
Abstract
Schizophrenia is a devastating mental illness with a worldwide prevalence of approximately 1 %. Although the clinical features of the disorder were described over one hundred years ago, its neurobiology is still largely elusive despite several decades of research. Schizophrenia is associated with marked sleep disturbances and memory impairment. Above and beyond altered sleep architecture, sleep rhythms including slow waves and spindles are disrupted in schizophrenia. In the healthy brain, these rhythms reflect and participate in plastic processes during sleep. This chapter discusses evidence that schizophrenia patients exhibit dysfunction of sleep-mediated plasticity on a behavioral, cellular, and molecular level and offers suggestions on how the study of sleeping brain activity can shed light on the pathophysiological mechanisms of the disorder.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Schizophrenia is a chronic mental illness, characterized by psychosis and other symptoms that result in significant cognitive, social, and occupational dysfunction. The lifetime prevalence of schizophrenia has been estimated at about 1 % of the population worldwide, with males and females equally affected. In addition to its positive (e.g., hallucinations, delusions) and negative (e.g., apathy, anhedonia) symptoms, schizophrenia is associated with marked sleep disturbances and cognitive deficits. Because diagnosis is based solely on clinical criteria, it is likely that schizophrenia is not a single disease entity but rather may include a group of disorders resulting in similar clinical symptomatology. Sleep may be of particular relevance to schizophrenia, since abnormal brain connectivity and plasticity are thought to be core pathological features of the disorder and both are reflected in the sleep electroencephalogram (EEG ) (Stephan et al. 2006). Furthermore, electrophysiological measurements during sleep eliminate many factors that confound waking assessments of brain activity and cognition, including movement, attention, motivation, and comprehension of instructions. Consequently, sleep provides a unique modality for investigating neuropathological mechanisms, biological markers, and therapeutic targets for schizophrenia. This chapter discusses the evidence that schizophrenia patients exhibit dysfunction of sleep-mediated plasticity on a behavioral, cellular, and molecular level and offers suggestions on how further study of sleeping brain activity can shed light on the pathophysiological mechanisms of the disorder.
2 Description of the Disorder
Schizophrenia tends to appear in late adolescence or early adulthood, with earlier onset in men than in women. Diagnosis of schizophrenia is based on the presence of two or more characteristic positive and negative symptoms that persist for at least 1 month, in conjunction with significant dysfunction in work, interpersonal relations and/or self-care, and continuous signs of illness that are present for at least 6 months (DSM V, (American Psychiatric Association 2013). Diagnostic criteria are summarized in Table 1. The vast majority of individuals afflicted with the disorder remain at least partially ill, with either a stable course or progressive deterioration; full remission is uncommon. Schizophrenia is associated with a 10- to 25-year reduction in life expectancy; this is related both to elevated rates of suicides and accidents, which are closely associated with the illness itself, and higher rates of medical disorders (e.g., cardiovascular disease and cancer), which are related to comorbid factors such as increased substance use, poor diet, side effects of antipsychotic medications, and suboptimal medical care (Laursen et al. 2012).
Whereas the clinical features of schizophrenia have been described for over one hundred years (Bleuler 1911), the neurobiology of the disorder is still largely elusive despite decades of research. A large body of evidence now exists identifying biological abnormalities that characterize schizophrenia and many competing theories have been put forward to explain the pathophysiological mechanisms that might explain these observations (MacDonald and Schulz 2009). Some key theories are summarized in Table 2. A number of studies indicate that abnormalities in neurotransmitter pathways, gray and white matter structures, and cortical circuits that are involved in both sleep and brain plasticity may underlie specific symptoms and neurocognitive deficits in schizophrenia. Dysfunction of thalamocortical circuits is of particular interest, as these connections are thought to play a central role in coordinating sleep-mediated plasticity.
2.1 Sleep Patterns in Schizophrenia
After the neurophysiological identification of rapid eye movement (REM) sleep in the early 1950s, schizophrenia was one of the first disorders to be studied using polysomnography, motivated by the apparent similarities between dreaming and the positive symptoms of schizophrenia (hallucinations, delusions). Although schizophrenia did not turn out to be a disorder of REM sleep intrusion into wakefulness (Dement 1955; Rechtschaffen et al. 1964), sleep disturbance can be a significant clinical symptom and objective sleep EEG abnormalities have been identified across numerous studies. During periods of acute psychosis, sleep is often severely affected, with profound insomnia, reduced total sleep time, and fragmented sleep. In terms of sleep architecture, sleep recordings in schizophrenic patients have fairly consistently documented objective evidence of insomnia, including prolonged latency to sleep, reduced sleep efficiency, and reduced total sleep amount (Benca et al. 1992; Chouinard et al. 2004). Decrements in the amounts of visually scored deep stage 3 sleep (N3), also called slow wave sleep (SWS), have also been reported in some (Hiatt et al. 1985; Keshavan et al. 1998; Sekimoto et al. 2007; Zarcone et al. 1987), but not in all studies (Kempenaers et al. 1988; Lauer et al. 1997; Tandon et al. 1992). REM sleep abnormalities in schizophrenia have not been demonstrated consistently, although a number of early studies suggested that schizophrenics, like patients with mood disorders, had reduced latency to REM sleep (Kupfer et al. 1970). However, several meta-analyses have failed to support any consistent changes in latency to REM sleep or REM sleep amounts (Benca et al. 1992; Chouinard et al. 2004).
2.2 Memory Deficits in Schizophrenia
One of the most debilitating manifestations of disrupted brain plasticity in schizophrenia is impaired memory in several domains. Verbal declarative memory deficits are the most consistently found impairments in schizophrenia, confirmed by several meta-analyses (Cirillo and Seidman 2003; Sitskoorn et al. 2004; Snitz et al. 2006). Verbal declarative memory is memory for facts, ideas, stories, or events that can be consciously recalled. Impairments are present throughout the course of the illness, and in high-risk subjects and non-psychotic relatives of individuals with schizophrenia, suggesting that these deficits are partly associated with a genetic vulnerability to the disorder (Gur et al. 2007; Sitskoorn et al. 2004). Working memory is the ‘scratch pad of the mind,’ used for holding and manipulating information online during a cognitive task (Baddeley 1992). Schizophrenia patients have impaired working memory (Horan et al. 2008) and in unaffected offspring reduced performance predicted the later development of psychosis (Erlenmeyer-Kimling et al. 2000). Procedural learning is non-declarative memory for the performance of perceptual or motor tasks. Most studies report normal procedural learning in schizophrenia patients (Clare et al. 1993; Weickert 2002). However, these reports were derived from testing within a single session. Since healthy individuals show improvement in procedural memory after a night of sleep, Manoach et al. (2004) investigated the influence of sleep on learning in patients with schizophrenia. Despite normal learning within a training session, schizophrenics failed to show an improvement after a night of sleep, in contrast to healthy individuals, indicating a failure of sleep-mediated procedural memory consolidation in schizophrenia.
3 Neurochemical Abnormalities and Sleep-Dependent Plasticity
Neurotransmitter systems implicated in the pathogenesis of schizophrenia include dopamine, gamma-aminobutyric acid (GABA), acetylcholine, glutamate, and neuromodulators such as melatonin and orexin. These systems play important roles in neuroplasticity. Furthermore, sleep–wake regulation is achieved through a complex interaction of neurotransmitters in distributed brain regions; thus, abnormalities in these systems in schizophrenia are likely to impinge upon sleep and sleep-related plasticity.
3.1 Dopamine
Numerous studies show that dopamine (DA) neurotransmission is reduced in the cortex and elevated in the striatum in schizophrenia (Abi-Dargham et al. 2012; Fusar-Poli and Meyer-Lindenberg 2013; Howes et al. 2012). Dopaminergic circuits regulate both neuroplasticity and sleep–wake cycles. DA is a powerful modulator of synaptic plasticity via complex signaling that varies by receptor type and neural circuit. DA exerts its effects via G-protein-coupled receptors that may be excitatory (D1) or inhibitory (D2), with downstream effects on membrane excitability, synaptic transmission, protein trafficking, and gene transcription (Tritsch and Sabatini 2012). DA promotes wake and suppresses REM and non-REM (NREM) sleep, acting through projections from the ventral tegmental area, substantia nigra pars compacta, and ventral periaqueductal gray matter to inhibit sleep-promoting neurons in the basal forebrain, midbrain, brainstem, and hypothalamus (Monti and Jantos 2008). Although the impact of DA dysfunction on sleep-dependent plasticity has not yet been examined in schizophrenia, several lines of evidence suggest that it deserves further attention.
