
Abstract
From embryonic neuronal migration to adolescent circuit refinement, the immune system plays an essential role throughout central nervous system (CNS) development. Immune signaling molecules serve as a common language between the immune system and CNS, allowing them to work together to modulate brain function both in health and disease. As the resident CNS macrophage, microglia comprise the majority of immune cells in the brain. Much like their peripheral counterparts, microglia survey their environment for pathology, clean up debris, and propagate inflammatory responses when necessary. Beyond this, recent studies have highlighted that microglia perform a number of complex tasks during neural development, from directing neuronal and axonal positioning to pruning synapses, receptors, and even whole cells. In this chapter, we discuss this literature within the framework that immune activation during discrete windows of neural development can profoundly impact brain function long-term, and thus the risk of neurodevelopmental and neuropsychiatric disorders. In this chapter, we review three sensitive developmental periods – embryonic wiring, early postnatal synaptic pruning, and adolescent circuit refinement – in order to highlight the diversity of functions that microglia perform in building a brain. In reviewing this literature, it becomes obvious that timing matters, perhaps more so than the nature of the immune activation itself; largely conserved patterns of microglial response to diverse insults result in different functional impacts depending on the stage of brain maturation at the time of the challenge.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Adolescence
- Circuit refinement
- Embryonic
- Inhibitory
- Microglia
- Neurodevelopment
- Neuroimmunology
- Neuroinflammation
- Sensitive periods
- Synaptic pruning
1 Introduction
Our appreciation of the immune system’s power to impact brain maturation is not new. Some of the strongest foundational evidence in support of the claim that the immune system can profoundly impact brain development comes from epidemiological studies. Such studies identified maternal infection with a variety of pathogens during pregnancy, including influenza, streptococcus, and toxoplasma, as a strong risk factor for neurodevelopmental disorders such as autism spectrum disorder (ASD) and schizophrenia in their offspring (Atladóttir et al. 2010; Brown 2012; Knuesel et al. 2014). Indeed, initiating maternal immune activation (MIA) by injection of a bacterial or viral compound during gestation in a rodent induces behavioral abnormalities in offspring consistent with human ASD and schizophrenia (Shi et al. 2003; Zuckerman et al. 2003; Malkova et al. 2012; Choi et al. 2016). The diversity of viral, bacterial, and parasitic contributions that influence similar neurological outcomes suggests that perturbation of offspring neural function is not due to the pathogens themselves, but rather the MIA that occurs in response to the pathogen (Estes and McAllister 2016). For example, administration of pro-inflammatory cytokines IL-6 or IL-17a in the absence of any pathogen is sufficient to induce behavioral and neurobiological abnormalities in offspring (Smith et al. 2007; Choi et al. 2016). Further, blocking the activity of these cytokines rescues many MIA-induced deficits (Smith et al. 2007; Choi et al. 2016; Wu et al. 2017; Shin Yim et al. 2017). These findings provide strong evidence that immune molecules must have some avenue through which they can impact the developing brain.
Immune cells that primarily reside in the periphery, such as lymphocytes and myeloid cells, can be found in low densities in the CNS in the absence of illness or disease (Banks and Erickson 2010). The CNS was once considered entirely immune-privileged, although neuroimmune discoveries in recent years have challenged that idea (Louveau et al. 2015). Skin grafting experiments from the 1940s where the rabbit brain parenchyma was found to be immunologically tolerant to allogenic skin grafts served as the foundational evidence for the brain being a unique immunological site (Medawar 1948). The CNS’ residence behind the physical protection of the blood-brain barrier supports this claim; in a healthy adult, both potential pathogens and circulating immune cells are generally blocked from entry by a tight system of endothelial cells, pericytes, and astrocytic endfeet (Daneman and Prat 2015). Despite this, there are still a variety of immunocompetent cells, both peripheral and resident, that are present and play active roles in building the brain. In the absence of immune challenge, very sparse numbers of lymphocytes, such as T cells and B cells, and other myeloid cells, such as dendritic cells and mast cells, can be found in the rodent brain during embryonic development (Tanabe and Yamashita 2018; Lenz et al. 2018; Bulloch et al. 2008). They continue their residency postnatally, performing broad functions ranging from B1a cells supporting the proliferation of oligodendrocyte precursor cells to mast cells mediating the masculinization of sex-specific brain regions and behaviors (Tanabe and Yamashita 2018; Lenz et al. 2018).
Like other organs in the body, the brain has its own tissue-resident macrophage: microglia. Comprising approximately 80% of the immune cells in the adult rodent CNS, microglia are the primary immune sentinel of the brain (Mrdjen et al. 2018). Once thought to be “resting” in the absence of a threat to homeostasis, we now recognize microglia as highly active cells, constantly surveilling their environment for changes in homeostasis (Nimmerjahn et al. 2005; Davalos et al. 2005; Wake et al. 2009; Carrier et al. 2020). Microglia rapidly respond to injury or pathogens by changing their morphology, moving to sites of damage, phagocytosing debris, and releasing factors to escalate an immune response (Davalos et al. 2005; Nimmerjahn et al. 2005; Haynes et al. 2006; Wolf et al. 2017; Eyo et al. 2018; Galloway et al. 2019). Although long recognized as critical players in neurodegeneration and trauma, the specific evidence outlining the wide range of microglial contributions across development is relatively underexplored. Here we chose to focus on the roles of microglia across brain maturation, focusing in on three periods when the impact of immune perturbations is particularly strong, and in many cases, long-lasting: embryonic wiring, postnatal experience-dependent pruning, and circuit refinement during adolescence. Microglial activity during these three stages is both highly dynamic and developmentally critical, positioning these immune cells as major facilitators of disruption during key sensitive periods of brain development. The recognition of sex differences in both the specific timing of microglial functions and the mechanisms they employ across brain maturation is of critical importance. While not the focus of this review, we highlight key findings when relevant (see Bordt et al. 2020 and VanRyzin et al. 2020 for detailed reviews). Additionally, we focus primarily on the rodent literature, as this is where most of our mechanistic knowledge comes from, and refer to human evidence where available.
2 Embryonic Wiring
Both their keen surveillance abilities and non-CNS origins make microglia particularly sensitive to perturbations during gestation. Unlike the other glia, oligodendrocytes and astrocytes, which are born from neural stem cells beginning around mouse embryonic day (E)16, most microglia colonize the brain beginning around E9 after traveling there from the yolk sac, either directly through the blood or through the fetal liver and aorta-gonad-mesonephros (AGM) (Ginhoux et al. 2010; Hoeffel and Ginhoux 2015). This ontogeny is almost entirely unique to brain-resident macrophages, both in rodent and human embryos, as other macrophages throughout the body do not share it (Sheng et al. 2015; Bian et al. 2020). The necessary journey of embryonic microglia to the brain, as well as their early residence prior to the closing of the blood-brain barrier at E13.5, leaves microglia vulnerable to react to signals in the periphery (Profaci et al. 2020). In the absence of disease or injury, once the blood-brain barrier closes at E13.5, the microglia that originally seeded the brain will expand and self-renew throughout the lifespan of the organism, suggesting that even small changes to these early microglia could profoundly impact the future brain (Askew and Gomez-Nicola 2018; Thion and Garel 2020). There is also evidence that another, smaller subpopulation of microglia, homeobox (Hoxb8)-lineage microglia, colonize the brain a few days later, traveling from the yolk sac and through the fetal liver and AGM to appear in the brain around E12.5 (Chen et al. 2010; De et al. 2018). Interestingly, loss of Hoxb8+ microglia resulted in an overall reduction of microglia in the brain and a pathological over-grooming phenotype (Chen et al. 2010). While little is known about microglial heterogeneity and its relevance in health and disease, the identification of a developmentally and phenotypically distinct microglial population strongly supports that different subsets of microglia may be programmed to perform specific functions from very early in life and requires further investigation (see Box 1).
