triggers immune evasion by Neisseria meningitidis. Nature 502:237–240
Lomovskaya O, Warren MS, Lee A, Galazzo J, Fronko R, Lee M, Blais J, Cho D, Chamberland S, Renau T et al (2001) Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: novel agents for combination therapy. Antimicrob Agents Chemother 45:105–116
Lu TK, Collins JJ (2007) Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA 104:11197–11202
Lu C, Maurer CK, Kirsch B, Steinbach A, Hartmann RW (2014a) Overcoming the unexpected functional inversion of a PqsR antagonist in Pseudomonas aeruginosa: an in vivo potent antivirulence agent targeting pqs quorum sensing. Angew Chem Int Ed Engl 53:1109–1112
Lu C, Kirsch B, Maurer CK, de Jong JC, Braunshausen A, Steinbach A, Hartmann RW (2014b) Optimization of anti-virulence PqsR antagonists regarding aqueous solubility and biological properties resulting in new insights in structure-activity relationships. Eur J Med Chem 79:173–183
Lucchetti-Miganeh C, Burrowes E, Baysse C, Ermel G (2008) The post-transcriptional regulator CsrA plays a central role in the adaptation of bacterial pathogens to different stages of infection in animal hosts. Microbiology 154:16–29
Lynch SV, Wiener-Kronish JP (2008) Novel strategies to combat bacterial virulence. Curr Opin Crit Care 14:593–599
Ma JK, Hunjan M, Smith R, Lehner T (1989) Specificity of monoclonal antibodies in local passive immunization against Streptococcus mutans. Clin Exp Immunol 77:331–337
Madsen JS, Burmolle M, Hansen LH, Sorensen SJ (2012) The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol Med Microbiol 65:183–195
Manefield M, de Nys R, Kumar N, Read R, Givskov M, Steinberg P, Kjelleberg S (1999) Evidence that halogenated furanones from Delisea pulchra inhibit acylated homoserine lactone (AHL)-mediated gene expression by displacing the AHL signal from its receptor protein. Microbiology 145(2):283–291
Manina G, Dhar N, McKinney JD (2015) Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17:32–46
Maresso AW, Wu R, Kern JW, Zhang R, Janik D, Missiakas DM, Duban ME, Joachimiak A, Schneewind O (2007) Activation of inhibitors by sortase triggers irreversible modification of the active site. J Biol Chem 282:23129–23139
Margalit M, Ash N, Zimran A, Halkin H (2002) Enzyme replacement therapy in the management of longstanding skeletal and soft tissue salmonella infection in a patient with Gaucher’s disease. Postgrad Med J 78:564–565
Marshall NC, Finlay BB (2014) Targeting the type III secretion system to treat bacterial infections. Expert Opin Ther Targets 18:137–152
Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martinez-Solano L, Sanchez MB (2009) A global view of antibiotic resistance. FEMS Microbiol Rev 33:44–65
McHugh SM, Hill AD, Humphreys H (2010) Preventing healthcare-associated infection through education: have surgeons been overlooked? Surgeon 8:96–100
McShan AC, De Guzman RN (2015) The bacterial type III secretion system as a target for developing new antibiotics. Chem Biol Drug Des 85:30–42
Mellin JR, Tiensuu T, Becavin C, Gouin E, Johansson J, Cossart P (2013) A riboswitch-regulated antisense RNA in Listeria monocytogenes. Proc Natl Acad Sci USA 110:13132–13137
Mellin JR, Koutero M, Dar D, Nahori MA, Sorek R, Cossart P (2014) Riboswitches. Sequestration of a two-component response regulator by a riboswitch-regulated noncoding RNA. Science 345:940–943
Miethke M, Marahiel MA (2007) Siderophore-based iron acquisition and pathogen control. Microbiol Mol Biol Rev 71:413–451
