
Abstract
Chagas disease (CD) is a neglected disease that is endemic to Central and South America and is caused by the protozoan parasite Trypanosoma cruzi. The discovery of new drugs against CD has not made any significative progress, as the same two drugs have been in use since the 1960s. Benznidazole (BZN), the first-line treatment and Nifurtimox (NFX), the second-line treatment are both nitro-heterocyclic derivatives. Significant problems of resistance have emerged with both drugs. Although new drugs and new Trypanosoma cruzi targets are the focus of studies worldwide, their development and release onto the market remain unresolved. This chapter aims to review current drugs for Chagas disease and their targets, as well as to discuss the challenges that exist in the discovery of new drugs. Furthermore, the evidence that points to the need to strengthen a collaborative network between institutions is emphasized along with the importance of multi-omic studies to support the development of new drugs for Chagas disease.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
1.1 Chagas Disease: A Problem for Public Health
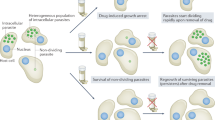
Chagas disease (CD) from among the group of neglected tropical diseases (NTDs) is still an important disease worldwide with a high morbimortality of about 50,000 deaths each year. CD is endemic to Central and South American countries but currently, non-endemic places such as Canada, USA, Europe, Australia, and Japan have been affected due to the increasing global migration from endemic countries to non-endemic areas [1, 2]. The prevalence is variable depending on the location but is highest in Bolivia and Argentina. In the United States, it is estimated that more than 30,0000 Latin American immigrants are currently infected with Chagas disease [3, 4]. The major route is vector-based transmission, but other transmission routes have been found (Fig. 1), including sexual transmission. In summary, the spread of CD is related to the migration of individuals with Chagas disease to previously non-endemic countries [1, 5, 6].

Main transmission routes for Chagas disease
CD is caused by infection with the protozoan Trypanosoma cruzi which has a complex life cycle with insect vectors of the subfamily Triatominae (Hemiptera: Reduviidae). T. cruzi diverged from other trypanosomatids about 200 million years ago; and it circulates in 120 species of mammals, including humans [7]. Historically, it has been established that Chagas’ disease existed as early as 7050 B.C. Exhumed mummies from archaeological sites in both Peru and Chile showed positive for T. cruzi’s kinetoplast DNA by polymerase chain reaction (PCR) [5, 8]; Approximately seven million people are infected by Trypanosoma cruzi, and 1.8–2.4 million of these infected people will develop severe clinical manifestations [9, 10]. In its life cycle, the parasite presents two evolutive forms: the first is the circulating infective but not replicative form known as trypomastigotes, which is the longest phase, and the second is the replicative and intracellular forms, known as amastigotes. The extracellular amastigotes have also been shown to be infective [11]. The metacyclic trypomastigotes and epimastigotes forms are found only in the insect vector. The former is infective and although epimastigotes were considered non-infective, some differentiated epimastigotes may be infective to mammalian hosts [12]. Besides the complex life cycle there is a genetic polymorphism in the parasite populations showing multiple genotypes and phenotypes. Currently the species is subdivided into seven genetically discrete typing units (DTUs): TcI to TcVI and TcB, an additional clade associated with bats [13, 14]. T. cruzi I is the DTU with the broadest geographical distribution and associated with severe cardiomyopathies. Whole genome sequencing results of several TcI isolates and the genetic subdivisions within TcI may be needed in the future [15]. Different Trypanosoma cruzi strains have been isolated from patients and it has been suggested that these parasite strains, regardless of the clinical presentation, reflect the principal DTU circulating in a particular region. However in several orally transmitted outbreaks, sylvatic strains are implicated [13]. All these factors regarding this genotypic and the phenotypic differences of T. cruzi strains and the different geographic distribution of DTUs increase the complexity making standardization of serological tests [16].
There are two distinct phases in the infection: (1) The acute phase – after vector-borne T. cruzi exposure. The acute phase begins after an incubation period of 1–2 weeks, and is characterized by a high parasitemia and circulating trypomastigotes are detectable by microscopy in fresh blood. This stage could be asymptomatic (90%) or the patient could present symptoms (10%) such as fever, anorexia, and tachycardia. These symptoms disappear in 90% of the cases, the mortality rate of the acute phases is 5%. (2) The chronic phase – 8 to 12 weeks after infection, parasitemia levels become undetectable by microscopy and, in the absence of treatment, some infected patients progress to the chronic phase – 60–70% of the infected individuals do not develop symptoms and are identified as a group with the indeterminate form of the disease. Up to 30% of the chronically infected people develop cardiac alterations and up to 10% develop digestive (megacolon or megaesophagus), neurological, or mixed alterations which may also require specific treatment [17]. The chronic chagasic cardiomyopathy is the most serious clinical manifestation of the disease [1, 2, 18]. The infection in an individual with immunodeficiency (HIV infection, organ transplantation, autoimmune disease, or oncological treatments) can be reactivated and generate great morbidity and mortality [19]. Another distinct difference between the two phases is that during the acute phase of CD the trypomastigotes are abundant in the peripheral blood, and during the chronic phase, the amastigotes are abundant in various tissues [12].
Recently in a study by McCall et al., the authors presented results that demonstrate that infection by T. cruzi modulates the fecal microbiome, suggesting host–microbe interaction in the CD [20]. In this work the authors applied metabolomics and sequencing of 16S rRNA during the acute and chronic stages of infection using a murine model of CD. Consequently, they were able to verify the microbial and chemical disturbances associated with the T. cruzi infection, highlighting the importance of multi-“omics” and poly-microbial studies in the area of parasitic diseases in general, and in CD in particular [20]. However, as there are no well-validated targets for CD, phenotypic screening of various compound collections is still considered the most useful and economical strategy to identify new leads or starting points [21].
At last, it is important to consider that Chagas disease and other neglected diseases are part of a group of illnesses for which a set of measures are being developed to prevent, control, and treat until the eradication of these diseases.
The World Health Organization established a current road map – WHO (2021–2030) coordinated by the public, private, the not-for-profit research Drugs for Neglected Disease Initiative (DNDi), and humanitarian medical organizations.
The Wellcome Trust, the Bill and Melinda Gates Foundation, and the NIH contribute with the research in this area, as well as world Research institutions, pharmaceutical companies such as GlaxoSmithKline, Tres Cantos Open Lab Foundation (TCOLF), Division of Biological Chemistry and Drug Discovery, School of Life Sciences, University of Dundee, The Novartis Institute for Tropical Disease (NITD), in cooperation with the Singapore Economic Development Board. All these efforts together bring a considerable development in the research of new drugs against these diseases [22,23,24].
2 Targets for the Treatment of Chagas Disease
Until this moment, there are no effective treatments against Trypanosoma cruzi. Currently, the research strategy is based on the biological differences between the parasite (Trypanosoma cruzi) and host cells (mammals) [25]. Intracellular parasites have high proteolytic activity, participating in various physiological and pathological events such as colonization, invasion, replication, differentiation, nutrition, dissemination, and evasion of the host’s immune system [26,27,28].
In the last 20 years, new strategies and tools had been used in the field of drug discovery, including new computational methodologies for omics analysis and medicinal chemistry, screening of new drugs, the study of the relation of three-dimensional structures and functions of biological molecules [16, 29]. Furthermore, studies into the basic biochemistry of the parasite have identified metabolic pathways in T. cruzi that provide or could provide novel targets for chemotherapy [30].
The inhibition of specific enzymes, metabolic pathways, or organelles exclusive to the parasite is an interesting strategy, called Target-Based Drug Discovery. Proteasomes is one of the major targets not only for Chagas disease but also for Leishmaniasis and sleeping sickness caused leishmania spp. and Trypanosoma brucei, respectively [31].
Other enzymes such as α-carbonic anhydrases from T. cruzi have also been studied [16, 32]. Several essential pathways, such as the glycolysis, pentose phosphate, and the Redox metabolism present in T. cruzi, have also been identified. Studies of the isoprenoid pathway have resulted in drugs that are being used in clinical trials. The toxicity and selectivity of the protozoan organelles is another crucial factor, since few compounds show such features. The characteristics of acidocalcisomes make these organelles potential targets for trypanocidal drugs [30]. Some of these targets are abandoned and are no longer of interest.
Despite the increase and urgency of research for new trypanocidal compounds against the infectious forms of T. cruzi, there is still no effective cure for Chagas disease. Most of these compounds being researched end up as inadequate due to the high toxicity to host cells. The selection of the therapeutic target is all-important, as it favors the rational search for compounds that induce a specific therapeutic response against T. cruzi [16, 30, 33,34,35]. An improved differentiation of the potential targets in the parasites that are absent in humans will help fight this neglected disease. Numerous questions arise related to the research of new drugs, and some of them were reported by [16, 23]. One recognized problem is the lack of standard methodology, different strains, different detection methods, and biomarkers to evaluate responses to therapy, diagnosis, and monitoring drug efficacy. In addition, according to Chatelain [36] the absence of standardization of animal models designed for Chagas disease drug discovery is directly involved in the translational process failure. All these factors constitute barriers in the development of new drugs.
In the next section the most promising and consolidated Trypanosoma cruzi targets are discussed.
2.1 Drug Targets
2.1.1 Cysteine Peptidase-Cruzipain
Some peptidases have been identified as possible targets for the development of new drugs, such as cysteine, serine, metallo- and threonine-peptidases. In general, peptidases are hydrolytic enzymes that have the capacity to break the peptide bonds of proteins and protein fragments with selectivity and specificity [37, 38].
Among the peptidases of T. cruzi, we can highlight cruzipain, a cysteine peptidase, which has a prominent proteolytic activity in all the developmental stages of T. cruzi. Thus, cruzipain is a therapeutic target that has been intensely studied for the treatment of Chagas disease [39, 40]. Also, according to Cazzulo [39], recombinant enzymes have been developed, and different drugs are being studied that specifically inhibit cysteine peptidase in vitro, blocking the proliferation of epimastigotes, amastigotes, and metacyclogenesis of the parasite.
Cruzipain inhibitors cause the parasite to die in vitro, probably due to the accumulation of proteins within the Golgi complex, which results in an osmotic shock within the endoplasmic reticulum of the parasite [41]. This enzyme is present in organelles related to lysosomes and is associated with the plasma membrane [42, 43]. The K777 is a peptide derivative of vinyl sulfone that inhibits cruzipain irreversibly [44]. It is worth mentioning that many of these inhibitors have shown a lack of selectivity and low bioavailability in the in vivo assays. In addition, some compounds are potent cruzipain inhibitors but with slight effectiveness against T. cruzi in cell cultures. Concerted efforts are being made to overcome these difficulties and obtain an inhibitor with the selectivity and safety required to treat Chagas disease [44, 45]. This project to perform the preclinical studies demonstrating safety pharmacology and toxicology was started in 2010 by DNDI. Still, it was stopped in 2013 due to poor tolerability findings at low doses in primates and dogs [46]. Another derivative, the Neq0682, a reversible covalent inhibitor, of cysteine peptidases obtained by replacing the K777 vinyl sulfone group with a nitrile moiety was synthesized and is under study [47]. Other inhibitors against cruzipain are being studied such as quinoline [48], thiophen-2-iminothiazolidine derivatives, and thiophene–thiazolidine hybrids [49].