Firstly, the efficacy of the psychostimulant modafinil and its effects on sleep EEG are modulated by the presence of a polymorphism in the catechol-o-methyl transferase (COMT) gene, which is more common in schizophrenia patients than in the general population (Bodenmann et al. 2009; Bodenmann and Landolt 2010). COMT is an enzyme that degrades dopamine, and the polymorphism predicts reduced cognitive performance (Weinberger et al. 2001). The genotype’s interaction with modafinil suggests that COMT may play a role in altered sleep–wake regulation in schizophrenia. Secondly, elevated levels of D4 receptors (members of the D2-like receptor family) have been reported in schizophrenia (Seeman et al. 1993). D4 receptors are present in the pineal gland, where they decrease melatonin release through binding of adrenergic receptors. The pineal gland regulates sleep–wake rhythms through the circadian release of melatonin; therefore, the increase of D4 receptors in schizophrenia could contribute to the irregular sleep–wake patterns observed in the disorder (González et al. 2012). Moreover, the thalamic reticular nucleus (TRN), the structure responsible for generating sleep spindles, is rich in D4 receptors. D4 receptors are present presynaptically on GABA-containing projections from the globus pallidus to the TRN (Gandia et al. 1993; García-Cabezas et al. 2007), and activation of the D4 receptors reduces GABA release onto the TRN (Govindaiah et al. 2010). Thus, an excess of D4 receptors in schizophrenia could disrupt TRN control of thalamocortical oscillations, which coordinate sleep-related plasticity. However, the origins of these projections and the effects of dopamine on TRN neurons are still controversial (Florán et al. 2004; Mrzljak et al. 1996). Finally, in schizophrenia patients, working memory impairment correlates with increased D1-receptor binding potential in prefrontal cortex, suggesting a compensatory upregulation of the receptor in response to cortical DA deficiencies (Abi-Dargham et al. 2002). Animal studies suggest that prefrontal D1 receptors sustain the activity of prefrontal neurons while a stimulus is held in memory, and this persistent activity is considered the neural correlate of working memory (Gao et al. 2001). At first glance, working memory might be considered a purely waking function; however, working memory performance is improved by prior sleep (Kuriyama et al. 2008) and impaired by sleep deprivation (Drummond et al. 2012; Hagewoud et al. 2010). Sleep may therefore be a useful tool for probing abnormal DA function and working memory in schizophrenia.
3.2 Acetylcholine
Reduced cholinergic activity and abnormal cholinergic transmission have been found consistently in schizophrenia (Crook et al. 2000; 2001; Raedler et al. 2003) and are related to cognitive symptoms in animal models (Sarter et al. 2012). Although acetylcholine (ACh) is excitatory, AChRs are located both pre- and post-synaptically, allowing for a complex role in fine-tuning neuronal excitability (Drever et al. 2011). ACh can both facilitate and inhibit long-term potentiation (LTP) through graded effects on the responsiveness of glutamatergic and GABAergic cells in the hippocampus that mediate memory formation (Drever et al. 2011) and DA neurons in the ventral tegmental area that mediate reward and addiction (Gao et al. 2001). ACh also supports longer-term changes in cortical circuits by promoting synchronous cortical and subcortical firing; this may facilitate long-term plasticity by increasing the baseline excitability of neurons, which are then more responsive to modulation by other transmitters (Picciotto et al. 2012). Cholinergic transmission in the brainstem is important for the initiation and coordination of REM sleep (Jones 2005; Marks and Birabil 1998). Administration of muscarinic agonists induces shorter REM latencies in healthy individuals. In schizophrenia patients, the effect is much more pronounced than in healthy controls, suggesting a muscarinic cholinergic supersensitivity in the disease (Riemann et al. 1994). Cholinergic tone is normally high during REM sleep and low during NREM sleep, and in healthy young men, pharmacologically lowering REM cholinergic tone impairs consolidation of motor memory (Rasch et al. 2009), while pharmacologically elevating NREM ACh tone impairs consolidation of verbal declarative memory (Gais and Born 2004). Therefore, the contribution of ACh dysfunction to impaired sleep-dependent memory in schizophrenia warrants further investigation.
3.3 Glutamate
Glutamate is the most prevalent excitatory neurotransmitter in the central nervous system, and dysfunctions of glutamatergic neurotransmission are hypothesized to be an important mechanism of schizophrenia pathophysiology (Table 2). In particular, abnormalities in the N-methyl-D-aspartate receptor (NMDAR), a glutamate-gated ion channel, are thought to contribute to disrupted synaptic plasticity in the disorder (Marsman et al. 2013). Multiple signaling pathways modulating sleep and synaptic plasticity converge on the NMDA receptor, for example, D1 agonists and D2 antagonists increase NMDAR-dependent LTP, whereas D2 agonists decrease LTP (Centonze et al. 2004; Tseng and O’Donnell 2004) and acetylcholine modulates NMDA-dependent LTP and long-term depression (LTD) in visual cortex (Kirkwood et al. 1999) and hippocampus (Yun et al. 2000). In mice, sleep deprivation altered the ratio between LTP and LTD, a key cellular mechanism of neural plasticity, and changed the subunit composition of NMDARs (Kopp et al. 2006). Low doses of NMDAR antagonists produce arousal, while high doses produce sedation (Siegel 2011). Ketamine, a NMDAR antagonist that induces schizophrenia-like symptoms, disrupts thalamocortical rhythmicity (Buzsáki 1991), which is important for sleep-dependent memory processes. The role of the thalamocortical system in sleep-dependent plasticity is discussed in further detail below.
4 Structural and Functional Abnormalities
Gray matter atrophy and disrupted white matter connectivity have been reported consistently in schizophrenia, especially in frontal and temporal areas and in thalamocortical networks, including the thalamic radiation which carries fibers from the thalamus to prefrontal areas (Borgwardt et al. 2008; Gaser et al. 2004; Heinrichs and Zakzanis 1998; Kawada et al. 2009; Kyriakopoulos and Frangou 2009; McIntosh et al. 2008; Nuechterlein 1983; Ren et al. 2013; Snitz et al. 2006). Functional magnetic resonance imaging (fMRI) has revealed abnormal patterns of activity in the default mode network, a highly interconnected brain system that subserves self-referential thought and interpretation of the external environment (Hasenkamp et al. 2011; Ren et al. 2013; Skudlarski et al. 2010). These findings are in keeping with the view that disordered brain connectivity is central to schizophrenia (Stephan et al. 2006) (Table 2). Interestingly, the functional findings did not co-localize with anatomical abnormalities in gray or white matter in the same subjects. Abnormal functional connectivity may reflect reorganization of functional networks in response to declining structural connections. Sleep EEG patterns reflect propagation of neural activity through structural and functional networks; therefore, the use of EEG systems with high spatial resolution could further characterize the relationship between altered anatomical and functional connectivity in schizophrenia. An advantage of EEG recordings is the ability to monitor activity on a timescale closer to neuronal signaling (milliseconds) than fMRI (seconds). For example, by performing source modeling analysis of scalp-recorded EEG slow waves, it is possible to track the origin and propagation of slow waves along a mesial cortical highway, and the propagation of this wave is thought to play a role in coordinating neuronal plastic activity (Massimini et al. 2004). This technique has yet to be applied to schizophrenia.
In task-related studies of schizophrenia, abnormal prefrontal cortical activation is the most established finding, often associated with impaired performance in cognitive paradigms involving working memory (Barch et al. 2012), reinforcement learning (Ragland et al. 2012), and executive functions (Carter et al. 2012). Increased prefrontal activity in schizophrenia patients has been interpreted as a failure of automation, the shift from controlled effortful cognitive performance to less attention-demanding strategies. Automation has been proposed to be accomplished by a system-level reorganization of memory traces during sleep, a process reflected in EEG waveforms including spindles and slow waves (Manoach and Stickgold 2009). Therefore, investigating sleep EEG in conjunction with learning tasks could shed light on the biological basis of dysfunctional memory processes in schizophrenia.
5 Genetic Factors
A strong familial risk for schizophrenia has long been recognized, and a meta-analysis of twin studies calculated that 80 % of the probability of developing schizophrenia could be attributed to genetic factors (Sullivan et al. 2003). Over 50 candidate genes have been identified, yet no gene or genetic factor is shared by all schizophrenia patients. Recent genome-wide association studies have found an increased frequency of single nucleotide polymorphisms (SNPs) in schizophrenia (Lee et al. 2012). SNPs are the most common form of genetic variation within the general population and are therefore not unique to schizophrenia. However, SNPs may confer additional risk on individuals already predisposed to the illness. Their increased incidence in schizophrenia may explain the complex and heterogeneous phenotypes encountered in the disorder, and support a polygenic inheritance pattern. One SNP associated with schizophrenia is a functional polymorphism in the COMT gene (Cross-Disorder Group of the Psychiatric Genomics Consortium et al. 2013). As described earlier, COMT is an enzyme that regulates extracellular dopamine concentration in prefrontal cortex and the polymorphism leads to dopamine deficiency, which could impair sleep-dependent plasticity via altered oscillatory activity in the prefrontal cortex and thalamic reticular nucleus. Indeed, lower performance on working memory and prefrontal executive tasks is predicted by the presence of the COMT val allele in healthy controls, schizophrenia patients, and relatives of patients (Weinberger et al. 2001). The COMT SNP also influences sleep–wake regulation. Specifically, carriers of the val allele required a much lower dose of the stimulant modafinil to maintain attention during sleep deprivation (Bodenmann et al. 2009). Non-schizophrenia subjects with the val/val variant showed increased EEG activity in the high slow wave range (3.0–6.75 Hz) and the beta range (>16.75 Hz) during recovery sleep following modafinil treatment (Bodenmann and Landolt 2010). The ability of the COMT SNP to explain phenotypic variation in sleep architecture and EEG has not yet been explored in schizophrenia.
A well-researched gene consistently associated with schizophrenia is disrupted-in-schizophrenia 1 (DISC1) , which encodes a multifunctional scaffold protein. It is implicated in neuronal migration and maturation during development, as well as in synaptic plasticity in the adult brain (Chubb et al. 2008). DISC1 also impacts sleep; transgenic flies expressing human DISC1 displayed increased duration of sleep bouts but normal circadian rhythms, suggesting a role for DISC1 in regulating sleep homeostasis (Sawamura et al. 2008). In the mouse brain, DISC1 is expressed only in some brain structures, including regions important for sleep and plasticity such as the hippocampus, parts of the neocortex, hyphothalamus, stria terminalis, and TRN. Furthermore, DISC1 is highly expressed in the TRN during development, in a period when thalamocortical connections take shape (Austin et al. 2004). Given their crucial role in sleep-dependent plasticity, abnormal development of thalamocortical circuits would have far-reaching effects on sleep and cognition, in keeping with the view of schizophrenia as a neurodevelopmental disorder.
