
Abstract
Over three decades of evidence indicate that dopamine (DA) D3 receptors (D3R) are involved in the control of drug-seeking behavior and may play an important role in the pathophysiology of substance use disorders (SUD). The expectation that a selective D3R antagonist/partial agonist would be efficacious for the treatment of SUD is based on the following key observations. First, D3R are distributed in strategic areas belonging to the mesolimbic DA system such as the ventral striatum, midbrain, and ventral pallidum, which have been associated with behaviors controlled by the presentation of drug-associated cues. Second, repeated exposure to drugs of abuse produces neuroadaptations in the D3R system. Third, the synthesis and characterization of highly potent and selective D3R antagonists/partial agonists have further strengthened the role of the D3R in SUD. Based on extensive preclinical and preliminary clinical evidence, the D3R shows promise as a target for the development of pharmacotherapies for SUD as reflected by their potential to (1) regulate the motivation to self-administer drugs and (2) disrupt the responsiveness to drug-associated stimuli that play a key role in reinstatement of drug-seeking behavior triggered by re-exposure to the drug itself, drug-associated environmental cues, or stress. The availability of PET ligands to assess clinically relevant receptor occupancy by selective D3R antagonists/partial agonists, the definition of reliable dosing, and the prospect of using human laboratory models may further guide the design of clinical proof of concept studies. Pivotal clinical trials for more rapid progression of this target toward regulatory approval are urgently required. Finally, the discovery that highly selective D3R antagonists, such as R-VK4-116 and R-VK4-40, do not adversely affect peripheral biometrics or cardiovascular effects alone or in the presence of oxycodone or cocaine suggests that this class of drugs has great potential in safely treating psychostimulant and/or opioid use disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- D3 receptor antagonist
- D3 receptor partial agonist
- Dopamine
- Opioids
- Psychostimulants
- Substance use disorders
1 Introduction: Brief Historical Perspective/Epidemiology
Meteorologists see perfect in strange things, and the meshing of three completely independent weather systems to form a hundred-year event is one of them. My God, thought Case, this is the perfect storm.
— Sebastian Junger, The Perfect Storm: A True Story of Men Against the Sea
1.1 The Perfect Storm
The COVID-19 pandemic has brought our world to its knees in a way that most of us never imagined. The SARS-CoV-2 virus has managed to infect and mutate, becoming more virulent over time, resulting in death and destruction of economies, livelihoods, and a way of life that no one could have predicted. While massive resources and biomedical research focused on vaccines and medications to save lives, two other crises were brewing. The opioid epidemic was just starting to see the beginning of a downward trend, at least in terms of death by overdose (Ahmad et al. 2021). And although the use of psychostimulants such as cocaine and methamphetamine was still a relevant health assailant, it had not yet reached the point of crisis. Nevertheless, the devastation, isolation, hopelessness, and fatigue brought on by the COVID-19 pandemic have exacerbated substance misuse, joining forces to reverse the upward trend of life longevity in the USA (Manchikanti et al. 2021) and resulting in >90,000 drug overdose deaths, an increase of >30% in 2020 over the year before (Ahmad et al. 2021; Volkow 2021). The decrease in health services, limited access to medical care, and increased access to highly potent opioids such as fentanyl, etonitazene, and their illicit analogues have been complemented by an increased supply of methamphetamine, the combinations of which were more deadly than either one alone or sometimes ingested without the user’s knowledge (Narayan and Balkrishnan 2021).
The challenges that the COVID-19 epidemic introduced to mental health cannot be underestimated. Isolation-related anxiety and depression are among the disorders that have increased and been exacerbated. Closely coupled to these is the management of pain, which has also been impaired by lack of access to medical care and is the leading reason patients take prescription opioids that for some can lead to dependence or addiction (Kibaly et al. 2021; Taquet et al. 2021a, b). Sheltering in place and restrictions in travel have impacted patients’ ability to obtain proper medical care and necessary medications. Patients in chronic pain become depressed and the vicious cycle is unrelenting, unless acutely mitigated by the use of illicit drugs – a solution that has devastating consequences.
People with substance use disorders (SUD) are at heightened risk for other life-threatening comorbidities including cardiovascular disease, mucociliary dysfunction, compromised immunity as well as multiple social factors that prevent proper treatment (Manchikanti et al. 2021). And indeed, those whose prescription opioid taking accelerates to illicit drug use and addiction, only enhances their chances of becoming infected with SARS-CoV-2 and succumbing to the virus, overdose, or both. Sadly, as with other crises, underserved populations receive the disproportionate impact of this trifecta of tragedy (Narayan and Balkrishnan 2021).
1.2 Opioid Crisis
By 2019, the opioid epidemic in the USA was noted as a health crisis that was continuing to escalate (Lyden and Binswanger 2019). Although illicit opioids such as heroin had been contributing to opioid-related deaths for decades before, the increase in prescribed opioids for the management of pain, and especially the over prescription of extended-release formulations of oxycodone (e.g., oxycontin) significantly escalated opioid dependence and addiction in the USA. As oxycontin was first marketed as less addictive than other opioid narcotics, a dramatic increase in its use for pain management ultimately resulted in escalated opioid overdoses in the last decade (Azadfard et al. 2021; Kibaly et al. 2021; Walker 2018). According to the Centers for Disease Control and Prevention (CDC), 96,779 people died from drug overdose in the 12-month period ending March 2021. Approximately 72,805 of these deaths were attributed to all opioids, and 61,230 were attributed to synthetic opioids such as fentanyl (Ahmad et al. 2021).
Studies that attempt to quantify the burden of opioid-related mortality conclude that premature deaths caused by opioid overdose has and undoubtedly will continue to impose an enormous health and economic burden on the USA (Gomes et al. 2018). In 2016, years of life lost (YLL) exceeded those attributed to hypertension, HIV, and pneumonia (Gomes et al. 2018). Sadly, 25–34 years of age was the demographic with the highest opioid overdose death rate. Young adults who had the potential to contribute so much to our society and may have left children behind – yet another tragic reality.
1.3 Psychostimulant Use Disorder: Cocaine and Methamphetamine
As if the COVID-19 pandemic and the opioid crisis were not enough to keep researchers and health care providers, legislators, and parents up at night, a new wave of drug abuse is now rolling through our cities and rural areas alike. Although cocaine continues to be a drug of high abuse potential and related death by overdose, methamphetamine has roared into our streets and communities (Compton et al. 2021; Fogger 2019; Jones et al. 2020). Methamphetamine is easily synthesized in home laboratories, has a longer half-life than cocaine, and is more easily accessible, likely contributing to its added popularity, which has increased during the COVID-19 pandemic.
1.4 Polysubstance Use Highlighting Opioids/Methamphetamine
The “old practice” of combining heroin with cocaine known as “speedball” has been replaced with the combination of methamphetamine and heroin or fentanyl, called “goofball” (Glick et al. 2021) with grave consequences. Some users of this combination of drugs claim that the addition of methamphetamine to the opioid reduces unpleasant sluggishness/lethargy and the opioid decreases the unpleasant intensity of methamphetamine (Ciccarone 2021; Glick et al. 2021). Clearly polysubstance use is prevalent and highly complex, leading to an increase in morbidity and poses further challenges for prevention and treatment. Although a decline in overdose deaths appeared in 2017–2018, the CDC reports an increased mortality that is alarming, driven by a dramatic increase in opioid-related deaths and now a “fourth wave” of high mortality involving cocaine and primarily methamphetamine (Ciccarone 2021).
1.5 Co-Morbidities with Other Neuropsychiatric Disorders
In addition to polysubstance use, prevailing public health problems that have been exacerbated by the COVID-19 pandemic are psychiatric disorders, including anxiety, major depressive disorder, and bipolar disorder. These disorders are complex and often difficult to treat. Equally alarming is the comorbidity between these disorders and SUD, a public health concern that emerged long before COVID-19 but has undoubtedly increased (Angarita et al. 2021a; Hellem et al. 2015; Murthy et al. 2019).
2 D3R Neurocircuitry and Relationship to SUD
2.1 Rationale of D3R-Based Medication Development for the Treatment of Psychostimulant and Opioid Use Disorders
2.1.1 Dopamine Hypothesis of Drug Reward
It is well documented that the mesolimbic and nigrostriatal dopamine (DA) systems are critically involved in psychostimulant and opioid reward (Galaj and Xi 2021; Lammel et al. 2014) (Fig. 1). These systems originate from DA neurons in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) in the midbrain and project to the nucleus accumbens (NAc), dorsal striatum (DST), ventral pallidum (VP), prefrontal cortex (PFC) and insula, as well as the amygdala (Amy). A great deal of evidence supports the importance of both the DA projection pathways in SUD. First, almost all addictive drugs, including cocaine, opioids, nicotine, and ethanol, increase extracellular DA in the NAc and DST (Koob and Bloom 1988; Self and Nestler 1995). Second, almost all addictive drugs can be self-administered by animals either intravenously or locally into the VTA or NAc, which can be blocked or attenuated by either chemical lesions of DA terminals or by pharmacological blockade of DA receptors (Bressan and Crippa 2005; Gardner 2000). And third, electrical or optical stimulation of brain DA loci maintains intracranial self-administration, which can be enhanced by drugs of abuse and attenuated by DA receptor antagonists (Wise 1996).

Schematic diagram of the mesolimbic and nigrostriatal reward pathways, illustrating the action sites (targets) of psychostimulants (cocaine, methamphetamine) and opioids in the brain. The mesolimbic DA circuit (RMTg → VTA → NAc) originates in the midbrain ventral tegmental area (VTA) and projects predominantly to the nucleus accumbens (NAc) and other forebrain regions (not shown). VTA DA neurons receive GABAergic inputs from local VTA GABA neurons and other brain regions including the NAc, ventral pallidum (VP), and rostromedial tegmental nucleus (RMTg), particularly from the RMTg. Psychostimulants elevate extracellular NAc DA by blocking DA transporters (DAT) (by cocaine) or reversing VMAT2 (by methamphetamine) on DA axon terminals in the NAc and dorsal striatum (DST). The nigrostriatal DA circuit (SNr → SNc → DST) originates from DA neurons in the substantia nigra pars compacta (SNc) and projects to the DST. SNc DA neurons receive dense GABAergic inputs from multiple brain regions including the SNr and RMTg, but mainly from SNr. Mu opioid receptors (MOR) are highly expressed in GABA neurons, particularly in the RMTg and SNr. Opioids bind to MORs and inhibit GABA neuron activity and GABA release, which subsequently disinhibits DA neurons in the VTA and SNc
Psychostimulants and opioids activate the mesolimbic and nigrostriatal DA systems by different molecular and cellular mechanisms. Cocaine elevates extracellular DA levels in the DA projection areas mainly by blockade of DA reuptake, while amphetamine or methamphetamine mainly promotes DA release from DA terminals by reversal of vesicular monoamine transporter 2 (VMAT2), which promotes DA exit from vesicles into cytoplasm and causes DA release from cytoplasm to extracellular space by reversal of membrane dopamine transporter (DAT) (Elkashef et al. 2008; Freyberg et al. 2016; Shen et al. 2021) (Fig. 1). These increases in synaptic or extracellular DA in the forebrain reward loci – especially in the NAc – are thought to underlie the euphoria associated with psychostimulant use (Wise 2005).
In contrast to psychostimulant reward, the neural mechanisms underlying opioid reward and abuse are still not fully understood. A classical hypothesis is that opioids initially bind to mu opioid receptors (MOR) located on GABAergic interneurons within the VTA and functionally inhibit GABAergic neuronal activity, which subsequently disinhibits neighboring DA neurons within the VTA (Galaj and Xi 2021; Xi and Stein 2002). This canonical two-neuron hypothesis, which was upheld for over half a century, has been challenged by recent findings suggesting that high density MORs are expressed in GABAergic neurons mainly in the rostromedial tegmental nucleus (RMTg, also called the tail of the VTA) and substantia nigra pars reticulata (SNr) in the midbrain (Galaj et al. 2020a; Galaj and Xi 2021; Matsui et al. 2014; Matsui and Williams 2011). It has been shown that DA neurons in the VTA and SNc receive intensive GABAergic inputs mainly from the RMTg and the SNr, respectively (Galaj et al. 2020a; Matsui and Williams 2011), suggesting that DA neurons in the VTA and SNc may be activated mainly by activation of MORs in GABAergic neurons in both the RMTg and SNr via a disinhibition mechanism. Thus, a two-pathway hypothesis (e.g., RTMg → VTA → NAc, SNr → SNc → DST) has been proposed to explain opioid reward and abuse (Fig. 1) (Galaj et al. 2020a; Galaj and Xi 2021).
Based on this DA hypothesis, one strategy to manipulate the downstream DA transmission in the brain reward circuitry is to target (block) DA receptors (D1, D2, D3, D4) for the treatment of SUD and another is to target the DAT specifically for the treatment of psychostimulant use disorder (PSUD) (Newman et al. 2021). The D3R is a major focus in the former strategy (Galaj et al. 2020b), which will be addressed extensively below, while developing various DAT inhibitors, particularly atypical DAT inhibitors, is the major focus in the latter strategy, which has recently been reviewed extensively elsewhere (Hersey et al. 2021; Tanda et al. 2021).
2.1.2 Unique Profiles of D3R
There are five G protein-coupled DA receptor subtypes identified, which are classified into D1-like (D1, D5) and D2-like (D2, D3, D4) groups based on their homology, pharmacology, and intracellular signaling properties (Beaulieu et al. 2015; Beaulieu and Gainetdinov 2011; Martel and Gatti McArthur 2020; Missale et al. 1998). The D1 and D5 receptors share 80% homology of their seven transmembrane domains, while the D2 receptors share 75% homology of their protein structure with D3R and only 53% homology with D4 receptors. The main structural differences among DA receptors are differences in size of the third intracellular loop connecting transmembrane domains and of the carboxyl-terminal intracellular segment. D1-like receptors stimulate intracellular cAMP signaling pathway through Gαs G-proteins, whereas D2-like receptors inhibit DA signaling through Gαi/o G-proteins.
2.1.2.1 High D3R Binding Affinity to DA
Each DA receptor binds endogenous ligand DA with affinities in the nM range. The D2-like receptor subtypes bind DA with higher affinities than the D1-like family with D3R binding DA with the highest affinity (Missale et al. 1998). Therefore, D3R has been described as being a major receptor underlying DA transmission in the brain reward system. Given that basal levels of extracellular DA (5–10 nM) and synaptic DA (~50 nM) (He and Shippenberg 2000; Ross 1991), it is expected that a fraction of D3R is constitutively activated, thus playing an essential role in both tonic and phasic DA signaling.
2.1.2.2 Restricted D3R Distribution
The human D3R was first cloned in 1990 (Giros et al. 1990), which was followed by the cloning and characterization of the rat D3R (Sokoloff et al. 1990). Since then, various radiolabeled ligands such as [3H]7-OH-DPAT), [3H]PD-128907, and [125I]epidepride were developed (Hall et al. 1996; Herroelen et al. 1994; Murray et al. 1994). A variety of techniques such as quantitative autoradiography and in situ mRNA hybridization have been used to map the distribution of D3R in the brain and periphery.
Using a polyclonal D3R antibody, Diaz et al. detected dense D3R-immunostaining mainly in the islands of Calleja and mammillary bodies, moderate to low signals in the shell of NAc, frontoparietal cortex, SNc, VTA, and lobules 9 and 10 of the cerebellum, but very low or no signal in other rat brain regions such as DST (Diaz et al. 2000; Lammers et al. 2000). However, due to the concerns of DA receptor antibody specificity, autoradiography and PET imaging have become the major techniques to map D3R distributions in the brain (Diaz et al. 2000; Lammers et al. 2000). Consistent with the findings by immunostaining, an autoradiogram study with [125I]7-OH-PIPAT also showed the restricted distributions of D3R in the rat brain with the highest level of D3R expression in the islands of Calleja, ventromedial shell of NAc, VP, and SN (Stanwood et al. 2000). Such a restricted distribution of D3R expression was also found in other species, such as mouse, guinea pig, and rabbit (Diaz et al. 1994, 1995; Levant 1998). Among these four species the mouse shows high density D3R expression in hippocampus and low expression in the frontal cortex (Levant 1998).
Subsequent PET imaging studies showed that [11C](+)-PHNO, a mixed D2R/D3R agonist (Narendran et al. 2006; Seeman et al. 2005) produces preferential uptake in the ventral pallidum and globus pallidus of humans and baboons in contrast to radiolabeled D2R antagonists (such as [11C]raclopride) or other D2R agonists (such as [11C]NPA) that show preferential uptake in the dorsal striatum (Gallezot et al. 2012; Ginovart et al. 2007; Graff-Guerrero et al. 2008; Kiss et al. 2011; Narendran et al. 2009; Rabiner and Laruelle 2010; Rabiner et al. 2009). The specific binding of [11C](+)-PHNO in the globus pallidus of baboons was inhibited by the partial D3R agonist BP-897 (N-[4-[4-(2-methoxyphenyl)-1-piperazinyl]butyl]naphthalene-2-carboxamide) suggesting that the D3R contribution to the specific binding signal of [11C](+)-PHNO is higher than that of [11C]raclopride (Doot et al. 2019).