2.1.2 Kinetoplast Proteasome
The proteasome is an enzymatic complex formed by threonine peptidases. They are responsible for numerous biological functions in eukaryotic cells, such as the turnover of short-lived, abnormal/damaged proteins, cell cycle regulation, cell differentiation, signal transduction pathways, stress signaling, inflammatory responses, and apoptosis among others [50]. The 26S proteasome was identified in the epimastigote stage of Trypanosoma as a high molecular weight complex involved in parasite cell differentiation. This effect is mediated by the 20S proteasomal degradation of oxidized proteins through an ATP/ubiquitin-independent mechanism [51, 52]. In this context, it has been demonstrated that inhibition is associated with significant defects in parasite proliferation, turning on the proteasome as an attractive target for Chagas disease and other trypanosomatids and Plasmodium falciparum [53]. The Genomics Institute of the Novartis Research Foundation (GNF) has identified an azabenzoxazole compound series (GNF5343 and the optimized GNF6702) [54, 55]. In addition, GlaxoSmithKline (GSK) and Dundee Drug Discovery have also identified a similar azabenzoxazole (GSK3494245). Azabenzoxazole (GNF6702), a non-competitive inhibitor of proteasome chymotrypsin-like activity, is a promising candidate for preclinical evaluation against neglected tropical diseases [22, 56]. This inhibitor was effective when tested against Trypanosoma cruzi as well as T. brucei and Leishmania donovani. Besides this, it does not inhibit the mammalian proteasome or growth of mammalian cells and is well tolerated in mice [16].
2.1.3 Carbonic Anhydrase
Carbonic anhydrases (CA, EC 4.2.1.1) are metalloenzymes with various physiological functions in all areas of life. They are involved in the pathological processes of human and prokaryotic/eukaryotic microorganisms such as bacteria, fungi, and protozoa. CAs catalyze the CO2 reversible hydration: CO2 + H2O→HCO3− + H+, which are involved in several physiological and pathological processes. CO2 homeostasis, biosynthetic reactions, calcification, and tumorigenicity are some examples, among others. In addition, these enzymes are related to the growth and virulence of microbial pathogens. In researching new medicines, this target was established to develop anticonvulsants, anti-obesity, anticancer, and anti-infective drugs. In this context, CAs belonging to the α- and β-class were recently identified, cloned, and recognized their potential as new enzymatic targets in T. cruzi (αTcCA) and from L. donovani chagasi (LdccCA), respectively [57, 58].
In Trypanosoma cruzi, an α-CA was identified, cloned, and characterized by Pan et al. [58]. The α-TcCA has a high catalytic activity for the CO2 hydration reaction. Inhibitors such as anions, sulfonamides, sulfamates, thiols, and hydroxamates were effective in low nanomolar in vitro tests [32, 59, 60]. One of the best inhibitors was hydroxamates which inhibited the growth of all three evolutive forms of the parasite at low concentrations (IC50 values from 7.0 μM to <1 μM) [61, 62]. Synthetic inhibitors such as sulfonamides have been tested against TcCA from T. cruzi with success. Bonardi et al. demonstrated that N-nitrosulfonamides and their salts inhibited the growth of the epimastigotes of T. cruzi, based on CA inhibition and are promising lead compounds for rational optimization of innovative agents for the treatment of Chagas disease [63]. All these results demonstrated the potential of the α-TcCA as a target yet underexplored for Chagas disease drugs [59]. The Cas is not a validated target for Chagas disease yet but is a possibility and further studies need to be performed to confirm this hypothesis.
2.1.4 Sirtuins
Silent-information regulator 2 (SIR2) proteins, or sirtuins, are a family of enzymes evolutionarily conserved and present in all kingdoms of life, from bacteria to higher eukaryotes [64]. According to Matutino Bastos et al., the inhibition of T. cruzi sirtuins by nicotinamide can cause growth arrest and morphologic alterations in the parasite, thus being a possible candidate for a drug against CD [65]. In the search for new inhibitors, these same authors [65] characterized human sirtuin inhibitors against T. cruzi sirtuins. As a result, they reported seven inhibitors of sirtuins, where all compounds prevented the proliferation of T. cruzi in mammalian cells.
2.1.5 Cyclophilin
Cyclophilins are enzymes that perform several biological functions such as protein folding, where Cyclophilin D (CyPD) is a mitochondrial isoform with a crucial role in opening the pores for mitochondrial permeability [66]. It is known that this enzyme activity is inhibited by the immunosuppressant Cyclosporin A. In a study by Búa et al., the authors demonstrated anti-T. cruzi activity with Cyclosporin A through the inhibition of Cyclophilins, suggesting that this may be a molecular target [67]. According to Jha et al., Cyclophilin 19 is an enzyme present in all stages of life of T. cruzi participating in several functions, among them the generation of ROS that increases the growth of the parasite. In the study carried out by the authors, a mutant knock-out parasite of Cyp19 was generated, which was unable to replicate in cell cultures or in immuno-competent mice [68]. The authors also performed repeated inoculation of knock-out parasites where they observed specific antibodies and T-cell responses. According to the authors, these results demonstrate a 100% effective immunization in preventing Chagas disease. This study generated a patent entitled “Live attenuated parasitic vaccine” (US20200147148A1) [69].
2.1.6 N-myristoyltransferase
N-Myristoyltransferase (NMT, EC 2.3.1.97) is an enzyme that catalyzes the co- and posttranslational addition of myristic acid (C14: 0) onto the N-terminal glycine of specific proteins [70]. Studies have shown that NMT is both essential and druggable in T. cruzi [71], where it was shown that the inhibitor DDD8564630 caused a reduction in parasite proliferation in the epimastigote stage [72]. In a study by Herrera et al., the authors demonstrated the effectiveness of inhibitors as antiproliferative agents, presenting very low toxicity against mammalian cells. Where, according to the authors, it was possible to demonstrate its specificity and validation of NMT as a drug target using inhibitors with potential for future explorations such as anti-CD [73].
2.1.7 Pentose Phosphate Pathway: Glucose-6-Phosphate Dehydrogenase and Trypanothione Reductase
Glucose is metabolized through two major pathways: glycolytic and pentose phosphate (PPP). The PPP produces the ribose 5-phosphate (R5P) required for nucleotide synthesis and reducing power in the form of NADPH. One branch of the pathway is the oxidative branch, involving glucose 6-phosphate dehydrogenase (G6PDH), among other enzymes. G6PDH is of central importance because it often has a high control coefficient for the PPP and can be considered a potential target for developing drugs for CD [74].
The studies with the PPP in Trypanosoma were scarce, and most of the pathway’s enzymes, their properties, and subcellular localization were unknown until 2004 [74, 75]. PPP is important for parasites, considering that oxidative burst forming reactive oxygen is the first line of host cell defense against infection. Maugeri and coworkers [76] demonstrated an increased flow of the PPP pathway in Trypanosoma cruzi in response to oxidative stress. The parasite is highly sensitive to oxidative stress. The primary protection against reactive oxygen (ROS) is the Trypanothione, which is kept reduced by trypanothione reductase, using NADPH as a cofactor [75]. In addition to the cytosolic localization, PPP is present in the organelle glycosomes from trypanosomatids. Dehydroepiandrosterone (DHEA), epiandrosterone (EA), and derived 16/-bromoepiandrosterone (16BrEA) are known to be uncompetitive inhibitors of mammalian and trypanosome G6PDH [77]. Trypanothione reductase is an attractive target for antitrypanosomal drug. Some compounds such as Quinoxaline and Clomipramine showed activity against T. cruzi TR [78, 79] and quebrachamine, cephalotaxine, cryptolepine, tomatidine, solanidine, and solasodine were detected as potent inhibitors [80].
In a study by Fredo Naciuk et al. [81], the authors synthesized and evaluated 26 steroid derivatives of epiandrosterone (nonselective inhibitors of G6PDH) in enzymatic assays. As a result, compounds 40, 15, 39, and 6 showed a certain degree of selectivity in cell assays was achieved, at least in terms of toxicity in the system used (intracellular T. cruzi forms and rat cardiomyocytes). Although these results are promising, further studies are necessary to get more selective inhibitors, which increases the knowledge about the interactive action mechanisms of the inhibitors with the host cell/parasite.
Ortiz et al. [82] demonstrated in Trypanosoma cruzi using immunofluorescence a cytoplasmic distribution of G6PDH and the absence of signal in major organelles. In addition cytochemical assays confirmed that parasitic G6PDH is the molecular target of derivative epiandrosterone. These results together demonstrated that glucose 6-phosphate dehydrogenase shows a potential for a drug target for Chagas disease and other diseases caused by trypanosomatids as already shown for the African Trypanosoma brucei [82].
2.1.8 Ergosterol Biosynthesis Inhibitors
As ergosterol is essential for T. cruzi, enzyme inhibitors of its biosynthesis pathway have become common targets in studies [83]. Several studies have focused on the trypanosome ergosterol biosynthesis pathway (Fig. 2) due to the availability of existing drugs (posaconazole or ravuconazole) capable of inhibiting sterol 14 α-demethylase (CYP51). CYP51 is an essential enzyme for this pathway and its inhibition reduces the capacity for invasion of heart cells by trypomastigotes and inhibits the multiplication of amastigotes [85, 86]. Posaconazole is not yet a viable alternative as it has a high cost and has a structure that is difficult to interact with T. cruzi CYP51 [86]. According to Osorio-Méndez and Cevallos [83], the evidence suggests that these inhibitors effectively fight infection in vitro and murine models; however, they have failed in clinical trials [23]. It is worth noting that the failure of CYP51 as a drug target is due to the cytostatic consequence of inhibition [23]. Consequently, assays have been developed to filter out compounds identified in phenotypic screens with this undesirable mode of action [87]. However, it may be possible to rescue posaconazole by combination therapy described recently by Rocha-Hasler et al. [88] demonstrating that Tomatidine improves the potency of posaconazole as antitrypanosomal agent.

Simplified ergosterol biosynthesis pathway in T. cruzi epimastigotes. Critical enzymes (blue color) of the ergosterol pathway. Adapted from [84]
2.1.9 Sphingolipids
Sphingolipids are biological molecules found in eukaryotes and prokaryotes. Their structure is composed by a sphingoid base with a fatty acid attached through an amide bond, forming the ceramide. A variable polar head-group is present (phosphocholine, inositol phosphate, or carbohydrates). In epimastigotes from Trypanosoma cruzi glycosphingolipids were detected [89]. The parasite synthesizes inositol phosphorylceramide (IPC) and sphingomyelin (SM) and its expression is modulated during parasite development. The IPC synthase enzyme catalyzes the transfer of inositol phosphate to ceramide moiety. Several glycoconjugates are attached to membranes by a glycosylphosphatidylinositol (GPI) anchor and in this context T. cruzi have different surface molecules including the glycoinositolphospholipids (GIPLs, in epimastigotes), trans-sialidase and Tc-85 glycoprotein in trypomastigotes, mucins in epimastigotes and metacyclic forms and complex GPI-anchored glycopeptide called NETNES in epimastigotes. All these molecules have important biological function on parasite related to the antigenicity, pathogenesis, cellular survival, and programmed cell death (PCD) and in cellular survival [90,91,92].
Landoni et al. [93] studied the effect of tamoxifen (TAM) on the sphingolipid pathway of T. cruzi. TAM is an anti-estrogen used for the treatment of breast cancer. This drug is involved in apoptosis mechanisms. Although there are few studies about the effect of TAM in the sphingolipid pathway, the authors tested it in Trypanosoma cruzi. The results demonstrated a dose-dependent inhibition according to the evolutive stage of the parasite. Lipid extracts from epimastigotes were analyzed by MALDI-TOF and HPLC-ESI mass spectrometry. The results showed that after TAM treatment, a high and discrete increase in the level of ceramide/ceramide-1P and sphingosine, respectively, indicates the involvement of TAM in the breakdown of ceramide. Microscopy analysis and flow cytometry of the treated parasite demonstrated an apoptotic-like death process. Previous studies of Miguel et al. [94] showed that TAM was ineffective in the treatment of the acute phase of Chagas disease in mice. These results suggest that the sphingolipid pathway could be a target for drug development in Chagas disease. More studies are needed, including tests with new concentrations of the drug and evaluation of toxicity in host cells.