6 Sleep-Specific EEG Rhythms in Schizophrenia
Whereas early studies of sleep in schizophrenia focused on visually scored sleep architecture in 30-s epochs, modern automated analyses allow a more detailed assessment of sleep-specific rhythms by measuring power density and waveform counts. High-density EEG recordings from 256 channels, as opposed to 6 or less in conventional polysomnography, afford sufficient spatial resolution to examine regional variation. Slow waves and spindles, the characteristic waveforms of NREM sleep, play important roles in neuroplasticity and have been studied extensively in schizophrenia. Interestingly, intracortical recordings in humans have shown that rather than being global brain phenomena, both slow waves and spindles can occur in local circuits as temporally and spatially independent phenomena (Nir et al. 2011). Furthermore, localized, use-dependent changes suggest that spindles and slow waves reflect the ability of local circuits to be plastic (Fisher and Vyazovskiy 2014; Goel et al. 2014; Huber et al. 2004). Therefore, sleep EEG could provide a powerful tool for testing the functionality of distinct circuits in schizophrenia.
6.1 Slow Wave Abnormalities in Schizophrenia
During the deepest stage of NREM sleep, the EEG is dominated by large slow waves in the delta frequency range (0.5–4 Hz) as detailed in Chap. 1 of this volume. At the cellular level, slow waves reflect synchronized transitions in large populations of cortical pyramidal neurons between a depolarized upstate (a state of increased firing) and a hyperpolarized downstate (a state of decreased firing) (Steriade et al. 1993). This slow oscillation is cortically generated and maintained (Amzica and Steriade 1995; Timofeev et al. 2000), but is shaped by the thalamus (Steriade et al. 1993). High-density EEG recordings show that cortical slow waves typically originate in frontal regions (the insula and cingulate gyrus) and travel in an anterior–posterior direction along the cingulate gyrus and neighboring regions (Murphy et al. 2009). This coincides with a major white fiber pathway identified by diffusion spectrum imaging (DSI) (Hagmann et al. 2008). Studies of sleep and learning demonstrate that slow waves reflect plastic changes. For example, when sleep was recorded following training on a visuomotor task, the cortex showed an increase in slow wave activity in task-relevant areas, which correlated with improved task performance the following morning (Huber et al. 2004). SWS also positively correlated with overnight improvement on tests of visual (Stickgold et al. 2000) and verbal declarative memory (Plihal and Born 1997). Moreover, selectively suppressing slow waves without shortening sleep time was detrimental to motor sequence learning (Landsness et al. 2011).
Slow wave deficits in schizophrenia have been frequently but inconsistently reported. Several studies have found that patients spend less time in visually scored stage N3. Automated analyses showed reduced power in the slow wave frequency range (0.5–4 Hz) and a lower count of slow waves across the total sleep period (Keshavan et al. 1998; Sekimoto et al. 2007). Differences in slow wave topography have also been reported. Whereas healthy controls showed significantly higher slow wave counts in right frontal and central regions, this asymmetry was lacking in patients with schizophrenia (Sekimoto et al. 2011). Slow wave deficits have been reported in medicated (Göder et al. 2004; Sekimoto et al. 2007), unmedicated (Feinberg et al. 1969), first-episode neuroleptic naïve (Poulin et al. 2003), and remitted (Kupfer et al. 1970; Traub 1972) patients. Furthermore, a reduced slow wave count has been identified in non-psychotic first-degree relatives of schizophrenia patients, suggesting it may be a trait linked to predisposition to the disease (Keshavan et al. 2004). However, a number of studies have found no deficits in visually scored N3 or in automated measures of slow wave incidence, amplitude, or slope in medicated patients (Ferrarelli et al. 2010a; Manoach et al. 2010), or neuroleptic naïve patients (Forest et al. 2007). A meta-analysis of unmedicated patients also failed to confirm abnormalities in SWS as a percentage of total sleep time in schizophrenics (Chouinard et al. 2004). Inconsistencies between reports could be due to factors including sample size, medication status, and methodological differences such as slow wave detection algorithms. Alternatively, given the heterogeneous nature of the disorder, it may be that slow wave deficits are present only in a subset of schizophrenic patients.
Göder et al. (2004) examined the relationship between SWS and plasticity in chronically medicated schizophrenia patients and healthy controls matched for age, sex, and education level. Compared to controls, patients exhibited significantly shorter SWS duration and poorer visuospatial memory performance. SWS duration was positively correlated with memory performance in patients but not in controls (the lack of correlation in controls may have been due to a ceiling effect on performance). A subsequent study found that greater SWS duration was associated with better verbal declarative memory the following morning in chronically medicated schizophrenia patients (Göder et al. 2008). Furthermore, enhancing slow waves using transcranial direct current stimulation during NREM sleep improved verbal memory retention in medicated schizophrenia patients (Göder et al. 2013). These studies establish that the correlation of SWS with memory performance that was previously demonstrated in healthy individuals is also present in schizophrenia patients. Furthermore, reduced SWS in the disorder correlates with reduced cognitive performance, suggesting that in schizophrenia, learning deficits may be related to dysfunction in SWS-mediated plasticity. The studies described above relied on visually scored measures of SWS. Automated analyses that may be useful in future are the slope and amplitude of slow waves, which reflect synaptic strength (Esser et al. 2007; Vyazovskiy et al. 2009). Such measures could provide more detailed insight into the mechanism by which slow wave deficits contribute to impaired learning in schizophrenia.
Slow wave power is high at birth, increases steeply in the first few years of life, and then decreases dramatically during adolescence (Buchmann et al. 2011; Feinberg and Campbell 2010; Tarokh and Carskadon 2010). This inverted u-shaped pattern parallels other markers of neural development throughout childhood and adolescence, including synaptic density (Huttenlocher and Dabholkar 1997), the size of pyramidal neurons and the size and complexity of dendritic trees in pre-frontal cortex (Petanjek et al. 2008), brain energy consumption measured by PET (Chugani et al. 1987), and microstructure of white matter tracts measured by diffusion tensor imaging (DTI) (Lebel et al. 2012). Healthy cortical maturation appears to follow a general pattern of early overproliferation followed by elimination, with adolescence being a critical period of reorganization (Feinberg and Campbell 2010). Gray matter matures in a regionally and temporally complex pattern (Gogtay et al. 2004) and a recent cross-sectional study demonstrated that healthy developmental declines in SWA were correlated with gray matter maturation in a regionally specific manner (Buchmann et al. 2011). A longitudinal study of gray matter volumes in early-onset schizophrenia has shown accelerated gray matter losses during adolescence that correlated with symptom severity (Thompson et al. 2001). The dramatic decline in slow wave activity during adolescence likely reflects a reduction in synaptic strength and in the number of neurons available to participate in the synchronous slow oscillation. Given that slow wave amplitude depends on the ability of cortical neuronal populations to fire synchronously (Vyazovskiy et al. 2009), impaired neuronal integration could result in less coordinated activity, detectable as slow wave deficits. The reduction in slow waves seen in adults with schizophrenia may therefore be a product of disrupted neurodevelopment. The relationship between gray matter development and slow wave activity has not yet been examined in schizophrenia patients, however. Beyond being a marker of cortical maturation, slow waves may actively participate in the pruning of synapses (Maret et al. 2011). Neural proliferation and pruning during adolescence is a critical period for integrating complex neural circuits, and errors during this process would have severe cognitive and neural consequences.
6.2 Spindle Abnormalities in Schizophrenia
Sleep spindles are characteristic of NREM sleep and define stage N2. Spindles are waxing and waning bursts of highly synchronized 12–16 Hz oscillations, generated in the TRN and propagated throughout the cortex (Steriade 2003). In healthy humans and animals, an increase in the number and duration of spindles correlates with overnight improvements in declarative and procedural memory (Fogel and Smith 2011). Neuroimaging studies using simultaneous functional MRI and EEG showed that networks subserving memory function were activated during spindle activity, including thalamus, hippocampus, midbrain, anterior cingulate, and prefrontal cortex (Schabus et al. 2007), and spindle amplitude correlated with activity in the putamen and with gains in performance on a motor task (Doyon et al. 2011).
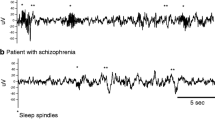
Several studies have found marked deficits in spindle activity in schizophrenia. Older studies using small sample sizes, manual techniques, and only 2 EEG leads (C3 and C4) had inconsistent spindle findings (Hiatt et al. 1985; Poulin et al. 2003; Van Cauter et al. 1991). Recent studies using automated detection methods, larger sample sizes, and more electrodes show more consistent deficits in schizophrenia. In the most comprehensive studies to date, medicated schizophrenia patients exhibited deficits in several spindle parameters compared to healthy controls, including power in the spindle range (13.75–15 Hz), spindle amplitude, duration, number, density (spindles/minute), and integrated spindle activity (ISA, calculated by integrating spindle activity over time) (Ferrarelli et al. 2007, 2010a). Furthermore, the spindle deficits were not present in non-schizophrenia patients receiving antipsychotics, including major depressive and bipolar patients; therefore, the effect is not explained by medication, not found in patients with other psychiatric disorders, and could be specific to the disorder. ISA was inversely correlated with positive symptoms and distinguished schizophrenia patients from psychiatric and healthy controls; therefore, spindle parameters could be a useful biological marker for schizophrenia (Ferrarelli 2013).