Box 1 Microglial Heterogeneity in Development
Microglia are specialized cells that continually sense, interpret, and react to environmental cues. Therefore, the traditional view that microglial functional heterogeneity, or distinct microglial “subtypes,” would develop from signals in their surroundings is logical. In the context of disease, microglia display distinct transcriptional patterns that seem to be in response to extrinsic cues (Keren-Shaul et al. 2017; Mrdjen et al. 2018). However, evidence is mounting that microglial heterogeneity may also have some intrinsic predetermination with great relevance for brain development. Hoxb8-lineage microglia display unique early postnatal dynamics relative to non-Hoxb8 cells, and the loss of the Hoxb8+ population results in an overall decrease in microglia in the brain (Chen et al. 2010; De et al. 2018). This is in spite of the fact that both populations have very similar transcriptional profiles (Chen et al. 2010; De et al. 2018). Studies investigating the two ligands for microglial CSF1R reveal a similar requirement for both populations; loss of CSF1 resulted in white-matter microglial density and functional reduction, while IL-34 loss impacted microglial density and function mostly in gray-matter regions (Kondo and Duncan 2009; Wang et al. 2012). Additionally, microglia populate the brain through self-renewal after traveling there during healthy embryonic development, so one study sought to determine how microglia are transcriptionally and epigenetically synchronized to perform all their necessary functions from these original seeding cells. Results from chromatin accessibility and histone modification investigation revealed specific hallmarks across three phases: early (until E14), pre- (until P0), and adult microglia (Matcovitch-Natan et al. 2016). A later single-cell transcriptomic study that sequenced mouse microglia across development revealed nine distinct subtypes of microglia from E14.5 to adulthood (Hammond et al. 2019). Interestingly, there was the greatest diversity of microglial subtypes in the earliest stages of development, E14.5 and P5 (Hammond et al. 2019). These subtypes were confirmed by localization of distinct microglial populations to particular brain regions, illustrated by single-molecule fluorescent in situ hybridization (smFISH) experiments (Hammond et al. 2019). Together, these studies support that microglial heterogeneity exists in early brain development, although the consequences of disrupting subpopulations of microglia are mostly still unknown. Recent work characterizing the impacts of a combined environmental stress MIA model on the early postnatal anterior cingulate cortex of male offspring found that MIA induced greater microglial heterogeneity, measured by protein expression, compared to controls (Block et al. 2020). Strikingly, the different populations of microglia had varied phagocytic capacity, and therefore their unbalanced presence resulted in a loss of characteristic early postnatal synaptic elimination (Block et al. 2020). This study provides some of the first evidence that microglial heterogeneity changes, such as downregulation of a critical microglial subtype and its functions, may contribute to neurodevelopmental disorder etiology. These and other outstanding questions about microglial heterogeneity highlight the need for more precise tools to modulate microglial function. Promising new work utilizing a Cre-dependent DREADD (designer receptors exclusively activated by designer drugs) system demonstrated that microglial activity can be bi-directionally modulated in a specific brain region through chemogenetic manipulation (Klawonn et al. 2021). This is especially important because we know that microglia in different brain regions vary in a number of ways, including their gene expression patterns, homeostatic signals, phagocytic function, density, physical shape, and, particularly relevant to this discussion, the timing of their colonization, expansion, and maturation (Tan et al. 2020). The ability to change the functional properties of microglia in a region-specific manner opens many doors for future work characterizing microglial heterogeneity.
As immune challenges during this perinatal period fundamentally alter neurobiology for the remainder of the lifespan (for example, in cases of MIA), it could be theorized that dampening microglial function during embryonic development may help prevent damage. However, mice lacking colony-stimulated factor 1 receptor (CSF1R), a receptor necessary for microglial growth and survival, have no microglia and die shortly after birth (Nandi et al. 2012; Erblich et al. 2011; Chitu et al. 2016). CSF1R−/− mice also display anatomical deformities, such as reduced brain size, cortical thickness, and corpus callosum axonal crossing, suggesting that microglia are indispensable for embryonic brain development (Nandi et al. 2012; Erblich et al. 2011; Chitu et al. 2016). Recent examination of the first identified human patients with homozygous CSF1R mutations, individuals almost entirely without microglia, confirms their necessity. One living patient with a homozygous missense mutation in the CSF1R gene did not show severe developmental regression until 12 years old, while an individual with a more severe splice mutation in the CSF1R gene lived to only 10 months old (Oosterhof et al. 2019). Robust white matter irregularities, epilepsy, and incorrect accumulations of neurons in both these patients’ brains support that microglia are of particular importance for both the development of oligodendrocytes and myelination, as well as proper interneuron migration in the cortex (Oosterhof et al. 2019). Interestingly, interneuron dysfunction and white matter abnormalities are two of the primary neuropathological hallmarks of schizophrenia, identified in both rodent models and human studies (Lewis et al. 2012; Kochunov and Elliot Hong 2014; Gonzalez-Burgos et al. 2015; Cetin-Karayumak et al. 2020). Together, paired with the strong evidence of MIA risk factors, these studies support immune involvement in the building of the embryonic brain, specifically through some action of microglia.
Once migrated from the yolk sac, microglia localization in the developing embryonic brain is precisely timed. For example, at E12.5, around when microglia are first colonizing the mouse brain, they distribute fairly evenly throughout the forebrain (Squarzoni et al. 2014). However, at embryonic day E14.5, microglia become localized around migrating axonal tract “decision” regions, avoiding the cortical plate until E16.5 (Squarzoni et al. 2014). Around birth, microglia become most associated with progenitor zones and areas of cell death in both rodents and non-human primates (Cunningham et al. 2013). This localization aligns with the critical roles microglia play in embryonic brain development. Using two different microglial depletion methods, Squarzoni and colleagues found that functional microglia are necessary for proper dopaminergic axon extension at E14.5 in mice, and in the absence of microglia, there was an overgrowth of tyrosine-hydroxylase (TH) + axons into the subpallium (Squarzoni et al. 2014). In contrast, an E13.5 lipopolysaccharide (LPS) injection MIA model to “activate” microglia reduced dopaminergic axon outgrowth at E14.5 to below control levels (Squarzoni et al. 2014). The discovery of TH+ fragments of dopaminergic axons inside of microglia in control mice provided further evidence that perturbing microglia in this window impacts their ability to direct, perhaps through phagocytosis, dopaminergic circuit organization during embryonic development (Squarzoni et al. 2014). Beyond this, both depletion and MIA models impacted the organization of neocortical inhibitory neurons, resulting in both altered migration timing and ultimately defective localization in the somatosensory cortex (Squarzoni et al. 2014).
Microglial involvement in the development of inhibitory neurons has long been suspected based on dysfunction consistently identified in both cell types across several neuropsychiatric disorders, in particular those with proposed early developmental origins (Thion and Garel 2020). Both postmortem analysis and live imaging of brains of human patients suffering from schizophrenia point towards aberrant GABAergic interneurons in psychopathology, such as through reduced gene expression of general GABAergic genes in the frontal cortices of schizophrenia patients compared to controls (Nakazawa et al. 2012; Hoftman et al. 2015; de Jonge et al. 2017). Administration of poly I:C, a synthetic double-stranded RNA to mimic viral infection, to mouse dams at E9 to induce MIA reduced parvalbumin (PV) interneuron GABAergic transmission onto pyramidal neurons in brain regions important for schizophrenia pathology in offspring, consistent with human postmortem literature (Canetta et al. 2016; Kaar et al. 2019). In 2019, Thion et al. dissected the embryonic impact of both E13.5 LPS injection and transient macrophage depletion (from E7-birth) on PV interneurons in the somatosensory barrel cortex. Both microglial perturbations revealed a two-phase alteration to PV firing (Thion et al. 2019). In juveniles (P20), there was a mild increase in PV density and an excess of inhibition onto principal excitatory neurons in the barrel cortex in both MIA and depletion offspring. By adulthood, however, the increase in interneuron density was resolved and PV cells were instead hypoactive, exhibiting reduced inhibition onto layer 4 cortical neurons (Thion et al. 2019). These findings are particularly interesting for two reasons. First, although inhibitory dysfunction is a hallmark of neurodevelopmental disorders, this is the first work to directly implicate developmental alterations in microglial activity to a change in inhibitory firing. Second, due to the consistent phenotype across both depletion and MIA, this finding supports the idea that the effect of MIA on microglia may not just be excessive reactivity, but instead a “distraction” from the critical developmental processes performed by microglia during specific periods. These findings also lend support to the hypothesis that the nature of the microglial perturbation may be less important than the specific period during which it occurs, as it is the timing of the challenge that reveals which microglial function and, therefore, which critical aspect of development, is altered.
Microglia are critical for regulating both the precursor cell pool and the functional capabilities of neurons that survive in both rodents and primate species. During the later half of cortical neurogenesis, E80-E100 in a rhesus macaque or E20-P0 in a rat, microglia eliminate excess neural and glial precursor cells through a phagocytosis mechanism in the proliferative zones of the brain (Cunningham et al. 2013). LPS injections administered to pregnant rats on E15 and E16 resulted in a significant reduction in proliferative zone neurons in offspring at E19 and P2 (Cunningham et al. 2013). Interestingly, concurrent exposure of rat offspring to MIA and doxycycline to suppress microglial activation resulted in a persistent increase in neural precursor cells (Cunningham et al. 2013). These findings suggest that conditions that may decrease microglial activation prenatally may result in the long-term survival of excess neurons at a detriment to the developing brain.