The distribution of the D3R gene in rats and mice is well established. Figure 2 shows the overall brain distribution of D1, D2, and D3 transcripts in mice using in situ hybridization (ISH) assays. D1R mRNA is highly expressed in the basal ganglia, including the DST, NAc, and olfactory tubercle (Monsma et al. 1990). D2R mRNA displays similar regional distribution as the one of D1R mRNA (Gerfen et al. 1990). In addition, D2R mRNAs were found in dopaminergic cell bodies within the SNc and VTA (Bunzow et al. 1988). In contrast, the highest level of D3R mRNA was seen in the islands of Calleja, the NAc, hippocampus (Hipp), and insular cortex in rats (Fig. 3) (Bouthenet et al. 1991; Landwehrmeyer et al. 1993a; Sokoloff et al. 1990). The levels of D4R and D5R mRNA in the striatum are very low (Meador-Woodruff et al. 1992; O'Malley et al. 1992).

D3 mRNA expression in rat brain as assessed by ISH at the level of the NAc (up panels) and thalamus (lower panel). DST dorsal striatum, NAc nucleus accumbens, S1 primary sensory cortex, CPu caudate putamen, LGP lateral globus pallidus, MGP medial globus pallidus. From the public (NIH) database at: https://www.ncbi.nlm.nih.gov/probe/docs/projgensat/

RNAscope in situ hybridization results, illustrating that low density D3R mRNA is expressed in a subpopulation of dopaminergic neurons in the VTA (a, red—DAT-positive dopamine neurons; yellow—D3 mRNA signal; blue—DAPI-labeled nuclei), while high density D3R mRNA is expressed in NAc D1-MSNs (red) (b, red—D1-MSNs; yellow—D3 mRNA signal; blue—DAPI-labeled nuclei) and insular glutamate neurons (red) in mice (c, red—VgluT1-positive glutamate neurons; yellow—D3 mRNA signal; blue—DAPI-labeled nuclei) (Xi ZX et al., unpublished data)
In the post-mortem human brain, D3R mRNA expression was found on principal cells of the prefrontal cortex (PFC) and abundant in basal ganglia, but low level of expression was also evident in cingulate cortex and subcortical regions (including thalamus, Amy, locus coeruleus, raphe nuclei, etc.) (Gurevich and Joyce 1999; Landwehrmeyer et al. 1993b; Larson and Ariano 1995; Suzuki et al. 1998). In contrast to the rat, in human no D3R mRNA was detected in the VTA (Gurevich and Joyce 1999). Combination of [11C]-(+)-PHNO PET imaging results with brain D3R and D2R mRNA expression demonstrated highest level of [11C]-(+)-PHNO binding in the VP, globus pallidus, NAc. There is strong correlation between [11C]-(+)-PHNO binding and D3R mRNA, but not D2R mRNA, expression (Komorowski et al. 2020). In addition, using [3H]-PD128907, high densities of D3R binding were also observed in the superficial layers of the dorsal horn at cervical and lumbar levels followed by the pars centralis and dorsal horn (Levant and McCarson 2001).
2.1.2.3 Cellular Distribution of D3R
To understand which neural substrates underlie D3R function, it is critical to understand which types of cells express D3R. Using a polyclonal D3R antibody, Diaz et al. detected D3R-immunostaining in all tyrosine hydroxylase (TH)-positive DA neurons in the VTA, SNc, and A8 retrorubral fields, suggesting that D3R may act as a functional autoreceptor regulating DA neuron activity and DA release from their projection terminals (Diaz et al. 2000). Using double-staining ISH methods to examine D3 mRNA expression in the NAc (Le Moine and Bloch 1996), the D3R mRNA is detected in a subpopulation of D1- or D2-expressing medium-spiny neurons (MSN)s, and also, in substance P- or enkephalin-expressing neurons, implying that DA may act on each population of postsynaptic neurons in the NAc, producing DA-dependent effects. Given that commercially available DA receptor antibodies (including anti-D3 antibodies) display poor receptor specificity (Bodei et al. 2009) and classical ISH images display poor mRNA signal resolution at cellular levels, the above findings regarding the D3R cellular distribution could not be conclusive. Recently, using more specific and sensitive fluorescent D3R reporter mice, Clarkson and colleagues identified D3R signal in a small population of pyramidal neurons in the layer 5 of the PFC (Clarkson et al. 2017). We have recently used a highly sensitive and specific RNAscope ISH assays to detect the cellular distribution of D3R mRNA. We found that D3R mRNA is expressed only in a subpopulation of dopamine neurons in the VTA, while high density D3R mRNA is detected in dopamine D1R-expressing medium-spiny neurons (D1-MSNs) in the NAc-shell and vesicular glutamate transporter 1 (VgluT1)-positive glutamate neurons in the insular cortex of mice (Fig. 3), indicating that such new advanced techniques are highly valuable in identifying the cellular distributions of D3R genes or protein in different brain regions and tissues.
2.2 Relationship to Neural Targets and Therapeutic Potential
The exact loci and neural substrates that D3R antagonists/partial agonists target in the brain are not fully understood. Based on the restricted regional and cellular distributions of D3R described above, it is reasonable to predict that both presynaptic and postsynaptic D3R mechanisms may underlie the therapeutic effects of D3R antagonists/partial agonists in animal models of drug addiction (Fig. 4).

Schematic diagram of the mesolimbic DA projection system in rat brain, illustrating where high densities of D3R binding or mRNA are found or upregulated by chronic use of psychostimulants or opioids, which may constitute important targets that D3R antagonists or partial agonists act
2.2.1 Presynaptic D3R Mechanism
As stated above, both systemic administration of psychostimulants and opioids produce an initial increase in extracellular DA in the NAc and DST, whereas prolonged withdrawal or abstinence seems to trigger a “hypodopaminergic state” in the mesolimbic DA system, which is closely associated with craving and relapse to drug seeking (Blum et al. 2021a, b; Luscher and Pascoli 2021; Salin et al. 2021; Samaha et al. 2021; Sanna et al. 2021). One may therefore hypothesize that normalization of decreased DA transmission in the reward circuits may decrease drug craving and relapse to drug-seeking behavior. Growing evidence indicates that activation of D3R by the agonist PD-128907 inhibits DA release in the NAc and PFC possibly via presynaptic D3 autoreceptors on DA terminals (Millan et al. 2010), while D3R antagonists/partial agonists produce an increase in extracellular DA levels in the NAc, PFC, or ventral hippocampus possibly by presynaptic D3 autoreceptor disinhibition (Gobert et al. 1996; Huang et al. 2019; Lacroix et al. 2003; Millan et al. 2000). Thus, we propose that blockade of presynaptic D3R may in part normalize (restore) the hypodopaminergic state, and therefore, contribute to the therapeutic effects of D3R antagonists in preventing relapse to drug seeking after abstinence.
In addition, previous studies have shown that D1R or D2R agonism improves various aspects of cognitive performance in rodents as well as primates (Cai and Arnsten 1997; Marino and Levy 2019; Nakako et al. 2013). Accordingly, elevated extracellular DA after D3R antagonism may in turn stimulate D1R and D2R in both the NAc and PFC (Clarkson et al. 2017), producing pro-cognitive and pro-social behavioral changes. Thus, indirect D1R or D2R activation following presynaptic D3R antagonism may also in part contribute to D3R antagonists’ effects on cognition and motivation for drug-seeking behavior.
2.2.2 Postsynaptic D3R Mechanisms
In addition to the presynaptic D3R mechanism, blockade of postsynaptic D3R in the brain reward circuits may also underlie D3R antagonists’ action in reducing drug-taking and drug-seeking behavior.
2.2.2.1 NAc D3R Mechanism
Recent optogenetic studies indicate that activation of D1-MSNs in the NAc is associated with positive reinforcement, while activation of D2-MSNs is mostly associated with aversion (Kravitz et al. 2012; Lobo et al. 2010). Accordingly, it was hypothesized that the acute rewarding effects of psychostimulants or opioids are most likely mediated by activation of D1-MSNs via Gs-coupled D1R and inhibition of D2-MSNs via Gi-coupled D2R (Hikida et al. 2013; Kravitz et al. 2012; Smith et al. 2013; Yawata et al. 2012). As stated above (Fig. 3), D3R appears to be co-expressed mainly in D1-MSNs and less in D2-MSNs in the NAc-shell. Thus, we hypothesize that blockade of D3R in D1-MSNs would cause D1-MSN disinhibition and increase their excitability, which may normalize the hypodopaminergic state observed in chronic drug users, and therefore, decrease craving and motivation for drug-seeking behavior. In contrast, blockade of D3R in D2-MSNs would also disinhibit D2-MSNs and increase their excitability, and therefore, potentiate D2-MSN-mediated aversive effects. However, D3R expression in D2-MSNs is very low (Fig. 3), and therefore, D3R antagonist action in D2-MSNs should be minimal. Thus, the final net effect of D3R antagonism on brain reward function would be mediated mainly by blockade of D3R on postsynaptic D1-MSNs. Furthermore, blockade of D3R directly counteracts DA action after acute drug administration.
2.2.2.2 VP D3R Mechanism
The VP is a key hub within the reward system that mediates drug-taking and drug-seeking behaviors (Creed et al. 2016; Heinsbroek et al. 2020). Previous studies have shown that drugs of abuse enhance DA release within the VP and produce reinforcing effects (Panagis and Spyraki 1996). As stated above, high density D3R is expressed in the VP. Thus, blockade of VP D3R may also in part explain how D3R antagonists attenuate the rewarding effects produced by psychostimulants or opioids under certain experimental conditions. In addition, Pribiag et al. (2021) recently reported that 2 weeks of forced abstinence from cocaine self-administration upregulates D3R expression in VP GABAergic neurons, which project to the lateral habenula (LHb). Activation of D3R in VP GABAergic neurons underlie contextual cue-induced cocaine-seeking behavior in rats via a VP-LHb circuit (Campbell and Lobo 2021; Pribiag et al. 2021). In the LHb, glutamatergic neurons project to the RMTg, where GABAergic neurons project to DA neurons in the VTA and functionally modulate DA neuron activity (Jhou et al. 2009). These findings suggest that D3R in VP GABA neurons may regulate VTA DA neuron activity via a VP-LHb-RMTg-VTA circuit, and therefore, modulate cocaine-seeking behavior. Accordingly, blockade of VP D3R may also explain how D3R antagonists attenuate drug- or cue-induced drug-seeking behavior.
2.2.2.3 PFC D3R Mechanism
An early study indicated low levels of D3R are expressed in the PFC (Larson and Ariano 1995), suggesting possible involvement of cortical D3R in the cognitive effects of D3R ligands (Nakajima et al. 2013). This is supported by a recent finding that a unique population of PFC principal neuron in layer 5 expresses D3R (Clarkson et al. 2017). Notably, such D3R-expressing cortical neurons lack expression of D1 or D2 receptor and activation of D3R in PFC neurons inhibits low-voltage-activated CaV3.2 calcium channels at the axon initial segment, causing a reduction in action potential (AP) firing. Importantly, the D3R-expressing PFC neurons send axonal projections to the contralateral cortex, NAc, and basolateral amygdala (BLA), thereby possibly modulating drug-taking and drug-seeking behavior via PFC-NAc and PFC-BLA circuits (Clarkson et al. 2017).
2.2.2.4 Insula D3R Mechanism
The insula is another node involved in the networks underlying SUD (Naqvi and Bechara 2009). The general notion emerging from recent studies is that drug craving and cue-induced urges could be complex interoceptive emotions that are processed in the insular cortex, particularly in its anterior part. Several studies in humans and experimental animals indicated insula lesions diminished drug-seeking behaviors (Contreras et al. 2007; Naqvi et al. 2007), an effect that was even more pronounced by combined damage of the insula and putamen (Gaznick et al. 2014), suggesting an abnormal connectivity of these two regions in SUD. This is further supported by a recent finding that alcoholism is associated with a loss of insula gray matter (Senatorov et al. 2015), and decreased functional connectivity between the NAc and insula was observed in alcohol-dependent rats (Scuppa et al. 2020) and aversion-resistant alcohol intake in rodents (Seif et al. 2013; Sullivan et al. 2013). The mechanisms and significance of this action remain unclear. Given that a hypodopaminergic state within the brain reward circuitry is a hallmark of an addicted state and that D3R mRNA is detected in presynaptic DA neurons in the VTA and postsynaptic glutamate neurons in the insular cortex (Fig. 3C), we predict that presynaptic D3R antagonism in the insula may also contribute to the normalization of the hypodopaminergic status, and therefore, improve the insula-NAc functional connectivity. Similarly, blockade of postsynaptic D3R in the insula would also counteract the action produced by elevated DA after acute cocaine or opioid administration.
2.2.2.5 Amygdala (Amy) D3R Mechanism
In addition to the above brain regions, the Amy is also involved in drug-taking and drug-seeking behavior. The Amy receives dopaminergic innervation (Asan 1997) and has high D3R expression (Herroelen et al. 1994; Murray et al. 1994; Suzuki et al. 1998; Tupala et al. 2001). Cocaine injections or exposure to cocaine-associated cues activates the Amy in animals and humans as assessed by neuroimaging and c-fos expression studies (Grant et al. 1996; Neisewander et al. 2000) and increase D3R expression in the Amy (Guerrero-Bautista et al. 2021). Amy lesions or microinjections of D3R receptor antagonists inhibit cocaine self-administration and contextual cue-induced cocaine seeking (McGregor and Roberts 1993; Xi et al. 2013). Microinjections of psychostimulants into the central amygdala (CeA), but not the BLA, produce a conditioned place preference, whereas selective lesions of the BLA do not affect cocaine self-administration (Meil and See 1997; Yun and Fields 2003), suggesting dissociable roles for the CeA and BLA in cocaine-related behavior (Li et al. 2008; Lu et al. 2005; O'Dell et al. 1999). The D3R expression and function in the CeA vs. BLA in psychostimulants or opioid action remain to be determined.
2.3 D3R Neuroadaptations Due to SUD
A growing body of evidence suggests that aberrant D3R signaling contributes to several brain disorders. Consequently, D3R has emerged as a potential therapeutic target in the treatment of major neurological and neuropsychiatric disorders such as schizophrenia, Parkinson’s disease, and SUD. However, the mechanisms underlying D3R signaling are poorly understood, either in healthy or diseased brain. Therefore, unraveling the unknown downstream signaling pathways activated by D3R in both the healthy and the diseased brain is likely to reveal new therapeutic strategies toward DA-associated disorders.
2.3.1 Development Changes in D3R Expression
Brain D3R mRNA is detected early in development and continually expressed during the postnatal period (Araki et al. 2007; Gurevich et al. 1999; Levant 1997). ISH assays indicate that D3R mRNA expression is restricted, almost entirely to the ventricular neuroepithelium during the whole prenatal ontogeny and that the neuronal expression of the D3R appears during the first postnatal week (after the DA innervation) (Diaz et al. 1997; Stanwood et al. 1997), suggesting that the increase in D3R mRNA expression in adults is likely to reflect functional changes in the dopaminergic innervation of the ventral striatum (Shafer and Levant 1998). This is supported by the findings that a lesion of DA neurons, impairment of axonal transport, or reduction of DA neuron firing causes a reduction in D3R gene expression (Levesque et al. 1995) and repeated treatment with levodopa rescued D3R mRNA expression in the NAc and induced an ectopic expression within the dorsal striatum (Bordet et al. 1997).
2.3.2 Tolerance and Desensitization after D3R Activation
DA induces only a marginal fraction of D3R to translocate from cell surface to intracellular vesicles, in stark contrast to D2R (Kim et al. 2001; Min et al. 2013), suggesting that D3R undergoes limited agonist-induced internalization. However, recent studies indicate that D3R agonists are able to induce D3R desensitization and internalization (Xu et al. 2019) via multiple intracellular signal mechanisms, including protein kinase C (PKC)- and Ca2+/calmodulin-dependent protein kinase II (CaMKII). Desensitization may occur at homodimeric and heterodimeric D3R. For example, D1R-D3R heterodimers can be internalized in response to the paired stimulation of both D1R and D3R via a β-arrestin-dependent mechanism in human embryonic kidney 293 cells (Fiorentini et al. 2008; Westrich et al. 2010). PKC-mediated phosphorylation of D3R can also induce clathrin-mediated D3R endocytosis and lysosomal D3R degradation (Zhang et al. 2016b). CaMKII-mediated D3R desensitization is intracellular Ca++-dependent, and therefore, is associated with neuronal activity (Liu et al. 2009). Palmitoylation is another post-translational modification that can regulate D3R activity. Palmitoylation is essential for cell surface expression, PKC-mediated endocytosis, and agonist-induced tolerance of D3R (Zhang et al. 2016a). Compared with D2R, D3R undergoes a more extensive palmitoylation on its cysteine residues at the carboxyl terminus tail.