2.1.10 Intracellular Calcium Homeostasis
Ca2+ is an essential signaling messenger in eukaryotic cells, including the Trypanosoma cruzi and other trypanosomatids [95,96,97]. The function of Ca2+ as a signaling messenger in trypanosomatids is well documented in several biological functions like flagellar movements, cellular differentiation, invasion of the host cell, osmoregulation, and nitric oxide transduction pathway [95, 98,99,100,101,102,103,104,105,106,107,108]. Ca2+ concentration in T. cruzi is regulated by mechanisms present in the plasma membrane and by intracellular organelles such as mitochondria, endoplasmic reticulum, and acidocalcisomes. The maintenance of calcium homeostasis is very important considering that Ca2+ is involved in the apoptotic process [109, 110]. In this context the disruption of intracellular calcium homeostasis has been proposed by Benaim et al. as a possible therapeutic target for drug development in Chagas disease [25]. The amiodarone and derivatives, for instance, an antiarrhythmic drug showed trypanocidal effects through disrupting the parasite Ca2+.
2.1.11 Acidocalcisomes
Acidocalcisomes are organelles that play an important role in osmoregulation which are rich in polyphosphate bound to calcium and different cations such as magnesium, calcium, sodium, and zinc [111, 112]. The matrix contains enzymes related to poly P metabolism and the membrane of the acidocalcisomes has several pumps and transporters including a Ca2+ -ATPase for Ca2+ uptake. The Ca2+ release is controlled by the inositol 1,4,5-trisphosphate receptor (IP3R) that is located in acidocalcisomes [95, 113]. Chiurillo et al. demonstrated that the remotion of the IP3R gene by CRISPR/Cas9 genome editing inhibits host cell invasion by trypomastigotes [114]. In contrast, TcIP3R overexpressing parasites showed decreased metacyclogenesis, trypomastigote host cell invasion, and intracellular amastigote replication. Summarizing Ca2+ signaling is important for the T cruzi differentiation for host cell invasion and to maintain cellular bioenergetics [95, 114]. Based on these facts it had been suggested that the acidocalcisomes and their components could be potential targets for chemotherapy [115, 116].
2.1.12 Kinetoplast
Trypanosomes have a single mitochondria that has the peculiar characteristic of having a circular DNA network known as kinetoplast or kDNA [117, 118]. The kDNA is composed of circular and interconnected DNA molecules forming a single network, therefore, knowledge of this topological architecture is fundamental for understanding the replication and segregation of the kDNA circles [119]. In a study by Zuma et al. [120] the authors analyzed the effects of Berenil on the ultrastructure and replication of T. cruzi kDNA. As a result, the authors demonstrated that Berenil caused significant changes in the kDNA arrangement and a reduction in the growth of T. cruzi, but cell viability was not affected. The authors suggest that Berenil mainly affects kDNA topology and replication, demonstrating, according to them, that Kinetoplast represents a potential target against trypanosomatids [120].
According to Cavalcanti and de Souza [119], previous studies used atomic force microscopy to analyze the effect of acriflavine, an intercalating drug [121] in the T. cruzi kDNA network. As a result, it was possible to evaluate the structure of kdna and investigate the topology changes caused by drugs, and the authors proposed that kdna can be an interesting target because it is affected by DNA-binding drugs, intercalating agents, and topoisomerase inhibitors [119].
3 Current Treatments for Chagas Disease
Only two drugs have been used to treat CD, nifurtimox or (RS)-3-methyl-N-[(1E)-(5-nitro-2-furyl)methylene] thiomorpholin-4-amine 1,1-dioxide (NFX – Tables 1, 1) and benznidazole or N-benzyl-2-(2-nitro-1H-imidazol-1-yl)acetamide, which is a 2-nitroimidazole derivative (BZN – Tables 1, 2). Both were discovered more than 50 years ago, showing that there is a continued lack of investment in research and development (R&D). The justification for this is that the predominance of CD is in emerging countries; consequently, the pharmaceutical industries are not interested in investing in new antichagasic drugs and their excessive costs for research in developing countries [122].
NFX was the first drug developed for this purpose and has been used since 1965 and BZN since 1971. BZN was first produced by the pharmaceutical company Roche (Rochagan® and Radanil®), now it is manufactured by the Pharmaceutical Laboratory of the State of Pernambuco (Lafepe), Brazil, and by the private laboratory in Elea (Abarax®), Argentina. NFX is free of charge through a WHO-Bayer agreement [17, 123, 124].
Studies of the mechanisms of both drugs are not fully understood yet. NFX and BZN are prodrugs and require activation by enzymatic activity. Wilkinson and coworkers [125] proposed that nitroreductase is the main enzyme involved in the activation of nitro-heterocyclic drugs in T. cruzi. In general, nitroreductases reduce the nitro group present in both the nitro-heterocyclic compounds, generating metabolites with different reactivities for each drug, such as molecules considered free radicals (e.g., R–NO-) and electrophilic metabolites (e.g., R–NHOH) [3, 126].
However, NFX and BZN require distinct enzymes for biological activation. For NFX, the proposed mechanism is through the one-electron route (type II nitroreductase). A re-oxidation leads to reactive oxygen species formation in cells (oxidative stress); the production of highly toxic oxygen metabolites and oxidative stress to the parasite by generating the nitro radical, redox cycling, and production of O2− and H2O2 [3, 122, 127, 128]. In addition, some studies have proposed that the reduction of NFX by nitroreductase can open the furan ring producing an unsaturated open-chain nitrile that is as cytotoxic as the parent compound. This suggests that NFX trypanocidal activity does not necessarily involve oxidative stress [129, 130].
For the benznidazole the two-electron route (type I nitroreductase) in the mechanism forming reactive nitroso and hydroxylamine intermediates (chemical stress). The hydroxylamine derivative (R–NHOH) converts to glyoxal, a highly cytotoxic and mutagenic compound [129]. Some studies indicate that the reactive species generated in BZN metabolites can improve phagocytosis, increase trypanosome death through IFN-γ induction, and inhibit T. cruzi NADH fumarate reductase (thus) inducing mitochondrial DNA damage. The T. cruzi cytotoxic activity of these metabolites may involve covalent modification of macromolecules such as DNA, proteins, and lipids [131, 132].
The chemical species generated by these drugs are highly reactive and can affect other molecules, especially in the vertebrate host. Consequently, this low specificity of action on the parasite contributes to the cytotoxic effects observed in treatments with patients. T. cruzi is probably deficient in the metabolic detoxification mechanisms for oxygen and that makes it very susceptible to partial reduction products of oxygen, consequently, it is more sensitive to oxidation than the vertebrate cells [3, 127, 128].
NFX and BZN are almost 100% effective in curing CD if given soon after infection, at the onset of the acute phase, in cases of congenital transmission and for those in whom the infection has been reactivated due to immunosuppression. However, the efficacy of both reduces as time passes [17]. BZN is usually the first-line treatment in most countries because it has a better safety record and efficacy profile than Nifurtimox, mainly for adult patients that require prolonged administration [129]. In the acute and undetermined chronic phases, treatment with BZN has proven to be highly effective and about 80% of patients have no sign of parasites in the blood after 12 months of treatment. The other benefits of treatment with BZN are the high cure rates in infants with congenital infection and in children with chronic infection [122].
In 2011, a partnership between DNDi and Lafepe laboratory (Brazil) allowed the development of the first pediatric formulation of benznidazole for the treatment of children with CD. Following partnerships with Chemo Research, Exeltis USA, Mundo Sano Foundation and Laboratory ELEA PHOENIX were set up aiming to enable a broader registration in endemic countries, with a commitment to make the pediatric formulation more widely available. In 2017, the FDA granted approval for the use of benznidazole in children with CD aged between 2 and 12 years old [17, 123, 133].
The treatment of the chronic phases of CD in adults with BZN on the growth rates of negative parasitemia increases with treatment time but there are no significant modifications with increased drug dosages or modifications in the drug formulation. Studies have demonstrated that negative parasitemia in patients with chronic CD has been observed after treatment from 55.97% after 2 months to 62.59% after 8–16 months and 72.81% after 9–11 years [122, 123, 133, 134]. Although BZN is able to reduce serum parasite detection in chronic CD, it does not significantly reduce cardiac clinical deterioration [135, 136].
NFX treatment studies of 114 adults with chronic Chagas were conducted. These studies demonstrated positive serology after an average of 6.6 years for all of them; and T. cruzi was not detected in 93.9% of the cases after treatment. However, in these cases, according to the current cure criterion, it is not possible to interpret the obtained results as a parasitological cure [137]. In treatment with both drugs, BZN and NFX, therapeutic failures are common for reasons that include influences of the parasite and host genetics, and the effects of toxicity on adherence to the treatment. The long-term therapy increases the chances of NFX presenting more intense adverse effects than BZN. The adverse reactions are more frequently reported with older aged patients with both NFX and BZN. In general, NFX has shown higher toxicity and adverse effects than BZN, including increased oxidative stress in rat pancreas and heart [135].
Problems related to suffering from adverse drug reactions (ADRs) are reported in more than 30% of the patients treated with BZN, especially those in the chronic phase. These effects involve dermatitis, disorders in vision, myelosuppression, and peripheral polyneuropathy, as well as gastrointestinal system disorders [16, 122, 138, 139].
The main contraindications to treatment with BZN are liver or kidney failure and pregnancy. NFX is contraindicated in patients with a history of psychiatric or neurological disorders (Table 1) [140, 141].
Additionally, significant problems of resistance have emerged with both drugs [17, 123]. The mechanism of BZN and NFX resistance has been associated with the deletion of copies of genes encoding two different nitroreductases, namely old yellow enzyme (TcOYE) or prostaglandin synthase, and trypanosomal type I nitroreductase [122].
4 New Drugs and Clinical Trials
4.1 Drugs in Clinical Trials
Since the introduction of pharmacotherapy for CD with BZN and NFX, only allopurinol and some azoles, such as: itraconazole, fluconazole, ketoconazole, posaconazole (POSA), E1224/ravuconazole (RAV) and fexinidazole have been tested for the treatment of CD [3].
4.1.1 Fexinidazole: First Global Approval
Fexinidazole (FXN) is a promising broad-spectrum antiparasitic agent with clinical trials already taking place. FXN was first discovered by Sanofi (former Hoechst AG) and was identified by the DNDi in 2005 as having activity against Trypanosoma brucei gambiense, T. b. rhodesiense, and T. cruzi [142]. This compound has a nitro group in its composition that is metabolized by the parasite nitroreductases, forming reactive species that inhibits DNA synthesis [124].
Clinical studies with FXN have been performed with both CD and visceral leishmaniasis patients [143]. Dose-finding studies over a maximum treatment time of 8 weeks were conducted with chronic indeterminate Chagas disease patients. A Phase II Proof of Concept (PoC) study of fexinidazole was initiated in 2014 in Cochabamba and Tarija, Bolivia. However, the study was interrupted due to safety and tolerability problems. Further analysis of the results showed high efficacy findings were at the lowest dose tested for all treatment durations. The higher doses of the drug were considered unsafe after only 14 days. However, the clinical study follow-up was extended to 12 months [140]. FXN has demonstrated its therapeutic potential against CD, since it has been approved by the European Medicines Agency (EMA) for the treatment of African trypanosomiasis, in both adults and children [124, 142].
4.1.2 E1224/Ravuconazole
E1224 (fosravuconazole or fosravuconazole l-lysine ethanolate) is a prodrug of ravuconazole (RVZ), which is a potent inhibitor of ergosterol biosynthesis with activity against a wide range of fungal species [144,145,146]. RVZ is a potent CYP51 inhibitor, but studies have shown that it has a short terminal half-life, where in studies with murine models it presented limited in vivo activity due to unfavorable pharmacokinetic properties [147, 148]. In studies with canine models, the results were suppressive but not curative due to the short terminal half-life [147]. In a clinical study with humans led by DNDi, the drug E1224 presented a rapid, but transient, parasite clearance effect – suggesting that this drug has a static rather than a parasiticidal mechanism of action [145, 146]. It was also observed that the combination of RVZ with BZN did not significantly affect efficacy [144]. According to Spósito et al. [148] the authors suggest that therapeutic failure may be related to inadequate azole levels in tissues and suboptimal drug exposure [148].
