
Abstract
GABAB receptors (GBRs), the G protein-coupled receptors for the inhibitory neurotransmitter γ-aminobutyric acid (GABA), activate Go/i-type G proteins that regulate adenylyl cyclase, Ca2+ channels, and K+ channels. GBR signaling to enzymes and ion channels influences neuronal activity, plasticity processes, and network activity throughout the brain. GBRs are obligatory heterodimers composed of GB1a or GB1b subunits with a GB2 subunit. Heterodimeric GB1a/2 and GB1b/2 receptors represent functional units that associate in a modular fashion with regulatory, trafficking, and effector proteins to generate receptors with distinct physiological functions. This review summarizes current knowledge on the structure, organization, and functions of multi-protein GBR complexes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Heterodimers Are the Minimal Functional Receptor Units
The GB1a and GB1b subunit isoforms were cloned in 1997 using a radioligand binding approach (Kaupmann et al. 1997). This showed that GBRs belong to class C GPCRs, which include metabotropic glutamate receptors, Ca2+-sensing receptors, and taste receptors (Kaupmann et al. 1997). As for other class C GPCRs, GB1 subunits contain a large extracellular venus fly trap domain (VFTD), the typical heptahelical transmembrane domain (7TMD), and an intracellular C-terminal domain (Fig. 1). When expressed in heterologous cells, GB1 subunits exhibited ~tenfold lower affinity for GABA than native GBRs (Kaupmann et al. 1997, 1998; White et al. 1998). In addition, GB1 subunits failed to exit the endoplasmic reticulum (Couve et al. 1998) and to efficiently inhibit adenylyl cyclase or activate Kir3 channels (Kaupmann et al. 1997). This showed that GB1 subunits do not form functional receptors by themselves. Soon after cloning of GB1 subunits, cDNA homology searches and yeast-two-hybrid screens identified the sequence-related GB2 subunit (Kaupmann et al. 1998; Kuner et al. 1999; Ng et al. 1999; White et al. 1998). GB2 was again non-functional when expressed in heterologous cells (Galvez et al. 2001). In situ hybridization studies showed that neurons generally co-express GB1 and GB2 transcripts, which indicated that GB1 and GB2 subunits might function together in a heterodimeric receptor. Co-expression of GB1 with GB2 subunits indeed generated receptors with tenfold higher affinity for GABA that also efficiently signaled to neuronal effectors, including adenylyl cyclase, Kir3 channels, and P/Q/N-type Ca2+ channels (Kaupmann et al. 1998; Kuner et al. 1999; Ng et al. 1999; White et al. 1998). This finding represented the earliest demonstration of an obligatory heterodimeric G protein-coupled receptor (Marshall et al. 1999). The GB1a and GB1b subunit isoforms derive from the same gene by differential promoter usage and exhibit distinct expression patterns in the central nervous system (Bischoff et al. 1999). Structurally, GB1a differs from GB1b by the presence of two sushi domains in the N-terminal domain (SD1, SD2) (Blein et al. 2004) (Fig. 1). The N-terminal SD1 has an intrinsically disordered structure, while SD2 is more compactly folded. The SDs function as axonal trafficking signals (Biermann et al. 2010) and stabilize the receptor at the cell surface (Hannan et al. 2012). Accordingly, axons predominantly express GB1a/2 receptors, while the somatodendritic compartment expresses GB1b/2 receptors. However, GB1a/2 receptors are also present in the dendrites but excluded from the spine heads, in contrast to GB1b/2 receptors (Biermann et al. 2010; Dinamarca et al. 2019; Vigot et al. 2006).

Structural model of the GBR heterodimer. The model is based on the published structures of SD1 [PDB ID: 6HKC (Rice et al. 2019)], SD2 [1SRZ (Blein et al. 2004)], baclofen-bound VFTDs [4MS4 (Geng et al. 2013)], active and inactive mGlu5 [6N51 and 6N52 (Koehl et al. 2019)], the coiled-coil domain of GBRs [4PAS (Burmakina et al. 2014)] and the heterotrimeric G protein complex [3SN6 (Rasmussen et al. 2011)]. GB1 and GB2 are colored in green and slate, respectively. The active and inactive conformations of the 7TMDmGlu5 were used in GB1 and GB2, respectively. The boxes in the C-terminal domain of GB1 indicate the retention motifs RSRR and EKSRLL that control heterodimer assembly during biosynthesis
GBRs have evolved quality control signals that prevent unfolded or unassembled subunits from exiting the endoplasmic reticulum (ER) and the Golgi apparatus. The intracellular C-terminal domain of GB1 subunits encodes the ER retention signal RSRR, which is located distal to a coiled-coil heterodimerization domain (Margeta-Mitrovic et al. 2000; Pagano et al. 2001) (Fig. 1). Prenylated rab acceptor family 2 (PRAF2) protein, an ER-resident molecule, binds to the ER retention signal and prevents exit of the GB1 subunit from the ER (Doly et al. 2016). Coiled-coil heteromerization of GB1 with GB2 subunits competitively displaces PRAF2 from its binding motif and enables forward trafficking of the GB1/2 heterodimer in the biosynthetic pathway. The coat protein complex I (COPI) also binds to the ER retention signal and shuttles unassembled GB1 subunits from the cis-Golgi back to the ER (Brock et al. 2005). An additional signal within the coiled-coil domain of GB1, the di-leucine motif EKSRLL (Fig. 1), controls release of receptors from the trans-Golgi network (Restituito et al. 2005). Msec7-1, a guanine-exchange factor protein of the ARF family of GTPases, binds to this di-leucine motif and prevents exit of unassembled GB1 subunits from the Golgi apparatus. Structural data show that heterodimerization of GB1 with GB2 subunits occludes the di-leucine signal and prevents Msec7-1 from binding (Burmakina et al. 2014).