Given the role of spindles in learning and memory, spindle deficits could be expected to correlate with cognitive impairment in schizophrenia. Indeed, Göder et al. (2008) found that in chronically medicated patients, greater spindle density was associated with better verbal declarative memory the following morning. The study did not include a non-schizophrenia control group as it was investigating the effect of olanzapine treatment. Wamsley et al. (2012) later tested the question directly. Compared to healthy controls, medicated patients with chronic schizophrenia showed less overnight improvement on a finger tapping motor task. Spindle number, density, and interelectrode coherence were reduced in the patient group and predicted overnight performance improvement. This impairment of sleep-dependent learning in schizophrenia is also present in daytime nap sleep (Seeck-Hirschner et al. 2010). Following a 40-min afternoon nap, visuomotor memory (mirror tracing) improved in healthy individuals and depressed patients. By contrast, medicated schizophrenia patients failed to improve and showed significantly reduced spindle density during their naps, although the correlation with performance was not significant. Manoach et al. (2010) compared the sleep and motor learning of medicated schizophrenia patients with demographically matched controls. They confirmed previous findings of spindle deficits in the patient group, reporting reductions in spindle power (13.5–15 Hz) and spindle density during the last quartile of sleep. Moreover, the patient group showed no overnight improvement on the motor task, in contrast to the control group which showed significant improvements that correlated with stage N2 duration.
Of note, spindles are also thought to block transmission of sensory information from lower brain structures to the cortex during sleep, resulting in decreased reactivity to stimuli, thereby promoting sleep continuity (Steriade et al. 1993). A failure of this gating mechanism would lead to a lower arousal threshold and more fragmented sleep. Therefore, in addition to impaired plasticity, spindle abnormalities may contribute to the common complaint of poor sleep quality and low sleep efficiency in schizophrenia.
7 The Thalamocortical Network in Sleep and Plasticity
Spindles and slow waves do not occur independently but rather participate in an important rhythmic network spanning cortical and subcortical structures. During sleep, almost all cortical neurons undergo a slow oscillation in membrane potential of <1 Hz (Steriade 2003). This underlying oscillation is a fundamental characteristic of NREM sleep and temporally groups slow waves, spindles, and hippocampal ripples, supporting synchronized activity in distant brain regions. Coordinated activity across thalamocortical networks during NREM sleep plays a crucial role in brain plasticity. One key mechanism thought to underlie hippocampal-dependent memory is the reactivation of firing patterns in hippocampal and neocortical neurons. In this view of memory consolidation, a newly encoded memory trace is initially stored in the hippocampus, and then during sleep, the temporary trace is reactivated and transferred to the cortex where enduring plastic changes occur to form long-term memories. Animal and human studies suggest that this ‘replaying’ of memory traces is mediated by hippocampal ripples which are temporally correlated with sleep spindles, and that spindles, in turn, are typically phase-locked with slow waves. This temporal coordination may facilitate synaptic plasticity by aligning hippocampal replay activity with periods of high cortical excitability during the depolarized upstate of slow waves (Mölle and Born 2011).
The slow wave and spindle deficits observed in schizophrenia therefore point to a fundamental dysfunction of the thalamocortical network. This is corroborated by findings of disrupted functional and structural thalamocortical connectivity in MRI studies. Further support for this notion comes from a recent study in a rat model of schizophrenia. Administration of mitotoxin methylazoxymethanol acetate to pregnant rats results in offspring that exhibit physiological and cognitive dysfunction reminiscent of schizophrenia, including impaired spatial working memory. Phillips et al. (2012) reported that these animals also showed disruption of thalamocortical coordination during NREM sleep. Slow wave synchrony and propagation across the cortex were impaired, spindle phase-locking to delta waves was disrupted and hippocampal ripples were temporally uncoupled from spindles. This loss of coordination may underlie the cognitive and memory deficits seen in this rat model and in schizophrenia patients.
One possible cause of thalamocortical dyscoordination in schizophrenia is the thalamic reticular nucleus , a structural and functional hub of thalamocortical connectivity. In addition to its role as the spindle generator, the TRN is ideally placed to modulate bottom-up and top-down processes underlying higher cognitive function. It directly innervates the thalamus and receives excitatory inputs from thalamocortical and cortico-thalamic projections (Jones 2007). The TRN acts as an integrator of multiple functional modalities, amplifying relevant information and attenuating irrelevant or distracting information (Pinault 2011); as a result, the TRN has been central to theories of attention regulation (Crick 1984), sensory gating (Yingling and Skinner 1977), and self-representation (Vukadinovic 2011), all of which are impaired in schizophrenia. Evidence from molecular, animal, and human studies indicates a neurobiological dysfunction in the TRN in schizophrenia (for recent reviews, see Ferrarelli and Tononi 2011; Vukadinovic 2011).
TRN interneurons and outputs to thalamic relay nuclei are entirely GABAergic (Houser et al. 1980). The mechanism of synchronization across TRN cells is still largely unknown. GABAergic synapses between TRN neurons are one possibility; GABA receptors give rise to depolarizing currents in TRN neurons, which generate persistent spindle oscillations in these cells (Bazhenov et al. 1999; Golomb et al. 1994). Both dopamine and glutamate have been proposed to contribute to GABA hypofunction in the TRN, and the NMDAR-antagonist ketamine, which produces schizophrenia-like symptoms in humans and animals, has been shown to block thalamocortical rhythmicity (Buzsáki 1991). The GABA hypofunction observed in schizophrenia could therefore contribute to decreased spindle synchronicity, resulting in uncoordinated thalamocortical rhythms and consequently impaired plasticity.
The TRN also plays a role in embryonic development, providing axonal guidance for thalamocortical and cortical thalamic neurons (Pinault 2011). Animal studies indicate that aberrant TRN-dependent processes during critical periods of development could lead to higher-order cognitive impairments. Normally developing human and rat neonates exhibit spontaneous muscle twitches and jerks. In rats, sensory feedback from these movements triggers spindles in a somatotopic manner in primary somatosensory cortex (S1) and similar EEG bursts are observed in preterm infants (Khazipov et al. 2004); the authors hypothesize that coordinated motor-spindle activity establishes a map of the muscular system in S1 and strengthens developing thalamocortical and motor-sensory connections. Thus, spindle activity during fetal development may contribute to the development of neuronal self-representation. Importantly, an inability to correctly identify self-generated actions and thoughts may result in misattribution of mental experience to an external source and this can manifest itself as psychosis, including auditory hallucinations. Disruption of spindle-mediated plasticity during sensorimotor development could therefore contribute to positive symptoms of schizophrenia such as hearing voices (Vukadinovic 2011).
8 Impact of Medication on Sleep and Plasticity
There is a need for new drugs to treat schizophrenia, as many of the cognitive symptoms are not improved by medication (Reichenberg and Harvey 2007). The strong link between memory and spindles and slow waves makes them an attractive target for therapy. Most medications used to treat schizophrenia are thought to exert their antipsychotic effects through antagonism of dopamine receptors, but they may also interact with acetylcholine, serotonin, and histamine receptors. A drug’s relative affinity for these different receptors gives rise to a variety of side effects, including sedation (via serotonergic and histaminergic pathways), movement abnormalities (via dopaminergic pathways), and disruption of REM sleep (via anticholinergic and serotonergic actions) (Krystal et al. 2008). Both spindles and slow waves are modulated by these neurotransmitter systems and are therefore sensitive to pharmacological manipulation by antipsychotics, with the potential for cognitive benefits as well as undesirable side effects.
Animal studies suggest that current antipsychotics interact with the TRN. Phencyclidine (PCP) is an NMDA-receptor antagonist that induces schizophrenia-like behavioral symptoms in humans and animals; therefore, it is used as a pharmacological model of the disease. PCP administration in rats induced metabolic hypofunction within the TRN, which was reversed after coadministration of typical (haloperidol) or atypical (clozapine) antipsychotics with PCP (Cochran et al. 2002, 2003). PCP also induced excitotoxic lesions in the posterior cingulate and retrosplenial cortices of rats following injections into the anterior thalamus. These lesions were likely due to reduced inhibitory control of the TRN on anterior thalamic nuclei, which led to excessive activation (excitotoxicity) of corticolimbic areas. Importantly, these excitotoxic effects on the rat brain could be prevented by administering antipsychotic medications, such as haloperidol and clozapine (Olney et al. 1991; Sharp et al. 2001; Tomitaka et al. 2000). However, despite the interaction of current anti-psychotics with the TRN, they do not appear to normalize spindles in cross-sectional samples (Ferrarelli et al. 2010b). Within-subject trials are needed to confirm the effects of medication on spindle parameters.
Some recent studies have investigated the utility of drugs that influence spindle activity, with mixed success. A study in healthy individuals used zolpidem (Ambien) to increase the number of spindles during a daytime nap. The spindle increase was associated with improved verbal memory but not motor learning and was detrimental to perceptual learning (Mednick et al. 2013). Whether the decrement in perceptual learning related to increased spindles or some other effect of the drug is unclear. A recent study of medicated schizophrenic patients suggests a potential benefit of spindle enhancement, although the study may have been underpowered. Eszopiclone (Lunesta), a non-benzodiazepine hypnotic, was used to increase spindle number and density (Wamsley et al. 2013). The eszopiclone group showed significant overnight improvement on a motor learning task and the placebo group did not; however, the difference between the two groups was not significant. When the groups were combined, spindle number and density correlated with overnight motor task improvement, as has been shown in non-intervention studies.