Montecucco C, Tonello F, Zanotti G (2004) Stop the killer: how to inhibit the anthrax lethal factor metalloprotease. Trends Biochem Sci 29:282–285
Mu Y, Shen Z, Jeon B, Dai L, Zhang Q (2013) Synergistic effects of anti-CmeA and anti-CmeB peptide nucleic acids on sensitizing Campylobacter jejuni to antibiotics. Antimicrob Agents Chemother 57:4575–4577
Muschiol S, Bailey L, Gylfe A, Sundin C, Hultenby K, Bergstrom S, Elofsson M, Wolf-Watz H, Normark S, Henriques-Normark B (2006) A small-molecule inhibitor of type III secretion inhibits different stages of the infectious cycle of Chlamydia trachomatis. Proc Natl Acad Sci USA 103:14566–14571
Nagai H, Kubori T (2011) Type IVB secretion systems of Legionella and other Gram-negative bacteria. Front Microbiol 2:136
Negrea A, Bjur E, Ygberg SE, Elofsson M, Wolf-Watz H, Rhen M (2007) Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar typhimurium. Antimicrob Agents Chemother 51:2867–2876
Neres J, Labello NP, Somu RV, Boshoff HI, Wilson DJ, Vannada J, Chen L, Barry CE 3rd, Bennett EM, Aldrich CC (2008) Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J Med Chem 51:5349–5370
Nestorovich EM, Bezrukov SM (2014) Designing inhibitors of anthrax toxin. Expert Opin Drug Discov 9:299–318
Ng WL, Bassler BL (2009) Bacterial quorum-sensing network architectures. Annu Rev Genet 43:197–222
Nikaido H, Pages JM (2012) Broad-specificity efflux pumps and their role in multidrug resistance of Gram-negative bacteria. FEMS Microbiol Rev 36:340–363
Nishikawa K, Matsuoka K, Watanabe M, Igai K, Hino K, Hatano K, Yamada A, Abe N, Terunuma D, Kuzuhara H et al (2005) Identification of the optimal structure required for a Shiga toxin neutralizer with oriented carbohydrates to function in the circulation. J Infect Dis 191:2097–2105
Nordfelth R, Kauppi AM, Norberg HA, Wolf-Watz H, Elofsson M (2005) Small-molecule inhibitors specifically targeting type III secretion. Infect Immun 73:3104–3114
Nuss AM, Heroven AK, Waldmann B, Reinkensmeier J, Jarek M, Beckstette M, Dersch P (2015) Trans-criptomic profiling of Yersinia pseudotuberculosis reveals reprogramming of the Crp regulon by temperature and uncovers Crp as a master regulator of small RNAs. PLoS Genet 11:e1005087
O’Connell KM, Hodgkinson JT, Sore HF, Welch M, Salmond GP, Spring DR (2013) Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew Chem Int Ed Engl 52:10706–10733
Ofek I, Hasty DL, Sharon N (2003) Anti-adhesion therapy of bacterial diseases: prospects and problems. FEMS Immunol Med Microbiol 38:181–191
Okshevsky M, Regina VR, Meyer RL (2015) Extracellular DNA as a target for biofilm control. Curr Opin Biotechnol 33:73–80
Oliva G, Sahr T, Buchrieser C (2015) Small RNAs, 5′ UTR elements and RNA-binding proteins in intracellular bacteria: impact on metabolism and virulence. FEMS Microbiol Rev 39:331–349
Palzkill T (2013) Metallo-beta-lactamase structure and function. Ann N Y Acad Sci 1277:91–104
Pan NJ, Brady MJ, Leong JM, Goguen JD (2009) Targeting type III secretion in Yersinia pestis. Antimicrob Agents Chemother 53:385–392
Panchal RG, Hermone AR, Nguyen TL, Wong TY, Schwarzenbacher R, Schmidt J, Lane D, McGrath C, Turk BE, Burnett J et al (2004) Identification of small molecule inhibitors of anthrax lethal factor. Nat Struct Mol Biol 11:67–72
Pannek S, Higgins PG, Steinke P, Jonas D, Akova M, Bohnert JA, Seifert H, Kern WV (2006) Multidrug efflux inhibition in Acinetobacter baumannii: comparison between 1-(1-naphthylmethyl)-piperazine and phenyl-arginine-beta-naphthylamide. J Antimicrob Chemother 57:970–974