4.1.3 New Drugs Discovery
The knowledge of parasite biology has, over the last decades, discovered small molecules that could be potential chemotherapeutic targets in the parasite for CD treatment, such as: enzymes that participate in the synthesis of ergosterol in T. cruzi; enzymes of the trypanothione metabolism such as trypanothione reductase; Cruzipain; Carbonic anhydrases; among others [84].
The potent activity of azole antifungal drugs against T. cruzi were reported more than 30 years ago. Since then, several azole antifungal drugs such as ketoconazole, fluconazole, itraconazole, posaconazole, D0870 and others, have been reported as sterol 14 α-demethylase inhibitors (CYP51) [149]. Triazole compounds such as posaconazole and D0870 have been shown to be effective at curing mice with chronic CD. On the other hand, clinical studies with ketoconazole or itraconazole in humans with chronic CD have not presented any significant cure of the disease. However, studies have been published demonstrating the synergistic activity of azole drugs with various other compounds, indicating that a combination of chemotherapies may be an effective strategy as this field moves ahead [149]. VNI is the first nonantifungal compound CYP51 inhibitor to target the 14 α-demethylase activity of T. cruzi. This compound demonstrated a cure for both the acute and chronic phases of infection with the Tulahuen T. cruzi strain in mice but failed to cure mice infected with the Y and Colombian T. cruzi strains in both phases of the infection [150].
Several compounds have been shown to inhibit T. cruzi trypanothione reductase and affect parasite growth in vitro or in vivo, such as nitrofurans and naphthoquinones, phenothiazines and crystal violet, diphenyl sulfide derivatives, polyamine derivatives, dibenzazepines, bisbenzylisoquinoline alkaloids, ajoene, acridines, terpyridine platinum complexes, and Mannich bases as well as some natural products [84, 151]. Buthionine sulfoximine (BSO) has been shown to be a potential drug candidate against T. cruzi alone, or jointly with free radical-producing drugs such as nitrofurazone and benzimidazole [84]. K777, a vinyl sulfone derivative, is a potentially irreversible inhibitor of cruzipain. Nevertheless, preclinical studies demonstrated poor tolerability even at low doses in primates and dogs. Novel scaffolds for the inhibition of cruzipain were identified from high-throughput screening (HTS) of GlaxoSmithKline HAT (Human African Trypanosomes) and Chagas chemical boxes [84].
Several aromatic/heterocyclic sulfonamides have been studied as carbonic anhydrases of T. cruzi (TcCA) inhibitors. The studies were performed against mammalian enzymes (hCA I and hCA II) and the inhibitors showed a greater inhibition of the T. cruzi TcCA indicating a positive selectivity [58]. Additionally, a series of the 4,5-dihydroisoxazoles incorporating hydroxamate moieties showed that they could also act as inhibitors of the T. cruzi α-CA and of peptidases from this pathogen, such as the cysteine and metallo-peptidase. These series were assayed in vitro against the tree forms of T. cruzi and in vivo. These studies showed that a leading compound had potential values for the growth inhibition of all three developmental forms of the Y strain of T. cruzi at relatively low concentrations [61].
5 Omics Platforms in Chagas Disease
Even after more than 100 years of the discovery of Chagas disease, and despite an enormous scientific research effort, this disease is still a major threat in several Latin American countries and an emerging global health problem [16, 152]. In work carried out by Alonso-Padilla et al. on strategies to diagnose and treat patients with CD, the authors cite the lack of public policies and funding as factors that make facing the problem even more challenging [10]. The authors attest that when there is no treatment ~30% of those chronically infected will develop cardiac and/or digestive disorders. According to the authors, a deeper understanding of the parasite’s biology and its interactions with the host is of fundamental importance for the discovery of safer drugs or vaccines. Therefore, studies on the genomics of the parasite and the host combined with other omics are essential to determine the factors that lead to the development of CD [10]. Research into the biology of parasites requires a sophisticated and integrated computational platform that allows the analysis of large volumes of data [153].
Platforms with large amounts of omic data (proteomics, genomics, transcriptomics, metabolomics, etc.) have already been created and continue to be developed at an ever faster rate, but the databases have heterogeneous formats that are often difficult to integrate with experimental data [153]. According to Wooden et al., transcriptomic, genomic, and omic data (proteome, metabolome, kinome, methylome, acetylome, lipidome, microbiome, phenome, exposome, meta-genome, and interactome) have been increasingly deposited for public use, however, this excess of information has made the analysis of these omic data sets a challenging task [152].
In relation to CD, other types of genetic studies, such as transcriptomics and epigenetics, are needed to expand and integrate the genomic data already studied, in addition to the need to understand how environmental factors imply susceptibility to the disease [154]. According to Talavera-López and Andersson [155], integrative omic approaches will be able to treat, on a large scale, biological information from the most diverse areas such as drug resistance, epidemiology, genetic exchange, immune evasion mechanisms, genetic function, etc. However, according to these authors, additional work is also needed for a complete characterization of individual targets and families of genes, and yet other aspects of parasitic biology are still relatively unexplored. Therefore, they emphasize that a lot of basic research is needed before these issues can be effectively addressed [155]. In summary, Talavera-López and Andersson highlighted some points that make parasitic research problematic: (a) Sampling that is difficult to perform. When they are carried out, they are often not collected in a standardized manner, limited metadata and often there is no sampling strategy guided by a specific biological issue. (b) Requirement for field studies with visits to remote locations that implies complex and costly logistics. (c) Some parasites such as Leishmania, T. cruzi, T. rangeli, etc. can only grow in vitro using culture media and even cloning by limiting dilution. In this way, a level of uncertainty is introduced in the experiment, considering that the parasites can change according to the culture medium in which they grow. And often the clones that dominate the culture may not be the same ones that occur in the host [155].
According to Sánchez-Ovejero et al., the integration of omic data is essential to understand the host–parasite relationship [156]. However, as demonstrated by Vermelho et al. [16], the technological advances to date have not resulted in the discovery of new drugs since the 1960s, nor in the cure of CD. However, with the advancement of metagenomics and metabolomics technologies, discoveries pertinent to various human conditions have been made, and for this reason, new light has begun to be shed on neglected tropical diseases [157]. Following this direction, in a review carried out by Kules et al. [158], the authors claim that high-throughput technologies, such as whole-genome sequencing, omics (metagenomics and meta-proteomics), and mass spectrometry (MS), open new opportunities for the diagnosis of Vector-Borne Diseases. According to Kratz [159], even with all the recent advances associated with the discovery of drugs for CD, the likelihood of a new ideal treatment emerging in the coming years is still uncertain. The author claims that success in this discovery does not depend only on new tools and technologies, but also on the availability of financing and collaborative R&D models, in addition to a deepening of the understanding of the pathophysiology of the disease [159]. Financial support is needed for the initial stages of research and development of medication for CD [160]. In a work carried out by Calogeropoulou et al., the authors attest that many research programs are successful and have initial leads, however, they fall prematurely into the “valley of death” of candidate compounds. Therefore, they suggest that drug discovery for CD should become collaborative and include academic research, national and international organizations, government initiatives and private research centers as well as pharmaceutical companies [160].
Currently, modern drug design and discovery is based on computational methods that predict and evaluate binding of ligands to receptors related to various pathologies [161]. Therefore, the methods that are capable of promoting correct data acquisition, mining, and analysis are crucial in order to be able to obtain reliable results [162]. Traditional drug discovery and development is known to be time consuming and cost-intensive to both biotechnology and pharmaceutical companies [163]. According to Shaker et al. computer-aided drug design (CADD) offers methods to discover and optimize potent drugs in silico, aiming to screen millions of substances in order to identify chemical compounds that can geometrically and chemically bind to a specific cavity on a target protein [164].
In a study for drug repositioning, Sayé et al. used computational methods to discover substances with a structure similar to crystal violet (CV) [165]. CV was chosen because it has been used in blood banks for several years to eliminate the parasite Trypanosoma cruzi. The authors claim that the mechanism of action of CV is the inhibition of proline uptake by the parasite. Thus, the in-silico drug repurposing strategy through a similarity-based virtual screening protocol was able to identify compounds structurally related to CV (loratadine, cyproheptadine, olanzapine, and clofazimine). As a result, they observed that loratadine, cyproheptadine, and clofazimine inhibit the proline transporter TcAAAP069 and also they had a trypanocidal effect against all stages of T. cruzi [165].
2D QSAR is a tool that seeks to explain the relationships between chemical structures and experimental observations so that predictions of new compounds with certain desired properties can be made [166]. In a recent review, Halder and Dias Soeiro Cordeiro [168] analyzed different in silico approaches that were successfully applied in the discovery of anti-leishmaniasis (anti-LM) and anti-trypanosomiasis (anti-TP) drugs. After reviewing these works, the authors [168] identified two in silico approaches that have been little explored in recent years, but that had a high potential in the development of anti-LM and anti-TP agents, which are QSARs with.
In the search for molecules with action against Trypanosoma cruzi, [169] used the quantitative structure-activity relationship (QSAR) approach to investigate how molecular physical-chemical characteristics affect biological activity. These authors assumed that there are compounds that at some point show activity against T. cruzi but occasionally fail when tested in “whole-cell phenotypic assays.” However, according to the authors, this result may be due to several factors such as inadequate physical-chemical and pharmacokinetic properties, resulting in molecules with little capacity to cross cell membranes. In this work, the authors applied artificial neural networks (ANNs) and kernel-based partial least squares regression (KPLS) to anti-T. cruzi activity data (Fig. 3). Through the analysis of atomic contribution maps, the authors found that fluorine and, in general, the heterocyclic aromatic rings and piperazine rings contributed positively to anti-T activity. Therefore, the integration of the ANN and KPLS analyses enabled the generation of a collection of key fragments strongly correlated with anti-T. cruzi compounds, providing valuable information to guide the design of new antichagasic agents with enhanced properties [169].

Proposed workflow for the design of new anti-Trypanosoma cruzi compounds based on the generation of contribution maps, prediction of trypanocidal activity using artificial neural networks and analysis of physical-chemical properties [169]
In 2012, Vincent et al. demonstrated the use of the metabolomic platform to elucidate the mode of action of an anti-trypanosomal drug [170]. According to Trochine et al., the first option for the treatment of CD is the administration of BZN, although its mode of action is not yet fully understood [171]. In order to analyze the metabolic response of T. cruzi to BZN, the authors [171] used a non-targeted MS-based metabolomics approach. Thus, the global changes in the metabolites that occur when BZN enters the parasite could be monitored. As a result, the authors concluded that treatment with BZN mainly affects molecules containing thiol in T. cruzi, therefore, this interference in thiol metabolism contributes to the action of the drug [171]. Later, the same group monitored, using the same metabolomic strategy, changes of low-mass metabolites in the epimastigote forms of T. cruzi treated with Bestatin [172]. Bestatin is a natural product with a broad-spectrum of inhibitory action on metalloaminopeptidases. Their results showed that Bestatin did not have a toxic effect directly on the parasite, but it had a substantial effect on the dipeptide pool, demonstrating its action as an inhibitor compound of dipeptidase enzymes [172]. In conclusion, the authors attested that the metabolomic platform has great potential for in situ analysis of enzymatic inhibition by pharmacological agents, and is an alternative for the evaluation of metabolic changes that occur after the exposure to a compound [172].