Together, these studies highlight the long-term impacts of microglial perturbations during the gestational period, as either dampening or “over-activating” microglial function both result in negative outcomes for developing neurons. Interestingly, these studies do not directly address any sex differences that may emerge in the response to either kind of microglial perturbation. This is despite the strong male sex-bias in disorders that we suspect become programmed during this period, as well as the male-specific androgen surge around P0 and subsequent critical period for sexual differentiation (McCarthy et al. 2018; VanRyzin et al. 2018; Bordt et al. 2020). Additionally, many questions remain about how and why some neuronal populations are differentially impacted by microglial dysfunction. What external cues or intrinsic factors direct microglia to populate certain zones during specific time windows? How do embryonic microglia differentiate between specific subtypes of neurons, such as in their preferential directing of dopaminergic axons or diverse impacts on excitatory- vs inhibitory-fated neuronal populations? These questions highlight just how much there is left to uncover about the heterogeneous roles that microglia play in shaping the embryonic brain.
3 Postnatal Synaptic Pruning and Neuronal Function
The discovery that microglia are constantly mobile and surveying their environment during homeostasis raises questions regarding what kinds of functions they must be performing to justify such a heavy metabolic need (Nimmerjahn et al. 2005; Davalos et al. 2005; Wake et al. 2009). We now know microglia to be attentive gardeners, carefully pruning damage and overgrowth across development in order to shape and encourage healthy and resilient maturation. More than a decade ago, the first evidence that microglial contact with synapses was indeed experience-driven was identified in the primary visual cortex (V1) by modulating light exposure during the visual critical period (Tremblay et al. 2010). Two-photon in vivo imaging revealed that following 6 days of dark adaptation, microglia were less mobile, had more process phagocytic structures, and changed their preference for contacting specific subsets of dendritic elements (Tremblay et al. 2010). Some of these changes could be partially reversed by 2 days of light re-exposure (Tremblay et al. 2010). Soon after, the role of microglia in the engulfment of synaptic material was supported by a study where chemokine fractalkine receptor (CX3CR1), primarily expressed by microglia in the CNS, was knocked out in mice. In the absence of this receptor, microglia still engulfed PSD-95+ puncta in early postnatal synapse refinement, but the overall density of synaptic material and spines was significantly greater in the knockouts at P15 in the hippocampus (Paolicelli et al. 2011). Additional experiments revealed that these extra connections were electrophysiologically immature, strongly pointing to a deficit in synaptic pruning by microglia during this period (Paolicelli et al. 2011). By the time of this discovery, the involvement of the classical complement cascade in developmental pruning of synapses in the dorsal lateral geniculate nucleus (dLGN) had been established (Stevens et al. 2007). Early in development, this region of the thalamus receives overlapping projections from the retinal ganglion cells of both eyes. Eventually the mature pattern of non-overlapping inputs develops following activity-dependent selection of projections from a singular eye, making it a great system to study synapse elimination (Shatz 1990). The complement component 1q (C1q) protein was found to be present on synaptic elements early postnatally, and knockout of C1q resulted in deficient elimination of overlapping inputs from both eyes in the normally developmentally-segregated input areas at P30 (Stevens et al. 2007). Again utilizing the early postnatal developing mouse dLGN system, Schafer and colleagues found that microglia prune presynaptic inputs by engulfment in a complement receptor 3 (CR3)/C3 dependent manner (Schafer et al. 2012). Modulation of neural activity by intraocular injection of tetrodotoxin to weaken inputs from one eye resulted in an increase in microglial phagocytosis of those inputs, while forskolin injection to enhance activity resulted in greater pruning of inputs from the other eye (Schafer et al. 2012). This was one of the first hints that microglia may work to shape circuits in health and disease, and while our understanding of these processes has grown immensely, some of the questions raised almost a decade ago still remain unanswered. For example, do microglia detect the activity levels of nearby neurons in order to “decide” which synaptic elements require removal? Could microglia instead be responding to inputs that have already been identified through a different, unknown mechanism for elimination?
Although the extent of microglial involvement in postnatal developmental synaptic pruning remains unknown, several potential pathways have been identified. We now recognize that the innate immune triggering receptor expressed on myeloid cells 2 (TREM2) is a necessary part of the synaptic pruning process in mice. When expressed on microglia, TREM2 primarily recognizes LPS and other danger molecules and generally performs anti-inflammatory functions (Daws et al. 2003; Wang et al. 2015). TREM2−/− mice showed a CA1-specific decrease in microglia density and activation markers, as well as an increase in excitatory synaptic markers and electrophysiological activity from P18–20 (Filipello et al. 2018). Further, both in vitro and in vivo at P18–20, TREM2−/− microglia were insufficient at phagocytosing synapses, strengthening the finding that these mutants had reduced brain region connectivity, increased repetitive behaviors, and social behavior deficits at P90 (Filipello et al. 2018). Interestingly, a separate study found that TREM2−/− animals had a reduction in synapses in both the cortex and hippocampus at 1 month of age and that KO microglia contained more internalized synaptic elements (Jay et al. 2019). Beyond this, loss of TREM2 in microglia seemed to enhance synapse element phagocytosis by astrocytes in brain regions with reduced microglial density (Jay et al. 2019). This finding is not entirely surprising; astrocytes, while only briefly discussed here, play an extremely important role in synaptic construction and maintenance (see Box 2). While the exact contribution of microglial TREM2 in postnatal synaptic refinement is still up for debate, these studies highlight microglia as complicated, interconnected players in the developing brain environment.
Box 2 Astrocytic Involvement in Synaptic Development
Although not from immune cell origins and therefore highly abundant in the CNS, astrocytes are necessary immunocompetent contributors to circuit construction and refinement. Astrocytes are intimately involved in synaptic transmission, where one rodent astrocyte can contact over 100,000 synaptic elements to monitor and modulate signaling, both by releasing their own factors and through regulation of those released into the synaptic cleft (Bushong et al. 2002; Chung et al. 2015). Therefore, it is unsurprising that astrocytes are also intimately involved in the creation and modulation of synapses across development (Farhy-Tselnicker and Allen 2018). Astrocyte-derived signals, such as thrombospondins, glypicans, and Hevin/Sparcl1, contribute to the proper construction of a synapse, and in some cases, specific signals are necessary for the development of certain kinds of synapses (Risher et al. 2014; Allen and Eroglu 2017). Additionally, evidence that supports the importance of cross-talk between astrocytes and microglia in circuit refinement continues to grow. For example, the presence of astrocytes is necessary for retinal ganglion cells (RGCs) to upregulate C1q, even though it is microglia that respond to the complement call (Stevens et al. 2007; Schafer et al. 2012). More recently, this is highlighted by findings such as the identification of astrocyte-produced cytokine interleukin (IL)-33 as important for modulation of microglial phagocytosis during the early postnatal period (Vainchtein et al. 2018).
Microglia are exposed to and need to interpret many signals from other cells in their environment. For example, neuron-derived CX3CL1 signals through microglial CX3CR1. In order to effectively target the correct synaptic elements for removal, microglia also recognize both “don’t eat me” signals, such as CD47, as well as contrasting “eat me” signals (Arcuri et al. 2017; Lehrman et al. 2018). One such recently identified signal is exposed phosphatidylserine (ePS), which is displayed by neurons as an “eat me” signal for microglia to recognize (Scott-Hewitt et al. 2020). Across brain regions, ePS is most present during critical windows of pruning, and in the dLGN this expression is co-localized with C1q labeling (Scott-Hewitt et al. 2020). In vitro, synaptic pruning of ePS by microglia was TREM2-dependent, while in vitro, in C1q-KO mice, there was both an increase in ePS tagged presynaptic elements and a reduction of microglial engulfment without C1q present (Scott-Hewitt et al. 2020). This study highlights one neuronal “tag” that initiates two different microglial pruning mechanisms. Interestingly, astrocytes can also recognize phosphatidylserine through phagocytic receptors MERTK and MEGF10 to eliminate synapses in the dLGN in an activity-dependent manner, both during the first week of life and throughout adulthood (Chung et al. 2013). This alternate phagocytosis pathway is not unique to mammals, suggesting it may have some important function independent of those in microglia (MacDonald et al. 2006; Chung et al. 2013). Further, microglia can also signal through the cannabinoid system. Van Ryzin and colleagues discovered that microglia phagocytose newborn cells in the rat amygdala in the first few days of life, a process that is regulated by androgen and endocannabinoids (VanRyzin et al. 2019). When engulfment of these cells, identified as primarily newborn astrocytes, by microglia is blocked through the administration of an anti-CD11b antibody, dramatic sex differences that are well-characterized in juvenile rat play behaviors are lost (VanRyzin et al. 2019). Even though microglial engulfment of whole cells early in brain development is not new or specific to rodents, this study was one of the first studies to directly implicate microglia in the development of social behavior (Cunningham et al. 2013; VanRyzin et al. 2019).