2.3.3 Neuroadaptations after Exposure to Drugs of Abuse
In vitro and in vivo studies in experimental animals suggest that drugs of abuse may cause D3R signaling abnormalities. In vitro, cocaine increases dendritic arborization and soma area in cultured dopaminergic neurons from mouse via D3R-dependent activation of ERK and Akt (Collo et al. 2012). In rats, nicotine upregulates D3R, but reduces D3nf mRNA levels in the NAc, and therefore, increasing the D3R/D3nf ratio (Smith et al. 2015). In humans, chronic drug use induces long-lasting neuroadaptations in D3R expression, although some of the findings are conflicting (Richtand 2006). PET imaging studies with the D3R-preferring radioligand [11C](+)PHNO have shown higher number of available D3R in the SN, hypothalamus, and Amy of patients who are addicted to cocaine, compared with healthy controls (Matuskey et al. 2014). Notably, SN D3R levels correlated with years of cocaine use. Consistent with this finding, a six-fold increase in D3R mRNA levels was found in the NAc of cocaine overdose victims, as compared with age-matched and drug-free control subjects (Segal et al. 1997). Similarly, increased [11C](+)PHNO binding is also observed in the SN of methamphetamine users (Boileau et al. 2016), and in the hypothalamus of alcohol-dependent patients (Erritzoe et al. 2014). Furthermore, the functionally enhanced D3R-Gly-9 variant was associated with the development of early-onset heroin dependence in a Chinese population (Kuo et al. 2014).
The neural mechanisms underlying D3R upregulation after chronic drug abuse are unclear. As stated above, almost all drugs of abuse increase extracellular DA and subjects with chronic drug use display hypodopaminergic states in the mesolimbic system (Leyton and Vezina 2014; Luscher and Pascoli 2021; Ron and Jurd 2005; Samaha et al. 2021). These findings suggest that the changes in D3R signaling (desensitization vs. upregulation) could be adaptative or compensatory responses to changes in extracellular DA. This is supported by the finding that DA depletion induces compensatory increases in the number and the affinity of D3R to endogenous DA or exogenous DA receptor ligands (Avalos-Fuentes et al. 2015; Prieto et al. 2011). A better understanding of how drugs of abuse alter D3R activity may uncover pathophysiologic mechanisms underlying SUD and lead to discovery of novel molecular targets for pharmacotherapeutic treatment.
2.4 Relationship of D3R to Pain
Previous studies have explored the role of DA receptors in opioid analgesia and tolerance. The majority focused on the D2R and showed that nonspecific D2-like receptor ligands (agonists or antagonists) are able to prevent morphine tolerance (Dai et al. 2016; Gomaa et al. 1989; Ozdemir et al. 2013). To dissect the role of different D2-like receptor subtypes in this action, the D3-preferring agonists 7-OH-DPAT and pramipexole were also tested. It was found that both the compounds can prevent tolerance to opioids (Cook et al. 2000; Rodgers et al. 2020; Zarrindast et al. 2002), suggesting that D3R mechanisms may be also involved in opioid analgesia. This is supported by our recent finding that both the highly selective D3R antagonists/partial agonists (VK4-116 and VK4-40) attenuate oxycodone self-administration and reinstatement to drug seeking, but without compromising oxycodone’s antinociceptive effects in rats (Jordan et al. 2019b; You et al. 2019). In fact, a potentiation effect on oxycodone analgesia was observed at higher doses.
However, the neural mechanisms underlying this D3R modulation of opioid analgesia are poorly understood. Early studies indicate that intra-NAc or VTA microinjections of a DA receptor antagonist blocks noxious stimuli-induced antinociception (Altier and Stewart 1998; Gear et al. 1999), suggesting that the mesolimbic DA system could be one of the major loci that D3R antagonists modulate pain and opioid analgesia (Schmidt et al. 2002). In addition, the spinal cord could be another important location underlying DA and opioid interactions as DA (D1, D3) and MOR receptors are detected in the dorsal horn (Abbadie et al. 2001; Levant and McCarson 2001). This is further supported by the finding that genetic deletion of D3R in D3-mutant mice altered pain-associated responses and morphine-induced antinociception at the spinal cord (Brewer et al. 2014; Clemens and Hochman 2004; Keeler et al. 2012). Furthermore, considerable evidence suggests an interaction between the D1R and D3R or between D3R and MOR receptors. The D3R has been shown to colocalize with D1R or form D1-D3 heterodimers in the striatum (Fiorentini et al. 2008, 2010), which has been reported to modulate opioid analgesia and reward (Rodgers et al. 2019). In addition, D3R and MOR are colocalized with D1R in NAc D1-MSNs (Galaj et al. 2020a), suggesting the possible presence of functional D3-MOR heterodimers. Given that both D3R and MOR modulate intracellular adenylate cyclase and cAMP levels, it is suggested that the D3R and MOR interaction may occur at intracellular cAMP/PKA level (Zarrindast et al. 2002; Zhang et al. 2008, 2012).
2.5 Relationship to Other Comorbid Neuropsychiatric Disorders
There is significant comorbidity between neuropsychiatric and SUD, which may be particularly evident in women (Chander and McCaul 2003). Persons living with affective and anxiety disorders are more likely to use alcohol or drugs of abuse. Recognition for both psychiatric and SUD comorbidity is important for improving treatment outcomes for these co-occurring conditions.
SUD and major depressive disorder (MDD) are prevalent and frequently co-occur (Volkow 2004). Comorbidity between bipolar disorder (BPD) and SUD is also highly prevalent (Post and Kalivas 2013; Salloum and Brown 2017). Lifetime prevalence estimates of depression are 30 ~ 50% among persons with cocaine use disorder (CUD) (Conway et al. 2006). The presence of depressive symptoms is associated with poorer outcomes in CUD (Leventhal et al. 2006; Raby et al. 2014). Anhedonia is a core symptom of MDD and characterized by reduced experiencing of pleasure. Anhedonia has been linked to DA dysfunction in the mesolimbic system (Der-Avakian and Markou 2012). In rodents, lower DA concentrations in the NAc have been associated with fewer attempts to work for rewards (Manduca et al. 2016). In humans, decreased DA response to psychostimulants, decreased availability of striatal D2/3 receptors, and increased availability of DA transporters have been observed and associated with a “reward deficiency” state in patients with MDD (Koob 2013). This hypodopaminergic state may in part explain such negative symptoms experienced during abstinence as dysphoria, anhedonia, and craving, which may lead to higher reward pursuits and motivation for using illicit drugs or precipitating relapse. Accordingly, prescription stimulants such as dextroamphetamine have been proposed to address such reward deficiency in a way similar as methadone for OUD (Angarita et al. 2021b) and antidepressants have been used for the treatment depression and SUD comorbidity (Zhou et al. 2015). However, a major concern with stimulants, such as amphetamine, is their abuse potential. An alternative strategy to minimize this potential risk involves the development and use of atypical DAT inhibitors (Newman et al. 2021). In addition, the D3R antagonists/partial agonist could be promising for the treatment of the CUD and MDD comorbidity (Keck et al. 2015; Newman et al. 2012) since blocking presynaptic D3R may facilitate DA release and normalize the hypodopaminergic status, while activation of postsynaptic DA receptors by DA or D3R partial agonists may not only mitigate withdrawal effects during abstinence but also improve dysphoria and anhedonia in patients with SUD and MDD comorbidity.
Anxiety disorders (AD) are characterized by excessive fear, anxiety, and related behavioral disturbances (Craske et al. 2017). Epidemiological studies revealed striking rates of co-occurring anxiety and SUD (Compton et al. 2007; Rogers et al. 2021). It is well documented that Amy directly modulates anxiety (Kalin et al. 2004; Lesscher et al. 2008). Early research emphasized a role of DA in the pathophysiology of anxiety (Taylor et al. 1982), which recently has been reinvigorated (Dedic et al. 2018; Kienast et al. 2008) as the Amy provides the main input to midbrain DA neurons (Fudge and Haber 2000). A recent study investigated the relationships between AD and brain D2/3 functional activity and functional connectivity. It was found that higher DA release in the Amy was associated with lower trait anxiety and lower cingulate–amygdala functional connectivity, suggesting that a negative relationship between DA functional activity and anxiety levels and a hypodopaminergic state may also exist in AD (Berry et al. 2019). Accordingly, we hypothesize here that D3R antagonists/partial agonists may also be useful for the treatment of SUD and AD comorbidity since blockade of presynaptic D3R may increase DA release and activation of postsynaptic D3R by DA or partial D3R agonist may normalize decreased DA transmission, thereby producing anxiolytic effects.
3 D3R Antagonists and Partial Agonists Currently under Preclinical Investigation for SUD
3.1 Past D3R Preferential and Selective Antagonists that Were Tested in Clinical Trials
To the best of our knowledge, there have been only a few selective D3R antagonists (GSK598809) or preferential D3R partial agonists (buspirone, cariprazine) that have been available clinically and only buspirone has been directly tested in a clinical study for CUD (Bergman et al. 2013).
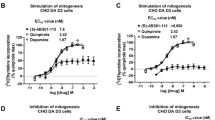
3.1.1 GSK598809
GSK598809 (Fig. 5) is a selective D3R antagonist with ~120-fold selectivity for D3R (Ki = 6.2 nM) over D2R (Ki = 740 nM) (Micheli et al. 2010; Searle et al. 2010). In a clinical study focusing on craving in smokers, a single dose of GSK598809 produced 72% to 89% D3R occupancy and transiently alleviated craving for cigarette smoking after overnight abstinence (Mugnaini et al. 2013). In addition, GSK598809 effectively reduced appetitive responses to food cues in overweight and obese individuals (Mogg et al. 2012; Nathan et al. 2012). In nonclinical studies in dogs and rats, GSK598809 was reported to increase blood pressure especially in the presence of cocaine (Appel et al. 2015), which dampened enthusiasm for conducting a clinical trial in patients with CUD.

Chemical structures of lead D3R antagonists/partial agonists
3.1.2 Buspirone
Buspirone is an FDA-approved medication for the treatment of anxiety. Its therapeutic effects are believed to be mediated mainly by its partial agonist action at 5-HT1A receptors Ki-High (19.2 nM) and Ki-Low (111 nM) (Noël et al. 2014). However, buspirone also binds to D3R (Ki = 98 nM) (Bergman et al. 2013; Kula et al. 1994) and it was therefore proposed but failed to be effective for a clinical population with CUD (Bergman et al. 2013; Newman et al. 2012). Paradoxically, clinical studies indicate that buspirone is effective for the treatment of anxiety in individuals with alcohol use disorder (Malec et al. 1996) but not in those with OUD (McRae et al. 2004). Buspirone is also ineffective in prevention of relapse for cigarette smoking (Schneider et al. 1996) or in reductions of drug (cocaine, cannabis) and alcohol consumption (Malec et al. 1996; McRae-Clark et al. 2015; Winhusen et al. 2014). The mechanisms underlying these negative findings are unclear; however, this may be related to its non-selectivity and low occupancy at the D3R in human brain (Le Foll et al. 2016) at doses used clinically. So far, there has been no clinical trial to evaluate the effectiveness of buspirone in controlling opioid intake and relapse. However, buspirone has been shown to reduce withdrawal symptoms in heroin addicted individuals (Buydens-Branchey et al. 2005; Rose et al. 2003).
3.1.3 Cariprazine
Cariprazine (Fig. 5; RGH-188) is a D3R-preferring partial agonist (Citrome 2013; Gyertyan et al. 2007; Kiss et al. 2010), showing approximately ten-fold higher affinity for human D3R (pKi = 10.07; Ki = 0.085 nM) over human D2L (pKi = 9.16; Ki = 0.49 nM) and D2S receptors (pKi = 9.31; Ki = 0.69 nM). In addition, it is an antagonist with high affinity at human 5-HT2B receptors (pKi = 9.24; Ki = 0.58 nM). Cariprazine has been recently approved for the treatment of schizophrenia and manic or mixed episodes associated with bipolar I disorder, by the FDA. Preclinical studies indicate that cariprazine is able to reduce the rewarding effect of cocaine and relapse to cocaine-seeking behavior with half maximal effective dose (ED50 values of 0.2 mg/kg) (Gyertyan et al. 2007; Roman et al. 2013). In addition, a recent case report indicates that cariprazine is able to improve both psychotic and addictive symptoms in subjects with persistent methamphetamine use (Ricci et al. 2022). Notably, a patient reported an abrupt decrease in substance craving and use and an improvement in positive and negative psychotic symptoms. These findings suggest that cariprazine deserves further research as an antipsychotic candidate for the treatment of SUD with bipolar disorder. Indeed, a new clinical trial has recently begun to assess the effectiveness of cariprazine for treatment of comorbid CUD and OUD (Kampman 2021).
3.2 New and Promising D3R Selective Antagonists/Partial Agonists for SUD
Although developing highly selective D3R antagonists/partial agonists with improved bioavailability and pharmacokinetics profiles is challenging (Heidbreder and Newman 2010; Keck et al. 2015; Leggio et al. 2016; Pich and Collo 2015), significant progress in medicinal chemistry has been made. High D3R selectivity maybe essential to minimize D2R-mediated extrapyramidal and motor side effects that would undoubtedly reduce compliance. Improved bioavailability and pharmacokinetics (PK) profiles are also critical to future translational studies toward the development of novel treatment modalities. Several D3R antagonists/partial agonists have been developed and tested in experimental animal models and have been recently reviewed systematically elsewhere (Galaj et al. 2020b; Keck et al. 2015; Newman et al. 2021). Although there are several groups who are continuing to pursue this class of agents toward application to SUD (Ewing et al. 2021; Lv et al. 2019; Thomsen et al. 2017), herein we highlight just a few promising D3R antagonists/partial agonists for the treatment of OUD and possibly PSUD based on their favorable receptor binding and PK profiles and their pharmacological efficacy in reducing drug-taking and drug-seeking behavior (Fig. 5).
3.2.1 (±)-VK4-116 and its R-Enantiomer
Racemic (±)-VK4-116 is a highly selective D3R antagonist with ~1700-fold binding selectivity for D3R (Ki = 6.84 nM) over D2R (Ki = 11,400 nM) and is also highly selective across >70 receptors, enzymes, and transporters (Kumar et al. 2016, NIDA Treatment Discovery Program). It also showed very high metabolic stability and half-life (t1/2 = 250, 116 and 102 min in rat, human, and monkey liver microsomes, respectively) (Kumar et al. 2016). (±)-VK4-116 displayed excellent brain penetration, after oral administration (You et al. 2019) and thus was identified as a lead compound with translational potential.
Preclinical studies in rodents with (±)-VK4-116 showed promising results. For example, pretreatment with (±)-VK4-116 dose-dependently reduced the acquisition of oxycodone-induced CPP, oxycodone self-administration under FR2 and PR reinforcement schedules in rats (You et al. 2019). In addition, pretreatment with (±)-VK4-116 decreased the escalation of oxycodone self-administration in male and female rats with extended access to drug (de Guglielmo et al. 2019), facilitated extinction of drug seeking, and reduced oxycodone-primed reinstatement of drug seeking in rats (You et al. 2019). It also reduced oxycodone-induced hyperactivity and repeated oxycodone-induced locomotor sensitization in mice (Kumar et al. 2016). Furthermore, pretreatment with (±)VK4-116 dose-dependently reduced naloxone-precipitated conditioned place aversion in rats (You et al. 2019) and withdrawal-induced hyperalgesia and irritability-like behaviors (de Guglielmo et al. 2019), suggesting that (±)-VK4-116 has the ability to attenuate opioid withdrawal symptoms, a critical aspect for therapeutic utility (Koob 2021). Notably, (±)-VK4-116 has been shown to potentiate the analgesic effects of oxycodone, as assessed in a hot plate assay (You et al. 2019). This unique characteristic of (±)-VK4-116 not only supports its potential utility in the treatment of opioid use disorders (OUD) but also suggests its coadministration with prescription opioids in pain management as lower doses of prescription opioids could be used to mitigate pain when combined with (±)-VK4-116, and thus reduce the risk of abuse and the development of dependence. Of note, R-VK4-116 (Fig. 5) is also a highly D3R-selective antagonist (Shaik et al. 2019) and is currently under development for treatment of OUD.