Papenfort K, Vogel J (2014) Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front Cell Infect Microbiol 4:91
Park S, Kim HS, Ok K, Kim Y, Park HD, Byun Y (2015) Design, synthesis and biological evaluation of 4-(alkyloxy)-6-methyl-2H-pyran-2-one derivatives as quorum sensing inhibitors. Bioorg Med Chem Lett 25:2913–2917
Parker P, Sando L, Pearson R, Kongsuwan K, Tellam RL, Smith S (2010) Bovine Muc1 inhibits binding of enteric bacteria to Caco-2 cells. Glycoconj J 27:89–97
Parsek MR, Val DL, Hanzelka BL, Cronan JE Jr, Greenberg EP (1999) Acyl homoserine-lactone quorum-sensing signal generation. Proc Natl Acad Sci USA 96:4360–4365
Paschos A, den Hartigh A, Smith MA, Atluri VL, Sivanesan D, Tsolis RM, Baron C (2011) An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect Immun 79:1033–1043
Paulasova P, Pellestor F (2004) The peptide nucleic acids (PNAs): a new generation of probes for genetic and cytogenetic analyses. Ann Genet 47:349–358
Pearson JS, Zhang Y, Newton HJ, Hartland EL (2015) Post-modern pathogens: surprising activities of translocated effectors from E. coli and Legionella. Curr Opin Microbiol 23:73–79
Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274:8405–8410
Peselis A, Serganov A (2014) Themes and variations in riboswitch structure and function. Biochim Biophys Acta 1839:908–918
Pinkner JS, Remaut H, Buelens F, Miller E, Aberg V, Pemberton N, Hedenstrom M, Larsson A, Seed P, Waksman G et al (2006) Rationally designed small compounds inhibit pilus biogenesis in uropathogenic bacteria. Proc Natl Acad Sci USA 103:17897–17902
Putrins M, Kogermann K, Lukk E, Lippus M, Varik V, Tenson T (2015) Phenotypic heterogeneity enables uropathogenic Escherichia coli to evade killing by antibiotics and serum complement. Infect Immun 83:1056–1067
Qin Z, Ou Y, Yang L, Zhu Y, Tolker-Nielsen T, Molin S, Qu D (2007) Role of autolysin-mediated DNA release in biofilm formation of Staphylococcus epidermidis. Microbiology 153:2083–2092
Rasko DA, Sperandio V (2010) Anti-virulence strategies to combat bacteria-mediated disease. Nat Rev Drug Discov 9:117–128
Rasko DA, Moreira CG, de Li R, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M et al (2008) Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080
Renau TE, Leger R, Flamme EM, Sangalang J, She MW, Yen R, Gannon CL, Griffith D, Chamberland S, Lomovskaya O et al (1999) Inhibitors of efflux pumps in Pseudomonas aeruginosa potentiate the activity of the fluoroquinolone antibacterial levofloxacin. J Med Chem 42:4928–4931
Rice LB (2008) Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197:1079–1081
Rogers MS, Cryan LM, Habeshian KA, Bazinet L, Caldwell TP, Ackroyd PC, Christensen KA (2012) A FRET-based high throughput screening assay to identify inhibitors of anthrax protective antigen binding to capillary morphogenesis gene 2 protein. PLoS ONE 7:e39911
Romeo T, Vakulskas CA, Babitzke P (2013) Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems. Environ Microbiol 15:313–324
Romling U, Balsalobre C (2012) Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med 272:541–561
Saar-Dover R, Bitler A, Nezer R, Shmuel-Galia L, Firon A, Shimoni E, Trieu-Cuot P, Shai Y (2012) D-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B streptococcus by increasing the cell wall density. PLoS Pathog 8:e1002891
Salyers AA, Gupta A, Wang Y (2004) Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol 12:412–416