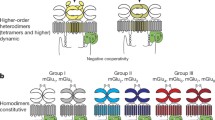
At high cell surface density, GB1/2 heterodimers assemble by random collision into higher-order oligomers of two or more heterodimers (Calebiro et al. 2013; Comps-Agrar et al. 2011; Maurel et al. 2008; Schwenk et al. 2010; Stewart et al. 2018). It appears that in higher-order oligomers GB1 subunits arrange in a line via the opposite sides of their 7TMDs, while GB2 subunits are on the side (Xue et al. 2019). Mutation of either E380 + L382, T410 + E412, or E413 in the VFTD of rat GB1a (VFTDGB1a) disrupts the formation of higher-order oligomers (Comps-Agrar et al. 2011; Stewart et al. 2018). Molecular modeling indicates that two G proteins can couple to one GBR tetramer (Xue et al. 2019). Interestingly, however, higher-order oligomerization limits the capacity of GBRs to activate G proteins, presumably because only one of the agonist binding sites in the two neighboring GB1 subunits of a GBR tetramer can be occupied (Stewart et al. 2018). It is unknown whether suppression of G protein signaling in higher-order oligomers is of regulatory significance or not.
2 Signal Transduction in the Receptor Heterodimer
GB1 and GB2 subunits fulfill distinct functions in the receptor heterodimer. Only GB1 contains a GABA binding site (Galvez et al. 1999), whereas GB2 couples to the G protein (Duthey et al. 2002; Havlickova et al. 2002). GB2 additionally allosterically increases GABA affinity at GB1 (Kaupmann et al. 1998; White et al. 1998). After binding of GABA at GB1, multiple allosteric interactions between subunit domains are necessary to activate the G protein at GB2. X-ray structures of the extracellular dimerization interface are now available (Geng et al. 2012, 2013). The VFTDGB1/VFTDGB2 dimer structure shows that GB1 and GB2 subunits interact sideways, facing opposite directions (Geng et al. 2013) (Fig. 2). Each VFTD has a bi-lobed structure, where lobe 1 (LB1) is positioned more distant from the plasma membrane than LB2. A peptide hinge connects LB1 and LB2, enabling LB2 to move in relation to LB1. In the heterodimer, VFTDGB1 and VFTDGB2 bind to each other via LB1. The LB1/LB1 interaction is stabilized by multiple hydrophobic contacts (Y113/Y117 in GB1b; Y118/W149 in GB2, amino acid numbering refers to human sequences in the remainder of the manuscript), salt bridges (R141 in GB1b, D109 in GB2), hydrogen bonds (E138 in GB1b, N110 in GB2), and multiple van der Waals contacts (Geng et al. 2013). The hydrophobic patch is located at the center of the interface and flanked by sites forming hydrogen bonds and water-mediated contacts.

Conformational changes in VFTDs during receptor activation. Structures of the VFTDs in the absence of a ligand [PDB ID: 4MQE, top] and with GABA bound to VFTDGB1 [4MS3, bottom (Geng et al. 2013)] are shown. Upon GABA binding, LB2 of VFTDGB1 (green) rotates by 29° and moves toward VFTDGB2 (slate) by ~10 Å. In contrast, LB2 of VFTDGB2 rotates only by 9° upon activation. Amino acid residues in VFTDGB1 involved in the binding of GABA are shown on the right, with numbering of residues according to human GB1b (Geng et al. 2013). Y250, W278, and S131 (water mediated contact) on LB2 interact with GABA and subsequently close the interface between the LB domains. The 2F(o)-F(c) electron density map of GABA is shown as mesh at σ = 1.5 (right bottom)
Mutagenesis and X-ray crystallography studies show that agonists and antagonists bind to a large crevice between LB1 and LB2 in VFTDGB1 (Galvez et al. 1999; Geng et al. 2013). Agonists and antagonists form multiple interactions with residues in LB1, including hydrogen bonds with S130, S153, H170, and E349, van der Waals contacts with W65, and water-mediated contacts with S131 (GB1b numbering) (Geng et al. 2013) (Fig. 2). Agonists additionally bind to Y250 and W278 in LB2. The antagonists CGP54626 and SCH50911 also bind to W278 in LB2, which increases antagonist-binding affinity (Geng et al. 2013). The large substituents at either side of antagonists physically prevent VFTDGB1 closure, which stabilizes VFTDGB1 in an open conformation (Geng et al. 2013). Conversely, agonists induce VFTDGB1 closure (Geng et al. 2012; Kniazeff et al. 2004). Mutations that stabilize VFTDGB1 closure therefore lead to constitutive activity. Upon agonist binding, LB2 in VFTDGB1 rotates 29° about a nearly horizontal axis, bringing LB2 close to LB1 and closing VFTDGB1 (Fig. 2). This rotation additionally moves VFTDGB1 closer to LB2 of VFTDGB2 that remains in a constitutively open conformation stabilized by hydrogen bonds between LB1 and LB2 (Geng et al. 2012). LB2 of VFTDGB2 twists by ~9° about a nearly vertical axis, moving it toward LB2 of VFTDGB1. As a result, a new LB2/LB2 interface forms that stabilizes VFTDGB1 in the closed conformation and increases agonist affinity. The LB2/LB2 interface is essential for receptor activation, as disruption of the interface by insertion of a glycan wedge precludes receptor activation (Rondard et al. 2008). Conversely, a covalent disulfide bridge linking the LB2 lobes locks the receptor in a constitutively active state (Geng et al. 2013). While agonist binding promotes VFTDGB1/VFTDGB2 interaction, it simultaneously causes a spatial reorientation of 7TMDGB1 and 7TMDGB2 that enables activation of the G protein at 7TMDGB2 (Matsushita et al. 2010; Monnier et al. 2011). Allosteric activation of 7TMDGB2 occurs in cis and in trans via VFTDGB2 and 7TMDGB1, respectively (Monnier et al. 2011). Receptor activation disrupts an ionic lock at the intracellular side of 7TMDGB2 (Binet et al. 2007). The ionic lock is formed by a salt bridge between D688 in TM6 and K574 in TM3, which prevents outward movement of TM6 and stabilizes the inactive closed conformation of the 7TMDGB2. Disruption of the ionic lock by mutation allosterically increases agonist affinity. Recent studies show that crosslinking of the TM6-TM6 interaction between GB1 and GB2 is sufficient for receptor activation and leads to constitutive activity (Xue et al. 2019). Interestingly, GBRs also exhibit constitutive activity in the absence of agonists (Galvez et al. 2001; Grunewald et al. 2002). This suggests that GBRs exhibit high intrinsic conformational flexibility and spontaneously oscillate between inactive and active states, similar as shown for the isolated VFTDs of metabotropic glutamate receptor 2 (Olofsson et al. 2014).