Olanzapine and risperidone, although primarily antagonists of the D2 receptor, are thought to increase SWS through antagonism of serotonin-2 receptors (Krystal et al. 2008). Given the benefit of SWS to learning in healthy individuals, these agents might be expected to improve memory in schizophrenia. However, this hypothesis was not supported when olanzapine was administered to schizophrenia patients who were also chronically medicated with amisulpride, another atypical antipsychotic (Göder et al. 2008). Before bedtime, patients were trained on visuomotor and verbal memory tasks and then took a single dose of either olanzapine or placebo. SWS was increased in the olanzapine group as expected, but memory performance the following morning did not differ between groups. Spindle density was significantly decreased in the olanzapine group, which could have negated any putative benefits of increased SWS.
Manipulating sleeping brain activity to improve cognitive function in schizophrenia is a promising avenue of research, but it remains to be seen which sleep parameters should be targeted and which interventions are most effective. A better understanding of the cellular dynamics and neuropharmacology of sleep spindles and slow waves could provide new insights into the mechanisms of current drug actions and more focused targets for drug development.
9 Conclusion
Brain activity during sleep reveals important clues to the biological underpinnings of schizophrenia, a widespread mental illness with devastating consequences for individuals and society. Disrupted sleep appears to contribute to cognitive deficits including memory impairment and psychotic symptoms. Abnormalities of sleep EEG draw attention to dysfunction of plastic processes including neurodevelopment, neurotransmitter pathways, and specific neuronal structures such as the thalamic reticular nucleus and thalamocortical circuits. Further investigation of sleeping brain activity in schizophrenia has the potential to uncover mechanisms and biomarkers of the disease as well as provide promising targets for drug development.
References
Abi-Dargham A, Mawlawi O, Lombardo I, Gil R, Martinez D, Huang Y, Hwang DR, Keilp J, Kochan L, Van Heertum R, Gorman JM, Laruelle M (2002) Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci Official J Soc Neurosci 22(9), 3708–3719. doi:20026302
Abi-Dargham A, Xu X, Thompson JL, Gil R, Kegeles LS, Urban N, Hwang DR, Laruelle M, Slifstein M (2012) Increased prefrontal cortical D1 receptors in drug naive patients with schizophrenia: a PET study with [11C]NNC112. J Psychopharmacol 26(6):794–805. doi:10.1177/0269881111409265
American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, 5th edn. American Psychiatric Publishing, Arlington
Amzica F, Steriade M (1995) Disconnection of intracortical synaptic linkages disrupts synchronization of a slow oscillation. J Neurosci Official J Soc Neurosci 15(6):4658–4677
Austin CP, Ky B, Ma L, Morris JA, Shughrue PJ (2004) Expression of disrupted-In-schizophrenia-1, a schizophrenia-associated gene, is prominent in the mouse hippocampus throughout brain development. Neuroscience 124(1):3–10. doi:10.1016/j.neuroscience.2003.11.010
Baddeley A (1992) Working memory. Science 255(5044):556–559
Barch DM, Moore H, Nee DE, Manoach DS, Luck SJ (2012) CNTRICS imaging biomarkers selection: Working memory. Schizophrenia Bull 38(1):43–52. doi:10.1093/schbul/sbr160
Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ (1999) Self-sustained rhythmic activity in the thalamic reticular nucleus mediated by depolarizing GABAA receptor potentials. Nature Neurosc 2(2):168–174. doi:10.1038/5729
Benca RM, Obermeyer WH, Thisted RA, Gillin JC (1992) Sleep and psychiatric disorders. A meta-analysis. Arch Gen Psychiatr 49(8):651–668; discussion 669–670
Bleuler E (1911) Dementia Praecox or the Group of Schizophrenias
Bodenmann S, Landolt H-P (2010) Effects of modafinil on the sleep EEG depend on Val158Met genotype of COMT. Sleep 33(8):1027–1035
Bodenmann S, Xu S, Luhmann UFO, Arand M, Berger W, Jung HH, Landolt H-P (2009) Pharmacogenetics of modafinil after sleep loss: catechol-O-methyltransferase genotype modulates waking functions but not recovery sleep. Clin Pharmacol Ther 85(3):296–304. doi:10.1038/clpt.2008.222
Borgwardt SJ, McGuire PK, Aston J, Gschwandtner U, Pflüger MO, Stieglitz RD, Radue EW, Riecher-Rössler A (2008) Reductions in frontal, temporal and parietal volume associated with the onset of psychosis. Schizophr Res 106(2–3):108–114. doi:10.1016/j.schres.2008.08.007
Buchmann A, Ringli M, Kurth S, Schaerer M, Geiger A, Jenni OG, Huber R (2011) EEG sleep slow-wave activity as a mirror of cortical maturation. Cereb Cortex 21(3):607–615. doi:10.1093/cercor/bhq129
Buzsáki G (1991) The thalamic clock: emergent network properties. Neuroscience 41(2–3):351–364
Carter CS, Minzenberg M, West R, Macdonald A 3rd (2012) CNTRICS imaging biomarker selections: executive control paradigms. Schizophr Bull 38(1):34–42. doi:10.1093/schbul/sbr114
Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, Calabresi P (2004) Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci Official J Soc Neurosci 24(38):8214–8222. doi:10.1523/JNEUROSCI.1274-04.2004
Chouinard S, Poulin J, Stip E, Godbout R (2004) Sleep in untreated patients with schizophrenia: a meta-analysis. Schizophr Bull 30(4):957–967
Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK (2008) The DISC locus in psychiatric illness. Mol Psychiatry 13(1):36–64. doi:10.1038/sj.mp.4002106
Chugani HT, Phelps ME, Mazziotta JC (1987) Positron emission tomography study of human brain functional development. Ann Neurol 22(4):487–497. doi:10.1002/ana.410220408
Cirillo MA, Seidman LJ (2003) Verbal declarative memory dysfunction in schizophrenia: from clinical assessment to genetics and brain mechanisms. Neuropsychol Rev 13(2):43–77
Clare L, McKenna PJ, Mortimer AM, Baddeley AD (1993) Memory in schizophrenia: what is impaired and what is preserved? Neuropsychologia 31(11):1225–1241. doi:10.1016/0028-3932(93)90070-G
Cochran SM, Fujimura M, Morris BJ, Pratt JA (2002) Acute and delayed effects of phencyclidine upon mRNA levels of markers of glutamatergic and GABAergic neurotransmitter function in the rat brain. Synapse 46(3):206–214. doi:10.1002/syn.10126
Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ (2003) Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. Official Publ Am Coll Neuropsychopharmacol 28(2):265–275. doi:10.1038/sj.npp.1300031
Crick F (1984) Function of the thalamic reticular complex: the searchlight hypothesis. Proc Nat Acad Sci USA 81(14):4586–4590
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B (2000) Decreased muscarinic receptor binding in subjects with schizophrenia: a study of the human hippocampal formation. Biol Psychiatry 48(5):381–388. doi:10.1016/S0006-3223(00)00918-5
Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B (2001) Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: a study of Brodmann’s areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am J Psychiatry 158(6):918–925
Cross-Disorder Group of the Psychiatric Genomics Consortium, Smoller JW, Craddock N, Kendler K, Lee PH, Neale BM, Sullivan PF, et al (2013) Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381(9875):1371–1379. doi:10.1016/S0140-6736(12)62129-1
Dement W (1955) Dream recall and eye movements during sleep in schizophrenics and normals. J Nerv Ment Dis 122(3):263–269
Doyon J, Orban P, Barakat M, Debas K, Lungu O, Albouy G, Benali H et al (2011) Plasticité du Cerveau Associée à l’Apprentissage Moteur. Méd Sci M/S 27(4):413–420. doi:10.1051/medsci/2011274018
Drever BD, Riedel G, Platt B (2011) The cholinergic system and hippocampal plasticity. Behav Brain Res 221(2):505–514. doi:10.1016/j.bbr.2010.11.037
Drummond SPA, Anderson DE, Straus LD, Vogel EK, Perez VB (2012) The effects of two types of sleep deprivation on visual working memory capacity and filtering efficiency. PLoS ONE 7(4). doi:10.1371/journal.pone.0035653
Erlenmeyer-Kimling L, Rock D, Roberts SA, Janal M, Kestenbaum C, Cornblatt B, Adamo UH, Gottesman II (2000) Attention, memory, and motor skills as childhood predictors of schizophrenia-related psychoses: the New York high-risk project. Am J Psychiatry 157(9):1416–1422
Esser SK, Hill SL, Tononi G (2007) Sleep homeostasis and cortical synchronization: i. modeling the effects of synaptic strength on sleep slow waves. Sleep 30(12):1617–1630