In a study on the metabolic changes that occur during the phases of exponential and stationary growth in T. cruzi epimastigotes, Barisón et al. studied the 47 metabolic intermediates of the most important pathways for energy metabolism and oxidative imbalance [173]. As a result, the authors showed for the first time that T. cruzi epimastigotes exhibit an adaptive metabolic mechanism that allows alternating the consumption of glucose to amino acids, in the transition from the exponential to the stationary phase. Mosquillo et al. carried out the first work that combined massive data from transcriptome and translatome, aiming to unravel the mechanism of action of two organometallic compounds (Pd-dppf-mpo and Pt-dppf-mpo) with trypanocidal activity [174]. As a result, the authors identified modified and/or metabolic enzymes present in the parasite, but absent in the mammalian host, which may become targets for rational drug planning. In a study conducted by Zrein and Chatelain on monitoring the status of CD in infected patients, the authors state that despite the great effort in basic research that seeks to help in the management of CD, to date there has been little translation into available products [175]. The authors pointed out that disease control becomes even more challenging due to the lack of methods that allow the effectiveness of treatments to be evaluated safely. Currently, patients infected with T. cruzi are diagnosed using antibody detection methods in serological assays [175]. However, the authors claim that it is essential to change the paradigm of serological methods in order to be able to monitor the elimination of the parasites correctly. Thus, they argued that analyzing the diversity of antibodies is more informative of the clinical status than the conventional serological tests that are designed for global detection of antibodies [175].
Therefore, to date, there are relatively few validated drug targets for CD. As seen in this section, the combination of different types of omics data (proteomics, genomics, transcriptomics, metabolomics, etc.) is necessary for a correct understanding of the functioning of the disease. As well as the resolution of how environmental factors and culture media can change the characteristics of the parasite under study. Thus, understanding the host–parasite relationship is fundamental for extracting useful information for the development of an effective drug against CD.
6 Perspectives: Challenges in New Drugs Discovery
Several recent reviews in the literature discuss this important point about the challenges present in new drugs discovery [16, 30, 56, 176,177,178,179,180].
According to the roadmap of Echeverría et al., prevention, diagnosis, and treatment are the three major levels of intervention for which it is necessary to seek solutions [1]. Progress has been made in the purpose of new drug discovery for treatment of CD. Parameters that need to be improved have been defined and joint actions bringing companies, support entities, and academies together have been established [181]. A successful drug discovery campaign typically takes 10–15 years [30, 56].
However challenges are still present such as a drug effective against the acute and chronic phase, difficulties in the translation process, genetic diversity of T. cruzi, development of standard tests to monitor the course of treatment with drugs to certify the cure of CD among other factors [16, 182]. New potential and promising targets, new methodologies including animal models of tropical disease infections that represent human disease are needed. Evolution in the standardization, as well as innovative strategies, is currently being developed to improve all process related to drug discovery [183]. Proteomic studies provide information on the pathogenic mechanisms of CD and other neglected tropical diseases, identifying molecular targets for drug discovery and development promising biomarkers to detect both stages of the disease and for potential use in diagnosis [184].
We can consider that despite all the difficulties, the panorama of discovering new drugs has improved in these years. A better understanding of the problems and challenges and greater integration between the pharmaceutical industries, academies, governmental and non-governmental intuitions are accelerating all the processes involved in developing new drugs to find the best treatment for Chagas disease and other neglected diseases.
Change history
24 April 2022
The original version of the chapter was inadvertently published with errors in the names of these author names: Giseli Capaci Rodrigues and Igor Almeida Rodrigues. However, the author names have now been corrected.
References
Echeverría LE, Marcus R, Novick G et al (2020) WHF IASC roadmap on chagas disease. Glob Heart 15:26. https://doi.org/10.5334/gh.484
Lidani KCF, Andrade FA, Bavia L et al (2019) Chagas disease: from discovery to a worldwide health problem. Front Public Heal 7. https://doi.org/10.3389/fpubh.2019.00166
Sales Junior PA, Molina I, Fonseca Murta SM et al (2017) Experimental and clinical treatment of chagas disease: a review. Am J Trop Med Hyg 97:1289–1303. https://doi.org/10.4269/ajtmh.16-0761
Zemore ZM, Wills BK (2020) Kissing bug bite. In: StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK554472/. Accessed 4 Nov 2020
Chao C, Leone JL, Vigliano CA (2020) Chagas disease: historic perspective. Biochim Biophys Acta Mol basis Dis 1866:165689. https://doi.org/10.1016/j.bbadis.2020.165689
Gomes C, Almeida AB, Rosa AC et al (2019) American trypanosomiasis and Chagas disease: sexual transmission. Int J Infect Dis 81:81–84. https://doi.org/10.1016/j.ijid.2019.01.021
Stevens JR, Noyes HA, Dover GA, Gibson WC (1999) The ancient and divergent origins of the human pathogenic trypanosomes, Trypanosoma brucei and T. cruzi. Parasitology 118:107–116. https://doi.org/10.1017/S0031182098003473
Aufderheide AC, Salo W, Madden M et al (2004) A 9,000-year record of Chagas’ disease. Proc Natl Acad Sci 101:2034–2039. https://doi.org/10.1073/pnas.0307312101
Naghavi M, Abajobir AA, Abbafati C et al (2017) Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390:1151–1210. https://doi.org/10.1016/S0140-6736(17)32152-9
Alonso-Padilla J, Cortés-Serra N, Pinazo MJ et al (2019) Strategies to enhance access to diagnosis and treatment for Chagas disease patients in Latin America. Expert Rev Anti-Infect Ther 17:145–157. https://doi.org/10.1080/14787210.2019.1577731
Ferreira ÉR, Bonfim-Melo A, Mortara RA, Bahia D (2012) Trypanosoma cruzi extracellular amastigotes and host cell signaling: more pieces to the puzzle. Front Immunol 3. https://doi.org/10.3389/fimmu.2012.00363
Kessler RL, Contreras VT, Marliére NP et al (2017) Recently differentiated epimastigotes from Trypanosoma cruzi are infective to the mammalian host. Mol Microbiol 104:712–736. https://doi.org/10.1111/mmi.13653
Zingales B (2018) Trypanosoma cruzi genetic diversity: something new for something known about Chagas disease manifestations, serodiagnosis and drug sensitivity. Acta Trop 184:38–52. https://doi.org/10.1016/j.actatropica.2017.09.017
Lima L, Espinosa-Álvarez O, Ortiz PA et al (2015) Genetic diversity of Trypanosoma cruzi in bats, and multilocus phylogenetic and phylogeographical analyses supporting Tcbat as an independent DTU (discrete typing unit). Acta Trop 151:166–177. https://doi.org/10.1016/j.actatropica.2015.07.015
Ramírez JD, Hernández C (2018) Trypanosoma cruzi I: towards the need of genetic subdivision?, part II. Acta Trop 184:53–58. https://doi.org/10.1016/j.actatropica.2017.05.005
Vermelho AB, Rodrigues GC, Supuran CT (2020) Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin Drug Discov 15:145–158. https://doi.org/10.1080/17460441.2020.1681394
WHO (2020) Chagas disease (also known as American trypanosomiasis). https://www.who.int/en/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis). Accessed 18 Dec 2020
López-Vélez R, Norman FF, Bern C (2020) American trypanosomiasis (Chagas disease). In: Hunter’s tropical medicine and emerging infectious diseases. Elsevier, pp 762–775
Lescure F-X, Le Loup G, Freilij H et al (2010) Chagas disease: changes in knowledge and management. Lancet Infect Dis 10:556–570. https://doi.org/10.1016/S1473-3099(10)70098-0
McCall L-I, Tripathi A, Vargas F et al (2018) Experimental Chagas disease-induced perturbations of the fecal microbiome and metabolome. PLoS Negl Trop Dis 12:e0006344. https://doi.org/10.1371/journal.pntd.0006344
Thompson AM, O’Connor PD, Marshall AJ et al (2020) Re-evaluating pretomanid analogues for Chagas disease: hit-to-lead studies reveal both in vitro and in vivo trypanocidal efficacy. Eur J Med Chem 207:112849. https://doi.org/10.1016/j.ejmech.2020.112849
Foodborne, D.E.; African, H.; Lymphatic, L.L.; Scabies, O.R.; Soil-transmitted, S.; Yaws, T. Chagas Disease Echinococcosis Foodborne Trematodiases Human African Trypanosomiasis Leishmaniasis Leprosy Rabies Yaws Ending the Neglect to Attain the Sustainable Development Goals a Sustainability Framework for Action against Neglected Tropical Diseases; World Health Organization: Geneva, Switzerland, 2020; ISBN 978-92-4-001035-2
Kourbeli V, Chontzopoulou E, Moschovou K et al (2021) An overview on target-based drug design against kinetoplastid protozoan infections: human African trypanosomiasis. Chagas Dis Leishmaniases Mol 26:4629. https://doi.org/10.3390/molecules26154629
MacLean LM, Thomas J, Lewis MD et al (2018) Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Negl Trop Dis 12:e0006612. https://doi.org/10.1371/journal.pntd.0006612
Wall RJ, Moniz S, Thomas MG et al (2018) Antitrypanosomal 8-hydroxy-naphthyridines are chelators of divalent transition metals. Antimicrob Agents Chemother 62. https://doi.org/10.1128/AAC.00235-18
Benaim G, Paniz-Mondolfi AE, Sordillo EM, Martinez-Sotillo N (2020) Disruption of intracellular calcium homeostasis as a therapeutic target against Trypanosoma cruzi. Front Cell Infect Microbiol 10. https://doi.org/10.3389/fcimb.2020.00046
de Almeida Rodrigues I, Alcântara da Silva B, Souza dos Santos AL et al (2010) Erratum to: a new experimental culture medium for cultivation of Leishmania amazonensis: its efficacy for the continuous in vitro growth and differentiation of infective promastigote forms. Parasitol Res 107:249–249. https://doi.org/10.1007/s00436-010-1894-y
Vermelho AB (2010) Trypanosoma cruzi peptidases: an overview. Open Parasitol J 4:120–131. https://doi.org/10.2174/1874421401004010120
Vermelho AB, Capaci GR, Rodrigues IA et al (2017) Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg Med Chem 25:1543–1555. https://doi.org/10.1016/j.bmc.2017.01.034
Mansoldo FRP, Carta F, Angeli A et al (2020) Chagas disease: perspectives on the past and present and challenges in drug discovery. Molecules 25:5483. https://doi.org/10.3390/molecules25225483