Almost all known allosteric modulators of GBRs bind to 7TMDGB2 (Binet et al. 2004; Dupuis et al. 2006; Sun et al. 2016). The only exception is Ca2+, which binds to S269 in LB1 of GB1a (S153 in GB1b) and thereby increases affinity for GABA (Galvez et al. 2000; Wise et al. 1999) (Fig. 2). Positive allosteric modulators (PAMs) at GBRs generally increase agonist potency and efficacy (Urwyler et al. 2005). Some PAMs also have agonistic properties and activate the receptor in the absence of orthosteric agonists, presumably by stabilizing the active conformation of 7TMDGB2. The negative allosteric modulator (NAM) CLH304 has inverse agonist properties, suppressing basal activity as well as agonist-induced receptor activation, likely by preventing 7TMDGB2 from reaching the active conformation (Chen et al. 2014; Sun et al. 2016). The binding sites of allosteric modulators in 7TMDGB2 are unknown. Crystal structures of other class C G protein-coupled receptors suggest that allosteric modulators enter a cavity located between transmembrane domains 3, 5, 6, and 7 (Christopher et al. 2015; Dore et al. 2014; Gregory et al. 2011; Wu et al. 2014). A GB2 homology model predicts a hydrophobic binding pocket in 7TMDGB2 and identified potential amino acid residues involved in binding of allosteric modulators (Freyd et al. 2017).
3 Auxiliary KCTD Subunits
Several observations pointed to native GBRs being composed of more than just a GB1 and a GB2 subunit. For example, GBR complexes isolated from brain tissue had molecular masses of 0.6–1.1 MDa, while the heterodimer only accounts for 240 kDa (Schwenk et al. 2010). Moreover, the kinetic properties of native GBR responses varied and differed from those of GB1/2 heterodimers expressed in heterologous cells (Turecek et al. 2014). Quantitative proteomic approaches identified approximately 30 proteins that interact with GB1 or GB2 in the brain (Dinamarca et al. 2019; Schwenk et al. 2010, 2016; Turecek et al. 2014). These proteins provide a molecular basis to explain the functional diversity of native GBRs. Known interactions between components of GBR complexes have been summarized recently (Fritzius and Bettler 2020).
Abundant GBR-interacting proteins are the K+ channel tetramerization domain (KCTD) proteins KCTD8, KCTD12, KCTD12b, and KCTD16 (herein collectively referred to as the KCTDs) (Schwenk et al. 2010). The KCTDs are part of a larger family of KCTD proteins comprising 26 members with sequence similarity to the cytoplasmic tetramerization (T1, also known as BTB or POZ) domain of voltage-gated K+ channels (Correale et al. 2013; Zheng et al. 2019). The KCTDs are composed of the N-terminal T1 domain and a H1 domain, with both isolated domains capable of forming oligomers (Correale et al. 2013; Fritzius et al. 2017). KCTD8 and KCTD16 additionally encode a C-terminal H2 domain that scaffolds effector channels and other receptor-associated proteins (see below). Structural studies demonstrate that T1KCTD12 and T1KCTD16 form homopentamers (Pinkas et al. 2017; Smaldone et al. 2016; Zheng et al. 2019; Zuo et al. 2019) (Figs. 3a and 5). The T1KCTD16 pentamer is open, with a gap of 8–16 Å at its narrowest and widest points. Since one T1KCTD16 monomer in the pentamer occupies 25 Å, the gap in the pentamer is too small to accommodate a sixth T1KCTD16 monomer. Multiple electrostatic interactions and nonpolar associations stabilize adjacent T1KCTD16 domains. Most of the conserved amino acid residues are involved in T1KCTD16 interactions, supporting that all four KCTDs assemble as pentamers. Co-crystallization of T1KCTD16 with a C-terminal GB2 peptide shows that the T1KCTD16 pentamer wraps around the peptide (Fig. 3a, b). The GB2 peptide loops inside the central opening of the pentamer, entering and leaving the pentamer at its N-terminal surface. The apex of the GB2 peptide loop forms a short helix that contains the Y903 residue critical for KCTD binding (Correale et al. 2013; Schwenk et al. 2010; Zheng et al. 2019) (Fig. 3a). X-ray crystallography reveals that Y903 is located in the middle of an extensive interaction interface (Zuo et al. 2019). The interaction takes place off center in the central pore of the pentamer, opposite of the gap. In each T1KCTD16 domain of the pentamer, the F80 residue protrudes into the central pore. A slight offset due to a tilt of each T1KCTD16 monomer forms a spiraling ladder of F80 residues in the inner wall of the pentamer (Fig. 3b). This arrangement allows F80 residues to bind many side chains in a GB2 peptide of 25 amino acid residues (Zheng et al. 2019). Consistent with the X-ray data, the F80A mutation completely abrogates KCTD16 binding to GBRs (Zuo et al. 2019). Interestingly, binding of GB2 results in a compaction of the T1KCTD16 pentamer (Fig. 3a).