Recently, a new molecular pathway utilized by microglia to decrease synapses in the absence of phagocytosis was proposed. In 2018, Cheadle et al. discovered that following eye-opening at P7, fibroblast growth factor-inducible 14 (Fn14), a member of the tumor necrosis factor receptor family, was selectively expressed in excitatory thalamocortical neurons (TC) in the dLGN (Cheadle et al. 2018). Its presence was important for the maturation of TC neuron dendritic spines and synaptic refinement during the vision-sensitive period (Cheadle et al. 2020). Tumor necrosis factor-like weak inducer of apoptosis (TWEAK), which is strongly upregulated by microglia only following visual experience, binds postsynaptic Fn14 and contributes to spine elimination. This elimination is not phagocytosis-mediated, as microglia in TWEAK-KO mice do not show phagocytosis deficits at P7 or P27, but instead depends on the proximity of spines to TWEAK-expressing microglia (Cheadle et al. 2020). Interestingly, this novel refinement mechanism is only present after P20, prompting the authors to suggest a 2-phase model. In phase 1, microglia work via complement signaling to engulf presynapses. After eye-opening and sensory experience, there is a transition to phase 2, where postsynaptic refinement happens through the experience-dependent TWEAK-Fn14 mechanism (Cheadle et al. 2020). Perhaps showcasing exactly how much we have left to learn, the mechanisms by which microglia perform this second phase of elimination, as well as whether these are the same microglia that perform phase 1 engulfment or if this is a result of heterogeneity, is still unknown.
What are the long-term consequences for adult behavior when these microglia functions early in postnatal life are perturbed by an immune challenge? Genetic tools have allowed us to look closely at the loss of functional pathways to elucidate their critical functions, but may not elucidate broader disease mechanisms. Despite the clinical relevance of understanding the impacts of early postnatal/neonatal infection on brain development, such as the prevalence of postnatal illness and the potential for increased vulnerability to neuropsychiatric disorders, the animal studies of this nature are limited in comparison to the MIA literature. Those studies that have looked show that early postnatal immune challenges can have long-term consequences on adult behavior (Bilbo and Schwarz 2012). Interestingly, a “second-hit,” such as a subsequent immune challenge, seems to be consistently required in order to reveal these long-term changes. For example, administering E. coli to rats at P4 primes the system such that if they are exposed to another immune challenge in adulthood, their long-term memory becomes impaired (Bilbo et al. 2006). This effect is mediated by a CD11b + −derived IL-1β response, which becomes sensitized following the initial immune “hit” (Williamson et al. 2011). Indeed, even two LPS injections early in life, administered at P3 and P5 to male pups, are enough to induce spatial memory deficits in adulthood, measured by Morris water maze performance (Peng et al. 2019). Other studies highlight that the second hit in this two-hit model does not need to be a direct immune challenge to the pups. The long-term impact of in utero exposure to maternal high fructose diet was revealed following a P7 LPS injection in rats, where the authors observed aberrant anxiety-like behaviors both during adolescence and adulthood (Bukhari et al. 2018). Interestingly, recent work has shown that a single P4 LPS induced a female-specific decrease in both sociability and social discrimination, which was maintained even in the absence of microglial MyD88-dependent pro-inflammatory signaling (Smith et al. 2020). This finding provides strong support for the investigation of both males and females across brain development, as it seems some microglial mechanisms are not necessarily utilized by both sexes.
Studies such as these also serve as a reminder that changes at single-cell levels in spine density or synaptic refinement may have long-term behavioral consequences despite those perturbations happening early in development. However, immune system perturbations happen throughout the lifespan. From birth to old age, we all suffer illness and stress, which we know impact microglial function (Niraula et al. 2017; Mariani and Kielian 2009). However, not all microglial challenges result in long-term functional changes in our behavior. For example, depletion of microglia in the CNS in adulthood via an antagonist to CSF1R, the loss of which is lethal in development, has no impact on either viability or behavioral function in adult mice (Elmore et al. 2014). This supports the view that there are sensitive periods when microglia are particularly vulnerable to challenges, perhaps due to special windows of rigorous and concentrated actions. Beyond the obvious highly active periods of brain construction detailed above, the literature suggests there is another sensitive window for microglia that extends beyond the initial weeks of life: adolescence.
4 Adolescent Circuit Refinement
The adolescent period, usually defined as around 10–20 years of age in humans, is behaviorally characterized by increased risk-taking behaviors and sensitivity to social cues and stress (Andersen and Teicher 2008; Steinberg 2008; Blakemore and Mills 2014). These changes are neuroanatomically mirrored by increased myelination and synaptic pruning in cortical and limbic areas (Giedd et al. 1999; Paus et al. 2008; Tamnes et al. 2013; Blakemore and Mills 2014). Adolescence is also the time when connectivity between diverse brain regions is refined and strengthened, resulting in changes in synaptic communication (Toga et al. 2006; Whitford et al. 2007). Analogous behavior and brain maturation also occurs in rodents, where adolescence is considered to be from around the time of weaning until sexual maturity, although the precise window is debated and varies by sex (Schneider 2013). The significance of the adolescent period as an important developmental stage is underscored by its identification as the primary age window for the appearance of schizophrenia, some affective disorders, and risk-taking behaviors that increase vulnerability to substance use disorders (Kessler et al. 2007; Paus et al. 2008; Steinberg 2008; Casey and Jones 2010). While the neuroimmune contribution to each of these disorders is not new, the literature investigating the homeostatic functions of microglia during the adolescent period of circuit refinement is still in its infancy.
Despite the relatively young body of adolescent microglia literature, it is clear that the actions of microglia across developmental periods have profound impacts on the brain and behavior during adolescence. Again, the long history of MIA studies reveal that embryonic perturbations of microglial function can culminate in eventual disease onset in adolescence (Brown 2012). As discussed in the previous section, perinatal perturbations of microglia can also profoundly impact the adolescent brain and behavior (VanRyzin et al. 2019). While important to our understanding of microglial roles in circuit refinement and the long-lasting effects of early perturbations of these processes, what these studies involving early life manipulations do not tell us is what may be different about microglia and their functions during the actual adolescent period. In the few transcriptional studies aiming to characterize microglial-specific gene expression changes across development, the adolescent period is entirely excluded (Matcovitch-Natan et al. 2016; Hanamsagar et al. 2017).
Across species, perturbations during the sensitive adolescent period have profound impacts on long-term behavior. It is well recognized that the brain at this time is highly neurobiologically vulnerable to addiction and social stress in humans (Chambers et al. 2003; Crews et al. 2007; Blakemore and Mills 2014). In rats, exposure to morphine during adolescence persistently impacted microglial TLR4 signaling and increased susceptibility to robust relapse of drug-seeking behavior later in life (Schwarz et al. 2011). This was not true for rats that were exposed to the same initial morphine paradigm as adults (Schwarz et al. 2011). Similarly, rodents that are exposed to chronic stressors as adolescents have increased anxiety-like behaviors weeks after exposure compared to those that went through the same stressors as adults (Yohn and Blendy 2017; Cotella et al. 2019). This is also true in the case of an acute, intense stressor, though this effect was specific to males (Lovelock and Deak 2019). Stress-based immune-activating stimuli during this period not only change behavior, but also have chronic effects on microglial function and activation (Schwarz and Bilbo 2013; McClain et al. 2011). Interestingly, administration of minocycline, a microglial activation inhibitor, can prevent the development of schizophrenia-like behaviors following an adolescent stressor in mice, providing more support for the critical role of microglia in both the initial wiring and the refinement of circuits up through the adolescent period (S. Giovanoli et al. 2016).
What are microglia actually doing during this time in the brain? Just as synaptic connections are refined early postnatally, pruning continues into rodent and human adolescence in brain structures known to continue to mature later in life, such as the prefrontal cortex (Petanjek et al. 2011; Delevich et al. 2018). While the quantification of this phenomena in the healthy brain is well observed and fairly consistent, only in 2019 were microglia first directly identified as a mechanism through which this reduction in synapses occurs during this period (Premachandran et al. 2020; Markham et al. 2013; Koss et al. 2014; Drzewiecki et al. 2016). Four days following the peak density of spines at P35 in the rat PFC, Mallya and colleagues observed a drastic increase in microglial engulfment of both dendritic spines and presynaptic elements. Interestingly, at P50, while engulfment of spines dropped below levels observed at P24, microglial engulfment of presynaptic elements continued to climb (Mallya et al. 2019). While the precise timing of these observations suggests microglia are at least partially responsible for the pruning of synapses in the PFC in adolescence, whether or not microglia are necessary for this process and the functional implications of their dysfunction remains unanswered.