Docampo R, Moreno SNJ (2017) Biochemistry of Trypanosoma cruzi. In: American trypanosomiasis chagas disease. Elsevier, pp 371–400
Wyllie S, Brand S, Thomas M et al (2019) Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc Natl Acad Sci 116:9318–9323. https://doi.org/10.1073/pnas.1820175116
Vermelho AB, da Silva Cardoso V, Ricci Junior E et al (2018) Nanoemulsions of sulfonamide carbonic anhydrase inhibitors strongly inhibit the growth of Trypanosoma cruzi. J Enzyme Inhib Med Chem 33:139–146. https://doi.org/10.1080/14756366.2017.1405264
Ortiz C, Moraca F, Medeiros A et al (2016) Binding mode and selectivity of steroids towards glucose-6-phosphate dehydrogenase from the pathogen Trypanosoma cruzi. Molecules 21:368. https://doi.org/10.3390/molecules21030368
de Faria TRB (2016) Quimioterapia contra doença de Chagas: proposição de modelo não patogênico, teste de novos compostos, otimização e padronização de nova metodologia. Instituto Nacional de Metrologia, Qualidade e Tecnologia
D’Antonio EL, Deinema MS, Kearns SP et al (2015) Structure-based approach to the identification of a novel group of selective glucosamine analogue inhibitors of Trypanosoma cruzi glucokinase. Mol Biochem Parasitol 204:64–76. https://doi.org/10.1016/j.molbiopara.2015.12.004
Chatelain E, Konar N (2015) Translational challenges of animal models in Chagas disease drug development: a review. Drug Des Devel Ther 4807. https://doi.org/10.2147/DDDT.S90208
Sajid M, McKerrow JH (2002) Cysteine proteases of parasitic organisms. Mol Biochem Parasitol 120:1–21. https://doi.org/10.1016/S0166-6851(01)00438-8
Soeiro MNC, de Castro SL (2009) Trypanosoma cruzi targets for new chemotherapeutic approaches. Expert Opin Ther Targets 13:105–121. https://doi.org/10.1517/14728220802623881
Cazzulo J (2002) Proteinases of Trypanosoma cruzi: potential targets for the chemotherapy of Chagas disease. Curr Top Med Chem 2:1261–1271. https://doi.org/10.2174/1568026023392995
McKerrow JH (2018) Update on drug development targeting parasite cysteine proteases. PLoS Negl Trop Dis 12:e0005850. https://doi.org/10.1371/journal.pntd.0005850
McKerrow J, Doyle P, Engel J et al (2009) Two approaches to discovering and developing new drugs for Chagas disease. Mem Inst Oswaldo Cruz 104:263–269. https://doi.org/10.1590/S0074-02762009000900034
Alvarez VE, Niemirowicz GT, Cazzulo JJ (2012) The peptidases of Trypanosoma cruzi: digestive enzymes, virulence factors, and mediators of autophagy and programmed cell death. Biochim Biophys Acta - Proteins Proteomics 1824:195–206. https://doi.org/10.1016/j.bbapap.2011.05.011
San Francisco J, Barría I, Gutiérrez B et al (2017) Decreased cruzipain and gp85/trans-sialidase family protein expression contributes to loss of Trypanosoma cruzi trypomastigote virulence. Microbes Infect 19:55–61. https://doi.org/10.1016/j.micinf.2016.08.003
Chen YT, Brinen LS, Kerr ID et al (2010) In vitro and in vivo studies of the trypanocidal properties of WRR-483 against Trypanosoma cruzi. PLoS Negl Trop Dis 4:e825. https://doi.org/10.1371/journal.pntd.0000825
McKerrow JH, Caffrey C, Kelly B et al (2006) Proteases in parasitic diseases. Annu Rev Pathol Mech Dis 1:497–536. https://doi.org/10.1146/annurev.pathol.1.110304.100151
DNDi (2010) K777 (Chagas)
Silva JRA, Cianni L, Araujo D et al (2020) Assessment of the Cruzain cysteine protease reversible and irreversible covalent inhibition mechanism. J Chem Inf Model 60:1666–1677. https://doi.org/10.1021/acs.jcim.9b01138
Yepes AF, Quintero-Saumeth J, Cardona-G W (2020) Chalcone-quinoline conjugates as potential T. cruzi cruzipain inhibitors: docking studies, molecular dynamics and evaluation of drug-likeness. ChemistrySelect 5:7104–7112. https://doi.org/10.1002/slct.202000777
Silva-Júnior EF, Silva EPS, França PHB et al (2016) Design, synthesis, molecular docking and biological evaluation of thiophen-2-iminothiazolidine derivatives for use against Trypanosoma cruzi. Bioorg Med Chem 24:4228–4240. https://doi.org/10.1016/j.bmc.2016.07.013
Huang L, Chen CH (2009) Proteasome regulators: activators and inhibitors. Curr Med Chem 16:931–939. https://doi.org/10.2174/092986709787581860
Cardoso J, Soares MJ, Menna-Barreto RFS et al (2008) Inhibition of proteasome activity blocks Trypanosoma cruzi growth and metacyclogenesis. Parasitol Res 103:941–951. https://doi.org/10.1007/s00436-008-1081-6
Gupta I, Aggarwal S, Singh K et al (2018) Ubiquitin proteasome pathway proteins as potential drug targets in parasite Trypanosoma cruzi. Sci Rep 8:8399. https://doi.org/10.1038/s41598-018-26532-z
Alvarez VE, Iribarren PA, Niemirowicz GT, Cazzulo JJ (2021) Update on relevant trypanosome peptidases: validated targets and future challenges. Biochim Biophys Acta - Proteins Proteomics 1869:140577. https://doi.org/10.1016/j.bbapap.2020.140577
Khare S, Nagle AS, Biggart A et al (2016) Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537:229–233. https://doi.org/10.1038/nature19339
Field MC, Horn D, Fairlamb AH et al (2017) Anti-trypanosomatid drug discovery: an ongoing challenge and a continuing need. Nat Rev Microbiol 15:217–231. https://doi.org/10.1038/nrmicro.2016.193
Landis MS, Bhattachar S, Yazdanian M, Morrison J (2018) Commentary: why pharmaceutical scientists in early drug discovery are critical for influencing the design and selection of optimal drug candidates. AAPS PharmSciTech 19:1–10. https://doi.org/10.1208/s12249-017-0849-3
Capasso C, Supuran CT (2015) An overview of the alpha-, beta- and gamma-carbonic anhydrases from Bacteria: can bacterial carbonic anhydrases shed new light on evolution of bacteria? J Enzyme Inhib Med Chem 30:325–332. https://doi.org/10.3109/14756366.2014.910202
Pan P, Vermelho AB, Capaci Rodrigues G et al (2013) Cloning, characterization, and sulfonamide and thiol inhibition studies of an α-carbonic anhydrase from Trypanosoma cruzi, the causative agent of Chagas disease. J Med Chem 56:1761–1771. https://doi.org/10.1021/jm4000616
Supuran CT (2016) Inhibition of carbonic anhydrase from Trypanosoma cruzi for the management of Chagas disease: an underexplored therapeutic opportunity. Future Med Chem 8:311–324. https://doi.org/10.4155/fmc.15.185
da Silva Cardoso V, Vermelho AB, Ricci Junior E et al (2018) Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzyme Inhib Med Chem 33:850–857. https://doi.org/10.1080/14756366.2018.1463221
Rodrigues GC, Feijó DF, Bozza MT et al (2014) Design, synthesis, and evaluation of hydroxamic acid derivatives as promising agents for the management of Chagas disease. J Med Chem 57:298–308. https://doi.org/10.1021/jm400902y
D’Ambrosio K, Supuran CT, De Simone G (2019) Are carbonic anhydrases suitable targets to fight protozoan parasitic diseases? Curr Med Chem 25:5266–5278. https://doi.org/10.2174/0929867325666180326160121
Bonardi A, Vermelho AB, da Silva CV et al (2019) N-nitrosulfonamides as carbonic anhydrase inhibitors: a promising chemotype for targeting Chagas disease and Leishmaniasis. ACS Med Chem Lett 10:413–418. https://doi.org/10.1021/acsmedchemlett.8b00430
Matutino Bastos T, Mannochio Russo H, Silvio Moretti N et al (2019) Chemical constituents of anacardium occidentale as inhibitors of Trypanosoma cruzi sirtuins. Molecules 24:1299. https://doi.org/10.3390/molecules24071299
Matutino Bastos T, Botelho Pereira Soares M, Haddad Franco C et al (2020) Identification of inhibitors to Trypanosoma cruzi sirtuins based on compounds developed to human enzymes. Int J Mol Sci 21:3659. https://doi.org/10.3390/ijms21103659
Milduberger N, Bustos PL, González C et al (2021) Trypanosome cruzi infection in Cyclophilin D deficient mice. Exp Parasitol 220:108044. https://doi.org/10.1016/j.exppara.2020.108044
Búa J, Ruiz AM, Potenza M, Fichera LE (2004) In vitro anti-parasitic activity of Cyclosporin A analogs on Trypanosoma cruzi. Bioorg Med Chem Lett 14:4633–4637. https://doi.org/10.1016/j.bmcl.2004.07.003
Jha B, Varikuti S, Bishop N et al (2020) An effective live vaccine strain of Trypanosoma cruzi prevents Chagas disease in the mouse model. https://doi.org/10.21203/rs.3.rs-92241/v1
McGwire B (2020) Live attenuated parasitic vaccine. 22
Corpas-Lopez V, Moniz S, Thomas M et al (2019) Pharmacological validation of N-myristoyltransferase as a drug target in Leishmania donovani. ACS Infect Dis 5:111–122. https://doi.org/10.1021/acsinfecdis.8b00226
Roberts AJ, Torrie LS, Wyllie S, Fairlamb AH (2014) Biochemical and genetic characterization of Trypanosoma cruzi N-myristoyltransferase. Biochem J 459:323–332. https://doi.org/10.1042/BJ20131033
Roberts AJ, Fairlamb AH (2016) The N-myristoylome of Trypanosoma cruzi. Sci Rep 6:31078. https://doi.org/10.1038/srep31078
Herrera LJ, Brand S, Santos A et al (2016) Validation of N-myristoyltransferase as potential chemotherapeutic target in mammal-dwelling stages of Trypanosoma cruzi. PLoS Negl Trop Dis 10:e0004540. https://doi.org/10.1371/journal.pntd.0004540
Kovářová J, Barrett MP (2016) The pentose phosphate pathway in parasitic trypanosomatids. Trends Parasitol 32:622–634. https://doi.org/10.1016/j.pt.2016.04.010
Igoillo-Esteve M, Maugeri D, Stern AL et al (2007) The pentose phosphate pathway in Trypanosoma cruzi: a potential target for the chemotherapy of Chagas disease. An Acad Bras Cienc 79:649–663. https://doi.org/10.1590/S0001-37652007000400007
Maugeri DA, Cazzulo JJ (2004) The pentose phosphate pathway in Trypanosoma cruzi. FEMS Microbiol Lett 234:117–123. https://doi.org/10.1111/j.1574-6968.2004.tb09522.x
Cordeiro AT, Thiemann OH (2010) 16-Bromoepiandrosterone, an activator of the mammalian immune system, inhibits glucose 6-phosphate dehydrogenase from Trypanosoma cruzi and is toxic to these parasites grown in culture. Bioorg Med Chem 18:4762–4768. https://doi.org/10.1016/j.bmc.2010.05.008
Ioset J-R, Chatelain E (2011) Drug discovery and development for neglected diseases: the DNDi model. Drug Des Devel Ther 5:175. https://doi.org/10.2147/DDDT.S16381
Fauro R, Lo Presti S, Bazan C et al (2013) Use of clomipramine as chemotherapy of the chronic phase of Chagas disease. Parasitology 140:917–927. https://doi.org/10.1017/S0031182013000103
Argüelles AJ, Cordell GA, Maruenda H (2016) Molecular docking and binding mode analysis of plant alkaloids as in vitro and in silico inhibitors of trypanothione reductase from Trypanosoma cruzi. Nat Prod Commun 11:1934578X1601100. https://doi.org/10.1177/1934578X1601100118
Fredo Naciuk F, do Nascimento Faria J, Gonçalves Eufrásio A et al (2020) Development of selective steroid inhibitors for the glucose-6-phosphate dehydrogenase from Trypanosoma cruzi. ACS Med Chem Lett 11:1250–1256. https://doi.org/10.1021/acsmedchemlett.0c00106
Ortíz C, Moraca F, Laverriere M et al (2021) Glucose 6-phosphate dehydrogenase from trypanosomes: selectivity for steroids and chemical validation in bloodstream Trypanosoma brucei. Molecules 26:358. https://doi.org/10.3390/molecules26020358