Santa Maria JP Jr, Sadaka A, Moussa SH, Brown S, Zhang YJ, Rubin EJ, Gilmore MS, Walker S (2014) Compound-gene interaction mapping reveals distinct roles for Staphylococcus aureus teichoic acids. Proc Natl Acad Sci USA 111:12510–12515
Schmitt CK, Meysick KC, O’Brien AD (1999) Bacterial toxins: friends or foes? Emerg Infect Dis 5:224–234
Schroeder GN, Hilbi H (2008) Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev 21:134–156
Schumacher A, Steinke P, Bohnert JA, Akova M, Jonas D, Kern WV (2006) Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Enterobacteriaceae other than Escherichia coli. J Antimicrob Chemother 57:344–348
Scobie HM, Thomas D, Marlett JM, Destito G, Wigelsworth DJ, Collier RJ, Young JA, Manchester M (2005) A soluble receptor decoy protects rats against anthrax lethal toxin challenge. J Infect Dis 192:1047–1051
Serganov A, Nudler E (2013) A decade of riboswitches. Cell 152:17–24
Shakhnovich EA, Hung DT, Pierson E, Lee K, Mekalanos JJ (2007) Virstatin inhibits dimerization of the transcriptional activator ToxT. Proc Natl Acad Sci USA 104:2372–2377
Shoop WL, Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichny JV, Felcetto T, Michael BF, Bansal A, Cummings RT et al (2005) Anthrax lethal factor inhibition. Proc Natl Acad Sci USA 102:7958–7963
Silberstein C, Lucero MS, Zotta E, Copeland DP, Lingyun L, Repetto HA, Ibarra C (2011) A glucosylceramide synthase inhibitor protects rats against the cytotoxic effects of shiga toxin 2. Pediatr Res 69:390–394
Skredenske JM, Koppolu V, Kolin A, Deng J, Kettle B, Taylor B, Egan SM (2013) Identification of a small-molecule inhibitor of bacterial AraC family activators. J Biomol Screen 18:588–598
Slepenkin A, Enquist PA, Hagglund U, de la Maza LM, Elofsson M, Peterson EM (2007) Reversal of the antichlamydial activity of putative type III secretion inhibitors by iron. Infect Immun 75:3478–3489
Smith KM, Bu Y, Suga H (2003a) Induction and inhibition of Pseudomonas aeruginosa quorum sensing by synthetic autoinducer analogs. Chem Biol 10:81–89
Smith KM, Bu Y, Suga H (2003b) Library screening for synthetic agonists and antagonists of a Pseudomonas aeruginosa autoinducer. Chem Biol 10:563–571
Smits WK, Kuipers OP, Veening JW (2006) Phenotypic variation in bacteria: the role of feedback regulation. Nat Rev Microbiol 4:259–271
Soheili V, Bazzaz BS, Abdollahpour N, Hadizadeh F (2015) Investigation of Pseudomonas aeruginosa quorum-sensing signaling system for identifying multiple inhibitors using molecular docking and structural analysis methodology. Microb Pathog 89:73–78
Stephenson K, Yamaguchi Y, Hoch JA (2000) The mechanism of action of inhibitors of bacterial two-component signal transduction systems. J Biol Chem 275:38900–38904
Sturm A, Heinemann M, Arnoldini M, Benecke A, Ackermann M, Benz M, Dormann J, Hardt WD (2011) The cost of virulence: retarded growth of Salmonella Typhimurium cells expressing type III secretion system 1. PLoS Pathog 7:e1002143
Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS et al (2014) Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog 10:e1004174
Suzuki N, Ohtaguro N, Yoshida Y, Hirai M, Matsuo H, Yamada Y, Imamura N, Tsuchiya T (2015) A compound inhibits biofilm formation of Staphylococcus aureus from Streptomyces. Biol Pharm Bull 38:889–892
Svensson M, Frendeus B, Butters T, Platt F, Dwek R, Svanborg C (2003) Glycolipid depletion in antimicrobial therapy. Mol Microbiol 47:453–461