Binding of the GB2 C-terminus to the T1KCTD16 pentamer. (a) Structure of a GB2 C-terminal peptide bound to the T1KCTD16 pentamer [PDB ID: 6M8R (Zheng et al. 2019)]. The F80 (red) and Y903 (yellow) residues in T1KCTD16 and GB2, respectively, are highlighted. T1KCTD16 domains form an open pentamer with C6 symmetry. A twist of the ring prevents the sixth subunit from being inserted. Upon binding to the GB2 C-terminal peptide, the open pentamer contracts by roughly 4–5 Å and creates a tight channel for the peptide (right). The orientation of the complex is indicated by the N- and C-terminus of KCTD16. (b) The twisted ring structure enables each T1KCTD16 subunit to form a distinct binding interface with the peptide. A cross-section of the pentamer shows the interaction of each of the five T1KCTD16 domains with the GB2 peptide. The F80 residues (red) and the T1KCTD16 domains are aligned vertically according to the position of GB2 peptide
Reverse affinity purification experiments using KCTD-specific antibodies revealed that KCTD8, KCTD12, KCTD12b, and KCTD16 not only bind to GB2 but also to the Gβγ subunits of the G protein (Turecek et al. 2014; Zheng et al. 2019). Co-crystallization studies show that each H1KCTD12 pentamer binds five Gβγ subunits in a near perfect C5 rotational symmetry (Zheng et al. 2019) (Fig. 4). The five Gβγ molecules form a tightly packed outer ring in which every Gβ subunit directly contacts neighboring Gβ subunits as well as two adjacent H1 domains of the pentamer. H1KCTD12 folds into a β sheet made up by five antiparallel β strands (β1-5) interspersed with two short α helices (α1 and α2). The amino acids at and around the loops between β1/β2 as well as β3/α2 bind to an acidic patch at the top of the Gβ propeller (interface I) and a groove between the N-terminal helix and the β propeller of Gβ (interface II) (Fig. 4). In H1KCTD12, R232 (contacting interface I on Gβ) and R257 (contacting interface II on Gβ) are particularly important for the interaction, as mutation of either residue completely abolishes Gβγ binding and modulation of G protein signaling by KCTD12. The Gγ subunit is located peripherally and does not interact with the H1 domain. However, Gγ allows anchoring of the complex at the plasma membrane (Figs. 4 and 5). When incubating KCTD12H1 with a substoichiometric amount of Gβγ, only full 5/5 complexes and free KCTD12 were observed, with no evidence of partial oligomers (Zheng et al. 2019). This suggests that binding of H1KCTD12 to Gβγ is highly cooperative. Supported by 3D reconstructions of electron microscopy images of the full-length KCTD12 protein in complex with Gβγ (Zheng et al. 2019), the picture of a large multi-protein complex emerges, in which KCTDs simultaneously bind via their T1 and H1 domains GB2 and Gβγ subunits, respectively (Fig. 5). Of note, Gβγ binding to the H1KCTD12 pentamer partially occludes the Gα binding-site on the surface of Gβγ, indicating that the trimeric G protein does not assemble with KCTD12 into a pentameric complex. This contrasts earlier biochemical findings that support that GBRs and KCTDs form a complex with the heterotrimeric G protein (Turecek et al. 2014). Co-crystallization of the H1 domain with Gβγ may therefore favor a structure that differs from the structure of the full-length KCTD protein assembled with receptor and the heterotrimeric G protein.

The H1KCTD12/Gβγ complex. Top: H1KCTD12 pentamers and five Gβ1γ2 subunits form together a complex with C5 symmetry [PDB ID: 6M8S, (Zheng et al. 2019)]. Each of the five Gβ1γ2 subunits binds two H1KCTD12 subunits. The cutout shows R232 (interface I) and R257 (interface II) that are crucial for Gβ1 recognition. Bottom: Due to lipidation of Gβγ subunits, the complex is expected to be tethered to the plasma membrane

Scheme of the multi-protein GBR/KCTD12/G protein signaling complex. The intracellular part of the GBR (dark pink), a pentamer of KCTD12 proteins (green), Gβ (blue), and Gγ (orange) subunits are depicted. The N-terminal T1 domain of the KCTDs forms an open pentamer that interacts with the cytoplasmic tail of GB2 (the GB2 peptide, containing the amino acids D888 to S913, co-crystallized with the T1KCTD16 is highlighted in bright pink). This part of GB2 loops inside the central opening of the T1 pentamer, entering and leaving it at its N-terminal surface. The amino acid Y903 (yellow circle) at the apex of the GB2 loop is critical for KCTD binding. A slight offset due to a tilt of each T1KCTD16 monomer allows the pentamer to bind a large number of amino acid side chains within the cytoplasmic tail of GB2. A short linker (35 Å) connects the N-terminal T1 domain with the C-terminal H1 domain of KCTDs, which binds to the Gβγ heterodimer of the G protein. The scheme depicts the closed H1KCTD12 pentamer bound to five copies of Gβγ. Anchoring of the Gγ subunit to the phospholipid bilayer tethers KCTDs to the plasma membrane. The expected location of the C-terminal H2 domain present in KCTD16 and KCTD8 is indicated for three of the KCTDs in the pentamer. Distances are derived from three-dimensional negative-stain electron microscopy reconstructions (Zheng et al. 2019)
Dual binding of the KCTDs to the receptor and the G protein enables KCTDs to regulate the kinetics of receptor signaling (Fritzius et al. 2017; Schwenk et al. 2010; Seddik et al. 2012; Turecek et al. 2014). Pre-assembly of the G protein via the KCTDs at the receptor significantly accelerates G protein signaling, most likely by overcoming slow diffusion-limited association of the G protein with the receptor (Turecek et al. 2014). When studying GBR-mediated K+ current responses, KCTDs shorten both the rise time and the delay between agonist binding and the onset of K+ currents. The KCTDs are therefore responsible for the fast kinetics observed with GBR-induced current responses in neurons (Schwenk et al. 2010; Turecek et al. 2014). While all KCTDs accelerate GBR signaling, selectively KCTD12 and KCTD12b induce a rapid desensitization of GBR-mediated K+ currents (Schwenk et al. 2010; Seddik et al. 2012) through an activity-dependent uncoupling of Gβγ from effector channels (Turecek et al. 2014; Zheng et al. 2019). Some neuronal populations simultaneously express multiple KCTDs, raising the possibility that the KCTDs form hetero-oligomers. Indeed, in heterologous cells, KCTD8, KCTD12, and KCTD16 form hetero-oligomers in all possible combinations (Balasco et al. 2019; Fritzius et al. 2017). Association of KCTD12/16 hetero-oligomers with GBRs in hippocampal pyramidal cells confers unique kinetic properties to GBR-induced K+ currents, showing that hetero-oligomers increase the kinetic repertoire of GBR signaling (Fritzius et al. 2017). Of note, the KCTDs exert little influence on allosteric and orthosteric binding sites of GBRs (Rajalu et al. 2015).
Reverse-affinity purification experiments support that the KCTDs do not bind to other GPCRs (Schwenk et al. 2016; Turecek et al. 2014). The KCTDs are non-obligatory GBR components, which, however, are expressed by most neurons (and some glial cells) in the vertebrate brain (Metz et al. 2011). Since the KCTDs stably associate with the receptor and control receptor kinetics and surface expression (Ivankova et al. 2013), they should be viewed as auxiliary receptor subunits.