Fatemi SH, Folsom TD (2009) The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull 35(3):528–548. doi:10.1093/schbul/sbn187
Feinberg I, Campbell IG (2010) Sleep EEG changes during adolescence: an index of a fundamental brain reorganization. Brain Cogn 72(1):56–65. doi:10.1016/j.bandc.2009.09.008
Feinberg I, Braun M, Koresko RL, Gottlieb F (1969) Stage 4 sleep in schizophrenia. Arch Gen Psychiatry 21(3):262–266
Ferrarelli F (2013) Endophenotypes and biological markers and of schizophrenia: from biological signs of illness to novel treatment targets. Curr Pharm Des
Ferrarelli F, Tononi G (2011) The thalamic reticular nucleus and schizophrenia. Schizophrenia Bull 37(2):306–315. doi:10.1093/schbul/sbq142
Ferrarelli F, Huber R, Peterson MJ, Massimini M, Murphy M, Riedner BA, Watson A, Bria P, Tononi G (2007) Reduced sleep spindle activity in schizophrenia patients. Am J Psychiatry 164(3):483–492. doi:10.1176/appi.ajp.164.3.483
Ferrarelli F, Peterson MJ, Sarasso S, Riedner BA, Murphy MJ, Benca RM, Bria P, Kalin NH, Tononi G (2010a) Thalamic dysfunction in schizophrenia suggested by whole-night deficits in slow and fast spindles. Am J Psychiatry 167(11):1339–1348. doi:10.1176/appi.ajp.2010.09121731
Ferrarelli F, Peterson MJ, Sarasso S, Riedner BA, Murphy MJ, Benca RM, Bria P, Kalin NH, Tononi G (2010b) Thalamic dysfunction in schizophrenia suggested by whole-night deficits in slow and fast spindles. Am J Psychiatry 167(11):1339–1348. doi:10.1176/appi.ajp.2010.09121731
Fisher SP, Vyazovskiy VV (2014) Local sleep taking care of high-maintenance cortical circuits under sleep restriction. Sleep 37(11):1727–1730. doi:10.5665/sleep.4156
Florán B, Florán L, Erlij D, Aceves J (2004) Activation of dopamine D4 receptors modulates [3H]GABA release in slices of the rat thalamic reticular nucleus. Neuropharmacology 46(4):497–503. doi:10.1016/j.neuropharm.2003.10.004
Fogel SM, Smith CT (2011) The function of the sleep spindle: a physiological index of intelligence and a mechanism for sleep-dependent memory consolidation. Neurosci Biobehav Rev 35(5):1154–1165. doi:10.1016/j.neubiorev.2010.12.003
Forest G, Poulin J, Daoust A-M, Lussier I, Stip E, Godbout R (2007) Attention and non-REM sleep in neuroleptic-naive persons with schizophrenia and control participants. Psychiatry Res 149:33–40. doi:10.1016/j.psychres.2005.11.005
Fusar-Poli P, Meyer-Lindenberg A (2013) Striatal presynaptic dopamine in schizophrenia, part II: meta-analysis of [(18)F/(11)C]-DOPA PET studies. Schizophr Bull 39(1):33–42. doi:10.1093/schbul/sbr180
Gais S, Born J (2004) Low acetylcholine during slow-wave sleep is critical for declarative memory consolidation. Proc Nat Acad Sci USA 101(7):2140–2144
Gandia JA, De Las Heras S, García M, Giménez-Amaya JM (1993) Afferent projections to the reticular thalamic nucleus from the globus pallidus and the substantia nigra in the rat. Brain Res Bull 32(4):351–358
Gao WJ, Krimer LS, Goldman-Rakic PS (2001) Presynaptic regulation of recurrent excitation by D1 receptors in prefrontal circuits. Proc Natl Acad Sci USA 98:295–300. doi:10.1073/pnas.011524298
García-Cabezas MA, Rico B, Sánchez-González MA, Cavada C (2007) Distribution of the dopamine innervation in the macaque and human thalamus. NeuroImage 34(3):965–984. doi:10.1016/j.neuroimage.2006.07.032
Gaser C, Nenadic I, Buchsbaum BR, Hazlett EA, Buchsbaum MS (2004) Ventricular enlargement in schizophrenia related to volume reduction of the thalamus, striatum, and superior temporal cortex. Am J Psychiatry 161(1):154–156
Gilmour G, Dix S, Fellini L, Gastambide F, Plath N, Steckler T, Talpos J, Tricklebank M (2012) NMDA receptors, cognition and schizophrenia—testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology 62(3):1401–1412. doi:10.1016/j.neuropharm.2011.03.015
Göder R, Boigs M, Braun S, Friege L, Fritzer G, Aldenhoff JB, Hinze-Selch D (2004) Impairment of visuospatial memory is associated with decreased slow wave sleep in schizophrenia. J Psychiatric Res 38(6):591–599. doi:10.1016/j.jpsychires.2004.04.005
Göder R, Fritzer G, Gottwald B, Lippmann B, Seeck-Hirschner M, Serafin I, Aldenhoff JB (2008) Effects of olanzapine on slow wave sleep, sleep spindles and sleep-related memory consolidation in schizophrenia. Pharmacopsychiatry 41(3):92–99. doi:10.1055/s-2007-1004592
Göder R, Baier PC, Beith B, Baecker C, Seeck-Hirschner M, Junghanns K, Marshall L (2013) Effects of transcranial direct current stimulation during sleep on memory performance in patients with schizophrenia. Schizophr Res 144(1–3):153–154. doi:10.1016/j.schres.2012.12.014
Goel N, Abe T, Braun ME, Dinges DF (2014) Cognitive workload and sleep restriction interact to influence sleep homeostatic responses. Sleep 37(11):1745–1756. doi:10.5665/sleep.4164
Gogtay N, Giedd JN, Lusk L, Hayashi KM, Greenstein D, Vaituzis AC, Thompson PM et al (2004) Dynamic mapping of human cortical development during childhood through early adulthood. Proc Nat Acad Sci USA 101(21):8174–8179. doi:10.1073/pnas.0402680101
Golomb D, Wang XJ, Rinzel J (1994) Synchronization properties of spindle oscillations in a thalamic reticular nucleus model. J Neurophysiol 72(3):1109–1126
González S, Moreno-Delgado D, Moreno E, Pérez-Capote K, Franco R, Mallol J, McCormick PJ et al (2012) Circadian-related heteromerization of adrenergic and dopamine D4 receptors modulates melatonin synthesis and release in the pineal gland. PLoS Biol 10(6):e1001347. doi:10.1371/journal.pbio.1001347
Govindaiah G, Wang T, Gillette MU, Crandall SR, Cox CL (2010) Regulation of inhibitory synapses by presynaptic D4 dopamine receptors in thalamus. J Neurophysiol 104(5):2757–2765. doi:10.1152/jn.00361.2010
Gur RE, Calkins ME, Gur RC, Horan WP, Nuechterlein KH, Seidman LJ, Stone WS (2007) The consortium on the genetics of schizophrenia: neurocognitive endophenotypes. Schizophr Bull 33(1):49–68. doi:10.1093/schbul/sbl055
Hagewoud R, Havekes R, Novati A, Keijser JN, Van der Zee EA, Meerlo P (2010) Sleep deprivation impairs spatial working memory and reduces hippocampal AMPA receptor phosphorylation. J Sleep Res 19(2):280–288. doi:10.1111/j.1365-2869.2009.00799.x
Hagmann P, Cammoun L, Gigandet X, Meuli R, Honey CJ, Wedeen VJ, Sporns O (2008) Mapping the structural core of human cerebral cortex. PLoS Biol 6(7):e159. doi:10.1371/journal.pbio.0060159
Hasenkamp W, James GA, Boshoven W, Duncan E (2011) Altered engagement of attention and default networks during target detection in schizophrenia. Schizophr Res 125(2–3):169–173. doi:10.1016/j.schres.2010.08.041
Heinrichs RW, Zakzanis KK (1998) Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology 12(3):426–445
Hiatt JF, Floyd TC, Katz PH, Feinberg I (1985) Further evidence of abnormal non-rapid-eye-movement sleep in schizophrenia. Arch Gen Psychiatry 42(8):797–802
Horan WP, Braff DL, Nuechterlein KH, Sugar CA, Cadenhead KS, Calkins ME, Green MF et al (2008) Verbal working memory impairments in individuals with schizophrenia and their first-degree relatives: findings from the Consortium on the Genetics of Schizophrenia. Schizophr Res 103(1–3):218–228. doi:10.1016/j.schres.2008.02.014
Houser CR, Vaughn JE, Barber RP, Roberts E (1980) GABA neurons are the major cell type of the nucleus reticularis thalami. Brain Res 200(2):341–354
Howes OD, Kapur S (2009) The dopamine hypothesis of schizophrenia: version III—the final common pathway. Schizophr Bull 35(3):549–562. doi:10.1093/schbul/sbp006
Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S (2012) The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry 69(8):776–786. doi:10.1001/archgenpsychiatry.2012.169
Huber R, Felice Ghilardi M, Massimini M, Tononi G (2004) Local sleep and learning. Nature 430(6995):78–81. doi:10.1038/nature02663
Huttenlocher PR, Dabholkar AS (1997) Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol 387(2):167–178
Jones BE (2005) From waking to sleeping: neuronal and chemical substrates. Trends Pharmacol Sci 26(11):578–586. doi:10.1016/j.tips.2005.09.009
Jones E (ed) (2007) The thalamus, 2nd edn. Cambridge University Press, Cambridge
Kawada R, Yoshizumi M, Hirao K, Fujiwara H, Miyata J, Shimizu M, Namiki C, Sawamoto N, Fukuyama H, Hayashi T, Murai T (2009) Brain volume and dysexecutive behavior in schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 33(7):1255–1260. doi:10.1016/j.pnpbp.2009.07.014