Microglia appear to perform a different critical function in the adolescent development of reward circuit pathways, which are of particular importance in the context of addiction. In the nucleus accumbens (NAc), dopamine receptors that receive dopaminergic input from the ventral tegmental area drive social play behaviors in adolescent rats (Manduca et al. 2016). In 2018, Kopec and colleagues found that the volume of dopamine D1 receptors (D1rs) in the NAc across adolescence is regulated by complement-dependent microglial phagocytosis in male rats, but not in females (Kopec et al. 2018). Blocking C3 receptors through lateralized NAc injection of a CD11b-subunit competitive antagonist (neutrophil inhibitor factor, NIF) around the peak of D1r expression only impacted male D1r expression (Kopec et al. 2018). Natural male juvenile play behavior peaks at the same time that D1rs peak in the NAc (P30), so a similar paradigm of NIF administration revealed that a decrease in microglial phagocytosis around the D1r peak prolonged male play behaviors beyond their normal timeline (Kopec et al. 2018). Administration of siRNA against D1r alongside NIF confirmed that these changes in social play were D1r-dependent, as the effect of NIF alone was blocked by the siRNA administration (Kopec et al. 2018). Interestingly, the timeline for female rat D1r volume peaks earlier than in males and did not seem to be associated with complement C3, once again showcasing the sex differences in microglial developmental mechanisms (Kopec et al. 2018; Smith et al. 2020).
A number of studies utilizing a pubertal LPS challenge provide evidence that adolescent perturbations have particularly sex-dependent effects. In CD1 mice, administration of an LPS injection at P42 is sufficient to impair learning in a Barnes maze task in both males and females (Kolmogorova et al. 2019). However, the same LPS administration at P42 impacts anxiety-like, depression-like, and sexual behaviors in a female-specific manner long-term (Ismail et al. 2011; Olesen et al. 2011; Ismail et al. 2013). Given that adolescent-emerging affective disorders such as anxiety and depression are female-biased, this female susceptibility to immune challenge hints at an immune-relevant mechanism, although the role of microglia is underexplored (Bekhbat and Neigh 2018). Further, the requirement of a second hit, discussed previously, is not unique to early postnatal challenges. There is a long history to the “two-hit hypothesis” in the context of adolescent-onset schizophrenia, where it was postulated that a genetic or environmental first hit early in brain development primes the system to synergize with a later insult to elicit schizophrenia onset (Bayer et al. 1999; Maynard et al. 2001). Indeed, this can also be modeled in mice, where a study showed a low dose of poly I:C on E9 was not sufficient to induce schizophrenia-relevant behavioral deficits in adult offspring until paired with a chronic variable stress paradigm from P30–40 (Giovanoli et al. 2013). Taken together, it is clear that our understanding of the role of microglia and the immune system during the adolescent period requires more attention.
5 Conclusions
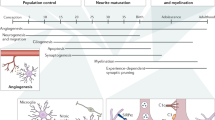
Across each stage of building the brain, microglia perform functions that are critical to the long-term successful functioning of neuronal circuitry and ultimately behavior (Fig. 1). The specific timing of a perturbation, perhaps more so than the nature of it, is most critical in predicting how it will ultimately impact the function of the organism. As the sentinels of the CNS, microglia are highly in tune with the brain environment and are quick to respond to changes, making their developmental functions highly sensitive to external stimuli. During the embryonic period, microglia are involved in directing axonal traffic to build the cortex, as well as ensuring that the correct number and type of neurons end up in the correct locations. Perturbations during this period, such as MIA, lead to persistent incorrect localization and maturation of neurons into adulthood in offspring. Interestingly, the same immune challenge administered to the early postnatal mouse seems to require a “second hit,” or subsequent immune challenge to elicit behavioral deficits later in life. Despite this difference, genetic studies confirm that microglia are indeed necessary for the critical somatosensory developmental period pruning of excess synapses, both through phagocytosis-dependent and -independent mechanisms. Finally, during the adolescent period, microglia work to refine circuits via synaptic and receptor phagocytosis mechanisms. Similarly, if unable to perform this receptor pruning, the stereotyped pattern of juvenile social behavior development is altered. While investigations into perturbations such as stress and drugs of abuse during adolescence are plentiful, there is little literature that allows us to compare the outcomes of the same challenge, such as microglial activation by LPS injection, across multiple sensitive periods in a continuous developmental timeline. Studies such as these would provide further insight into the functional changes of microglia across development, as well as continue to reveal the critical role of timing in the context of a single perturbation. Beyond this, new research continues to highlight how much work there is left to do to understand the mechanistic differences in microglial actions between the sexes. This work may prove to be critical to bettering our understanding of microglial dysfunction in the context of sex-biased neurological disorders.

Embryonic microglia (green) localize to “hotspots” at E14.5 and engulf TH+ dopaminergic fragments (magenta) to modulate and direct axon outgrowth (1). Both anti-CSF1R antibody administration and transgenic myeloid cell to deplete microglia result in an overgrowth of TH+ axons at E14.5 (2). In situ hybridization for lhx6 mRNA expressed in interneurons (blue) at E18.5 show that in the absence of microglia, specific laminar positioning in cortical layer V is lost (3, 4). In the early postnatal period, microglia (teal) recognize synaptic elements with weak inputs and phagocytose them in the complement-dependent manner (5). Loss of C1q, the complement cascade initiator, C3, or its receptor, CR3, results in deficient synaptic remodeling (6). Microglia engulf dopamine-1 receptors (D1Rs) in the adolescent rat nucleus accumbens (NAc) (7). After administration of a CD11b competitive antagonist to block microglia CR3 activation, there is a greater density of D1R (8). Data from Squarzoni et al. (2014), Stevens et al. (2007), Schafer et al. (2012), Kopec et al. (2018)
References
Allen NJ, Eroglu C (2017) Cell biology of astrocyte-synapse interactions. Neuron 96(3):697–708. https://doi.org/10.1016/j.neuron.2017.09.056
Andersen SL, Teicher MH (2008) Stress, sensitive periods and maturational events in adolescent depression. Trends Neurosci 31(4):183–191. https://doi.org/10.1016/j.tins.2008.01.004
Arcuri C, Mecca C, Bianchi R, Giambanco I, Donato R (2017) The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing CNS. Front Mol Neurosci 10. https://doi.org/10.3389/fnmol.2017.00191
Askew K, Gomez-Nicola D (2018) A story of birth and death: insights into the formation and dynamics of the microglial population. Brain Behav Immun 69:9–17. https://doi.org/10.1016/j.bbi.2017.03.009
Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET (2010) Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 40(12):1423–1430. https://doi.org/10.1007/s10803-010-1006-y
Banks WA, Erickson MA (2010) The blood–brain barrier and immune function and dysfunction. Neurobiol Dis 37(1):26–32. https://doi.org/10.1016/j.nbd.2009.07.031
Bayer TA, Falkai P, Maier W (1999) Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the ‘two hit hypothesis’. J Psychiatr Res 33(6):543–548. https://doi.org/10.1016/s0022-3956(99)00039-4
Bekhbat M, Neigh GN (2018) Sex differences in the neuro-immune consequences of stress: focus on depression and anxiety. Brain Behav Immun 67:1–12. https://doi.org/10.1016/j.bbi.2017.02.006
Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, Shi H et al (2020) Deciphering human macrophage development at single-cell resolution. Nature 582(7813):571–576. https://doi.org/10.1038/s41586-020-2316-7
Bilbo SD, Schwarz JM (2012) The immune system and developmental programming of brain and behavior. Front Neuroendocrinol 33(3):267–286. https://doi.org/10.1016/j.yfrne.2012.08.006