Tarr PI, Gordon CA, Chandler WL (2005) Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365:1073–1086
Thanassi DG, Bliska JB, Christie PJ (2012) Surface organelles assembled by secretion systems of Gram-negative bacteria: diversity in structure and function. FEMS Microbiol Rev 36:1046–1082
Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K et al (2009) The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956
Tonello F, Seveso M, Marin O, Mock M, Montecucco C (2002) Screening inhibitors of anthrax lethal factor. Nature 418:386
Totir MA, Helfand MS, Carey MP, Sheri A, Buynak JD, Bonomo RA, Carey PR (2007) Sulbactam forms only minimal amounts of irreversible acrylate-enzyme with SHV-1 beta-lactamase. Biochemistry 46:8980–8987
Trachtman H, Cnaan A, Christen E, Gibbs K, Zhao S, Acheson DW, Weiss R, Kaskel FJ, Spitzer A, Hirschman GH et al (2003) Effect of an oral Shiga toxin-binding agent on diarrhea-associated hemolytic uremic syndrome in children: a randomized controlled trial. JAMA 290:1337–1344
Tree JJ, Wang D, McInally C, Mahajan A, Layton A, Houghton I, Elofsson M, Stevens MP, Gally DL, Roe AJ (2009) Characterization of the effects of salicylidene acylhydrazide compounds on type III secretion in Escherichia coli O157: H7. Infect Immun 77:4209–4220
Trokter M, Felisberto-Rodrigues C, Christie PJ, Waksman G (2014) Recent advances in the structural and molecular biology of type IV secretion systems. Curr Opin Struct Biol 27:16–23
Tsou LK, Dossa PD, Hang HC (2013) Small molecules aimed at type III secretion systems to inhibit bacterial virulence. Medchemcomm 4:68–79
Turk BE, Wong TY, Schwarzenbacher R, Jarrell ET, Leppla SH, Collier RJ, Liddington RC, Cantley LC (2004) The structural basis for substrate and inhibitor selectivity of the anthrax lethal factor. Nat Struct Mol Biol 11:60–66
Vakulskas CA, Potts AH, Babitzke P, Ahmer BM, Romeo T (2015) Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev 79:193–224
Vanella de Cuetos EE, Fernandez RA, Bianco MI, Sartori OJ, Piovano ML, Luquez C, de Jong LI (2011) Equine botulinum antitoxin for the treatment of infant botulism. Clin Vaccine Immunol 18:1845–1849
Vanier G, Sekizaki T, Dominguez-Punaro MC, Esgleas M, Osaki M, Takamatsu D, Segura M, Gottschalk M (2008) Disruption of srtA gene in Streptococcus suis results in decreased interactions with endothelial cells and extracellular matrix proteins. Vet Microbiol 127:417–424
Veenendaal AK, Sundin C, Blocker AJ (2009) Small-molecule type III secretion system inhibitors block assembly of the Shigella type III secreton. J Bacteriol 191:563–570
Vimberg V, Kuzma M, Stodulkova E, Novak P, Bednarova L, Sulc M, Gazak R (2015) Hydnocarpin-type Flavonolignans: semisynthesis and inhibitory effects on Staphylococcus aureus biofilm formation. J Nat Prod 78:2095–2103
Vogel J, Luisi BF (2011) Hfq and its constellation of RNA. Nat Rev Microbiol 9:578–589
Vogt SL, Raivio TL (2012) Just scratching the surface: an expanding view of the Cpx envelope stress response. FEMS Microbiol Lett 326:2–11
Voth DE, Broederdorf LJ, Graham JG (2012) Bacterial type IV secretion systems: versatile virulence machines. Future Microbiol 7:241–257
Waksman G, Orlova EV (2014) Structural organisation of the type IV secretion systems. Curr Opin Microbiol 17:24–31
Wang D, Zetterstrom CE, Gabrielsen M, Beckham KS, Tree JJ, Macdonald SE, Byron O, Mitchell TJ, Gally DL, Herzyk P et al (2011) Identification of bacterial target proteins for the salicylidene acylhydrazide class of virulence-blocking compounds. J Biol Chem 286:29922–29931