Osorio-Méndez JF, Cevallos AM (2019) Discovery and genetic validation of chemotherapeutic targets for Chagas’ disease. Front Cell Infect Microbiol 8. https://doi.org/10.3389/fcimb.2018.00439
Villalta F, Rachakonda G (2019) Advances in preclinical approaches to Chagas disease drug discovery. Expert Opin Drug Discov 14:1161–1174. https://doi.org/10.1080/17460441.2019.1652593
Khare S, Roach SL, Barnes SW et al (2015) Utilizing chemical genomics to identify cytochrome b as a novel drug target for Chagas disease. PLoS Pathog 11:e1005058. https://doi.org/10.1371/journal.ppat.1005058
de Oliveira PIC, de Santana Miranda PH, Lourenço EMG et al (2020) Planning new Trypanosoma cruzi CYP51 inhibitors using QSAR studies. Mol Divers. https://doi.org/10.1007/s11030-020-10113-2
De Rycker M, Thomas J, Riley J et al (2016) Identification of trypanocidal activity for known clinical compounds using a new Trypanosoma cruzi hit-discovery screening cascade. PLoS Negl Trop Dis 10:e0004584. https://doi.org/10.1371/journal.pntd.0004584
Rocha-Hasler M, de Oliveira GM, da Gama AN et al (2021) Combination with tomatidine improves the potency of posaconazole against Trypanosoma cruzi. Front Cell Infect Microbiol 11. https://doi.org/10.3389/fcimb.2021.617917
Barreto-Bergter E, Vermelho AB, Hogge L, Gorin PAJ (1985) Glycolipid components of epimastigote forms of Trypanosoma cruzi. Comp Biochem Physiol Part B Comp Biochem 80:543–545. https://doi.org/10.1016/0305-0491(85)90287-1
Koeller CM, Heise N (2011) The sphingolipid biosynthetic pathway is a potential target for chemotherapy against Chagas disease. Enzyme Res 2011:1–13. https://doi.org/10.4061/2011/648159
Giorgi ME, de Lederkremer RM (2020) The Glycan structure of T. cruzi mucins depends on the host. Insights on the chameleonic galactose. Molecules 25:3913. https://doi.org/10.3390/molecules25173913
Booth L-A, Smith TK (2020) Lipid metabolism in Trypanosoma cruzi: a review. Mol Biochem Parasitol 240:111324. https://doi.org/10.1016/j.molbiopara.2020.111324
Landoni M, Piñero T, Soprano LL et al (2019) Tamoxifen acts on Trypanosoma cruzi sphingolipid pathway triggering an apoptotic death process. Biochem Biophys Res Commun 516:934–940. https://doi.org/10.1016/j.bbrc.2019.06.149
Miguel DC, Ferraz ML, Alves RO et al (2010) The anticancer drug tamoxifen is active against Trypanosoma cruzi in vitro but ineffective in the treatment of the acute phase of Chagas disease in mice. Mem Inst Oswaldo Cruz 105:945–948. https://doi.org/10.1590/S0074-02762010000700021
Docampo R, Huang G (2015) Calcium signaling in trypanosomatid parasites. Cell Calcium 57:194–202. https://doi.org/10.1016/j.ceca.2014.10.015
Benaim G, Garcia CRS (2011) Review paper targeting calcium homeostasis as the therapy of Chagas’ disease and leishmaniasis--a review. Trop Biomed 28:471–481
Schoijet AC, Sternlieb T, Alonso GD (2019) Signal transduction pathways as therapeutic target for Chagas disease. Curr Med Chem 26:6572–6589. https://doi.org/10.2174/0929867326666190620093029
Lammel EM, Barbieri MA, Wilkowsky SE et al (1996) Trypanosoma cruzi: involvement of intracellular calcium in multiplication and differentiation. Exp Parasitol 83:240–249. https://doi.org/10.1006/expr.1996.0070
Ruiz CR, Favoreto S, Dorta LM et al (1998) Infectivity of Trypanosoma cruzi strains is associated with differential expression of surface glycoproteins with differential Ca2+ signalling activity. Biochem J 330:505–511. https://doi.org/10.1042/bj3300505
Huang G, Bartlett PJ, Thomas AP et al (2013) Acidocalcisomes of Trypanosoma brucei have an inositol 1,4,5-trisphosphate receptor that is required for growth and infectivity. Proc Natl Acad Sci 110:1887–1892. https://doi.org/10.1073/pnas.1216955110
Rohloff P, Rodrigues CO, Docampo R (2003) Regulatory volume decrease in Trypanosoma cruzi involves amino acid efflux and changes in intracellular calcium. Mol Biochem Parasitol 126:219–230. https://doi.org/10.1016/S0166-6851(02)00277-3
Paveto C, Pereira C, Espinosa J et al (1995) The nitric oxide transduction pathway in Trypanosoma cruzi. J Biol Chem 270:16576–16579. https://doi.org/10.1074/jbc.270.28.16576
Cortez M, Neira I, Ferreira D et al (2003) Infection by Trypanosoma cruzi metacyclic forms deficient in gp82 but expressing a related surface molecule, gp30. Infect Immun 71:6184–6191. https://doi.org/10.1128/IAI.71.11.6184-6191.2003
Walker DM, Oghumu S, Gupta G et al (2014) Mechanisms of cellular invasion by intracellular parasites. Cell Mol Life Sci 71:1245–1263. https://doi.org/10.1007/s00018-013-1491-1
Misra S, Naskar K, Sarkar D, Ghosh D (1991) Role of Ca2+ ion on Leishmania -macrophage attachment. Mol Cell Biochem 102. https://doi.org/10.1007/BF00232154
Moreno SN, Silva J, Vercesi AE, Docampo R (1994) Cytosolic-free calcium elevation in Trypanosoma cruzi is required for cell invasion. J Exp Med 180:1535–1540. https://doi.org/10.1084/jem.180.4.1535
Yakubu MA, Majumder S, Kierszenbaum F (1994) Changes in Trypanosoma cruzi infectivity by treatments that affect calcium ion levels. Mol Biochem Parasitol 66:119–125. https://doi.org/10.1016/0166-6851(94)90042-6
Lu H-G, Zhong L, Chang K-P, Docampo R (1997) Intracellular Ca 2+ pool content and signaling and expression of a calcium pump are linked to virulence in Leishmania mexicana amazonesis amastigotes. J Biol Chem 272:9464–9473. https://doi.org/10.1074/jbc.272.14.9464
Docampo R (1993) Calcium homeostasis in Trypanosoma cruzi. Biol Res 26:189–196
Oz HS, Wittner M, Tanowitz HB et al (1992) Trypanosoma cruzi: mechanisms of intracellular calcium homeostasis. Exp Parasitol 74:390–399. https://doi.org/10.1016/0014-4894(92)90201-K
Docampo R, Moreno SNJ (2011) Acidocalcisomes. Cell Calcium 50:113–119. https://doi.org/10.1016/j.ceca.2011.05.012
Huang G, Moreno SNJ, Docampo R (2020) Isolation and characterization of acidocalcisomes from trypanosomatids. Methods Mol Biol 2116:673–688
Docampo R, Ulrich P, Moreno SNJ (2010) Evolution of acidocalcisomes and their role in polyphosphate storage and osmoregulation in eukaryotic microbes. Philos Trans R Soc B Biol Sci 365:775–784. https://doi.org/10.1098/rstb.2009.0179
Chiurillo MA, Lander N, Vercesi AE, Docampo R (2020) IP3 receptor-mediated Ca2+ release from acidocalcisomes regulates mitochondrial bioenergetics and prevents autophagy in Trypanosoma cruzi. Cell Calcium 92:102284. https://doi.org/10.1016/j.ceca.2020.102284
Docampo R, Moreno SNJ (2008) The acidocalcisome as a target for chemotherapeutic agents in protozoan parasites. Curr Pharm Des 14:882–888. https://doi.org/10.2174/138161208784041079
Menna-Barreto R, de Castro S (2017) Clear shot at primary aim: susceptibility of Trypanosoma cruzi organelles, structures and molecular targets to drug treatment. Curr Top Med Chem 17:1212–1234. https://doi.org/10.2174/1568026616666161025161858
Lander N, Chiurillo MA, Bertolini MS et al (2018) The mitochondrial calcium uniporter complex in trypanosomes. Cell Biol Int 42:656–663. https://doi.org/10.1002/cbin.10928
Docampo R, Vercesi AE, Huang G (2014) Mitochondrial calcium transport in trypanosomes. Mol Biochem Parasitol 196:108–116. https://doi.org/10.1016/j.molbiopara.2014.09.001
Cavalcanti DP, de Souza W (2018) The kinetoplast of trypanosomatids: from early studies of electron microscopy to recent advances in atomic force microscopy. Scanning 2018:1–10. https://doi.org/10.1155/2018/9603051
Zuma AA, Cavalcanti DP, Zogovich M et al (2015) Unveiling the effects of berenil, a DNA-binding drug, on Trypanosoma cruzi: implications for kDNA ultrastructure and replication. Parasitol Res 114:419–430. https://doi.org/10.1007/s00436-014-4199-8
Manchester T, Cavalcanti DP, Zogovich M et al (2013) Acriflavine treatment promotes dyskinetoplasty in Trypanosoma cruzi as revealed by ultrastructural analysis. Parasitology 140:1422–1431. https://doi.org/10.1017/S0031182013001029
Leite TOC (2019) Developments on treatment of Chagas disease--from discovery to current times. Eur Rev Med Pharmacol Sci 23:2576–2586
DNDi (2019) Chagas disease. https://dndi.org/diseases/chagas/. Accessed 1 Dec 2021
Ribeiro V, Dias N, Paiva T et al (2020) Current trends in the pharmacological management of Chagas disease. Int J Parasitol Drugs Drug Resist 12:7–17. https://doi.org/10.1016/j.ijpddr.2019.11.004
Wilkinson SR, Taylor MC, Horn D et al (2008) A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc Natl Acad Sci 105:5022–5027. https://doi.org/10.1073/pnas.0711014105
Maya JD, Cassels BK, Iturriaga-Vásquez P et al (2007) Mode of action of natural and synthetic drugs against Trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem Physiol Part A Mol Integr Physiol 146:601–620. https://doi.org/10.1016/j.cbpa.2006.03.004
Murta, Ropert, Alves et al (1999) In-vivo treatment with benznidazole enhances phagocytosis, parasite destruction and cytokine release by macrophages during infection with a drug-susceptible but not with a derived drug-resistant Trypanosoma cruzi population. Parasite Immunol 21:535–544. https://doi.org/10.1046/j.1365-3024.1999.00251.x
Turrens JF, Watts BP, Zhong L, Docampo R (1996) Inhibition of Trypanosoma cruzi and T. brucei NADH fumarate reductase by benznidazole and anthelmintic imidazole derivatives. Mol Biochem Parasitol 82:125–129. https://doi.org/10.1016/0166-6851(96)02722-3
Bermudez J, Davies C, Simonazzi A et al (2016) Current drug therapy and pharmaceutical challenges for Chagas disease. Acta Trop 156:1–16. https://doi.org/10.1016/j.actatropica.2015.12.017
Hall BS, Bot C, Wilkinson SR (2011) Nifurtimox activation by trypanosomal type I nitroreductases generates cytotoxic nitrile metabolites. J Biol Chem 286:13088–13095. https://doi.org/10.1074/jbc.M111.230847
Patterson S, Fairlamb AH (2019) Current and future prospects of nitro-compounds as drugs for Trypanosomiasis and Leishmaniasis. Curr Med Chem 26:4454–4475. https://doi.org/10.2174/0929867325666180426164352
Docampo R (1990) Sensitivity of parasites to free radical damage by antiparasitic drugs. Chem Biol Interact 73:1–27. https://doi.org/10.1016/0009-2797(90)90106-W
Morillo CA, Marin-Neto JA, Avezum A et al (2015) Randomized trial of benznidazole for chronic Chagas’ cardiomyopathy. N Engl J Med 373:1295–1306. https://doi.org/10.1056/NEJMoa1507574
Pécoul B (2016) Un modèle alternatif et innovant de Recherche et Développement pour garantir l’accès aux médicaments. Médecine/Sciences 32:1049–1050. https://doi.org/10.1051/medsci/20163212001
Petravicius PO, Costa-Martins AG, Silva MN et al (2019) Mapping benznidazole resistance in trypanosomatids and exploring evolutionary histories of nitroreductases and ABCG transporter protein sequences. Acta Trop 200:105161. https://doi.org/10.1016/j.actatropica.2019.105161