3.2.2 (±)-VK4-40 and its Enantiomers
Racemic (±)-VK4-40 (Fig. 5) is another newly developed and low efficacy D3R partial agonist with high affinity for D3R (Ki = 0.36 nM) over D2R (Ki = 151 nM) and ~ 400-fold selectivity (Kumar et al. 2016). The R-enantiomer (R-VK4-40) is a D3R antagonist, whereas the S-enantiomer is a partial agonist, like the racemate. R-VK4–40 displays high affinity for D3R (Ki = 0.29 nM) over D2R (Ki = 75.8 nM) and 261-fold selectivity for D3R over D2R (Shaik et al. 2019). The S-enantiomer is equally D3R-selective. (±)-VK4–40 was shown not only to attenuate cocaine-primed reinstatement and cocaine-enhanced brain-stimulation reward maintained by optical stimulation of VTA DA neurons, but also to reduce cocaine self-administration across multiple cocaine doses under an FR2 schedule (Jordan et al. 2020), suggesting that (±)-VK4-40 is a potential D3R partial agonist candidate for the treatment for PSUD.
R-VK4–40 is metabolically stable in the presence of NADPH with 86% remaining level in the plasma over 1 h and showed excellent brain penetration after oral administration in rats (Jordan et al. 2019b). In animal models of OUD, R-VK4-40 dose-dependently inhibited oxycodone self-administration maintained under FR1 and PR schedules of reinforcement in rats and attenuated oxycodone-enhanced ICSS maintained by optical activation of VTA DA neurons in mice (Jordan et al. 2019b), suggesting that R-VK4-40 can reduce the rewarding effects of opioids. Notably, S-VK4-40 displayed similar pharmacological efficacy, as R-VK4-40, in attenuation of cocaine-enhanced brain-stimulation reward in the optical intracranial self-stimulation (ICSS) assays (Galaj et al. 2020b; Newman et al. 2021). Pretreatment with R-VK4–40 did not compromise the analgesic effects of oxycodone and in fact, it increased latencies to emission of thermal nociceptive response, shifting the oxycodone-dose response curve upward (Jordan et al. 2019b), suggesting an additive analgesic effect to oxycodone. R-VK4-40 alone also produced analgesic effects without affecting locomotor activity or performance on the rotarod test (Jordan et al. 2019b). The neural mechanisms underlying R-VK4-40-induced analgesic effects are yet to be determined. A possible interaction between D3R and MOR may occur in the dorsal horn of the spinal cord (Abbadie et al. 2002; Levant and McCarson 2001; Ray and Wadhwa 2004), which may in part underlie the potentiation of opioid analgesia after D3R antagonism (Jordan et al. 2019b).
3.2.3 Dual-Target Mu Opioid Receptor (MOR): D3R Partial Agonists
The recognition of D3R antagonism/partial agonism as an alternative and nonopioid approach for treatment of OUD without compromising opioid analgesia, combined with the possible presence of D3R-MOR heterodimers prompted us to develop a novel class of dual-target ligands with MOR partial agonist and D3R antagonist/partial agonist profiles (Bonifazi et al. 2021). The idea was that these molecules would, on the one hand, block D3R, mitigating the reinforcing effects of opioids as reported previously (de Guglielmo et al. 2019; Jordan et al. 2019a; Kumar et al. 2016; You et al. 2017, 2019), while, on the other hand, partially activate MORs, producing additive or synergistic effects on opioid analgesia as D3R antagonism potentiates opioid analgesia. This drug design may lead to the development of safer dual target drugs, bridging the most promising pharmacological effects of two classes of molecules/targets previously developed independently.
3.3 Potential Challenges: Cardiovascular Toxicity in the Presence of Cocaine
Although D3R has long been a focus of medication development for addiction, translational potential of D3R-targeted ligands to clinical settings has, to date, been limited. One potential safety concern relates to cardiovascular effects after systemic administration. This is based on the finding that the D3R, in addition to their CNS expression, are also found in the kidney, regulating blood pressure. It was reported that blockade of peripheral D3R may cause sodium retention and possibly hypertension by antagonizing the inhibitory effects of DA on sodium transport (Zeng et al. 2004, 2008). Such effects were observed in mice with genetic deletion of D3R that developed elevated systolic blood pressure and diastolic hypertension (Jose et al. 1997). In addition, two older D3R antagonists SB277011A and GSK598809 were reported to produce an increase in blood pressure in dogs and rats, particularly in the presence of cocaine (Appel et al. 2015).
To further address this issue, we have recently examined the cardiovascular effects of the novel D3R compounds R-VK4-116 and R-VK4-40 in comparison with SB-277011A and L-741,626 (a selective D2R antagonist) as controls. In this study, we found that neither R-VK4-116 nor R-VK4-40 exhibited adverse cardiovascular effects (Jordan et al. 2019b), while both SB277011A and L-741,626 did. In rats implanted with telemetric devices, cocaine or oxycodone produced a small increase in blood pressure, heart rate, body temperature, and locomotor activity, while R-VK4-116 produced a reduction in body temperature when administered alone (Jordan et al. 2019b). However, pretreatment with R-VK4-116 significantly reduced oxycodone-induced increases in body temperature and blood pressure. Similarly, cocaine-induced increases in blood pressure and heart rate were also attenuated by R-VK4-116 (Jordan et al. 2019b). Moreover, R-VK4-40 also lacks these adverse cardiovascular effects. R-VK4-40 alone reduced blood pressure and heart rate in rats, while pretreatment with R-VK4-40 attenuated oxycodone-induced increases in blood pressure and oxycodone or cocaine-induced increases in heart rate and body temperature (Jordan et al. 2019b). Greater selectivity for D3R over other receptors (e.g., D1, D2, or 5-HT receptors) could be an important reason why R-VK4-116 and R-VK4-40 do not share the cardiovascular effects of other older D3R antagonists (SB-277011A and GSK598809) since many other receptors also regulate cardiovascular function (Alves et al. 2019; Cuevas et al. 2013; Yang et al. 2021). Nevertheless, these unique characteristics make both R-VK4-116 and R-VK4-40 attractive lead candidates in translational medicine for OUD and PSUD.
4 Perspective on Clinical Application as Treatments for SUD
4.1 Limitations and Advances in the Translational Value of Animal Models of SUD
Based upon their favorable preclinical safety profiles and overwhelming evidence of efficacy in animal models of reinstatement to drug-seeking behavior, selective D3R antagonists would be expected to reduce relapse to drug-, cue-, and stress-driven consumption post-abstinence and to produce some pro-cognitive effects. Before discussing potential clinical applications of selective D3R antagonists, one must first recognize the inherent limitations of preclinical models, hence limitations in the translational value they carry to clinical research.
To mimic real-world situations, drug delivery should be active (i.e., the subject must have full control over drug delivery), dose-response effects should be systematically observed, and drug exposure should be chronic or sub-chronic rather than acute. Several animal models are based on passive drug administration, systematic dose-response studies are inconsistent, and relatively low exposure to the drug is still observed. Furthermore, evaluations of potential pharmacotherapies for SUD in animal models most often use acute medication pretreatment paradigms. The predictive validity of those models would improve if they were to adopt protocols that include longer periods of medication treatment.
Most reinstatement models include extinction training. Although the latter isolates the influence of the conditioned stimuli on reinstatement from that of the context, response habit or stress, it reduces the face validity of the model given that humans rarely undergo extinction. Thus, models that assess drug seeking after a drug-free abstinence period as opposed to instrumental extinction training may better capture the nature of cue-induced relapse in humans. In the case of abstinence models, the fact that subjects do not undergo extinction training improves the face validity of this model but restricts data interpretation as drug-seeking may actually reflect response habit, novelty-induced stress, exploratory behavior, and/or innate motivation in addition to context-induced incentive motivation for drug.
Despite limitations, recent advances in nonclinical paradigms also show promise in modeling specific DSM-5 criteria for SUD. First, the concept of addiction as a progressive transition from a positive to a negative reinforcement process that drives the motivated behavior somehow reaffirms the importance of withdrawal in addiction. In that respect, measuring the degree of dysphoria produced by drug withdrawal is highly relevant. Second, the escalation in drug intake observed after long-access training and drug intake escalation mimic increased consumption over time. Third, the increased final ratios observed in progressive ratio paradigms appear to model the increased time and energy expended to obtain the drug. Fourth, the translational value of behavioral economics models to address the notion of discounting of delayed rewards may provide a readout of impulsivity and its related corollary of loss of control. Animal studies investigating the link between abnormal information processing in the mesocorticolimbic system and changes in responding for delayed or intermittent reinforcement are thus extremely valuable. Similarly, procedures examining choice responding under concurrent schedules of reinforcement may provide valuable insight into drug-seeking because the impact of competing reinforcers, and the work required to obtain each, can be measured simultaneously. Finally, significant work remains to be done to explore the mechanisms involved in animal models of craving and relapse and how to relate these mechanisms to vulnerability to SUD.
4.2 Key Translational Medicine Questions Relevant to Clinical Development
The translational value of nonclinical paradigms should be based upon a good understanding of what needs to be achieved for the target patient population and how pharmacodynamic (PD) data can be reliably linked to pharmacological kinetics (PK). This can only be done by answering the following questions: What exactly is the therapeutic indication for the D3R drug candidate (target product profile)? What is the proposed treatment response profile? What is the proposed clinical route and frequency of dosing? What is the expected efficacious concentration in a physiological fluid (i.e., concentration-effect relationship)? How long should that concentration be maintained to obtain the desired pharmacological response? What, if any, are the biological markers to monitor toxicity and/or therapeutic effects? Do changes in route or delivery rate alter the course of effect? Is response to treatment time-dependent (e.g., onset mechanism, disease progression)? If a valid PK/PD strategy is in place and if a strong PK/PD relationship is characterized, then efficacy and tolerability can be reliably predicted from the PK data and relevant scenarios can be simulated for decision-making or clinical purposes.
The availability of PET ligands as discussed in another chapter significantly strengthens this strategy by providing PK/PD combined with receptor occupancy (RO) estimates. In this case, the investigational D3R drug can be radiolabeled and its anatomical distribution and binding in the target tissue can be traced. Alternatively, one may assess the extent to which an unlabeled investigational D3R drug inhibits specific binding of a PET ligand with known receptor affinity. In the latter case, receptor occupancy at the target receptor can be quantified, thereby enabling a deep understanding of the relationship between dose, plasma concentration, occupancy, and pharmacodynamic or clinical effects of the investigational drug. This information, in turn, leads to invaluable information to optimally design clinical Phase 1 and Phase 2 proof of concept studies. For example, a randomized, double-blind, placebo-controlled, balanced two-way crossover study established a relationship between the occupancy of D3R in the brain using ex-vivo [125I]7OH-PIPAT autoradiography in the rat and [11C](+)-PHNO PET in human, the pharmacokinetic exposure to GSK598809, the ability of GSK598809 to reduce nicotine-seeking behavior using a conditioned place preference paradigm in rats, and the effect of GSK598809 on cigarette craving in smokers (Mugnaini et al. 2013). In this study, a single dose of GSK598809, giving 72–89% levels of D3R occupancy, transiently alleviated craving in smokers after overnight abstinence. GSK598809 also partially reversed the attentional bias of abstinent smokers as assessed by the Stroop test, a model of selective attention and cognitive flexibility.
The combination of PET and resting-state functional magnetic resonance imaging (fMRI) is another example of translational medicine efforts to support the development of new molecules targeting the D3R. [11C](+)-PHNO binding combined with fMRI showed that high midbrain D3R availability is associated with reduced functional connectivity between the orbitofrontal cortex and neuronal networks implicated in cognitive control and salience processing (Cole et al. 2012). Furthermore, using a rat model of chronic intermittent exposure (CIE) to alcohol (i.e., daily cycles of alcohol intoxication and withdrawal over weeks or months to mimic a pattern of alcohol use typically seen in populations with alcohol use disorder), it was shown that a history of alcohol use produced weaker functional connectivity between the insular and the cingulate cortex, but stronger connectivity between the insula and components of the mesolimbic DA system. The selective D3R antagonist, SB-277011A, however, was shown to normalize the aberrant connectivity induced by CIE to alcohol (Scuppa et al. 2020).
Altogether, these examples emphasize the importance of a thorough PK/PD/RO strategy to determine reliable dosing in humans, and/or to design combined Phase IIb/III trials allowing for more rapid progression of the medication toward regulatory approval. Specifically, they suggest that D3R are upregulated in persons living with SUD, an effect that is opposite to that found for D2R. Second, they show that greater dopaminergic transmission at the D3R may contribute to motivation to use drugs of abuse. Third, they suggest that drug craving and relapse to drug-seeking behavior can be partly explained by disrupted connectivity within highly integrated neuronal networks that are relying on optimal D3R availability. One may therefore logically suggest that by modulating specific nodes in those networks, selective D3R antagonists have the potential to “normalize” functional connectivity to significantly reduce reinstatement of drug-seeking and drug-taking behaviors.
4.3 Most Suitable Clinical Paradigms for Medication Development Purpose
Based on the efficacy of selective D3R antagonists in a wide range of animal models of SUD and preliminary clinical Phase I data, three hypotheses could be tested in the clinic: (1) selective D3R antagonists enhance the ability to stop using the substance; (2) selective D3R antagonists have value in treating withdrawal symptoms; (3) selective D3R antagonists prevent relapse to drug-seeking and drug-taking after abstinence has been achieved (or relapse to heavy use after a reduction in use). Clinical endpoints depend upon which of these efficacy criteria are chosen and can therefore be quit rates, reduction in withdrawal symptoms, or relapse (conversely abstinence) rates over time.
There is no animal model for self-motivated stopping, little is known about the neurochemical substrates of readiness for change (stopping), and there are no data to suggest that selective D3R antagonists would enhance readiness to stop substance use. There is, however, some evidence to suggest that selective D3R antagonists would be effective for treating withdrawal symptoms. For example, (±)VK4-116 (You et al. 2018) and SB-277011A were shown to reduce conditioned place aversion (CPA) produced by naloxone-precipitated withdrawal from acute opioid administration (Rice et al. 2012) and SB-277011A also attenuated the expression of fear conditioning (Swain et al. 2008).
Based upon their efficacy in animal models of reinstatement to drug-seeking behavior, selective D3R antagonists might be considered optimal medications for the prevention of relapse in the newly abstinent substance-dependent individual across all SUD. If relapse prevention is the expected target endpoint of selective D3R antagonists, a clinical proof of concept study could be a design in which a withdrawal phase precedes randomization to either placebo vs. the new D3R antagonist in a blinded parallel design that would last 6–12 weeks. However, individuals who successfully quit during the withdrawal phase may decline to enter the randomization phase, and/or the rate of successful quitting (achieving abstinence) may be so small that large numbers of subjects must be enrolled for a relatively small number of subjects in the two arms of the randomization phase. These operational challenges translate into costly and unusually long trials for a proof of concept.
In contrast, human laboratory trials can model several aspects of SUD that are most relevant to selective D3R antagonists including cue-induced craving in abstinent individuals, choice or reward paradigms, progressive ratio paradigms, and assessments of how much a subject is willing to work for a given substance in the abstinent state. These paradigms are ideal for demonstrating clinical proof of concept since they require small sample sizes, rely primarily on crossover rather than parallel designs, and are relatively short. Although these human laboratory models have been studied with various substances, their predictive validity to demonstrate clinical efficacy of a new chemical entity remains to be established.
Craving has been described as a core feature of SUD, including those associated with opioids, alcohol, nicotine, cannabis, cocaine, and other psychoactive substances (Kakko et al. 2019). The importance of craving as both a symptom and driver of SUD has elevated the relevance of its reduction as a critical treatment target and has renewed research focus on its role in addiction treatment and relapse (Kleykamp et al. 2019). This need was recently reinforced by the US Food and Drug Administration (FDA) in a statement on the necessity for new approaches to treat OUD (Opioid-Use-Disorder 2020; Statement-from-FDA-Commissioner 2018). There is substantial evidence showing increased craving and signs of physiological arousal to drug-related vs. neutral cues in drug users. Cue-induced craving can be studied in the human laboratory and/or in combination with imaging assessments. For example, reproducible findings have been observed in cue-induced craving in newly abstinent alcoholics (Myrick et al. 2008; Wrase et al. 2008) and in abstinent smokers (Brody et al. 2007; Due et al. 2002). The effect of a new medication on these reproducible cue-induced fMRI signals could be relatively easily determined in either single or repeat dose, parallel or crossover design, using a small number of subjects and completing the trial in a relatively short time period. Recent preliminary evidence (Regier et al. 2021) also suggests that a sustained response to repeated cocaine cues within a single passive-viewing fMRI task, featuring novel evocative (cocaine, sexual, aversive) and neutral comparator cues which were repeated later, is a potential predictor of drug-use outcomes. One may therefore suggest that pharmacological interventions that would restore a normal (i.e., decreased) response to the repeated presentation of drug-associated cues in this paradigm may predict a reduction in future drug use. This hypothesis, however, warrants future studies with potential new investigational drug candidates such as selective D3R antagonists.