Bilbo SD, Rudy JW, Watkins LR, Maier SF (2006) A Behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res 169(1):39–47. https://doi.org/10.1016/j.bbr.2005.12.002
Blakemore S-J, Mills KL (2014) Is adolescence a sensitive period for sociocultural processing? Annu Rev Psychol 65(1):187–207. https://doi.org/10.1146/annurev-psych-010213-115202
Block CL, Eroglu O, Mague SD, Sriworarat C, Blount C, Malacon KE, Beben KA et al (2020) Prenatal environmental stressors impair postnatal microglia function and adult behavior in males. BioRxiv. https://doi.org/10.1101/2020.10.15.336669
Bordt EA, Ceasrine AM, Bilbo SD (2020) Microglia and sexual differentiation of the developing brain: a focus on ontogeny and intrinsic factors. Glia 68(6):1085–1099. https://doi.org/10.1002/glia.23753
Brown AS (2012) Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol 72(10):1272–1276. https://doi.org/10.1002/dneu.22024
Bukhari SHF, Clark OE, Williamson LL (2018) Maternal high fructose diet and neonatal immune challenge alter offspring anxiety-like behavior and inflammation across the lifespan. Life Sci 197:114–121. https://doi.org/10.1016/j.lfs.2018.02.010
Bulloch K, Miller MM, Gal-Toth J, Milner TA, Gottfried-Blackmore A, Waters EM, Kaunzner UW et al (2008) CD11c/EYFP transgene illuminates a discrete network of dendritic cells within the embryonic, neonatal, adult, and injured mouse brain. J Comp Neurol 508(5):687–710. https://doi.org/10.1002/cne.21668
Bushong EA, Martone ME, Jones YZ, Ellisman MH (2002) Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci 22(1):183–192. https://doi.org/10.1523/JNEUROSCI.22-01-00183.2002
Canetta S, Bolkan S, Padilla-Coreano N, Song LJ, Sahn R, Harrison NL, Gordon JA, Brown A, Kellendonk C (2016) Maternal immune activation leads to selective functional deficits in offspring parvalbumin interneurons. Mol Psychiatry 21(7):956–968. https://doi.org/10.1038/mp.2015.222
Carrier M, Robert M-È, Ibáñez FG, Desjardins M, Tremblay M-È (2020) Imaging the neuroimmune dynamics across space and time. Front Neurosci 14:903. https://doi.org/10.3389/fnins.2020.00903
Casey BJ, Jones RM (2010) Neurobiology of the adolescent brain and behavior: implications for substance use disorders. J Am Acad Child Adolesc Psychiatry 49(12):1189–1201. https://doi.org/10.1016/j.jaac.2010.08.017
Cetin-Karayumak S, Di Biase MA, Chunga N, Reid B, Somes N, Lyall AE, Kelly S et al (2020) White matter abnormalities across the lifespan of schizophrenia: a harmonized multi-site diffusion MRI study. Mol Psychiatry 25(12):3208–3219. https://doi.org/10.1038/s41380-019-0509-y
Chambers RA, Taylor JR, Potenza MN (2003) Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. Am J Psychiatry 160(6):1041–1052. https://doi.org/10.1176/appi.ajp.160.6.1041
Cheadle L, Tzeng CP, Kalish BT, Harmin DA, Rivera S, Ling E, Nagy MA, Hrvatin S, Hu L, Stroud H, Burkly LC, Chen C, Greenberg ME (2018) Visual experience-dependent expression of Fn14 is required for retinogeniculate refinement. Neuron 99(3):525–539.e10. https://doi.org/10.1016/j.neuron.2018.06.036
Cheadle L, Rivera SA, Phelps JS, Ennis KA, Stevens B, Burkly LC, Lee W-CA, Greenberg ME (2020) Sensory experience engages microglia to shape neural connectivity through a non-phagocytic mechanism. Neuron 108(3):451–468.e9. https://doi.org/10.1016/j.neuron.2020.08.002
Chen S-K, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G, Capecchi MR (2010) Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell 141(5):775–785. https://doi.org/10.1016/j.cell.2010.03.055
Chitu V, Gokhan Ş, Nandi S, Mehler MF, Richard Stanley E (2016) Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci 39(6):378–393. https://doi.org/10.1016/j.tins.2016.03.005
Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR (2016) The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351(6276):933–939. https://doi.org/10.1126/science.aad0314
Chung W-S, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J et al (2013) Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504(7480):394–400. https://doi.org/10.1038/nature12776
Chung W-S, Allen NJ, Eroglu C (2015) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol 7(9):a020370. https://doi.org/10.1101/cshperspect.a020370
Cotella EM, Gomez AS, Lemen P, Chen C, Fernández G, Hansen C, Herman JP, Paglini MG (2019) Long-term impact of chronic variable stress in adolescence versus adulthood. Prog Neuro-Psychopharmacol Biol Psychiatry 88:303–310. https://doi.org/10.1016/j.pnpbp.2018.08.003
Crews F, He J, Hodge C (2007) Adolescent cortical development: a critical period of vulnerability for addiction. Pharmacol Biochem Behav 86(2):189–199. https://doi.org/10.1016/j.pbb.2006.12.001
Cunningham CL, Martínez-Cerdeño V, Noctor SC (2013) Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci Off J Soc Neurosci 33(10):4216–4233. https://doi.org/10.1523/JNEUROSCI.3441-12.2013
Daneman R, Prat A (2015) The blood–brain barrier. Cold Spring Harb Perspect Biol 7(1):a020412. https://doi.org/10.1101/cshperspect.a020412
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan W-B (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8(6):752–758. https://doi.org/10.1038/nn1472
Daws MR, Sullam PM, Niemi EC, Chen TT, Tchao NK, Seaman WE (2003) Pattern recognition by TREM-2: binding of anionic ligands. J Immunol 171(2):594–599. https://doi.org/10.4049/jimmunol.171.2.594
de Jonge JC, Vinkers CH, Hulshoff Pol HE, Marsman A (2017) GABAergic mechanisms in schizophrenia: linking postmortem and in vivo studies. Front Psych 8. https://doi.org/10.3389/fpsyt.2017.00118
De S, Van Deren D, Peden E, Hockin M, Boulet A, Titen S, Capecchi MR (2018) Two distinct ontogenies confer heterogeneity to mouse brain microglia. Development 145(13). https://doi.org/10.1242/dev.152306
Delevich K, Wren Thomas A, Wilbrecht L (2018) Adolescence and ‘late blooming’ synapses of the prefrontal cortex. Cold Spring Harb Symp Quant Biol 83:37–43. https://doi.org/10.1101/sqb.2018.83.037507
Drzewiecki CM, Willing J, Juraska JM (2016) Synaptic number changes in the medial prefrontal cortex across adolescence in male and female rats: a role for pubertal onset. Synapse 70(9):361–368. https://doi.org/10.1002/syn.21909
Elmore MRP, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M et al (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82(2):380–397. https://doi.org/10.1016/j.neuron.2014.02.040
Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW (2011) Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6(10):e26317. https://doi.org/10.1371/journal.pone.0026317
Estes ML, McAllister AK (2016) Maternal immune activation: implications for neuropsychiatric disorders. Science 353(6301):772–777. https://doi.org/10.1126/science.aag3194
Eyo UB, Mo M, Yi M-H, Murugan M, Liu J, Yarlagadda R, Margolis DJ, Xu P, Long-Jun W (2018) P2Y12R-dependent translocation mechanisms gate the changing microglial landscape. Cell Rep 23(4):959–966. https://doi.org/10.1016/j.celrep.2018.04.001
Farhy-Tselnicker I, Allen NJ (2018) Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev 13(1):7. https://doi.org/10.1186/s13064-018-0104-y
Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, Erreni M et al (2018) The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity 48(5):979–991.e8. https://doi.org/10.1016/j.immuni.2018.04.016
Galloway DA, Phillips AEM, Owen DRJ, Moore CS (2019) Phagocytosis in the brain: homeostasis and disease. Front Immunol 10. https://doi.org/10.3389/fimmu.2019.00790
Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL (1999) Brain development during childhood and adolescence: a longitudinal MRI study. Nat Neurosci 2(10):861–863. https://doi.org/10.1038/13158
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330(6005):841–845. https://doi.org/10.1126/science.1194637
Giovanoli S, Engler H, Engler A, Richetto J, Voget M, Willi R, Winter C et al (2013) Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 339(6123):1095–1099. https://doi.org/10.1126/science.1228261
Giovanoli S, Engler H, Engler A, Richetto J, Feldon J, Riva MA, Schedlowski M, Meyer U (2016) Preventive effects of minocycline in a neurodevelopmental two-hit model with relevance to schizophrenia. Transl Psychiatry 6:e772. https://doi.org/10.1038/tp.2016.38
Gonzalez-Burgos G, Cho RY, Lewis DA (2015) Alterations in cortical network oscillations and parvalbumin neurons in schizophrenia. Biol Psychiatry 77(12):1031–1040. https://doi.org/10.1016/j.biopsych.2015.03.010
Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ et al (2019) Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50(1):253–271.e6. https://doi.org/10.1016/j.immuni.2018.11.004
Hanamsagar R, Alter MD, Block CS, Sullivan H, Bolton JL, Bilbo SD (2017) Generation of a microglial developmental index in mice and in humans reveals a sex difference in maturation and immune reactivity. Glia 65(9):1504–1520. https://doi.org/10.1002/glia.23176
Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan W-B, Julius D (2006) The P2Y 12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci 9(12):1512–1519. https://doi.org/10.1038/nn1805
Hoeffel G, Ginhoux F (2015) Ontogeny of tissue-resident macrophages. Front Immunol 6:486. https://doi.org/10.3389/fimmu.2015.00486
Hoftman GD, Volk DW, Holly Bazmi H, Li S, Sampson AR, Lewis DA (2015) Altered cortical expression of GABA-related genes in schizophrenia: illness progression vs developmental disturbance. Schizophr Bull 41(1):180–191. https://doi.org/10.1093/schbul/sbt178
Ismail N, Garas P, Blaustein JD (2011) Long-term effects of pubertal stressors on female sexual receptivity and estrogen receptor-α expression in CD-1 female mice. Horm Behav 59(4):565–571. https://doi.org/10.1016/j.yhbeh.2011.02.010
Ismail N, Kumlin AM, Blaustein JD (2013) A pubertal immune challenge alters the antidepressant-like effects of chronic estradiol treatment in inbred and outbred adult female mice. Neuroscience 249:43–52. https://doi.org/10.1016/j.neuroscience.2012.09.047
Jay TR, von Saucken VE, Muñoz B, Codocedo JF, Atwood BK, Lamb BT, Landreth GE (2019) TREM2 is required for microglial instruction of astrocytic synaptic engulfment in neurodevelopment. Glia 67(10):1873–1892. https://doi.org/10.1002/glia.23664
Kaar SJ, Angelescu I, Marques TR, Howes OD (2019) Pre-frontal Parvalbumin interneurons in schizophrenia: a meta-analysis of post-mortem studies. J Neural Transm (Vienna) 126(12):1637–1651. https://doi.org/10.1007/s00702-019-02080-2
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E et al (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169(7):1276–1290.e17. https://doi.org/10.1016/j.cell.2017.05.018
Kessler RC, Paul Amminger G, Aguilar-Gaxiola S, Alonso J, Lee S, Bedirhan Ustün T (2007) Age of onset of mental disorders: a review of recent literature. Curr Opin Psychiatry 20(4):359–364. https://doi.org/10.1097/YCO.0b013e32816ebc8c
Klawonn AM, Fritz M, Castany S, Pignatelli M, Canal C, Similä F, Tejeda HA et al (2021) Microglial activation elicits a negative affective state through prostaglandin-mediated modulation of striatal neurons. Immunity 54(2):225–234.e6. https://doi.org/10.1016/j.immuni.2020.12.016
Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, Toovey S, Prinssen EP (2014) Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 10(11):643–660. https://doi.org/10.1038/nrneurol.2014.187
Kochunov P, Elliot Hong L (2014) Neurodevelopmental and neurodegenerative models of schizophrenia: white matter at the center stage. Schizophr Bull 40(4):721–728. https://doi.org/10.1093/schbul/sbu070
Kolmogorova D, Paré C, Kostuck S, Hudson EC, Lebel N, Houlding E, Gregory JG, Ismail N (2019) Pubertal immune stress transiently alters spatial memory processes in adulthood. Psychoneuroendocrinology 102:261–272. https://doi.org/10.1016/j.psyneuen.2018.12.224
Kondo Y, Duncan ID (2009) Selective reduction in microglia density and function in the white matter of Colony-stimulating factor-1-deficient mice. J Neurosci Res 87(12):2686–2695. https://doi.org/10.1002/jnr.22096
Kopec AM, Smith CJ, Ayre NR, Sweat SC, Bilbo SD (2018) Microglial dopamine receptor elimination defines sex-specific nucleus accumbens development and social behavior in adolescent rats. Nat Commun 9(1):3769. https://doi.org/10.1038/s41467-018-06118-z
Koss WA, Belden CE, Hristov AD, Juraska JM (2014) Dendritic remodeling in the adolescent medial prefrontal cortex and the basolateral amygdala of male and female rats. Synapse 68(2):61–72. https://doi.org/10.1002/syn.21716
Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ et al (2018) CD47 protects synapses from excess microglia-mediated pruning during development. Neuron 100(1):120–134.e6. https://doi.org/10.1016/j.neuron.2018.09.017
Lenz KM, Pickett LA, Wright CL, Davis KT, Joshi A, McCarthy MM (2018) Mast cells in the developing brain determine adult sexual behavior. J Neurosci 38(37):8044–8059. https://doi.org/10.1523/JNEUROSCI.1176-18.2018
Lewis DA, Curley AA, Glausier JR, Volk DW (2012) Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 35(1):57–67. https://doi.org/10.1016/j.tins.2011.10.004
Louveau A, Harris TH, Kipnis J (2015) Revisiting the mechanisms of CNS immune privilege. Trends Immunol 36(10):569–577. https://doi.org/10.1016/j.it.2015.08.006
Lovelock DF, Deak T (2019) Acute stress imposed during adolescence yields heightened anxiety in Sprague Dawley rats that persists into adulthood: sex differences and potential involvement of the medial amygdala. Brain Res 1723:146392. https://doi.org/10.1016/j.brainres.2019.146392
MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR (2006) The drosophila cell corpse engulfment receptor draper mediates glial clearance of severed axons. Neuron 50(6):869–881. https://doi.org/10.1016/j.neuron.2006.04.028
Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH (2012) Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 26(4):607–616. https://doi.org/10.1016/j.bbi.2012.01.011
Mallya AP, Wang H-D, Lee HNR, Deutch AY (2019) Microglial pruning of synapses in the prefrontal cortex during adolescence. Cereb Cortex 29(4):1634–1643. https://doi.org/10.1093/cercor/bhy061
Manduca A, Servadio M, Damsteegt R, Campolongo P, Vanderschuren LJ, Trezza V (2016) Dopaminergic neurotransmission in the nucleus accumbens modulates social play behavior in rats. Neuropsychopharmacology 41(9):2215–2223. https://doi.org/10.1038/npp.2016.22
Mariani MM, Kielian T (2009) Microglia in infectious diseases of the central nervous system. J Neuroimmune Pharmacol 4(4):448–461. https://doi.org/10.1007/s11481-009-9170-6
Markham JA, Mullins SE, Koenig JI (2013) Periadolescent maturation of the prefrontal cortex is sex-specific and is disrupted by prenatal stress. J Comp Neurol 521(8):1828–1843. https://doi.org/10.1002/cne.23262
Matcovitch-Natan O, Winter DR, Giladi A, Aguilar SV, Spinrad A, Sarrazin S, Ben-Yehuda H et al (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science 353(6301):aad8670. https://doi.org/10.1126/science.aad8670
Maynard TM, Sikich L, Lieberman JA, LaMantia AS (2001) Neural development, cell-cell signaling, and the ‘two-hit’ hypothesis of schizophrenia. Schizophr Bull 27(3):457–476. https://doi.org/10.1093/oxfordjournals.schbul.a006887
McCarthy MM, Herold K, Stockman SL (2018) Fast, furious and enduring: sensitive versus critical periods in sexual differentiation of the brain. Physiol Behav 187:13–19. https://doi.org/10.1016/j.physbeh.2017.10.030
McClain JA, Morris SA, Ayumi Deeny M, Alex Marshall S, Hayes DM, Kiser ZM, Nixon K (2011) Adolescent binge alcohol exposure induces long-lasting partial activation of microglia. Brain Behav Immun 25:S120–S128. https://doi.org/10.1016/j.bbi.2011.01.006
Medawar PB (1948) Immunity to homologous grafted skin. III. The fate of skin homographs transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol 29(1):58–69
Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, Lelios I et al (2018) High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48(2):380–395.e6. https://doi.org/10.1016/j.immuni.2018.01.011
Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, Belforte JE (2012) GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 62(3):1574–1583. https://doi.org/10.1016/j.neuropharm.2011.01.022
Nandi S, Gokhan S, Dai X-M, Wei S, Enikolopov G, Lin H, Mehler MF, Richard Stanley E (2012) The CSF-1 receptor ligands IL-34 and CSF-1 exhibit distinct developmental brain expression patterns and regulate neural progenitor cell maintenance and maturation. Dev Biol 367(2):100–113. https://doi.org/10.1016/j.ydbio.2012.03.026
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. https://doi.org/10.1126/science.1110647
Niraula A, Sheridan JF, Godbout JP (2017) Microglia priming with aging and stress. Neuropsychopharmacology 42(1):318–333. https://doi.org/10.1038/npp.2016.185
Olesen KM, Ismail N, Merchasin ED, Blaustein JD (2011) Long-term alteration of anxiolytic effects of ovarian hormones in female mice by a peripubertal immune challenge. Horm Behav 60(4):318–326. https://doi.org/10.1016/j.yhbeh.2011.06.005