Kempenaers C, Kerkhofs M, Linkowski P, Mendlewicz J (1988) Sleep EEG variables in young schizophrenic and depressive patients. Biol Psychiatry 24(7):833–838
Keshavan MS, Reynolds CF 3rd, Miewald MJ, Montrose DM, Sweeney JA, Vasko RC Jr, Kupfer DJ (1998) Delta sleep deficits in schizophrenia: evidence from automated analyses of sleep data. Archiv Gen Psychiatry 55(5):443–448
Keshavan MS, Diwadkar VA, Montrose DM, Stanley JA, Pettegrew JW (2004) Premorbid characterization in schizophrenia: the Pittsburgh high risk study. World Psychiatry 3(3):163–168
Khazipov R, Sirota A, Leinekugel X, Holmes GL, Ben-Ari Y, Buzsáki G (2004) Early motor activity drives spindle bursts in the developing somatosensory cortex. Nature 432(7018):758–761. doi:10.1038/nature03132
Kim Y, Zerwas S, Trace SE, Sullivan PF (2011) Schizophrenia genetics: where next? Schizophr Bull 37(3):456–463. doi:10.1093/schbul/sbr031
Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF (1999) Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci Official J Soc Neurosci 19(5):1599–1609
Kopp C, Longordo F, Nicholson JR, Lüthi A (2006) Insufficient sleep reversibly alters bidirectional synaptic plasticity and NMDA receptor function. J Neurosci Official J Soc Neurosci 26(48):12456–12465. doi:10.1523/JNEUROSCI.2702-06.2006
Krystal AD, Goforth HW, Roth T (2008) Effects of antipsychotic medications on sleep in schizophrenia. Int Clin Psychopharmacol 23(3):150–160. doi:10.1097/YIC.0b013e3282f39703
Kupfer DJ, Wyatt RJ, Scott J, Snyder F (1970) Sleep disturbance in acute schizophrenic patients. Am J Psychiatry 126(9):1213–1223
Kuriyama K, Mishima K, Suzuki H, Aritake S, Uchiyama M (2008) Sleep accelerates the improvement in working memory performance. J Neurosci 28(40):10145–10150. doi:10.1523/JNEUROSCI.2039-08.2008
Kyriakopoulos M, Frangou S (2009) Recent diffusion tensor imaging findings in early stages of schizophrenia. Curr Opin Psychiatry 22(2):168–176. doi:10.1097/YCO.0b013e328325aa23
Landsness EC, Ferrarelli F, Sarasso S, Goldstein MR, Riedner BA, Cirelli C, Perfetti B, Moisello C, Ghilardi MF, Tononi G (2011) Electrophysiological traces of visuomotor learning and their renormalization after sleep. Clin Neurophysiol 122:2418–2425. doi:10.1016/j.clinph.2011.05.001
Lauer CJ, Schreiber W, Pollmächer T, Holsboer F, Krieg JC (1997) Sleep in schizophrenia: a polysomnographic study on drug-naive patients. Neuropsychopharmacology. Official Publ Am Coll Neuropsychopharmacol 16(1):51–60. doi:10.1016/S0893-133X(96)00159-5
Laursen TM, Munk-Olsen T, Vestergaard M (2012) Life expectancy and cardiovascular mortality in persons with schizophrenia. Curr Opin Psychiatry 25(2):83–88. doi:10.1097/YCO.0b013e32835035ca
Lebel C, Gee M, Camicioli R, Wieler M, Martin W, Beaulieu C (2012) Diffusion tensor imaging of white matter tract evolution over the lifespan. NeuroImage 60(1):340–352. doi:10.1016/j.neuroimage.2011.11.094
Lee SH, DeCandia TR, Ripke S, Yang J, Schizophrenia Psychiatric Genome-Wide Association Study Consortium (PGC-SCZ), International Schizophrenia Consortium (ISC), Wray NR, et al (2012) Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nature Genet 44(3):247–250. doi:10.1038/ng.1108
MacDonald AW, Schulz SC (2009) What we know: findings that every theory of schizophrenia should explain. Schizophr Bull 35(3):493–508. doi:10.1093/schbul/sbp017
Manoach DS, Stickgold R (2009) Does abnormal sleep impair memory consolidation in schizophrenia? Front Hum Neurosci 3:21. doi:10.3389/neuro.09.021.2009
Manoach DS, Cain MS, Vangel MG, Khurana A, Goff DC, Stickgold R (2004) A failure of sleep-dependent procedural learning in chronic, medicated schizophrenia. Biol Psychiatry 56(12):951–956. doi:10.1016/j.biopsych.2004.09.012
Manoach DS, Thakkar KN, Stroynowski E, Ely A, McKinley SK, Wamsley E, Djonlagic I, Vangel MG, Goff DC, Stickgold R (2010) Reduced overnight consolidation of procedural learning in chronic medicated schizophrenia is related to specific sleep stages. J Psychiatr Res 44(2):112–120. doi:10.1016/j.jpsychires.2009.06.011
Maret S, Faraguna U, Nelson AB, Cirelli C, Tononi G (2011) Sleep and waking modulate spine turnover in the adolescent mouse cortex. Nature Neurosci 14(11):1418–1420. doi:10.1038/nn.2934
Marks GA, Birabil CG (1998) Enhancement of rapid eye movement sleep in the rat by cholinergic and adenosinergic agonists infused into the pontine reticular formation. Neuroscience 86(1):29–37
Marsman A, Heuvel MP van den, Klomp DWJ, Kahn RS, Luijten PR, Pol HEH (2013) Glutamate in Schizophrenia: A Focused Review and Meta-Analysis of 1H-MRS Studies. Schizophr Bull 39:120–129. doi:10.1093/schbul/sbr069
Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G (2004) The sleep slow oscillation as a traveling wave. J Neurosci 24(31):6862–6870. doi:10.1523/JNEUROSCI.1318-04.2004
McIntosh AM, Muñoz Maniega S, Lymer GKS, McKirdy J, Hall J, Sussmann JED, Bastin ME, Clayden JD, Lawrie SM (2008) White matter tractography in bipolar disorder and schizophrenia. Biol Psychiatry 64(12):1088–1092. doi:10.1016/j.biopsych.2008.07.026
Mednick SC, McDevitt EA, Walsh JK, Wamsley E, Paulus M, Kanady JC, Drummond SPA (2013) The critical role of sleep spindles in hippocampal-dependent memory: a pharmacology study. J Neurosci Official J Soc Neurosci 33(10):4494–4504. doi:10.1523/JNEUROSCI.3127-12.2013
Mölle M, Born J (2011) Slow oscillations orchestrating fast oscillations and memory consolidation. Prog Brain Res 193:93–110. doi:10.1016/B978-0-444-53839-0.00007-7
Monti JM, Jantos H (2008) The roles of dopamine and serotonin, and of their receptors, in regulating sleep and waking. In: Esposito E, Di Matteo V, Di Giovanni G (eds) Progress in brain research, vol 172, pp 625–646. Elsevier, New York. Retrieved from http://www.sciencedirect.com/science/article/pii/S0079612308009291
Mrzljak L, Bergson C, Pappy M, Huff R, Levenson R, Goldman-Rakic PS (1996) Localization of dopamine D4 receptors in GABAergic neurons of the primate brain. Nature 381(6579):245–248. doi:10.1038/381245a0
Murphy M, Riedner BA, Huber R, Massimini M, Ferrarelli F, Tononi G (2009) Source modeling sleep slow waves. Proc Nat Acad Sci USA 106(5):1608–1613. doi:10.1073/pnas.0807933106
Nir Y, Staba RJ, Andrillon T, Vyazovskiy VV, Cirelli C, Fried I, Tononi G (2011) Regional slow waves and spindles in human sleep. Neuron 70(1):153–169. doi:10.1016/j.neuron.2011.02.043
Nuechterlein KH (1983) Signal detection in vigilance tasks and behavioral attributes among offspring of schizophrenic mothers and among hyperactive children. J Abnorm Psychology 92(1):4–28
Olney JW, Labruyere J, Wang G, Wozniak DF, Price MT, Sesma MA (1991) NMDA antagonist neurotoxicity: mechanism and prevention. Science 254(5037):1515–1518
Olney JW, Newcomer JW, Farber NB (1999) NMDA receptor hypofunction model of schizophrenia. J Psychiatric Res 33(6):523–533. doi:10.1016/S0022-3956(99)00029-1
Owen MJ (2012) Implications of genetic findings for understanding schizophrenia. Schizophre Bull 38(5):904–907. doi:10.1093/schbul/sbs103
Petanjek Z, Judas M, Kostović I, Uylings HBM (2008) Lifespan alterations of basal dendritic trees of pyramidal neurons in the human prefrontal cortex: a layer-specific pattern. Cereb Cortex 18(4):915–929. doi:10.1093/cercor/bhm124
Phillips KG, Bartsch U, McCarthy AP, Edgar DM, Tricklebank MD, Wafford KA, Jones MW (2012) Decoupling of sleep-dependent cortical and hippocampal interactions in a neurodevelopmental model of schizophrenia. Neuron 76(3):526–533. doi:10.1016/j.neuron.2012.09.016
Picciotto MR, Higley MJ, Mineur YS (2012) Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76:116–129. doi:10.1016/j.neuron.2012.08.036
Pinault D (2011) Dysfunctional thalamus-related networks in schizophrenia. Schizophr Bull 37(2):238–243. doi:10.1093/schbul/sbq165
Plihal W, Born J (1997) Effects of early and late nocturnal sleep on declarative and procedural memory. J Cogn Neurosci 9:534–547. doi:10.1162/jocn.1997.9.4.534
Poulin J, Daoust A-M, Forest G, Stip E, Godbout R (2003) Sleep architecture and its clinical correlates in first episode and neuroleptic-naive patients with schizophrenia. Schizophre Res 62(1–2):147–153
Raedler TJ, Knable MB, Jones DW, Urbina RA, Gorey JG, Lee KS, Egan MF, Coppola R, Weinberger DR (2003) In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am J Psychiatry 160(1):118–127
Ragland JD, Cohen NJ, Cools R, Frank MJ, Hannula DE, Ranganath C (2012) CNTRICS imaging biomarkers final task selection: long-term memory and reinforcement learning. Schizophr Bull 38(1):62–72. doi:10.1093/schbul/sbr168