Other surrogate markers might include abstinence-induced cognitive changes, such as interference on the Stroop task. For example, abstinent smokers may show altered reaction time to cigarette cues vs. neutral cues in the Stroop task, known as attentional bias induced by cues. If a compound, such as a selective D3R antagonist, is effective in preventing cue-induced relapse it would also be expected to prevent abstinence-induced cognitive changes, many of which are cue-induced. Medication effects have been demonstrated in this paradigm (Franken 2003) using either a single or repeat dose crossover study design (Patterson et al. 2009).
Ultimately, a more suitable model for SUD might be the one typically used for major depressive disorder (MDD). That model proposes acute treatment of 4–9 months post-clinical response for the first MDD episode, but even longer treatment for 2 or more episodes (Qaseem et al. 2008). Such a treatment paradigm is one for which selective D3R antagonists would be uniquely suited, perhaps providing long-term relapse prevention for the highly recurrent and relapsing disorders of substance dependence.
As extensively reviewed, D3R is highly expressed in several brain regions such as the NAc, Amy, and prefrontal cortex (including the insula) that are critically involved in reward, anxiety, and cognitive functions. A hypodopaminergic state may exist in persons living with SUD or comorbidity with MDD or AD. Thus, we propose that selective D3R antagonists or partial agonists may be ideal for the treatment of SUD, perhaps particularly for those with MDD or AD comorbidity (Fig. 6). On the one hand, blockade of presynaptic D3R in these brain regions may normalize the hypodopaminergic state, therefore relieving craving motivation for drug seeking, and improve withdrawal/negative affect and cognitive function. Conversely, blockade of postsynaptic D3R in these brain regions may reduce the rewarding effects produced by acute use of psychostimulants and/or opioids.

Schematic diagram illustrating the major brain regions that D3R antagonists or partial agonists may target, and the major pharmacological action produced by D3R antagonists or partial agonists based on recent findings from preclinical and clinical studies
4.4 From Monotherapy to Combination of Medications
The population of persons living with SUD has evolved considerably over time. Recent analyses suggest that among fatal opioid overdoses 78% involved another opioid, 21.6% involved cocaine, 11.1% involved alcohol, and 5.4% involved a psychostimulant other than cocaine (Jones et al. 2018). Polysubstance use of tobacco, psychostimulants, cannabis, or alcohol has also been observed in opioid-related emergency department visits (Liu and Vivolo-Kantor 2020), and the likelihood of these visits has been associated with the degree of severity of other SUD (John et al. 2019; Zale et al. 2015). Recent reports also indicate that methamphetamine use is associated with a discontinuation of buprenorphine treatment in people with an OUD (Tsui et al. 2020).
Polysubstance use is therefore a considerable challenge for translational medicine and medication development. The majority of research on SUD has indeed focused on single drugs in isolation, with a multiple drug use history often considered an exclusion criterion for pivotal clinical trials. Real-world settings, however, indicate that polysubstance use is associated with poorer treatment retention, higher rates of relapse, and a three-fold higher mortality rate compared to mono-substance use (Williamson et al. 2006). This is to say that pharmacotherapy may also require multipronged rather than monotherapeutic strategies. Therefore, studies examining the efficacy of pharmacotherapy alone vs. combined medication and psychosocial counselling are required to better understand the role each treatment modality may have. Preliminary data indicate that buprenorphine + naloxone, used in combination with an extended-release injectable formulation of naltrexone may be associated with reductions in cocaine use among people who met DSM-4 criteria for cocaine dependence and past or current opioid dependence or abuse (Ling et al. 2016). Similarly, adults with methamphetamine use disorder who received extended-release injectable naltrexone plus oral extended-release bupropion over a 12-week period seemed to show a reduction in use as well (Trivedi et al. 2021). The use of long-acting injectable formulations of well-established medications for OUD in combination with new investigational drug candidates such as a D3R antagonist/partial agonist may open new avenues to prevent reinstatement of drug-seeking and drug-taking behaviors. In addition, the D3R antagonist/partial agonist may allow the reduction in dose of the canonical monotherapies, such as methadone or buprenorphine, and thus reduce side effects (e.g., constipation) and potential overdose.
5 Summary
The prevalence and horrific loss of life from SUD has recently been highlighted by the opioid crisis. COVID-19 has further exacerbated this societal problem. Social isolation, devastation brought on by massive loss of life, and fatigue of lives disrupted have all contributed to an increase in SUD which has then translated into >90,000 drug overdose deaths in the past year, in the USA alone. The rapidity with which vaccines and medications have been developed to treat COVID-19 demonstrates that when a crisis is taken seriously, biomedical research can in fact be translated into clinically useful treatments quickly. And yet, this same fervor has never been directed toward SUD. The sad outcome is limited or indeed no medications available to treat PSUD. Although medications to treat OUD are clinically approved, they are not always effective (Strain et al. 2021) nor universally available, especially in this time of restricted access. The need for moving new medications forward through the pipeline is far overdue. Although admittedly complicated, SUD is a serious and life-ending disorder for many. The time for advancing medications such as the D3R antagonists/partial agonists as monotherapies or as part of a therapeutic regimen is now. Even if these medications are only effective for a subpopulation of patients, e.g., those who suffer from comorbidities with SUD, lives will be saved, and a perfect storm may be survived.
References
Abbadie C, Pasternak GW, Aicher SA (2001) Presynaptic localization of the carboxy-terminus epitopes of the mu opioid receptor splice variants MOR-1C and MOR-1D in the superficial laminae of the rat spinal cord. Neuroscience 106:833–842
Ahmad FB, Rossen LM, Sutton P (2021) Provisional drug overdose death counts. National Center for Health Statistics. https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm
Altier N, Stewart J (1998) Dopamine receptor antagonists in the nucleus accumbens attenuate analgesia induced by ventral tegmental area substance P or morphine and by nucleus accumbens amphetamine. J Pharmacol Exp Ther 285:208–215
Alves BB, Oliveira GP, Moreira Neto MG, Fiorilli RB, Cestario E (2019) Use of atypical antipsychotics and risk of hypertension: a case report and review literature. SAGE Open Med Case Rep 7:2050313X19841825
Angarita GA, Hadizadeh H, Cerdena I, Potenza MN (2021a) Can pharmacotherapy improve treatment outcomes in people with co-occurring major depressive and cocaine use disorders? Expert Opin Pharmacother 22:1669–1683
Angarita GA, Hadizadeh H, Cerdena I, Potenza MN (2021b) Can pharmacotherapy improve treatment outcomes in people with co-occurring major depressive and cocaine use disorders? Expert Opin Pharmacother 22(13):1669–1683
Appel NM, Li SH, Holmes TH, Acri JB (2015) Dopamine D3 receptor antagonist (GSK598809) potentiates the hypertensive effects of cocaine in conscious, freely-moving dogs. J Pharmacol Exp Ther 354:484–492
Araki KY, Sims JR, Bhide PG (2007) Dopamine receptor mRNA and protein expression in the mouse corpus striatum and cerebral cortex during pre- and postnatal development. Brain Res 1156:31–45
Asan E (1997) Ultrastructural features of tyrosine-hydroxylase-immunoreactive afferents and their targets in the rat amygdala. Cell Tissue Res 288:449–469
Avalos-Fuentes A, Albarran-Bravo S, Loya-Lopez S, Cortes H, Recillas-Morales S, Magana JJ, Paz-Bermudez F, Rangel-Barajas C, Aceves J, Erlij D, Floran B (2015) Dopaminergic denervation switches dopamine D3 receptor signaling and disrupts its ca(2+) dependent modulation by CaMKII and calmodulin in striatonigral projections of the rat. Neurobiol Dis 74:336–346
Azadfard M, Huecker MR, Leaming JM (2021) Opioid addiction. StatPearls, Treasure Island
Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217
Beaulieu JM, Espinoza S, Gainetdinov RR (2015) Dopamine receptors – IUPHAR review 13. Br J Pharmacol 172:1–23
Bergman J, Roof RA, Furman CA, Conroy JL, Mello NK, Sibley DR, Skolnick P (2013) Modification of cocaine self-administration by buspirone (buspar(R)): potential involvement of D3 and D4 dopamine receptors. Int J Neuropsychopharmacol 16:445–458
Berry AS, White RL 3rd, Furman DJ, Naskolnakorn JR, Shah VD, D'Esposito M, Jagust WJ (2019) Dopaminergic mechanisms underlying normal variation in trait anxiety. J Neurosci 39:2735–2744
Blum K, Cadet JL, Gold MS (2021a) Psychostimulant use disorder emphasizing methamphetamine and the opioid -dopamine connection: digging out of a hypodopaminergic ditch. J Neurol Sci 420:117252
Blum K, Khalsa J, Cadet JL, Baron D, Bowirrat A, Boyett B, Lott L, Brewer R, Gondre-Lewis M, Bunt G, Kazmi S, Gold MS (2021b) Cannabis-induced hypodopaminergic anhedonia and cognitive decline in humans: embracing putative induction of dopamine homeostasis. Front Psych 12:623403
Bodei S, Arrighi N, Spano P, Sigala S (2009) Should we be cautious on the use of commercially available antibodies to dopamine receptors? Naunyn Schmiedeberg’s Arch Pharmacol 379:413–415
Boileau I, Payer D, Rusjan PM, Houle S, Tong J, McCluskey T, Wilson AA, Kish SJ (2016) Heightened dopaminergic response to amphetamine at the D3 dopamine receptor in methamphetamine users. Neuropsychopharmacology 41:2994–3002
Bonifazi A, Battiti FO, Sanchez J, Zaidi SA, Bow E, Makarova M, Cao J, Shaik AB, Sulima A, Rice KC, Katritch V, Canals M, Lane JR, Newman AH (2021) Novel dual-target mu-opioid receptor and dopamine D3 receptor ligands as potential nonaddictive pharmacotherapeutics for pain management. J Med Chem 64:7778–7808
Bordet R, Ridray S, Carboni S, Diaz J, Sokoloff P, Schwartz JC (1997) Induction of dopamine D3 receptor expression as a mechanism of behavioral sensitization to levodopa. Proc Natl Acad Sci U S A 94:3363–3367
Bouthenet ML, Souil E, Martres MP, Sokoloff P, Giros B, Schwartz JC (1991) Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: comparison with dopamine D2 receptor mRNA. Brain Res 564:203–219
Bressan RA, Crippa JA (2005) The role of dopamine in reward and pleasure behaviour – review of data from preclinical research. Acta Psychiatr Scand Suppl (427):14–21
Brewer KL, Baran CA, Whitfield BR, Jensen AM, Clemens S (2014) Dopamine D3 receptor dysfunction prevents anti-nociceptive effects of morphine in the spinal cord. Front Neural Circuits 8:62
Brody AL, Mandelkern MA, Olmstead RE, Jou J, Tiongson E, Allen V, Scheibal D, London ED, Monterosso JR, Tiffany ST, Korb A, Gan JJ, Cohen MS (2007) Neural substrates of resisting craving during cigarette cue exposure. Biol Psychiatry 62:642–651
Bunzow JR, Van Tol HH, Grandy DK, Albert P, Salon J, Christie M, Machida CA, Neve KA, Civelli O (1988) Cloning and expression of a rat D2 dopamine receptor cDNA. Nature 336:783–787
Buydens-Branchey L, Branchey M, Reel-Brander C (2005) Efficacy of buspirone in the treatment of opioid withdrawal. J Clin Psychopharmacol 25:230–236
Cai JX, Arnsten AF (1997) Dose-dependent effects of the dopamine D1 receptor agonists A77636 or SKF81297 on spatial working memory in aged monkeys. J Pharmacol Exp Ther 283:183–189
Campbell RR, Lobo MK (2021) DRD3-dependent plasticity within the VP drives subcircuit activity critical for cocaine seeking. Neuron 109:2043–2044
Chander G, McCaul ME (2003) Co-occurring psychiatric disorders in women with addictions. Obstet Gynecol Clin N Am 30:469–481
Ciccarone D (2021) The rise of illicit fentanyls, stimulants and the fourth wave of the opioid overdose crisis. Curr Opin Psychiatry 34:344–350
Citrome L (2013) Cariprazine: chemistry, pharmacodynamics, pharmacokinetics, and metabolism, clinical efficacy, safety, and tolerability. Expert Opin Drug Metab Toxicol 9:193–206
Clarkson RL, Liptak AT, Gee SM, Sohal VS, Bender KJ (2017) D3 receptors regulate excitability in a unique class of prefrontal pyramidal cells. J Neurosci 37:5846–5860
Clemens S, Hochman S (2004) Conversion of the modulatory actions of dopamine on spinal reflexes from depression to facilitation in D3 receptor knock-out mice. J Neurosci 24:11337–11345
Cole DM, Beckmann CF, Searle GE, Plisson C, Tziortzi AC, Nichols TE, Gunn RN, Matthews PM, Rabiner EA, Beaver JD (2012) Orbitofrontal connectivity with resting-state networks is associated with midbrain dopamine D3 receptor availability. Cereb Cortex 22:2784–2793
Collo G, Bono F, Cavalleri L, Plebani L, Merlo Pich E, Millan MJ, Spano PF, Missale C (2012) Pre-synaptic dopamine D(3) receptor mediates cocaine-induced structural plasticity in mesencephalic dopaminergic neurons via ERK and Akt pathways. J Neurochem 120:765–778
Compton WM, Thomas YF, Stinson FS, Grant BF (2007) Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry 64:566–576
Compton WM, Valentino RJ, DuPont RL (2021) Polysubstance use in the U.S. opioid crisis. Mol Psychiatry 26:41–50
Contreras M, Ceric F, Torrealba F (2007) Inactivation of the interoceptive insula disrupts drug craving and malaise induced by lithium. Science 318:655–658
Conway KP, Compton W, Stinson FS, Grant BF (2006) Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry 67:247–257
Cook CD, Barrett AC, Syvanthong C, Picker MJ (2000) The dopamine D3/2 agonist 7-OH-DPAT attenuates the development of morphine tolerance but not physical dependence in rats. Psychopharmacology 152:93–104
Craske MG, Stein MB, Eley TC, Milad MR, Holmes A, Rapee RM, Wittchen HU (2017) Anxiety disorders. Nat Rev Dis Primers 3:17024
Creed M, Ntamati NR, Chandra R, Lobo MK, Luscher C (2016) Convergence of reinforcing and anhedonic cocaine effects in the ventral pallidum. Neuron 92:214–226
Cuevas S, Villar VA, Jose PA, Armando I (2013) Renal dopamine receptors, oxidative stress, and hypertension. Int J Mol Sci 14:17553–17572
Dai WL, Xiong F, Yan B, Cao ZY, Liu WT, Liu JH, Yu BY (2016) Blockade of neuronal dopamine D2 receptor attenuates morphine tolerance in mice spinal cord. Sci Rep 6:38746