4 SD-Interacting Proteins
Proteomic studies showed that the β-amyloid precursor protein (APP), the adherence junction-associated protein 1 (AJAP-1), and the PILRα-associated neural protein (PIANP) form three distinct complexes with GB1a/2 receptors (Dinamarca et al. 2019; Schwenk et al. 2016). NMR studies identified sequence-related epitopes in the extracellular domains of APP, AJAP-1, and PIANP that bind with nanomolar affinities to the N-terminal SD1 of GB1a, with a rank order of affinities AJAP-1 > PIANP >> APP (Dinamarca et al. 2019). APP is best known as the source of β-amyloid (Aβ) peptides in Alzheimer’s disease. Electrophysiological and biochemical experiments showed that binding of APP to GB1a is necessary for vesicular trafficking of GBRs to axon terminals (Dinamarca et al. 2019), consistent with the proposed role of the SDs in axonal trafficking (Biermann et al. 2010). Proteomic data show that APP associates with calsyntenins and c-Jun N-terminal kinase-interacting proteins (JIPs) that link the APP/GBR complex in cargo vesicles to the axonal kinesin-1 motor. Of potential relevance for Alzheimer’s disease, complex formation with GBRs stabilizes APP at the cell surface and reduces proteolysis of APP to Aβ (Dinamarca et al. 2019). A related study showed that binding of the soluble form of APP (sAPP) to the SD1 of GB1a inhibits neurotransmitter release, synaptic transmission and spontaneous neuronal activity (Rice et al. 2019). The fact that a GBR antagonist disinhibits sAPP-inhibited neurotransmitter release supports that sAPP acts as a GBR agonist or positive allosteric modulator. However, it was also reported that sAPP has no functional effects on GBR signaling in heterologous cells (Dinamarca et al. 2019). Therefore, additional studies need to confirm sAPP effects on GBR signaling. AJAP-1 and PIANP, the two other proteins binding to the SD of GB1a, do not play a role in axonal trafficking of GBRs (Dinamarca et al. 2019). These proteins localize to adherens junctions that stabilize cell-cell interactions (Winkler et al. 2019; Yamada and Nelson 2007) and may be important for anchoring GB1a/2 receptors at presynaptic sites, either in cis or through trans-synaptic interactions. In support of this hypothesis, PIANP knock-out mice exhibit a deficit in GBR-mediated inhibition of glutamate release in the hippocampus (Winkler et al. 2019).
Amyloid-like protein 2 (APLP2) and integral membrane protein 2B (ITM2B) and ITM2C are additional transmembrane proteins that selectively co-purify with GB1a/2 receptors (Dinamarca et al. 2019; Schwenk et al. 2016). These proteins associate with APP and are secondary interactors of GBRs. GBRs can therefore assemble with multi-protein APP complexes into super-complexes (complexes of complexes).
5 Effector Channels
The best-studied GBR functions in the central nervous system are the gating of voltage-sensitive Ca2+ (Cav) channels and inwardly rectifying Kir3-type K+ channels by the Gβγ subunits of the activated G protein (Gassmann and Bettler 2012). GBRs inhibit N- and P/Q-type Cav channels, which suppress neurotransmitter release at most synapses in the brain. GBR activation of Kir3 channels hyperpolarizes the membrane, shunts postsynaptic currents in the dendrites, and inhibits neuronal firing. The α1B, α2, δ1, and δ2 subunits of N-type CaV channels co-purify with the GB1, GB2, and KCTD16 subunits, supporting that these channels bind to GBRs via KCTD16 (Schwenk et al. 2016). Association of GBR with N-type CaV channels directly links the receptor to the presynaptic release machinery. Proteomic work did not support a physical association of native GBRs with Kir3 channels (Schwenk et al. 2016), in contrast to earlier studies in heterologous expression systems (Ciruela et al. 2010; David et al. 2006; Fowler et al. 2007). It is possible that proteomic approaches miss weak interactions of Kir3 channels with GBRs. Alternatively, overexpression of two membrane proteins in heterologous cells may lead to artificial aggregates detected in BRET and immunoprecipitation experiments. Proteomic work additionally identified novel effector channels of GBRs, such as the transient receptor potential vanilloid 1 (TRPV1) (Hanack et al. 2015) and HCN2 channels (Schwenk et al. 2016). Sensitization of TRPV1 channels is central to the initiation of pathological forms of pain. TRPV1 assembles in a complex with GB1 (Hanack et al. 2015). Since agonist activity at GB1 reverts the sensitized state of TRPV1 channels, it may be possible to exploit the TRPV1/GB1 complex for anti-pain therapy. HCN1 and HCN2, like N-type Cav channels, appear to associate with GBRs through KCTD16 (Schwenk et al. 2016). HCN channels are widely expressed in the heart and the central nervous system, where they are involved in the generation of rhythmic activity (Biel et al. 2009). GBRs activate HCN currents in dopaminergic neurons of the ventral tegmental area and thereby shorten the duration of inhibitory postsynaptic potentials (Schwenk et al. 2016). The mechanism of GBR-induced HCN channel activation is unknown but may include (1) membrane hyperpolarization via Kir3 channels, (2) allosteric gating of HCN channels by conformational changes in the receptor, and/or (3) dynamic interactions of HCN channels with G protein subunits or second messengers.
6 Additional Receptor-Associated Proteins
Additional components of native GBR signaling complexes are calnexin, reticulocalbin-2, inactive dipeptidyl-peptidases 6/10, 14-3-3 proteins, synaptotagmin-11, and neuroligin-3 (Schwenk et al. 2016). The anatomically and temporally restricted expression of these proteins in the brain limits the set of available receptor constituents in individual cells and further supports a modular GBR architecture. For some of these receptor components binding sites on GB1, GB2, or the KCTDs have been identified (Fritzius and Bettler 2020). Yeast-two-hybrid screens identified several additional proteins that potentially interact with GB1 or GB2 (Pin and Bettler 2016). These proteins may represent low-abundance or transiently interacting GBR components that escaped detection in proteomic approaches.
7 Concluding Remarks
During the past decade, numerous structural and biophysical studies have greatly improved our understanding of the sequence of allosteric events involved in the activation of heterodimeric GBRs. However, the structures of the full-length heterodimeric GBR at atomic resolution in its active and inactive state, with and without bound G protein or allosteric modulators, are still missing. Cryo-electron microscopy appears to be a promising approach to obtain such high-resolution structural information, which is necessary to validate and extend current concepts. The functional relevance of higher-order GBR complexes is still unclear and needs to be addressed in native tissue. The recognition that GBR heterodimers interact with an inventory of ~30 proteins to form a variety of multi-protein complexes with distinct kinetic properties, localizations, and functions represents a departure from earlier concepts based on receptor protomers working in isolation. For some GBR interacting proteins (KCTDs, APP, HCN channels), we have identified functional effects and/or obtained high-resolution structures in association with the receptor. However, we still lack functional and structural information for most of the receptor components identified in proteomic approaches. Furthermore, much effort needs to be devoted to the study of the structural dynamics in GBR complexes during physiological processes. Understanding the structure and function of identified GBR complexes in the brain hopefully will help to identify promising molecular targets for therapeutic intervention.