Oosterhof N, Chang IJ, Karimiani EG, Kuil LE, Jensen DM, Daza R, Young E et al (2019) Homozygous mutations in CSF1R cause a pediatric-onset leukoencephalopathy and can result in congenital absence of microglia. Am J Hum Genet 104(5):936–947. https://doi.org/10.1016/j.ajhg.2019.03.010
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333(6048):1456–1458. https://doi.org/10.1126/science.1202529
Paus T, Keshavan M, Giedd JN (2008) Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci 9(12):947–957. https://doi.org/10.1038/nrn2513
Peng L, Zhu M, Yang Y, Weng Y, Zou W, Zhu X, Guo Q, Zhong T (2019) Neonatal lipopolysaccharide challenge induces long-lasting spatial cognitive impairment and dysregulation of hippocampal histone acetylation in mice. Neuroscience 398:76–87. https://doi.org/10.1016/j.neuroscience.2018.12.001
Petanjek Z, Judaš M, Šimić G, Rašin MR, Uylings HBM, Rakic P, Kostović I (2011) Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci 108(32):13281–13286. https://doi.org/10.1073/pnas.1105108108
Premachandran H, Zhao M, Arruda-Carvalho M (2020) Sex differences in the development of the rodent corticolimbic system. Front Neurosci 14. https://doi.org/10.3389/fnins.2020.583477
Profaci CP, Munji RN, Pulido RS, Daneman R (2020) The blood-brain barrier in health and disease: important unanswered questions. J Exp Med 217(4). https://doi.org/10.1084/jem.20190062
Risher WC, Patel S, Kim IH, Uezu A, Bhagat S, Wilton DK, Pilaz L-J et al (2014) Astrocytes refine cortical connectivity at dendritic spines. eLife 3. https://doi.org/10.7554/eLife.04047
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74(4):691–705. https://doi.org/10.1016/j.neuron.2012.03.026
Schneider M (2013) Adolescence as a vulnerable period to alter rodent behavior. Cell Tissue Res 354(1):99–106. https://doi.org/10.1007/s00441-013-1581-2
Schwarz JM, Bilbo SD (2013) Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. J Neurosci Off J Soc Neurosci 33(3):961–971. https://doi.org/10.1523/JNEUROSCI.2516-12.2013
Schwarz JM, Hutchinson MR, Bilbo SD (2011) Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression. J Neurosci 31(49):17835–17847. https://doi.org/10.1523/JNEUROSCI.3297-11.2011
Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A et al (2020) Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. EMBO J 39(16):e105380. https://doi.org/10.15252/embj.2020105380
Shatz CJ (1990) Competitive interactions between retinal ganglion cells during prenatal development. J Neurobiol 21(1):197–211. https://doi.org/10.1002/neu.480210113
Sheng J, Ruedl C, Karjalainen K (2015) Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43(2):382–393. https://doi.org/10.1016/j.immuni.2015.07.016
Shi L, Hossein Fatemi S, Sidwell RW, Patterson PH (2003) Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci Off J Soc Neurosci 23(1):297–302
Smith SEP, Li J, Garbett K, Mirnics K, Patterson PH (2007) Maternal immune activation alters fetal brain development through Interleukin-6. J Neurosci Off J Soc Neurosci 27(40):10695–10702. https://doi.org/10.1523/JNEUROSCI.2178-07.2007
Smith CJ, Kingsbury MA, Dziabis JE, Hanamsagar R, Malacon KE, Tran JN, Norris HA, Gulino M, Bordt EA, Bilbo SD (2020) Neonatal immune challenge induces female-specific changes in social behavior and somatostatin cell number. Brain Behav Immun 90:332–345. https://doi.org/10.1016/j.bbi.2020.08.013
Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F, Garel S (2014) Microglia modulate wiring of the embryonic forebrain. Cell Rep 8(5):1271–1279. https://doi.org/10.1016/j.celrep.2014.07.042
Steinberg L (2008) A social neuroscience perspective on adolescent risk-taking. Dev Rev 28(1):78–106. https://doi.org/10.1016/j.dr.2007.08.002
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131(6):1164–1178. https://doi.org/10.1016/j.cell.2007.10.036
Tamnes CK, Walhovd KB, Dale AM, Østby Y, Grydeland H, Richardson G, Westlye LT et al (2013) Brain development and aging: overlapping and unique patterns of change. NeuroImage 68:63–74. https://doi.org/10.1016/j.neuroimage.2012.11.039
Tan Y-L, Yuan Y, Tian L (2020) Microglial regional heterogeneity and its role in the brain. Mol Psychiatry 25(2):351–367. https://doi.org/10.1038/s41380-019-0609-8
Tanabe S, Yamashita T (2018) B-1a lymphocytes promote oligodendrogenesis during brain development. Nat Neurosci 21(4):506–516. https://doi.org/10.1038/s41593-018-0106-4
Thion MS, Garel S (2020) Microglial ontogeny, diversity and neurodevelopmental functions. Curr Opin Genet Dev 65:186–194. https://doi.org/10.1016/j.gde.2020.06.013
Thion MS, Mosser C-A, Férézou I, Grisel P, Baptista S, Low D, Ginhoux F, Garel S, Audinat E (2019) Biphasic impact of prenatal inflammation and macrophage depletion on the wiring of neocortical inhibitory circuits. Cell Rep 28(5):1119–1126.e4. https://doi.org/10.1016/j.celrep.2019.06.086
Toga AW, Thompson PM, Sowell ER (2006) Mapping brain maturation. Focus 4(3):378–390. https://doi.org/10.1176/foc.4.3.378
Tremblay M-È, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8(11):e1000527. https://doi.org/10.1371/journal.pbio.1000527
Vainchtein ID, Chin G, Cho FS, Kelley KW, Miller JG, Chien EC, Liddelow SA et al (2018) Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science 359(6381):1269–1273. https://doi.org/10.1126/science.aal3589
VanRyzin JW, Pickett LA, McCarthy MM (2018) Microglia: driving critical periods and sexual differentiation of the brain. Dev Neurobiol 78(6):580–592. https://doi.org/10.1002/dneu.22569
VanRyzin JW, Marquardt AE, Argue KJ, Vecchiarelli HA, Ashton SE, Arambula SE, Hill MN, McCarthy MM (2019) Microglial phagocytosis of newborn cells is induced by endocannabinoids and sculpts sex differences in juvenile rat social play. Neuron 102(2):435–449.e6. https://doi.org/10.1016/j.neuron.2019.02.006
VanRyzin JW, Marquardt AE, Pickett LA, McCarthy MM (2020) Microglia and sexual differentiation of the developing brain: a focus on extrinsic factors. Glia 68(6):1100–1113. https://doi.org/10.1002/glia.23740
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci Off J Soc Neurosci 29(13):3974–3980. https://doi.org/10.1523/JNEUROSCI.4363-08.2009
Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13(8):753–760. https://doi.org/10.1038/ni.2360
Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S et al (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160(6):1061–1071. https://doi.org/10.1016/j.cell.2015.01.049
Whitford TJ, Rennie CJ, Grieve SM, Richard Clark C, Gordon E, Williams LM (2007) Brain maturation in adolescence: concurrent changes in neuroanatomy and neurophysiology. Hum Brain Mapp 28(3):228–237. https://doi.org/10.1002/hbm.20273
Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD (2011) Microglia and memory: modulation by early-life infection. J Neurosci 31(43):15511–15521. https://doi.org/10.1523/JNEUROSCI.3688-11.2011
Wolf SA, Boddeke HWGM, Kettenmann H (2017) Microglia in physiology and disease. Annu Rev Physiol 79:619–643. https://doi.org/10.1146/annurev-physiol-022516-034406
Wu W-L, Hsiao EY, Yan Z, Mazmanian SK, Patterson PH (2017) The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav Immun 62:11–23. https://doi.org/10.1016/j.bbi.2016.11.007
Yim S, Yeong AP, Berrios J, Lafourcade M, Pascual LM, Soares N, Kim JY et al (2017) Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature 549(7673):482–487. https://doi.org/10.1038/nature23909
Yohn NL, Blendy JA (2017) Adolescent chronic unpredictable stress exposure is a sensitive window for long-term changes in adult behavior in mice. Neuropsychopharmacology 42(8):1670–1678. https://doi.org/10.1038/npp.2017.11
Zuckerman L, Rehavi M, Nachman R, Weiner I (2003) Immune activation during pregnancy in rats leads to a PostPubertal emergence of disrupted latent inhibition, dopaminergic hyperfunction, and altered limbic morphology in the offspring: a novel neurodevelopmental model of schizophrenia. Neuropsychopharmacology 28(10):1778–1789. https://doi.org/10.1038/sj.npp.1300248
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Dziabis, J.E., Bilbo, S.D. (2021). Microglia and Sensitive Periods in Brain Development. In: Andersen, S.L. (eds) Sensitive Periods of Brain Development and Preventive Interventions. Current Topics in Behavioral Neurosciences, vol 53. Springer, Cham. https://doi.org/10.1007/7854_2021_242
Download citation
DOI: https://doi.org/10.1007/7854_2021_242
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-04472-4
Online ISBN: 978-3-031-04473-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)