de Guglielmo G, Kallupi M, Sedighim S, Newman AH, George O (2019) Dopamine D3 receptor antagonism reverses the escalation of oxycodone self-administration and decreases withdrawal-induced hyperalgesia and irritability-like behavior in oxycodone-dependent heterogeneous stock rats. Front Behav Neurosci 13:292
Dedic N, Kuhne C, Jakovcevski M, Hartmann J, Genewsky AJ, Gomes KS, Anderzhanova E, Pohlmann ML, Chang S, Kolarz A, Vogl AM, Dine J, Metzger MW, Schmid B, Almada RC, Ressler KJ, Wotjak CT, Grinevich V, Chen A, Schmidt MV, Wurst W, Refojo D, Deussing JM (2018) Chronic CRH depletion from GABAergic, long-range projection neurons in the extended amygdala reduces dopamine release and increases anxiety. Nat Neurosci 21:803–807
Der-Avakian A, Markou A (2012) The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci 35:68–77
Diaz J, Levesque D, Griffon N, Lammers CH, Martres MP, Sokoloff P, Schwartz JC (1994) Opposing roles for dopamine D2 and D3 receptors on neurotensin mRNA expression in nucleus accumbens. Eur J Neurosci 6:1384–1387
Diaz J, Levesque D, Lammers CH, Griffon N, Martres MP, Schwartz JC, Sokoloff P (1995) Phenotypical characterization of neurons expressing the dopamine D3 receptor in the rat brain. Neuroscience 65:731–745
Diaz J, Ridray S, Mignon V, Griffon N, Schwartz JC, Sokoloff P (1997) Selective expression of dopamine D3 receptor mRNA in proliferative zones during embryonic development of the rat brain. J Neurosci 17:4282–4292
Diaz J, Pilon C, Le Foll B, Gros C, Triller A, Schwartz JC, Sokoloff P (2000) Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci 20:8677–8684
Doot RK, Dubroff JG, Labban KJ, Mach RH (2019) Selectivity of probes for PET imaging of dopamine D3 receptors. Neurosci Lett 691:18–25
Due DL, Huettel SA, Hall WG, Rubin DC (2002) Activation in mesolimbic and visuospatial neural circuits elicited by smoking cues: evidence from functional magnetic resonance imaging. Am J Psychiatry 159:954–960
Elkashef A, Vocci F, Hanson G, White J, Wickes W, Tiihonen J (2008) Pharmacotherapy of methamphetamine addiction: an update. Subst Abus 29:31–49
Erritzoe D, Tziortzi A, Bargiela D, Colasanti A, Searle GE, Gunn RN, Beaver JD, Waldman A, Nutt DJ, Bani M, Merlo-Pich E, Rabiner EA, Lingford-Hughes A (2014) In vivo imaging of cerebral dopamine D3 receptors in alcoholism. Neuropsychopharmacology 39:1703–1712
Ewing ST, Dorcely C, Maidi R, Paker G, Schelbaum E, Ranaldi R (2021) Low-dose polypharmacology targeting dopamine D1 and D3 receptors reduces cue-induced relapse to heroin seeking in rats. Addict Biol 26:e12988
Fiorentini C, Busi C, Gorruso E, Gotti C, Spano P, Missale C (2008) Reciprocal regulation of dopamine D1 and D3 receptor function and trafficking by heterodimerization. Mol Pharmacol 74:59–69
Fiorentini C, Busi C, Spano P, Missale C (2010) Dimerization of dopamine D1 and D3 receptors in the regulation of striatal function. Curr Opin Pharmacol 10:87–92
Fogger SA (2019) Methamphetamine use: a new wave in the opioid crisis? J Addict Nurs 30:219–223
Franken IH (2003) Drug craving and addiction: integrating psychological and neuropsychopharmacological approaches. Prog Neuro-Psychopharmacol Biol Psychiatry 27:563–579
Freyberg Z, Sonders MS, Aguilar JI, Hiranita T, Karam CS, Flores J, Pizzo AB, Zhang Y, Farino ZJ, Chen A, Martin CA, Kopajtic TA, Fei H, Hu G, Lin YY, Mosharov EV, McCabe BD, Freyberg R, Wimalasena K, Hsin LW, Sames D, Krantz DE, Katz JL, Sulzer D, Javitch JA (2016) Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in drosophila brain. Nat Commun 7:10652
Fudge JL, Haber SN (2000) The central nucleus of the amygdala projection to dopamine subpopulations in primates. Neuroscience 97:479–494
Galaj E, Xi ZX (2021) Progress in opioid reward research: from a canonical two-neuron hypothesis to two neural circuits. Pharmacol Biochem Behav 200:173072
Galaj E, Han X, Shen H, Jordan CJ, He Y, Humburg B, Bi GH, Xi ZX (2020a) Dissecting the role of GABA neurons in the VTA versus SNr in opioid reward. J Neurosci 40:8853–8869
Galaj E, Newman AH, Xi ZX (2020b) Dopamine D3 receptor-based medication development for the treatment of opioid use disorder: rationale, progress, and challenges. Neurosci Biobehav Rev 114:38–52
Gallezot JD, Beaver JD, Gunn RN, Nabulsi N, Weinzimmer D, Singhal T, Slifstein M, Fowles K, Ding YS, Huang Y, Laruelle M, Carson RE, Rabiner EA (2012) Affinity and selectivity of [(1)(1)C]-(+)-PHNO for the D3 and D2 receptors in the rhesus monkey brain in vivo. Synapse 66:489–500
Gardner EL (2000) What we have learned about addiction from animal models of drug self-administration. Am J Addict 9:285–313
Gaznick N, Tranel D, McNutt A, Bechara A (2014) Basal ganglia plus insula damage yields stronger disruption of smoking addiction than basal ganglia damage alone. Nicotine Tob Res 16:445–453
Gear RW, Aley KO, Levine JD (1999) Pain-induced analgesia mediated by mesolimbic reward circuits. J Neurosci 19:7175–7181
Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr, Sibley DR (1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250:1429–1432
Ginovart N, Willeit M, Rusjan P, Graff A, Bloomfield PM, Houle S, Kapur S, Wilson AA (2007) Positron emission tomography quantification of [11C]-(+)-PHNO binding in the human brain. J Cereb Blood Flow Metab 27:857–871
Giros B, Martres MP, Sokoloff P, Schwartz JC (1990) Gene cloning of human dopaminergic D3 receptor and identification of its chromosome. C R Acad Sci III 311:501–508
Glick SN, Klein KS, Tinsley J, Golden MR (2021) Increasing heroin-methamphetamine (goofball) use and related morbidity among Seattle area people who inject drugs. Am J Addict 30:183–191
Gobert A, Lejeune F, Rivet JM, Cistarelli L, Millan MJ (1996) Dopamine D3 (auto) receptors inhibit dopamine release in the frontal cortex of freely moving rats in vivo. J Neurochem 66:2209–2212
Gomaa AA, Mohamed LH, Ahmed HN (1989) Modification of morphine-induced analgesia, tolerance and dependence by bromocriptine. Eur J Pharmacol 170:129–135
Gomes T, Tadrous M, Mamdani MM, Paterson JM, Juurlink DN (2018) The burden of opioid-related mortality in the United States. JAMA Netw Open 1:e180217
Graff-Guerrero A, Willeit M, Ginovart N, Mamo D, Mizrahi R, Rusjan P, Vitcu I, Seeman P, Wilson AA, Kapur S (2008) Brain region binding of the D2/3 agonist [11C]-(+)-PHNO and the D2/3 antagonist [11C]raclopride in healthy humans. Hum Brain Mapp 29:400–410
Grant S, London ED, Newlin DB, Villemagne VL, Liu X, Contoreggi C, Phillips RL, Kimes AS, Margolin A (1996) Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci U S A 93:12040–12045
Guerrero-Bautista R, Franco-Garcia A, Hidalgo JM, Fernandez-Gomez FJ, Ribeiro Do Couto B, Milanes MV, Nunez C (2021) Distinct regulation of dopamine D3 receptor in the basolateral amygdala and dentate gyrus during the reinstatement of cocaine CPP induced by drug priming and social stress. Int J Mol Sci 22:3100
Gurevich EV, Joyce JN (1999) Distribution of dopamine D3 receptor expressing neurons in the human forebrain: comparison with D2 receptor expressing neurons. Neuropsychopharmacology 20:60–80
Gurevich EV, Himes JW, Joyce JN (1999) Developmental regulation of expression of the D3 dopamine receptor in rat nucleus accumbens and islands of Calleja. J Pharmacol Exp Ther 289:587–598
Gyertyan I, Kiss B, Gal K, Laszlovszky I, Horvath A, Gemesi LI, Saghy K, Pasztor G, Zajer M, Kapas M, Csongor EA, Domany G, Tihanyi K, Szombathelyi Z (2007) Effects of RGH-237 [N-{4-[4-(3-aminocarbonyl-phenyl)-piperazin-1-yl]-butyl}-4-bromo-benzamide], an orally active, selective dopamine D(3) receptor partial agonist in animal models of cocaine abuse. J Pharmacol Exp Ther 320:1268–1278
Hall H, Halldin C, Dijkstra D, Wikstrom H, Wise LD, Pugsley TA, Sokoloff P, Pauli S, Farde L, Sedvall G (1996) Autoradiographic localisation of D3-dopamine receptors in the human brain using the selective D3-dopamine receptor agonist (+)-[3H]PD 128907. Psychopharmacology 128:240–247
He M, Shippenberg TS (2000) Strain differences in basal and cocaine-evoked dopamine dynamics in mouse striatum. J Pharmacol Exp Ther 293:121–127
Heidbreder CA, Newman AH (2010) Current perspectives on selective dopamine D(3) receptor antagonists as pharmacotherapeutics for addictions and related disorders. Ann N Y Acad Sci 1187:4–34
Heinsbroek JA, Bobadilla AC, Dereschewitz E, Assali A, Chalhoub RM, Cowan CW, Kalivas PW (2020) Opposing regulation of cocaine seeking by glutamate and GABA neurons in the ventral pallidum. Cell Rep 30:2018–2027.e2013
Hellem TL, Lundberg KJ, Renshaw PF (2015) A review of treatment options for co-occurring methamphetamine use disorders and depression. J Addict Nurs 26:14–23; quiz E11
Herroelen L, De Backer JP, Wilczak N, Flamez A, Vauquelin G, De Keyser J (1994) Autoradiographic distribution of D3-type dopamine receptors in human brain using [3H]7-hydroxy-N,N-di-n-propyl-2-aminotetralin. Brain Res 648:222–228
Hersey M, Bacon AK, Bailey LG, Coggiano MA, Newman AH, Leggio L, Tanda G (2021) Psychostimulant use disorder, an unmet therapeutic goal: can modafinil narrow the gap? Front Neurosci 15:656475
Hikida T, Yawata S, Yamaguchi T, Danjo T, Sasaoka T, Wang Y, Nakanishi S (2013) Pathway-specific modulation of nucleus accumbens in reward and aversive behavior via selective transmitter receptors. Proc Natl Acad Sci U S A 110:342–347
Huang M, He W, Kiss B, Farkas B, Adham N, Meltzer HY (2019) The role of dopamine D3 receptor partial agonism in cariprazine-induced neurotransmitter efflux in rat hippocampus and nucleus accumbens. J Pharmacol Exp Ther 371:517–525
Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC (2009) The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron 61:786–800
John WS, Zhu H, Mannelli P, Subramaniam GA, Schwartz RP, McNeely J, Wu LT (2019) Prevalence and patterns of opioid misuse and opioid use disorder among primary care patients who use tobacco. Drug Alcohol Depend 194:468–475
Jones CM, Einstein EB, Compton WM (2018) Changes in synthetic opioid involvement in drug overdose deaths in the United States, 2010-2016. JAMA 319:1819–1821
Jones CM, Bekheet F, Park JN, Alexander GC (2020) The evolving overdose epidemic: synthetic opioids and rising stimulant-related harms. Epidemiol Rev 42:154–166
Jordan CJ, Humburg B, Rice M, Bi GH, You ZB, Shaik AB, Cao J, Bonifazi A, Gadiano A, Rais R, Slusher B, Newman AH, Xi ZX (2019a) The highly selective dopamine D3R antagonist, R-VK4-40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology 158:107597
Jordan CJ, Humburg BA, Thorndike EB, Shaik AB, Xi ZX, Baumann MH, Newman AH, Schindler CW (2019b) Newly developed dopamine D3 receptor antagonists, R-VK4-40 and R-VK4-116, do not potentiate cardiovascular effects of cocaine or oxycodone in rats. J Pharmacol Exp Ther 371:602–614
Jordan CJ, He Y, Bi GH, You ZB, Cao J, Xi ZX, Newman AH (2020) (+/−)VK4-40, a novel dopamine D3 receptor partial agonist, attenuates cocaine reward and relapse in rodents. Br J Pharmacol 177:4796–4807
Jose PA, Drago J, Accili D, Eisner GM, Felder RA (1997) Transgenic mice to study the role of dopamine receptors in cardiovascular function. Clin Exp Hypertens 19:15–25
Kakko J, Alho H, Baldacchino A, Molina R, Nava FA, Shaya G (2019) Craving in opioid use disorder: from neurobiology to clinical practice. Front Psych 10:592
Kalin NH, Shelton SE, Davidson RJ (2004) The role of the central nucleus of the amygdala in mediating fear and anxiety in the primate. J Neurosci 24:5506–5515
Kampman K (2021) Cariprazine for comorbid cocaine and opioid use didorder. ClinicalTrials.Gov NCT05063201. https://clinicaltrials.gov/ct2/show/NCT05063201?term=cariprazine+and+cocaine&draw=05063202&rank=05063201
Keck TM, John WS, Czoty PW, Nader MA, Newman AH (2015) Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J Med Chem 58:5361–5380
Keeler BE, Baran CA, Brewer KL, Clemens S (2012) Increased excitability of spinal pain reflexes and altered frequency-dependent modulation in the dopamine D3-receptor knockout mouse. Exp Neurol 238:273–283
Kibaly C, Alderete JA, Liu SH, Nasef HS, Law PY, Evans CJ, Cahill CM (2021) Oxycodone in the opioid epidemic: high ‘liking’, ‘wanting’, and abuse liability. Cell Mol Neurobiol 41:899–926
Kienast T, Hariri AR, Schlagenhauf F, Wrase J, Sterzer P, Buchholz HG, Smolka MN, Grunder G, Cumming P, Kumakura Y, Bartenstein P, Dolan RJ, Heinz A (2008) Dopamine in amygdala gates limbic processing of aversive stimuli in humans. Nat Neurosci 11:1381–1382
Kim KM, Valenzano KJ, Robinson SR, Yao WD, Barak LS, Caron MG (2001) Differential regulation of the dopamine D2 and D3 receptors by G protein-coupled receptor kinases and beta-arrestins. J Biol Chem 276:37409–37414
Kiss B, Horvath A, Nemethy Z, Schmidt E, Laszlovszky I, Bugovics G, Fazekas K, Hornok K, Orosz S, Gyertyan I, Agai-Csongor E, Domany G, Tihanyi K, Adham N, Szombathelyi Z (2010) Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther 333:328–340
Kiss B, Horti F, Bobok A (2011) In vitro and in vivo comparison of [(3)H](+)-PHNO and [(3)H]raclopride binding to rat striatum and lobes 9 and 10 of the cerebellum: a method to distinguish dopamine D(3) from D(2) receptor sites. Synapse 65:467–478
Kleykamp BA, Weiss RD, Strain EC (2019) Time to reconsider the role of craving in opioid use disorder. JAMA Psychiat 76:1113–1114
Komorowski A, Weidenauer A, Murgas M, Sauerzopf U, Wadsak W, Mitterhauser M, Bauer M, Hacker M, Praschak-Rieder N, Kasper S, Lanzenberger R, Willeit M (2020) Association of dopamine D2/3 receptor binding potential measured using PET and [(11)C]-(+)-PHNO with post-mortem DRD2/3 gene expression in the human brain. NeuroImage 223:117270
Koob GF (2013) Addiction is a reward deficit and stress surfeit disorder. Front Psych 4:72
Koob GF (2021) Drug addiction: hyperkatifeia/negative reinforcement as a framework for medications development. Pharmacol Rev 73:163–201
Koob GF, Bloom FE (1988) Cellular and molecular mechanisms of drug dependence. Science 242:715–723
Kravitz AV, Tye LD, Kreitzer AC (2012) Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neurosci 15:816–818
Kula NS, Baldessarini RJ, Kebabian JW, Neumeyer JL (1994) S-(+)-aporphines are not selective for human D3 dopamine receptors. Cell Mol Neurobiol 14:185–191
Kumar V, Bonifazi A, Ellenberger MP, Keck TM, Pommier E, Rais R, Slusher BS, Gardner E, You ZB, Xi ZX, Newman AH (2016) Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: new leads for opioid dependence treatment. J Med Chem 59:7634–7650
Kuo SC, Yeh YW, Chen CY, Huang CC, Chang HA, Yen CH, Ho PS, Liang CS, Chou HW, Lu RB, Huang SY (2014) DRD3 variation associates with early-onset heroin dependence, but not specific personality traits. Prog Neuro-Psychopharmacol Biol Psychiatry 51:1–8
Lacroix LP, Hows ME, Shah AJ, Hagan JJ, Heidbreder CA (2003) Selective antagonism at dopamine D3 receptors enhances monoaminergic and cholinergic neurotransmission in the rat anterior cingulate cortex. Neuropsychopharmacology 28:839–849
Lammel S, Lim BK, Malenka RC (2014) Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology 76 Pt B:351–359