References
Balasco N, Smaldone G, Vitagliano L (2019) The structural versatility of the BTB domains of KCTD proteins and their recognition of the GABAB receptor. Biomol Ther 9(8). pii: E323
Biel M, Wahl-Schott C, Michalakis S, Zong X (2009) Hyperpolarization-activated cation channels: from genes to function. Physiol Rev 89:847–885
Biermann B, Ivankova-Susankova K, Bradaia A, Abdel Aziz S, Besseyrias V, Kapfhammer JP, Missler M, Gassmann M, Bettler B (2010) The sushi domains of GABAB receptors function as axonal targeting signals. J Neurosci 30:1385–1394
Binet V, Brajon C, Le Corre L, Acher F, Pin JP, Prezeau L (2004) The heptahelical domain of GABAB2 is activated directly by CGP7930, a positive allosteric modulator of the GABAB receptor. J Biol Chem 279:29085–29091
Binet V, Duthey B, Lecaillon J, Vol C, Quoyer J, Labesse G, Pin JP, Prezeau L (2007) Common structural requirements for heptahelical domain function in class A and class C G protein-coupled receptors. J Biol Chem 282:12154–12163
Bischoff S, Leonhard S, Reymann N, Schuler V, Shigemoto R, Kaupmann K, Bettler B (1999) Spatial distribution of GABABR1 receptor mRNA and binding sites in the rat brain. J Comp Neurol 412:1–16
Blein S, Ginham R, Uhrin D, Smith BO, Soares DC, Veltel S, McIlhinney RA, White JH, Barlow PN (2004) Structural analysis of the complement control protein (CCP) modules of GABAB receptor 1a: only one of the two CCP modules is compactly folded. J Biol Chem 279:48292–48306
Brock C, Boudier L, Maurel D, Blahos J, Pin JP (2005) Assembly-dependent surface targeting of the heterodimeric GABAB receptor is controlled by COPI but not 14-3-3. Mol Biol Cell 16:5572–5578
Burmakina S, Geng Y, Chen Y, Fan QR (2014) Heterodimeric coiled-coil interactions of human GABAB receptor. Proc Natl Acad Sci U S A 111:6958–6963
Calebiro D, Rieken F, Wagner J, Sungkaworn T, Zabel U, Borzi A, Cocucci E, Zurn A, Lohse MJ (2013) Single-molecule analysis of fluorescently labeled G-protein-coupled receptors reveals complexes with distinct dynamics and organization. Proc Natl Acad Sci U S A 110:743–748
Chen LH, Sun B, Zhang Y, Xu TJ, Xia ZX, Liu JF, Nan FJ (2014) Discovery of a negative allosteric modulator of GABAB receptors. ACS Med Chem Lett 5:742–747
Christopher JA, Aves SJ, Bennett KA, Dore AS, Errey JC, Jazayeri A, Marshall FH, Okrasa K, Serrano-Vega MJ, Tehan BG et al (2015) Fragment and structure-based drug discovery for a class C GPCR: discovery of the mGlu5 negative allosteric modulator HTL14242 (3-chloro-5-[6-(5-fluoropyridin-2-yl)pyrimidin-4-yl]benzonitrile). J Med Chem 58:6653–6664
Ciruela F, Fernandez-Duenas V, Sahlholm K, Fernandez-Alacid L, Nicolau JC, Watanabe M, Lujan R (2010) Evidence for oligomerization between GABAB receptors and GIRK channels containing the GIRK1 and GIRK3 subunits. Eur J Neurosci 32:1265–1277
Comps-Agrar L, Kniazeff J, Norskov-Lauritsen L, Maurel D, Gassmann M, Gregor N, Prezeau L, Bettler B, Durroux T, Trinquet E, Pin JP (2011) The oligomeric state sets GABAB receptor signalling efficacy. EMBO J 30:2336–2349
Correale S, Esposito C, Pirone L, Vitagliano L, Di Gaetano S, Pedone E (2013) A biophysical characterization of the folded domains of KCTD12: insights into interaction with the GABAB2 receptor. J Mol Recognit 26:488–495
Couve A, Filippov AK, Connolly CN, Bettler B, Brown DA, Moss SJ (1998) Intracellular retention of recombinant GABAB receptors. J Biol Chem 273:26361–26367
David M, Richer M, Mamarbachi AM, Villeneuve LR, Dupre DJ, Hebert TE (2006) Interactions between GABA-B1 receptors and Kir 3 inwardly rectifying potassium channels. Cell Signal 18:2172–2181
Dinamarca MC, Raveh A, Schneider A, Fritzius T, Fruh S, Rem PD, Stawarski M, Lalanne T, Turecek R, Choo M et al (2019) Complex formation of APP with GABAB receptors links axonal trafficking to amyloidogenic processing. Nat Commun 10:1331
Doly S, Shirvani H, Gata G, Meye FJ, Emerit MB, Enslen H, Achour L, Pardo-Lopez L, Yang SK, Armand V et al (2016) GABAB receptor cell-surface export is controlled by an endoplasmic reticulum gatekeeper. Mol Psychiatry 21:480–490
Dore AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B et al (2014) Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature 511:557–562
Dupuis DS, Relkovic D, Lhuillier L, Mosbacher J, Kaupmann K (2006) Point mutations in the transmembrane region of GABAB2 facilitate activation by the positive modulator N,N′-dicyclopentyl-2-methylsulfanyl-5-nitro-pyrimidine-4,6-diamine (GS39783) in the absence of the GABAB1 subunit. Mol Pharmacol 70:2027–2036
Duthey B, Caudron S, Perroy J, Bettler B, Fagni L, Pin JP, Prezeau L (2002) A single subunit (GB2) is required for G-protein activation by the heterodimeric GABAB receptor. J Biol Chem 277:3236–3241
Fowler CE, Aryal P, Suen KF, Slesinger PA (2007) Evidence for association of GABA(B) receptors with Kir3 channels and regulators of G protein signalling (RGS4) proteins. J Physiol 580:51–65
Freyd T, Warszycki D, Mordalski S, Bojarski AJ, Sylte I, Gabrielsen M (2017) Ligand-guided homology modelling of the GABAB2 subunit of the GABAB receptor. PLoS One 12:e0173889
Fritzius T, Bettler B (2020) The organizing principle of GABAB receptor complexes: physiological and pharmacological implications. Basic Clin Pharmacol Toxicol 126(Suppl 6):25–34