Weber GG, Kortmann J, Narberhaus F, Klose KE (2014) RNA thermometer controls temperature-dependent virulence factor expression in Vibrio cholerae. Proc Natl Acad Sci USA 111:14241–14246
Weigel WA, Demuth DR (2015) QseBC, a two component bacterial adrenergic receptor and global regulator of virulence in Enterobacteriaceae and Pasteurellaceae. Mol Oral Microbiol. doi:10.1111/omi.12138. [Epub ahead of print]
Westermann AJ, Gorski SA, Vogel J (2012) Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630
Wilke KE, Francis S, Carlson EE (2015) Inactivation of multiple bacterial histidine kinases by targeting the ATP-binding domain. ACS Chem Biol 10:328–335
Wolf K, Betts HJ, Chellas-Gery B, Hower S, Linton CN, Fields KA (2006) Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle. Mol Microbiol 61:1543–1555
Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL (2011) Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol 80:1420–1438
Worthington RJ, Blackledge MS, Melander C (2013) Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Med Chem 5:1265–1284
Wright KJ, Hultgren SJ (2006) Sticky fibers and uropathogenesis: bacterial adhesins in the urinary tract. Future Microbiol 1:75–87
Wright JS 3rd, Jin R, Novick RP (2005) Transient interference with staphylococcal quorum sensing blocks abscess formation. Proc Natl Acad Sci USA 102:1691–1696
Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichny JV, Tang W, Bansal A, Cummings RT, Cunningham BR, Friedlander AM et al (2006) The discovery of a potent and selective lethal factor inhibitor for adjunct therapy of anthrax infection. Bioorg Med Chem Lett 16:964–968
Zahid MS, Awasthi SP, Asakura M, Chatterjee S, Hinenoya A, Faruque SM, Yamasaki S (2015) Suppression of virulence of toxigenic Vibrio cholerae by anethole through the cyclic AMP (cAMP)-cAMP receptor protein signaling system. PLoS ONE 10:e0137529
Zambelloni R, Marquez R, Roe AJ (2015) Development of antivirulence compounds: a biochemical review. Chem Biol Drug Des 85:43–55
Zetterstrom CE, Hasselgren J, Salin O, Davis RA, Quinn RJ, Sundin C, Elofsson M (2013) The resveratrol tetramer (-)-hopeaphenol inhibits type III secretion in the gram-negative pathogens Yersinia pseudotuber-culosis and Pseudomonas aeruginosa. PLoS ONE 8:e81969
Zhang C, Zhang W (2010) Escherichia coli K88ac fimbriae expressing heat-labile and heat-stable (STa) toxin epitopes elicit antibodies that neutralize cholera toxin and STa toxin and inhibit adherence of K88ac fimbrial E. coli. Clin Vaccine Immunol 17:1859–1867
Zhang J, Liu H, Zhu K, Gong S, Dramsi S, Wang YT, Li J, Chen F, Zhang R, Zhou L et al (2014) Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc Natl Acad Sci USA 111:13517–13522
Acknowledgements
Work of P. Dersch is supported by the German Research Foundation and the German Center of Infection Research (DZIF, TTU-GI).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing AG
About this chapter
Cite this chapter
Mühlen, S., Dersch, P. (2015). Anti-virulence Strategies to Target Bacterial Infections. In: Stadler, M., Dersch, P. (eds) How to Overcome the Antibiotic Crisis . Current Topics in Microbiology and Immunology, vol 398. Springer, Cham. https://doi.org/10.1007/82_2015_490
Download citation
DOI: https://doi.org/10.1007/82_2015_490
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-49282-7
Online ISBN: 978-3-319-49284-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)