Lammers CH, Diaz J, Schwartz JC, Sokoloff P (2000) Dopamine D3 receptor gene expression in the shell of nucleus accumbens is increased by chronic antidepressant treatment. Mol Psychiatry 5:229
Landwehrmeyer B, Mengod G, Palacios JM (1993a) Differential visualization of dopamine D2 and D3 receptor sites in rat brain. A comparative study using in situ hybridization histochemistry and ligand binding autoradiography. Eur J Neurosci 5:145–153
Landwehrmeyer B, Mengod G, Palacios JM (1993b) Dopamine D3 receptor mRNA and binding sites in human brain. Brain Res Mol Brain Res 18:187–192
Larson ER, Ariano MA (1995) D3 and D2 dopamine receptors: visualization of cellular expression patterns in motor and limbic structures. Synapse 20:325–337
Le Foll B, Payer D, Di Ciano P, Guranda M, Nakajima S, Tong J, Mansouri E, Wilson AA, Houle S, Meyer JH, Graff-Guerrero A, Boileau I (2016) Occupancy of dopamine D3 and D2 receptors by buspirone: a [11C]-(+)-PHNO PET study in humans. Neuropsychopharmacology 41:529–537
Le Moine C, Bloch B (1996) Expression of the D3 dopamine receptor in peptidergic neurons of the nucleus accumbens: comparison with the D1 and D2 dopamine receptors. Neuroscience 73:131–143
Leggio GM, Bucolo C, Platania CB, Salomone S, Drago F (2016) Current drug treatments targeting dopamine D3 receptor. Pharmacol Ther 165:164–177
Lesscher HM, McMahon T, Lasek AW, Chou WH, Connolly J, Kharazia V, Messing RO (2008) Amygdala protein kinase C epsilon regulates corticotropin-releasing factor and anxiety-like behavior. Genes Brain Behav 7:323–333
Levant B (1997) The D3 dopamine receptor: neurobiology and potential clinical relevance. Pharmacol Rev 49:231–252
Levant B (1998) Differential distribution of D3 dopamine receptors in the brains of several mammalian species. Brain Res 800:269–274
Levant B, McCarson KE (2001) D(3) dopamine receptors in rat spinal cord: implications for sensory and motor function. Neurosci Lett 303:9–12
Leventhal AM, Mooney ME, DeLaune KA, Schmitz JM (2006) Using addiction severity profiles to differentiate cocaine-dependent patients with and without comorbid major depression. Am J Addict 15:362–369
Levesque D, Martres MP, Diaz J, Griffon N, Lammers CH, Sokoloff P, Schwartz JC (1995) A paradoxical regulation of the dopamine D3 receptor expression suggests the involvement of an anterograde factor from dopamine neurons. Proc Natl Acad Sci U S A 92:1719–1723
Leyton M, Vezina P (2014) Dopamine ups and downs in vulnerability to addictions: a neurodevelopmental model. Trends Pharmacol Sci 35:268–276
Li YQ, Li FQ, Wang XY, Wu P, Zhao M, Xu CM, Shaham Y, Lu L (2008) Central amygdala extracellular signal-regulated kinase signaling pathway is critical to incubation of opiate craving. J Neurosci 28:13248–13257
Ling W, Hillhouse MP, Saxon AJ, Mooney LJ, Thomas CM, Ang A, Matthews AG, Hasson A, Annon J, Sparenborg S, Liu DS, McCormack J, Church S, Swafford W, Drexler K, Schuman C, Ross S, Wiest K, Korthuis PT, Lawson W, Brigham GS, Knox PC, Dawes M, Rotrosen J (2016) Buprenorphine + naloxone plus naltrexone for the treatment of cocaine dependence: the cocaine use reduction with buprenorphine (CURB) study. Addiction 111:1416–1427
Liu S, Vivolo-Kantor A (2020) A latent class analysis of drug and substance use patterns among patients treated in emergency departments for suspected drug overdose. Addict Behav 101:106142
Liu XY, Mao LM, Zhang GC, Papasian CJ, Fibuch EE, Lan HX, Zhou HF, Xu M, Wang JQ (2009) Activity-dependent modulation of limbic dopamine D3 receptors by CaMKII. Neuron 61:425–438
Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, Mouzon E, Mogri M, Neve RL, Deisseroth K, Han MH, Nestler EJ (2010) Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330:385–390
Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y (2005) Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci 8:212–219
Luscher C, Pascoli V (2021) ‘Ups, downs, and sideways’ of dopamine in drug addiction. Trends Neurosci 44:593–594
Lv Y, Hu RR, Jing M, Zhao TY, Wu N, Song R, Li J, Hu G (2019) Selective dopamine D3 receptor antagonist YQA14 inhibits morphine-induced behavioral sensitization in wild type, but not in dopamine D3 receptor knockout mice. Acta Pharmacol Sin 40:583–588
Lyden J, Binswanger IA (2019) The United States opioid epidemic. Semin Perinatol 43:123–131
Malec E, Malec T, Gagne MA, Dongier M (1996) Buspirone in the treatment of alcohol dependence: a placebo-controlled trial. Alcohol Clin Exp Res 20:307–312
Manchikanti L, Vanaparthy R, Atluri S, Sachdeva H, Kaye AD, Hirsch JA (2021) COVID-19 and the opioid epidemic: two public health emergencies that intersect with chronic pain. Pain Ther 10:269–286
Manduca A, Servadio M, Damsteegt R, Campolongo P, Vanderschuren LJ, Trezza V (2016) Dopaminergic neurotransmission in the nucleus accumbens modulates social play behavior in rats. Neuropsychopharmacology 41:2215–2223
Marino RA, Levy R (2019) Differential effects of D1 and D2 dopamine agonists on memory, motivation, learning and response time in non-human primates. Eur J Neurosci 49:199–214
Martel JC, Gatti McArthur S (2020) Dopamine receptor subtypes, physiology and pharmacology: new ligands and concepts in schizophrenia. Front Pharmacol 11:1003
Matsui A, Williams JT (2011) Opioid-sensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J Neurosci 31:17729–17735
Matsui A, Jarvie BC, Robinson BG, Hentges ST, Williams JT (2014) Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron 82:1346–1356
Matuskey D, Gallezot JD, Pittman B, Williams W, Wanyiri J, Gaiser E, Lee DE, Hannestad J, Lim K, Zheng MQ, Lin SF, Labaree D, Potenza MN, Carson RE, Malison RT, Ding YS (2014) Dopamine D(3) receptor alterations in cocaine-dependent humans imaged with [(1)(1)C](+)PHNO. Drug Alcohol Depend 139:100–105
McGregor A, Roberts DC (1993) Dopaminergic antagonism within the nucleus accumbens or the amygdala produces differential effects on intravenous cocaine self-administration under fixed and progressive ratio schedules of reinforcement. Brain Res 624:245–252
McRae AL, Sonne SC, Brady KT, Durkalski V, Palesch Y (2004) A randomized, placebo-controlled trial of buspirone for the treatment of anxiety in opioid-dependent individuals. Am J Addict 13:53–63
McRae-Clark AL, Baker NL, Gray KM, Killeen TK, Wagner AM, Brady KT, DeVane CL, Norton J (2015) Buspirone treatment of cannabis dependence: a randomized, placebo-controlled trial. Drug Alcohol Depend 156:29–37
Meador-Woodruff JH, Mansour A, Grandy DK, Damask SP, Civelli O, Watson SJ Jr (1992) Distribution of D5 dopamine receptor mRNA in rat brain. Neurosci Lett 145:209–212
Meil WM, See RE (1997) Lesions of the basolateral amygdala abolish the ability of drug associated cues to reinstate responding during withdrawal from self-administered cocaine. Behav Brain Res 87:139–148
Micheli F, Arista L, Bonanomi G, Blaney FE, Braggio S, Capelli AM, Checchia A, Damiani F, Di-Fabio R, Fontana S, Gentile G, Griffante C, Hamprecht D, Marchioro C, Mugnaini M, Piner J, Ratti E, Tedesco G, Tarsi L, Terreni S, Worby A, Ashby CR Jr, Heidbreder C (2010) 1,2,4-Triazolyl azabicyclo[3.1.0]hexanes: a new series of potent and selective dopamine D(3) receptor antagonists. J Med Chem 53:374–391
Millan MJ, Gobert A, Newman-Tancredi A, Lejeune F, Cussac D, Rivet JM, Audinot V, Dubuffet T, Lavielle G (2000) S33084, a novel, potent, selective, and competitive antagonist at dopamine D(3)-receptors: I. Receptorial, electrophysiological and neurochemical profile compared with GR218,231 and L741,626. J Pharmacol Exp Ther 293:1048–1062
Millan MJ, Buccafusco JJ, Loiseau F, Watson DJ, Decamp E, Fone KC, Thomasson-Perret N, Hill M, Mocaer E, Schneider JS (2010) The dopamine D3 receptor antagonist, S33138, counters cognitive impairment in a range of rodent and primate procedures. Int J Neuropsychopharmacol 13:1035–1051
Min C, Zheng M, Zhang X, Caron MG, Kim KM (2013) Novel roles for beta-arrestins in the regulation of pharmacological sequestration to predict agonist-induced desensitization of dopamine D3 receptors. Br J Pharmacol 170:1112–1129
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG (1998) Dopamine receptors: from structure to function. Physiol Rev 78:189–225
Mogg K, Bradley BP, O'Neill B, Bani M, Merlo-Pich E, Koch A, Bullmore ET, Nathan PJ (2012) Effect of dopamine D(3) receptor antagonism on approach responses to food cues in overweight and obese individuals. Behav Pharmacol 23:603–608
Monsma FJ Jr, Mahan LC, McVittie LD, Gerfen CR, Sibley DR (1990) Molecular cloning and expression of a D1 dopamine receptor linked to adenylyl cyclase activation. Proc Natl Acad Sci U S A 87:6723–6727
Mugnaini M, Iavarone L, Cavallini P, Griffante C, Oliosi B, Savoia C, Beaver J, Rabiner EA, Micheli F, Heidbreder C, Andorn A, Merlo Pich E, Bani M (2013) Occupancy of brain dopamine D3 receptors and drug craving: a translational approach. Neuropsychopharmacology 38:302–312
Murray AM, Ryoo HL, Gurevich E, Joyce JN (1994) Localization of dopamine D3 receptors to mesolimbic and D2 receptors to mesostriatal regions of human forebrain. Proc Natl Acad Sci U S A 91:11271–11275
Murthy P, Mahadevan J, Chand PK (2019) Treatment of substance use disorders with co-occurring severe mental health disorders. Curr Opin Psychiatry 32:293–299
Myrick H, Anton RF, Li X, Henderson S, Randall PK, Voronin K (2008) Effect of naltrexone and ondansetron on alcohol cue-induced activation of the ventral striatum in alcohol-dependent people. Arch Gen Psychiatry 65:466–475
Nakajima S, Gerretsen P, Takeuchi H, Caravaggio F, Chow T, Le Foll B, Mulsant B, Pollock B, Graff-Guerrero A (2013) The potential role of dopamine D(3) receptor neurotransmission in cognition. Eur Neuropsychopharmacol 23:799–813
Nakako T, Murai T, Ikejiri M, Ishiyama T, Taiji M, Ikeda K (2013) Effects of a dopamine D1 agonist on ketamine-induced spatial working memory dysfunction in common marmosets. Behav Brain Res 249:109–115
Naqvi NH, Bechara A (2009) The hidden island of addiction: the insula. Trends Neurosci 32:56–67
Naqvi NH, Rudrauf D, Damasio H, Bechara A (2007) Damage to the insula disrupts addiction to cigarette smoking. Science 315:531–534
Narayan A, Balkrishnan R (2021) A health crisis within a health crisis: opioid access in the COVID-19 pandemic. Subst Abus 42:148–152
Narendran R, Slifstein M, Guillin O, Hwang Y, Hwang DR, Scher E, Reeder S, Rabiner E, Laruelle M (2006) Dopamine (D2/3) receptor agonist positron emission tomography radiotracer [11C]-(+)-PHNO is a D3 receptor preferring agonist in vivo. Synapse 60:485–495
Narendran R, Frankle WG, Mason NS, Laymon CM, Lopresti BJ, Price JC, Kendro S, Vora S, Litschge M, Mountz JM, Mathis CA (2009) Positron emission tomography imaging of D(2/3) agonist binding in healthy human subjects with the radiotracer [(11)C]-N-propyl-norapomorphine: preliminary evaluation and reproducibility studies. Synapse 63:574–584
Nathan PJ, O'Neill BV, Mogg K, Bradley BP, Beaver J, Bani M, Merlo-Pich E, Fletcher PC, Swirski B, Koch A, Dodds CM, Bullmore ET (2012) The effects of the dopamine D(3) receptor antagonist GSK598809 on attentional bias to palatable food cues in overweight and obese subjects. Int J Neuropsychopharmacol 15:149–161
Neisewander JL, Baker DA, Fuchs RA, Tran-Nguyen LT, Palmer A, Marshall JF (2000) Fos protein expression and cocaine-seeking behavior in rats after exposure to a cocaine self-administration environment. J Neurosci 20:798–805
Newman AH, Blaylock BL, Nader MA, Bergman J, Sibley DR, Skolnick P (2012) Medication discovery for addiction: translating the dopamine D3 receptor hypothesis. Biochem Pharmacol 84:882–890
Newman AH, Ku T, Jordan CJ, Bonifazi A, Xi ZX (2021) New drugs, old targets: tweaking the dopamine system to treat psychostimulant use disorders. Annu Rev Pharmacol Toxicol 61:609–628
O'Dell LE, Sussman AN, Meyer KL, Neisewander JL (1999) Behavioral effects of psychomotor stimulant infusions into amygdaloid nuclei. Neuropsychopharmacology 20:591–602
O'Malley KL, Harmon S, Tang L, Todd RD (1992) The rat dopamine D4 receptor: sequence, gene structure, and demonstration of expression in the cardiovascular system. New Biol 4:137–146
Opioid-Use-Disorder (2020) Opioid use disorder: endpoints for demonstrating effectiveness of drugs for treatment guidance for industry. https://www.fda.gov/media/114948/download.accessed
Ozdemir E, Bagcivan I, Gursoy S (2013) Role of D(1)/D(2) dopamin receptors antagonist perphenazine in morphine analgesia and tolerance in rats. Bosn J Basic Med Sci 13:119–125
Panagis G, Spyraki C (1996) Neuropharmacological evidence for the role of dopamine in ventral pallidum self-stimulation. Psychopharmacology 123:280–288
Patterson F, Jepson C, Strasser AA, Loughead J, Perkins KA, Gur RC, Frey JM, Siegel S, Lerman C (2009) Varenicline improves mood and cognition during smoking abstinence. Biol Psychiatry 65:144–149
Pich EM, Collo G (2015) Pharmacological targeting of dopamine D3 receptors: possible clinical applications of selective drugs. Eur Neuropsychopharmacol 25:1437–1447
Post RM, Kalivas P (2013) Bipolar disorder and substance misuse: pathological and therapeutic implications of their comorbidity and cross-sensitisation. Br J Psychiatry 202:172–176
Pribiag H, Shin S, Wang EH, Sun F, Datta P, Okamoto A, Guss H, Jain A, Wang XY, De Freitas B, Honma P, Pate S, Lilascharoen V, Li Y, Lim BK (2021) Ventral pallidum DRD3 potentiates a pallido-habenular circuit driving accumbal dopamine release and cocaine seeking. Neuron 109:2165–2182.e2110
Prieto GA, Perez-Burgos A, Palomero-Rivero M, Galarraga E, Drucker-Colin R, Bargas J (2011) Upregulation of D2-class signaling in dopamine-denervated striatum is in part mediated by D3 receptors acting on ca V 2.1 channels via PIP2 depletion. J Neurophysiol 105:2260–2274
Qaseem A, Snow V, Denberg TD, Forciea MA, Owens DK, Clinical Efficacy Assessment Subcommittee of American College of Physicians (2008) Using second-generation antidepressants to treat depressive disorders: a clinical practice guideline from the American College of Physicians. Ann Intern Med 149:725–733
Rabiner EA, Laruelle M (2010) Imaging the D3 receptor in humans in vivo using [11C](+)-PHNO positron emission tomography (PET). Int J Neuropsychopharmacol 13:289–290
Rabiner EA, Slifstein M, Nobrega J, Plisson C, Huiban M, Raymond R, Diwan M, Wilson AA, McCormick P, Gentile G, Gunn RN, Laruelle MA (2009) In vivo quantification of regional dopamine-D3 receptor binding potential of (+)-PHNO: studies in non-human primates and transgenic mice. Synapse 63:782–793
Raby WN, Rubin EA, Garawi F, Cheng W, Mason E, Sanfilippo L, Lord S, Bisaga A, Aharonovich E, Levin F, McDowell D, Nunes EV (2014) A randomized, double-blind, placebo-controlled trial of venlafaxine for the treatment of depressed cocaine-dependent patients. Am J Addict 23:68–75