Fritzius T, Turecek R, Seddik R, Kobayashi H, Tiao J, Rem PD, Metz M, Kralikova M, Bouvier M, Gassmann M, Bettler B (2017) KCTD hetero-oligomers confer unique kinetic properties on hippocampal GABAB receptor-induced K+ currents. J Neurosci 37:1162–1175
Galvez T, Parmentier ML, Joly C, Malitschek B, Kaupmann K, Kuhn R, Bittiger H, Froestl W, Bettler B, Pin JP (1999) Mutagenesis and modeling of the GABAB receptor extracellular domain support a venus flytrap mechanism for ligand binding. J Biol Chem 274:13362–13369
Galvez T, Urwyler S, Prezeau L, Mosbacher J, Joly C, Malitschek B, Heid J, Brabet I, Froestl W, Bettler B et al (2000) Ca2+ requirement for high-affinity g-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol Pharmacol 57:419–426
Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prezeau L, Pin JP (2001) Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO J 20:2152–2159
Gassmann M, Bettler B (2012) Regulation of neuronal GABAB receptor functions by subunit composition. Nat Rev Neurosci 13:380–394
Geng Y, Xiong D, Mosyak L, Malito DL, Kniazeff J, Chen Y, Burmakina S, Quick M, Bush M, Javitch JA et al (2012) Structure and functional interaction of the extracellular domain of human GABAB receptor GBR2. Nat Neurosci 15:970–978
Geng Y, Bush M, Mosyak L, Wang F, Fan QR (2013) Structural mechanism of ligand activation in human GABAB receptor. Nature 504:254–259
Gregory KJ, Dong EN, Meiler J, Conn PJ (2011) Allosteric modulation of metabotropic glutamate receptors: structural insights and therapeutic potential. Neuropharmacology 60:66–81
Grunewald S, Schupp BJ, Ikeda SR, Kuner R, Steigerwald F, Kornau HC, Kohr G (2002) Importance of the g-aminobutyric acidB receptor C-termini for G-protein coupling. Mol Pharmacol 61:1070–1080
Hanack C, Moroni M, Lima WC, Wende H, Kirchner M, Adelfinger L, Schrenk-Siemens K, Tappe-Theodor A, Wetzel C, Kuich PH et al (2015) GABA blocks pathological but not acute TRPV1 pain signals. Cell 160:759–770
Hannan S, Wilkins ME, Smart TG (2012) Sushi domains confer distinct trafficking profiles on GABAB receptors. Proc Natl Acad Sci U S A 109:12171–12176
Havlickova M, Prezeau L, Duthey B, Bettler B, Pin JP, Blahos J (2002) The intracellular loops of the GB2 subunit are crucial for G-protein coupling of the heteromeric g-aminobutyrate B receptor. Mol Pharmacol 62:343–350
Ivankova K, Turecek R, Fritzius T, Seddik R, Prezeau L, Comps-Agrar L, Pin JP, Fakler B, Besseyrias V, Gassmann M, Bettler B (2013) Up-regulation of GABAB receptor signaling by constitutive assembly with the K+ channel tetramerization domain-containing protein 12 (KCTD12). J Biol Chem 288:24848–24856
Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, McMaster G, Angst C, Bittiger H, Froestl W, Bettler B (1997) Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature 386:239–246
Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R et al (1998) GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature 396:683–687
Kniazeff J, Saintot PP, Goudet C, Liu J, Charnet A, Guillon G, Pin JP (2004) Locking the dimeric GABAB G-protein-coupled receptor in its active state. J Neurosci 24:370–377
Koehl A, Hu H, Feng D, Sun B, Zhang Y, Robertson MJ, Chu M, Kobilka TS, Laeremans T, Steyaert J et al (2019) Structural insights into the activation of metabotropic glutamate receptors. Nature 566:79–84
Kuner R, Kohr G, Grunewald S, Eisenhardt G, Bach A, Kornau HC (1999) Role of heteromer formation in GABAB receptor function. Science 283:74–77
Margeta-Mitrovic M, Jan YN, Jan LY (2000) A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron 27:97–106
Marshall FH, Jones KA, Kaupmann K, Bettler B (1999) GABAB receptors – the first 7TM heterodimers. Trends Pharmacol Sci 20:396–399
Matsushita S, Nakata H, Kubo Y, Tateyama M (2010) Ligand-induced rearrangements of the GABAB receptor revealed by fluorescence resonance energy transfer. J Biol Chem 285:10291–10299
Maurel D, Comps-Agrar L, Brock C, Rives ML, Bourrier E, Ayoub MA, Bazin H, Tinel N, Durroux T, Prezeau L et al (2008) Cell-surface protein-protein interaction analysis with time-resolved FRET and snap-tag technologies: application to GPCR oligomerization. Nat Methods 5:561–567
Metz M, Gassmann M, Fakler B, Schaeren-Wiemers N, Bettler B (2011) Distribution of the auxiliary GABAB receptor subunits KCTD8, 12, 12b, and 16 in the mouse brain. J Comp Neurol 519:1435–1454
Monnier C, Tu H, Bourrier E, Vol C, Lamarque L, Trinquet E, Pin JP, Rondard P (2011) Trans-activation between 7TM domains: implication in heterodimeric GABAB receptor activation. EMBO J 30:32–42
Ng GY, Clark J, Coulombe N, Ethier N, Hebert TE, Sullivan R, Kargman S, Chateauneuf A, Tsukamoto N, McDonald T et al (1999) Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J Biol Chem 274:7607–7610
Olofsson L, Felekyan S, Doumazane E, Scholler P, Fabre L, Zwier JM, Rondard P, Seidel CA, Pin JP, Margeat E (2014) Fine tuning of sub-millisecond conformational dynamics controls metabotropic glutamate receptors agonist efficacy. Nat Commun 5:5206
Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, Ristig D, Schuler V, Meigel I, Lampert C et al (2001) C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J Neurosci 21:1189–1202
Pin JP, Bettler B (2016) Organization and functions of mGlu and GABAB receptor complexes. Nature 540:60–68
Pinkas DM, Sanvitale CE, Bufton JC, Sorrell FJ, Solcan N, Chalk R, Doutch J, Bullock AN (2017) Structural complexity in the KCTD family of Cullin3-dependent E3 ubiquitin ligases. Biochem J 474:3747–3761