Rapoport JL, Giedd JN, Gogtay N (2012) Neurodevelopmental model of schizophrenia: update 2012. Mol Psychiatry 17(12):1228–1238. doi:10.1038/mp.2012.23
Rasch B, Gais S, Born J (2009) Impaired off-line consolidation of motor memories after combined blockade of cholinergic receptors during REM sleep-rich sleep. Neuropsychopharmacology 34(7):1843–1853. doi:10.1038/npp.2009.6
Rechtschaffen A, Schulsinger F, Mednick SA (1964) Schizophrenia and physiological indices of dreaming. Arch Gen Psychiatry 10:89–93
Reichenberg A, Harvey PD (2007) Neuropsychological impairments in schizophrenia: integration of performance-based and brain imaging findings. Psychol Bull 133(5):833–858. doi:10.1037/0033-2909.133.5.833
Ren W, Lui S, Deng W, Li F, Li M, Huang X, Wang Y, Li T, Sweeney JA, Gong Q (2013). Anatomical and functional brain abnormalities in drug-naive first-episode schizophrenia. Am J Psychiatry. doi:10.1176/appi.ajp.2013.12091148
Riemann D, Hohagen F, Bahro M, Lis S, Stadmüller G, Gann H, Berger M (1994) Cholinergic neurotransmission, REM sleep and depression. J Psychosom Res 38(Suppl 1):15–25
Sarter M, Lustig C, Taylor SF (2012) Cholinergic contributions to the cognitive symptoms of schizophrenia and the viability of cholinergic treatments. Neuropharmacology 62(3):1544–1553. doi:10.1016/j.neuropharm.2010.12.001
Sawamura N, Ando T, Maruyama Y, Fujimuro M, Mochizuki H, Honjo K, Sawa A et al (2008) Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Molecular Psychiatry 13(12):1138–1148. doi:10.1038/mp.2008.101
Schabus M, Dang-Vu TT, Albouy G, Balteau E, Boly M, Carrier J, Maquet P et al (2007) Hemodynamic cerebral correlates of sleep spindles during human non-rapid eye movement sleep. Proc Nat Acad Sci USA 104(32):13164–13169. doi:10.1073/pnas.0703084104
Seeck-Hirschner M, Baier PC, Sever S, Buschbacher A, Aldenhoff JB, Göder R (2010) Effects of daytime naps on procedural and declarative memory in patients with schizophrenia. J Psychiatr Res 44(1):42–47. doi:10.1016/j.jpsychires.2009.05.008
Seeman P, Guan HC, Van Tol HH (1993) Dopamine D4 receptors elevated in schizophrenia. Nature 365(6445):441–445. doi:10.1038/365441a0
Sekimoto M, Kato M, Watanabe T, Kajimura N, Takahashi K (2007) Reduced frontal asymmetry of delta waves during all-night sleep in schizophrenia. Schizophr Bull 33(6):1307–1311. doi:10.1093/schbul/sbl069
Sekimoto M, Kato M, Watanabe T, Kajimura N, Takahashi K (2011) Cortical regional differences of delta waves during all-night sleep in schizophrenia. Schizophr Res 126(1–3):284–290. doi:10.1016/j.schres.2010.11.003
Sharp FR, Tomitaka M, Bernaudin M, Tomitaka S (2001) Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci 24(6):330–334
Siegel JM (2011) REM sleep. In: Principles and practice of sleep medicine 5th edn. W.B. Saunders, Philadelphia, pp 92–111. Retrieved from http://www.sciencedirect.com/science/article/pii/B9781416066453000086
Sitskoorn MM, Aleman A, Ebisch SJH, Appels MCM, Kahn RS (2004) Cognitive deficits in relatives of patients with schizophrenia: a meta-analysis. Schizophr Res 71(2–3):285–295. doi:10.1016/j.schres.2004.03.007
Skudlarski P, Jagannathan K, Anderson K, Stevens MC, Calhoun VD, Skudlarska BA, Pearlson G (2010) Brain connectivity is not only lower but different in schizophrenia: a combined anatomical and functional approach. Biol Psychiatry 68(1):61–69. doi:10.1016/j.biopsych.2010.03.035
Snitz BE, Macdonald AW 3rd, Carter CS (2006) Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull 32(1):179–194. doi:10.1093/schbul/sbi048
Stephan KE, Baldeweg T, Friston KJ (2006) Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry 59(10):929–939. doi:10.1016/j.biopsych.2005.10.005
Stephan KE, Friston KJ, Frith CD (2009) Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull 35(3):509–527. doi:10.1093/schbul/sbn176
Steriade M (2003) The corticothalamic system in sleep. Front Biosci J Virtual Libr 8:d878–d899
Steriade M, McCormick DA, Sejnowski TJ (1993) Thalamocortical oscillations in the sleeping and aroused brain. Science 262(5134):679–685. doi:10.1126/science.8235588
Stickgold R, James L, Hobson JA (2000) Visual discrimination learning requires sleep after training. Nat Neurosci 3:1237–1238. doi:10.1038/81756
Sullivan PF, Kendler KS, Neale MC (2003) Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Archiv Gen Psychiatry 60(12):1187–1192. doi:10.1001/archpsyc.60.12.1187
Tandon R, Shipley JE, Taylor S, Greden JF, Eiser A, DeQuardo J, Goodson J (1992) Electroencephalographic sleep abnormalities in schizophrenia. Relationship to positive/negative symptoms and prior neuroleptic treatment. Arch Gen Psychiatry 49(3):185–194
Tarokh L, Carskadon MA (2010) Developmental changes in the human sleep EEG during early adolescence. Sleep 33(6):801–809
Thompson PM, Vidal C, Giedd JN, Gochman P, Blumenthal J, Nicolson R, Rapoport JL et al (2001) Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Nat Acad Sci 98(20):11650–11655. doi:10.1073/pnas.201243998
Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M (2000) Origin of slow cortical oscillations in deafferented cortical slabs. Cerebr Cortex 10(12):1185–1199
Tomitaka S, Tomitaka M, Tolliver BK, Sharp FR (2000) Bilateral blockade of NMDA receptors in anterior thalamus by dizocilpine (MK-801) injures pyramidal neurons in rat retrosplenial cortex. Eur J Neurosci 12(4):1420–1430
Traub AC (1972) Sleep stage deficits in chronic schizophrenia. Psychol Rep 31(3):815–820
Tritsch NX, Sabatini BL (2012) Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76(1):33–50. doi:10.1016/j.neuron.2012.09.023
Tseng KY, O’Donnell P (2004) Dopamine-glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J Neurosci Official J Soc Neurosci 24(22):5131–5139. doi:10.1523/JNEUROSCI.1021-04.2004
Van Cauter E, Linkowski P, Kerkhofs M, Hubain P, L’Hermite-Balériaux M, Leclercq R, Mendlewicz J et al (1991) Circadian and sleep-related endocrine rhythms in schizophrenia. Archives of General Psychiatry 48(4):348–356
Vukadinovic Z (2011) Sleep abnormalities in schizophrenia may suggest impaired trans-thalamic cortico-cortical communication: towards a dynamic model of the illness. Eur J Neurosci 34(7):1031–1039. doi:10.1111/j.1460-9568.2011.07822.x
Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, Tononi G et al (2009) Cortical firing and sleep homeostasis. Neuron 63(6):865–878. doi:10.1016/j.neuron.2009.08.024
Wamsley EJ, Tucker MA, Shinn AK, Ono KE, McKinley SK, Ely AV, Manoach DS et al (2012) Reduced Sleep Spindles and Spindle Coherence in Schizophrenia: Mechanisms of Impaired Memory Consolidation? Biological Psychiatry 71(2):154–161. doi:10.1016/j.biopsych.2011.08.008
Wamsley EJ, Shinn AK, Tucker MA, Ono KE, McKinley SK, Ely AV, Manoach DS et al (2013) The effects of eszopiclone on sleep spindles and memory consolidation in schizophrenia: a randomized placebo-controlled trial. Sleep 36(9):1369–1376. doi:10.5665/sleep.2968
Weickert TW (2002) Habit and skill learning in schizophrenia: evidence of normal striatal processing with abnormal cortical input. Learn Mem 9(6):430–442. doi:10.1101/lm.49102
Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, Goldberg TE et al (2001) Prefrontal neurons and the genetics of schizophrenia. Biological Psychiatry 50(11):825–844
Yingling C, Skinner J (1977) Gating of thalamic input to cerebral cortex by nucleus reticularis thalami. In: Desmedt J (ed) Attention, voluntary contraction and event-related cerebral potentials, vol 1. Karger, Basel, pp 70–96
Yun SH, Cheong MY, Mook-Jung I, Huh K, Lee C, Jung MW (2000) Cholinergic modulation of synaptic transmission and plasticity in entorhinal cortex and hippocampus of the rat. Neuroscience 97(4):671–676
Zarcone VP Jr, Benson KL, Berger PA (1987) Abnormal rapid eye movement latencies in schizophrenia. Arch Gen Psychiatry 44(1):45–48
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Sprecher, K.E., Ferrarelli, F., Benca, R.M. (2015). Sleep and Plasticity in Schizophrenia. In: Meerlo, P., Benca, R., Abel, T. (eds) Sleep, Neuronal Plasticity and Brain Function. Current Topics in Behavioral Neurosciences, vol 25. Springer, Berlin, Heidelberg. https://doi.org/10.1007/7854_2014_366
Download citation
DOI: https://doi.org/10.1007/7854_2014_366
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-46877-7
Online ISBN: 978-3-662-46878-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)