Regier PS, Jagannathan K, Franklin TR, Wetherill RR, Langleben DD, Gawyrsiak M, Kampman KM, Childress AR (2021) Sustained brain response to repeated drug cues is associated with poor drug-use outcomes. Addict Biol 26:e13028
Ricci V, Di Salvo G, Maina G (2022) Remission of persistent methamphetamine-induced psychosis after cariprazine therapy: presentation of a case report. J Addict Dis 40(1):145–148
Rice OV, Gardner EL, Heidbreder CA, Ashby CR Jr (2012) The acute administration of the selective dopamine D(3) receptor antagonist SB-277011A reverses conditioned place aversion produced by naloxone precipitated withdrawal from acute morphine administration in rats. Synapse 66:85–87
Richtand NM (2006) Behavioral sensitization, alternative splicing, and d3 dopamine receptor-mediated inhibitory function. Neuropsychopharmacology 31:2368–2375
Rodgers HM, Yow J, Evans E, Clemens S, Brewer KL (2019) Dopamine D1 and D3 receptor modulators restore morphine analgesia and prevent opioid preference in a model of neuropathic pain. Neuroscience 406:376–388
Rodgers HM, Lim SA, Yow J, Dinkins ML, Patton R, Clemens S, Brewer KL (2020) Dopamine D1 or D3 receptor modulators prevent morphine tolerance and reduce opioid withdrawal symptoms. Pharmacol Biochem Behav 194:172935
Rogers AH, Zvolensky MJ, Ditre JW, Buckner JD, Asmundson GJG (2021) Association of opioid misuse with anxiety and depression: a systematic review of the literature. Clin Psychol Rev 84:101978
Roman V, Gyertyan I, Saghy K, Kiss B, Szombathelyi Z (2013) Cariprazine (RGH-188), a D(3)-preferring dopamine D(3)/D(2) receptor partial agonist antipsychotic candidate demonstrates anti-abuse potential in rats. Psychopharmacology 226:285–293
Ron D, Jurd R (2005) The “ups and downs” of signaling cascades in addiction. Sci STKE 2005:re14
Rose JS, Branchey M, Wallach L, Buydens-Branchey L (2003) Effects of buspirone in withdrawal from opiates. Am J Addict 12:253–259
Ross SB (1991) Synaptic concentration of dopamine in the mouse striatum in relationship to the kinetic properties of the dopamine receptors and uptake mechanism. J Neurochem 56:22–29
Salin A, Lardeux V, Solinas M, Belujon P (2021) Protracted abstinence from extended cocaine self-administration is associated with hypodopaminergic activity in the VTA but not in the SNc. Int J Neuropsychopharmacol 24:499–504
Salloum IM, Brown ES (2017) Management of comorbid bipolar disorder and substance use disorders. Am J Drug Alcohol Abuse 43:366–376
Samaha AN, Khoo SY, Ferrario CR, Robinson TE (2021) Dopamine ‘ups and downs’ in addiction revisited. Trends Neurosci 44:516–526
Sanna A, Fattore L, Badas P, Corona G, Diana M (2021) The hypodopaminergic state ten years after: transcranial magnetic stimulation as a tool to test the dopamine hypothesis of drug addiction. Curr Opin Pharmacol 56:61–67
Schmidt BL, Tambeli CH, Barletta J, Luo L, Green P, Levine JD, Gear RW (2002) Altered nucleus accumbens circuitry mediates pain-induced antinociception in morphine-tolerant rats. J Neurosci 22:6773–6780
Schneider NG, Olmstead RE, Steinberg C, Sloan K, Daims RM, Brown HV (1996) Efficacy of buspirone in smoking cessation: a placebo-controlled trial. Clin Pharmacol Ther 60:568–575
Scuppa G, Tambalo S, Pfarr S, Sommer WH, Bifone A (2020) Aberrant insular cortex connectivity in abstinent alcohol-dependent rats is reversed by dopamine D3 receptor blockade. Addict Biol 25:e12744
Searle G, Beaver JD, Comley RA, Bani M, Tziortzi A, Slifstein M, Mugnaini M, Griffante C, Wilson AA, Merlo-Pich E, Houle S, Gunn R, Rabiner EA, Laruelle M (2010) Imaging dopamine D3 receptors in the human brain with positron emission tomography, [11C]PHNO, and a selective D3 receptor antagonist. Biol Psychiatry 68:392–399
Seeman P, Ko F, Willeit M, McCormick P, Ginovart N (2005) Antiparkinson concentrations of pramipexole and PHNO occupy dopamine D2(high) and D3(high) receptors. Synapse 58:122–128
Segal DM, Moraes CT, Mash DC (1997) Up-regulation of D3 dopamine receptor mRNA in the nucleus accumbens of human cocaine fatalities. Brain Res Mol Brain Res 45:335–339
Seif T, Chang SJ, Simms JA, Gibb SL, Dadgar J, Chen BT, Harvey BK, Ron D, Messing RO, Bonci A, Hopf FW (2013) Cortical activation of accumbens hyperpolarization-active NMDARs mediates aversion-resistant alcohol intake. Nat Neurosci 16:1094–1100
Self DW, Nestler EJ (1995) Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci 18:463–495
Senatorov VV, Damadzic R, Mann CL, Schwandt ML, George DT, Hommer DW, Heilig M, Momenan R (2015) Reduced anterior insula, enlarged amygdala in alcoholism and associated depleted von Economo neurons. Brain 138:69–79
Shafer RA, Levant B (1998) The D3 dopamine receptor in cellular and organismal function. Psychopharmacology 135:1–16
Shaik AB, Kumar V, Bonifazi A, Guerrero AM, Cemaj SL, Gadiano A, Lam J, Xi ZX, Rais R, Slusher BS, Newman AH (2019) Investigation of novel primary and secondary pharmacophores and 3-substitution in the linking chain of a series of highly selective and bitopic dopamine D3 receptor antagonists and partial agonists. J Med Chem 62:9061–9077
Shen H, Chen K, Marino RAM, McDevitt RA, Xi ZX (2021) Deletion of VGLUT2 in midbrain dopamine neurons attenuates dopamine and glutamate responses to methamphetamine in mice. Pharmacol Biochem Behav 202:173104
Smith RJ, Lobo MK, Spencer S, Kalivas PW (2013) Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways). Curr Opin Neurobiol 23:546–552
Smith LN, Bachus SE, McDonald CG, Smith RF (2015) Role of the D3 dopamine receptor in nicotine sensitization. Behav Brain Res 289:92–104
Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC (1990) Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature 347:146–151
Stanwood GD, McElligot S, Lu L, McGonigle P (1997) Ontogeny of dopamine D3 receptors in the nucleus accumbens of the rat. Neurosci Lett 223:13–16
Stanwood GD, Artymyshyn RP, Kung MP, Kung HF, Lucki I, McGonigle P (2000) Quantitative autoradiographic mapping of rat brain dopamine D3 binding with [(125)I]7-OH-PIPAT: evidence for the presence of D3 receptors on dopaminergic and nondopaminergic cell bodies and terminals. J Pharmacol Exp Ther 295:1223–1231
Statement-from-FDA-Commissioner (2018) Statement from FDA Commissioner Scott Gottlieb, M.D., on new steps to encourage more widespread innovation and development of new treatments for opioid use disorder. https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-new-steps-encourage-more-widespread-innovation-and.accessed
Strain EC, Kampman KM, Weiss RD (2021) Moving beyond medications that act at the mu receptor in the treatment of opioid use disorder. JAMA Psychiat 78:701–702
Sullivan EV, Muller-Oehring E, Pitel AL, Chanraud S, Shankaranarayanan A, Alsop DC, Rohlfing T, Pfefferbaum A (2013) A selective insular perfusion deficit contributes to compromised salience network connectivity in recovering alcoholic men. Biol Psychiatry 74:547–555
Suzuki M, Hurd YL, Sokoloff P, Schwartz JC, Sedvall G (1998) D3 dopamine receptor mRNA is widely expressed in the human brain. Brain Res 779:58–74
Swain SN, Beuk J, Heidbreder CA, Beninger RJ (2008) Role of dopamine D3 receptors in the expression of conditioned fear in rats. Eur J Pharmacol 579:167–176
Tanda G, Hersey M, Hempel B, Xi ZX, Newman AH (2021) Modafinil and its structural analogs as atypical dopamine uptake inhibitors and potential medications for psychostimulant use disorder. Curr Opin Pharmacol 56:13–21
Taquet M, Holmes EA, Harrison PJ (2021a) Depression and anxiety disorders during the COVID-19 pandemic: knowns and unknowns. Lancet 398(10312):1665–1666
Taquet M, Luciano S, Geddes JR, Harrison PJ (2021b) Bidirectional associations between COVID-19 and psychiatric disorder: retrospective cohort studies of 62 354 COVID-19 cases in the USA. Lancet Psychiatry 8:130–140
Taylor DP, Riblet LA, Stanton HC, Eison AS, Eison MS, Temple DL Jr (1982) Dopamine and antianxiety activity. Pharmacol Biochem Behav 17(Suppl 1):25–35
Thomsen M, Barrett AC, Butler P, Negus SS, Caine SB (2017) Effects of acute and chronic treatments with dopamine D2 and D3 receptor ligands on cocaine versus food choice in rats. J Pharmacol Exp Ther 362:161–176
Trivedi MH, Walker R, Ling W, Dela Cruz A, Sharma G, Carmody T, Ghitza UE, Wahle A, Kim M, Shores-Wilson K, Sparenborg S, Coffin P, Schmitz J, Wiest K, Bart G, Sonne SC, Wakhlu S, Rush AJ, Nunes EV, Shoptaw S (2021) Bupropion and naltrexone in methamphetamine use disorder. N Engl J Med 384:140–153
Tsui JI, Mayfield J, Speaker EC, Yakup S, Ries R, Funai H, Leroux BG, Merrill JO (2020) Association between methamphetamine use and retention among patients with opioid use disorders treated with buprenorphine. J Subst Abus Treat 109:80–85
Tupala E, Hall H, Bergstrom K, Sarkioja T, Rasanen P, Mantere T, Callaway J, Hiltunen J, Tiihonen J (2001) Dopamine D(2)/D(3)-receptor and transporter densities in nucleus accumbens and amygdala of type 1 and 2 alcoholics. Mol Psychiatry 6:261–267
Volkow ND (2004) The reality of comorbidity: depression and drug abuse. Biol Psychiatry 56:714–717
Volkow ND (2021) Drug overdose deaths in 2020 were horrifying. Radical change is needed to address the drug crisis. Scientific America. https://www.drugabuse.gov/about-nida/noras-blog/2021/2008/drug-overdose-deaths-in-2020-were-horrifying-radical-change-needed-to-address-drug-crisis
Walker G (2018) The opioid crisis: a 21st century pain. Drugs Today (Barc) 54:283–286
Westrich L, Gil-Mast S, Kortagere S, Kuzhikandathil EV (2010) Development of tolerance in D3 dopamine receptor signaling is accompanied by distinct changes in receptor conformation. Biochem Pharmacol 79:897–907
Williamson A, Darke S, Ross J, Teesson M (2006) The effect of persistence of cocaine use on 12-month outcomes for the treatment of heroin dependence. Drug Alcohol Depend 81:293–300
Winhusen TM, Kropp F, Lindblad R, Douaihy A, Haynes L, Hodgkins C, Chartier K, Kampman KM, Sharma G, Lewis DF, VanVeldhuisen P, Theobald J, May J, Brigham GS (2014) Multisite, randomized, double-blind, placebo-controlled pilot clinical trial to evaluate the efficacy of buspirone as a relapse-prevention treatment for cocaine dependence. J Clin Psychiatry 75:757–764
Wise RA (1996) Addictive drugs and brain stimulation reward. Annu Rev Neurosci 19:319–340
Wise RA (2005) Forebrain substrates of reward and motivation. J Comp Neurol 493:115–121
Wrase J, Makris N, Braus DF, Mann K, Smolka MN, Kennedy DN, Caviness VS, Hodge SM, Tang L, Albaugh M, Ziegler DA, Davis OC, Kissling C, Schumann G, Breiter HC, Heinz A (2008) Amygdala volume associated with alcohol abuse relapse and craving. Am J Psychiatry 165:1179–1184
Xi ZX, Stein EA (2002) GABAergic mechanisms of opiate reinforcement. Alcohol Alcohol 37:485–494
Xi ZX, Li X, Li J, Peng XQ, Song R, Gaal J, Gardner EL (2013) Blockade of dopamine D3 receptors in the nucleus accumbens and central amygdala inhibits incubation of cocaine craving in rats. Addict Biol 18:665–677
Xu W, Reith MEA, Liu-Chen LY, Kortagere S (2019) Biased signaling agonist of dopamine D3 receptor induces receptor internalization independent of beta-arrestin recruitment. Pharmacol Res 143:48–57
Yang J, Villar VAM, Jose PA, Zeng C (2021) Renal dopamine receptors and oxidative stress: role in hypertension. Antioxid Redox Signal 34:716–735
Yawata S, Yamaguchi T, Danjo T, Hikida T, Nakanishi S (2012) Pathway-specific control of reward learning and its flexibility via selective dopamine receptors in the nucleus accumbens. Proc Natl Acad Sci U S A 109:12764–12769
You ZB, Gao JT, Bi GH, He Y, Boateng C, Cao J, Gardner EL, Newman AH, Xi ZX (2017) The novel dopamine D3 receptor antagonists/partial agonists CAB2-015 and BAK4-54 inhibit oxycodone-taking and oxycodone-seeking behavior in rats. Neuropharmacology 126:190–199
You ZB, Bi GH, Galaj E, Kumar V, Cao J, Gadiano A, Rais R, Slusher BS, Gardner EL, Xi ZX, Newman AH (2019) Dopamine D3R antagonist VK4-116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology 44:1415–1424
Yun IA, Fields HL (2003) Basolateral amygdala lesions impair both cue- and cocaine-induced reinstatement in animals trained on a discriminative stimulus task. Neuroscience 121:747–757
Zale EL, Dorfman ML, Hooten WM, Warner DO, Zvolensky MJ, Ditre JW (2015) Tobacco smoking, nicotine dependence, and patterns of prescription opioid misuse: results from a nationally representative sample. Nicotine Tob Res 17:1096–1103
Zarrindast MR, Dinkoub Z, Homayoun H, Bakhtiarian A, Khavandgar S (2002) Dopamine receptor mechanism(s) and morphine tolerance in mice. J Psychopharmacol 16:261–266
Zeng C, Sanada H, Watanabe H, Eisner GM, Felder RA, Jose PA (2004) Functional genomics of the dopaminergic system in hypertension. Physiol Genomics 19:233–246
Zeng C, Asico LD, Yu C, Villar VA, Shi W, Luo Y, Wang Z, He D, Liu Y, Huang L, Yang C, Wang X, Hopfer U, Eisner GM, Jose PA (2008) Renal D3 dopamine receptor stimulation induces natriuresis by endothelin B receptor interactions. Kidney Int 74:750–759
Zhang D, Zhang H, Jin GZ, Zhang K, Zhen X (2008) Single dose of morphine produced a prolonged effect on dopamine neuron activities. Mol Pain 4:57
Zhang Y, Zhang F, Yang C, Jin H, Yang Y, Xu M (2012) Dopamine affects the change of pain-related electrical activity induced by morphine dependence. Neurochem Res 37:977–982
Zhang X, Le HT, Zhang X, Zheng M, Choi BG, Kim KM (2016a) Palmitoylation on the carboxyl terminus tail is required for the selective regulation of dopamine D2 versus D3 receptors. Biochim Biophys Acta 1858:2152–2162
Zhang X, Sun N, Zheng M, Kim KM (2016b) Clathrin-mediated endocytosis is responsible for the lysosomal degradation of dopamine D3 receptor. Biochem Biophys Res Commun 476:245–251
Zhou X, Qin B, Del Giovane C, Pan J, Gentile S, Liu Y, Lan X, Yu J, Xie P (2015) Efficacy and tolerability of antidepressants in the treatment of adolescents and young adults with depression and substance use disorders: a systematic review and meta-analysis. Addiction 110:38–48
Acknowledgements
The authors acknowledge all our colleagues and collaborators who have worked with us over the years to investigate the D3R and its role in SUD. A.H.N. and Z.-X.X. acknowledge support from the National Institute on Drug Abuse-Intramural Research Program (ZIADA000424).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply
About this chapter

Cite this chapter
Newman, A.H., Xi, ZX., Heidbreder, C. (2022). Current Perspectives on Selective Dopamine D3 Receptor Antagonists/Partial Agonists as Pharmacotherapeutics for Opioid and Psychostimulant Use Disorders. In: Boileau, I., Collo, G. (eds) Therapeutic Applications of Dopamine D3 Receptor Function. Current Topics in Behavioral Neurosciences, vol 60. Springer, Cham. https://doi.org/10.1007/7854_2022_347
Download citation
DOI: https://doi.org/10.1007/7854_2022_347
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23057-8
Online ISBN: 978-3-031-23058-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)