Urbina JA (2010) Specific chemotherapy of Chagas disease: Relevance, current limitations and new approaches. Acta Trop 115:55–68. https://doi.org/10.1016/j.actatropica.2009.10.023
Vergara C, Muñoz G, Martínez G et al (2019) Detection of Trypanosoma cruzi by PCR in adults with chronic Chagas disease treated with nifurtimox. PLoS One 14:e0221100. https://doi.org/10.1371/journal.pone.0221100
Sgambatti de Andrade ALS, Zicker F, de Oliveira RM et al (1996) Randomised trial of efficacy of benznidazole in treatment of early Trypanosoma cruzi infection. Lancet 348:1407–1413. https://doi.org/10.1016/S0140-6736(96)04128-1
Sperandio da Silva GM, Mediano MFF, Americano A, do Brasil PE et al (2014) A clinical adverse drug reaction prediction model for patients with chagas disease treated with benznidazole. Antimicrob Agents Chemother 58:6371–6377. https://doi.org/10.1128/AAC.02842-14
DNDi (2019) Fexinidazole for Chagas
WHO (2020) Treatment of Chagas disease. https://www.who.int/chagas/disease/treatment/en. Accessed 21 Jan 2021
Deeks ED (2019) Fexinidazole: first global approval. Drugs 79:215–220. https://doi.org/10.1007/s40265-019-1051-6
Watson JA, Strub-Wourgraft N, Tarral A et al (2019) Pharmacokinetic-pharmacodynamic assessment of the hepatic and bone marrow toxicities of the new trypanoside fexinidazole. Antimicrob Agents Chemother 63. https://doi.org/10.1128/AAC.02515-18
Torrico F, Gascón J, Barreira F et al (2021) New regimens of benznidazole monotherapy and in combination with fosravuconazole for treatment of Chagas disease (BENDITA): a phase 2, double-blind, randomised trial. Lancet Infect Dis 21:1129–1140. https://doi.org/10.1016/S1473-3099(20)30844-6
Ribeiro I, Blum B, Fernandes J et al (2021) Drug-drug interaction study of benznidazole and E1224 in healthy male volunteers. Antimicrob Agents Chemother 65. https://doi.org/10.1128/AAC.02150-19
Hata K (2021) Development of E1224 by leveraging a strategic partnership for the medicines creation against neglected tropical diseases. Parasitol Int 81:102278. https://doi.org/10.1016/j.parint.2020.102278
García-Huertas P, Cardona-Castro N (2021) Advances in the treatment of Chagas disease: promising new drugs, plants and targets. Biomed Pharmacother 142:112020. https://doi.org/10.1016/j.biopha.2021.112020
Spósito PÁ, Mazzeti AL, de Castro KCMP et al (2021) Higher oral efficacy of ravuconazole in self-nanoemulsifying systems in shorter treatment in experimental chagas disease. Exp Parasitol 228:108142. https://doi.org/10.1016/j.exppara.2021.108142
Buckner FS (2008) Sterol 14-demethylase inhibitors for Trypanosoma cruzi infections. In: Drug targets in kinetoplastid parasites. Springer, New York, pp 61–80
Soeiro MNC, de Souza EM, da Silva CF et al (2013) In vitro and in vivo studies of the antiparasitic activity of sterol 14α-demethylase (cyp51) inhibitor vni against drug-resistant strains of Trypanosoma cruzi. Antimicrob Agents Chemother 57:4151–4163. https://doi.org/10.1128/AAC.00070-13
Beltran-Hortelano I, Perez-Silanes S, Galiano S (2017) Trypanothione reductase and superoxide dismutase as current drug targets for Trypanosoma cruzi: an overview of compounds with activity against Chagas disease. Curr Med Chem 24. https://doi.org/10.2174/0929867323666161227094049
Wooden B, Goossens N, Hoshida Y, Friedman SL (2017) Using big data to discover diagnostics and therapeutics for gastrointestinal and liver diseases. Gastroenterology 152:53–67.e3. https://doi.org/10.1053/j.gastro.2016.09.065
Parikh PP, Minning TA, Nguyen V et al (2012) A semantic problem solving environment for integrative parasite research: identification of intervention targets for Trypanosoma cruzi. PLoS Negl Trop Dis 6:e1458. https://doi.org/10.1371/journal.pntd.0001458
Cortes-Serra N, Losada-Galvan I, Pinazo M-J et al (2020) State-of-the-art in host-derived biomarkers of Chagas disease prognosis and early evaluation of anti-Trypanosoma cruzi treatment response. Biochim Biophys Acta Mol basis Dis 1866:165758. https://doi.org/10.1016/j.bbadis.2020.165758
Talavera-López C, Andersson B (2017) Parasite genomics—time to think bigger. PLoS Negl Trop Dis 11:e0005463. https://doi.org/10.1371/journal.pntd.0005463
Sánchez-Ovejero C, Benito-Lopez F, Díez P et al (2016) Sensing parasites: proteomic and advanced bio-detection alternatives. J Proteome 136:145–156. https://doi.org/10.1016/j.jprot.2015.12.030
Preidis GA, Hotez PJ (2015) The newest “omics”—metagenomics and metabolomics—enter the battle against the neglected tropical diseases. PLoS Negl Trop Dis 9:e0003382. https://doi.org/10.1371/journal.pntd.0003382
Kuleš J, Potocnakova L, Bhide K et al (2017) The challenges and advances in diagnosis of vector-borne diseases: where do we stand? Vector-Borne Zoonotic Dis 17:285–296. https://doi.org/10.1089/vbz.2016.2074
Kratz JM (2019) Drug discovery for chagas disease: a viewpoint. Acta Trop 198:105107. https://doi.org/10.1016/j.actatropica.2019.105107
Calogeropoulou T, Magoulas GE, Pöhner I et al (2019) Hits and lead discovery in the identification of new drugs against the trypanosomatidic infections. In: Med chem neglected trop dis adv des synth antimicrob agents. CRC Press, pp 185–231
Lešnik S, Konc J (2020) In silico laboratory: tools for similarity-based drug discovery. Methods Mol Biol 2089:1–28
Kanakaveti V, Shanmugam A, Ramakrishnan C et al (2020) Computational approaches for identifying potential inhibitors on targeting protein interactions in drug discovery. Elsevier
Schaduangrat N, Lampa S, Simeon S et al (2020) Towards reproducible computational drug discovery. J Cheminform 12:1–30. https://doi.org/10.1186/s13321-020-0408-x
Shaker B, Yu MS, Lee J et al (2020) User guide for the discovery of potential drugs via protein structure prediction and ligand docking simulation. J Microbiol 58:235–244. https://doi.org/10.1007/s12275-020-9563-z
Sayé M, Gauna L, Valera-Vera E et al (2020) Crystal violet structural analogues identified by in silico drug repositioning present anti-Trypanosoma cruzi activity through inhibition of proline transporter TcAAAP069. PLoS Negl Trop Dis 14:e0007481. https://doi.org/10.1371/journal.pntd.0007481
Lewis RA, Wood D (2014) Modern 2D QSAR for drug discovery. Wiley Interdiscip Rev Comput Mol Sci 4:505–522. https://doi.org/10.1002/wcms.1187
Halder AK, Dias Soeiro Cordeiro MN (2020) Advanced in silico methods for the development of anti- leishmaniasis and anti-trypanosomiasis agents. Curr Med Chem 27:697–718. https://doi.org/10.2174/0929867325666181031093702
de Souza AS, Ferreira LLG, de Oliveira AS, Andricopulo AD (2019) Quantitative structure–activity relationships for structurally diverse chemotypes having anti-Trypanosoma cruzi activity. Int J Mol Sci 20:2801. https://doi.org/10.3390/ijms20112801
Vincent IM, Creek DJ, Burgess K et al (2012) Untargeted metabolomics reveals a lack of synergy between nifurtimox and eflornithine against Trypanosoma brucei. PLoS Negl Trop Dis 6:e1618. https://doi.org/10.1371/journal.pntd.0001618
Trochine A, Creek DJ, Faral-Tello P et al (2014) Benznidazole biotransformation and multiple targets in Trypanosoma cruzi revealed by metabolomics. PLoS Negl Trop Dis 8:e2844. https://doi.org/10.1371/journal.pntd.0002844
Trochine A, Creek DJ, Faral-Tello P et al (2015) Bestatin induces specific changes in Trypanosoma cruzi dipeptide pool. Antimicrob Agents Chemother 59:2921–2925. https://doi.org/10.1128/AAC.05046-14
Barisón MJ, Rapado LN, Merino EF et al (2017) Metabolomic profiling reveals a finely tuned, starvation-induced metabolic switch in Trypanosoma cruzi epimastigotes. J Biol Chem 292:8964–8977. https://doi.org/10.1074/jbc.M117.778522
Mosquillo MF, Smircich P, Ciganda M et al (2020) Comparative high-throughput analysis of the Trypanosoma cruzi response to organometallic compounds. Metallomics. https://doi.org/10.1039/D0MT00030B
Zrein M, Chatelain E (2020) The unmet medical need for Trypanosoma cruzi-infected patients: monitoring the disease status. Biochim Biophys Acta Mol basis Dis 1866:165628. https://doi.org/10.1016/j.bbadis.2019.165628
Chatelain E, Scandale I (2020) Animal models of Chagas disease and their translational value to drug development. Expert Opin Drug Discov:1–22. https://doi.org/10.1080/17460441.2020.1806233
Chatelain E, Ioset J-R (2018) Phenotypic screening approaches for Chagas disease drug discovery. Expert Opin Drug Discov 13:141–153. https://doi.org/10.1080/17460441.2018.1417380
Liu Q, Chen J, Zhou X-N (2020) Preparedness for Chagas disease spreading worldwide. Infect Dis Poverty 9:44. https://doi.org/10.1186/s40249-020-00658-7
Domagalska MA, Dujardin J-C (2020) Next-generation molecular surveillance of TriTryp diseases. Trends Parasitol 36:356–367. https://doi.org/10.1016/j.pt.2020.01.008
Lascano F, García Bournissen F, Altcheh J (2020) Review of pharmacological options for the treatment of Chagas disease. Br J Clin Pharmacol. https://doi.org/10.1111/bcp.14700
Alonso-Padilla J, Abril M, Alarcón de Noya B et al (2020) Target product profile for a test for the early assessment of treatment efficacy in Chagas disease patients: an expert consensus. PLoS Negl Trop Dis 14:e0008035. https://doi.org/10.1371/journal.pntd.0008035
Moraes CB, Giardini MA, Kim H et al (2015) Nitroheterocyclic compounds are more efficacious than CYP51 inhibitors against Trypanosoma cruzi: implications for Chagas disease drug discovery and development. Sci Rep 4:4703. https://doi.org/10.1038/srep04703
Chatelain E (2017) Chagas disease research and development: is there light at the end of the tunnel? Comput Struct Biotechnol J 15:98–103. https://doi.org/10.1016/j.csbj.2016.12.002
Saviola AJ, Negrão F, Yates JR (2020) Proteomics of select neglected tropical diseases. Annu Rev Anal Chem 13:315–336. https://doi.org/10.1146/annurev-anchem-091619-093003
Acknowledgements
The authors would like to thank the Brazilian agencies FAPERJ, CAPES, and CNPq for the financial support.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Ethics declarations
Conflict of Interest: The authors declare that they have no conflict of interest.
Funding: This work was financed in part by: the Coordenação de Aperfeiçoamento Pessoal de Nível Superior- Brasil (CAPES), Grant code 001; the Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) the Conselho Nacional de Desenvolvimento Científico e Tecnológico (MCTI-CNPq).
Ethical Approval: This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Vermelho, A.B. et al. (2022). Chagas Disease: Drug Development and Parasite Targets. In: Vermelho, A.B., Supuran, C.T. (eds) Antiprotozoal Drug Development and Delivery. Topics in Medicinal Chemistry, vol 39. Springer, Cham. https://doi.org/10.1007/7355_2021_143
Download citation
DOI: https://doi.org/10.1007/7355_2021_143
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-06849-2
Online ISBN: 978-3-031-06850-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)