Rajalu M, Fritzius T, Adelfinger L, Jacquier V, Besseyrias V, Gassmann M, Bettler B (2015) Pharmacological characterization of GABAB receptor subtypes assembled with auxiliary KCTD subunits. Neuropharmacology 88:145–154
Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D et al (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477:549–555
Restituito S, Couve A, Bawagan H, Jourdain S, Pangalos MN, Calver AR, Freeman KB, Moss SJ (2005) Multiple motifs regulate the trafficking of GABAB receptors at distinct checkpoints within the secretory pathway. Mol Cell Neurosci 28:747–756
Rice HC, de Malmazet D, Schreurs A, Frere S, Van Molle I, Volkov AN, Creemers E, Vertkin I, Nys J, Ranaivoson FM et al (2019) Secreted amyloid-b precursor protein functions as a GABABR1a ligand to modulate synaptic transmission. Science 363(6423). pii: eaao4827
Rondard P, Huang S, Monnier C, Tu H, Blanchard B, Oueslati N, Malhaire F, Li Y, Trinquet E, Labesse G et al (2008) Functioning of the dimeric GABAB receptor extracellular domain revealed by glycan wedge scanning. EMBO J 27:1321–1332
Schwenk J, Metz M, Zolles G, Turecek R, Fritzius T, Bildl W, Tarusawa E, Kulik A, Unger A, Ivankova K et al (2010) Native GABAB receptors are heteromultimers with a family of auxiliary subunits. Nature 465:231–235
Schwenk J, Perez-Garci E, Schneider A, Kollewe A, Gauthier-Kemper A, Fritzius T, Raveh A, Dinamarca MC, Hanuschkin A, Bildl W et al (2016) Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat Neurosci 19:233–242
Seddik R, Jungblut SP, Silander OK, Rajalu M, Fritzius T, Besseyrias V, Jacquier V, Fakler B, Gassmann M, Bettler B (2012) Opposite effects of KCTD subunit domains on GABAB receptor-mediated desensitization. J Biol Chem 287:39869–39877
Smaldone G, Pirone L, Pedone E, Marlovits T, Vitagliano L, Ciccarelli L (2016) The BTB domains of the potassium channel tetramerization domain proteins prevalently assume pentameric states. FEBS Lett 590:1663–1671
Stewart GD, Comps-Agrar L, Norskov-Lauritsen LB, Pin JP, Kniazeff J (2018) Allosteric interactions between GABAB1 subunits control orthosteric binding sites occupancy within GABAB oligomers. Neuropharmacology 136:92–101
Sun B, Chen L, Liu L, Xia Z, Pin JP, Nan F, Liu J (2016) A negative allosteric modulator modulates GABAB-receptor signalling through GB2 subunits. Biochem J 473:779–787
Turecek R, Schwenk J, Fritzius T, Ivankova K, Zolles G, Adelfinger L, Jacquier V, Besseyrias V, Gassmann M, Schulte U et al (2014) Auxiliary GABAB receptor subunits uncouple G protein bg subunits from effector channels to induce desensitization. Neuron 82:1032–1044
Urwyler S, Gjoni T, Koljatic J, Dupuis DS (2005) Mechanisms of allosteric modulation at GABAB receptors by CGP7930 and GS39783: effects on affinities and efficacies of orthosteric ligands with distinct intrinsic properties. Neuropharmacology 48:343–353
Vigot R, Barbieri S, Brauner-Osborne H, Turecek R, Shigemoto R, Zhang YP, Lujan R, Jacobson LH, Biermann B, Fritschy JM et al (2006) Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron 50:589–601
White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH (1998) Heterodimerization is required for the formation of a functional GABAB receptor. Nature 396:679–682
Winkler M, Biswas S, Berger SM, Küchler M, Enkel T, Preisendörfer L, Choo M, Früh S, Rem PD, Enkel T et al (2019) Pianp deficiency links GABAB receptor signaling and hippocampal and cerebellar neuronal cell composition to autism-like behavior. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0519-9
Wise A, Green A, Main MJ, Wilson R, Fraser N, Marshall FH (1999) Calcium sensing properties of the GABAB receptor. Neuropharmacology 38:1647–1656
Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V et al (2014) Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science 344:58–64
Xue L, Sun Q, Zhao H, Rovira X, Gai S, He Q, Pin JP, Liu J, Rondard P (2019) Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat Commun 10:2765
Yamada S, Nelson WJ (2007) Synapses: sites of cell recognition, adhesion, and functional specification. Annu Rev Biochem 76:267–294
Zheng S, Abreu N, Levitz J, Kruse AC (2019) Structural basis for KCTD-mediated rapid desensitization of GABAB signalling. Nature 567:127–131
Zuo H, Glaaser I, Zhao Y, Kurinov I, Mosyak L, Wang H, Liu J, Park J, Frangaj A, Sturchler E et al (2019) Structural basis for auxiliary subunit KCTD16 regulation of the GABAB receptor. Proc Natl Acad Sci U S A 116:8370–8379
Acknowledgments
We thank M. Gassmann for helpful discussions. This work was supported by grants of the Swiss Science Foundation (31003A-172881) and the National Center for Competences in Research (NCCR) “Synapsy, Synaptic Basis of Mental Health Disease” (to B.B).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Fritzius, T., Stawarski, M., Isogai, S., Bettler, B. (2020). Structural Basis of GABAB Receptor Regulation and Signaling. In: Vlachou, S., Wickman, K. (eds) Behavioral Neurobiology of GABAB Receptor Function. Current Topics in Behavioral Neurosciences, vol 52. Springer, Cham. https://doi.org/10.1007/7854_2020_147
Download citation
DOI: https://doi.org/10.1007/7854_2020_147
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91334-2
Online ISBN: 978-3-030-91335-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)