
Abstract
Fungi are eukaryotic, single cell or multicellular organisms which cause a wide range of human diseases ranging from superficial skin to invasive life-threatening infections. Over the last couple of decades the incidence of life-threatening fungal infections has increased seriously as the patients of AIDS, cancer, organ transplant and immune-compromised population have increased. Though a significant progress has been made in the discovery of antifungal agents in the form of polyenes, azoles and allylamines yet the antifungal therapy poses severe challenge because of the side effects, narrow spectrum of activity and recently development resistance among patients against the present antifungals. This chapter deals with the current antifungal agents, their spectrum of activity, mode of action, limitations, current challenges in antifungal therapy, and new avenues for future developments.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Allylamines
- Antifungal therapy
- Azole
- Cell membrane
- Ergosterol
- Immunocompromise
- Monoclonal antibodies
- Pathogenic fungi
- Polyenes
1 Introduction
Fungi are one of the extensively spread organisms on earth and have great environmental and medical importance. The kingdom fungi contains about 1.5 million [1] different species which are either unicellular or multicellular eukaryotic, heterotrophic organisms that can be divided into biotrophs: which obtain their nutrients from a living host (plant or animal), saprotrophs: which obtained their nutrients from animals or dead plants, and necrotrophs: which infect a living host and kill host cells to obtain their nutrients [2].
Besides being beneficial organisms for humans in bio-production of alcohol and bakery, fungal species like Aspergillus sp., Penicillium sp., and Acremonium sp. are associated with the production of enzymes and antibiotics. Along with the above positive impacts certain species adversely affect the crops and humans by producing diseases. A number of fungi have been reported as causal agents of human and animal infections and the first published record of infection in human is a case of oral manifestation of Candida albicans infection that was recorded in 1665 as a fatal disease [3].
In the atmosphere fungi are present from temperate to subtropical and tropical areas, and these organisms are mostly non-pathogenic and can cause infection under certain compromised conditions like immune suppression which may be due to various factors [4]. The fungi can cause infection of any part of the body starting from the hair of the scalp to nails of the toe web. However these infections are opportunistic in nature and the fungi causing these infections are categorized as opportunistic pathogens. The true pathogenic fungi are only four in number and these are Coccidioides, Paracoccidiodes, Blastomyces, and Histoplasma [5]. Fortunately the geographic distribution of these fungi is known to restricted area [6]. In case of superficial fungal infections the value is more of cosmetic in nature and man hour loss in terms and public nuisance. However the systemic infections pose a serious challenge in the form of early and accurate diagnosis as well as treatment [7].
A number of antifungal agents as described in this chapter are available in the market. Barring amphotericin B almost all the known antifungal agents are fungistatic in nature. Amphotericin B considered to be the gold standard of the antifungal agents is fungicidal; however its use is very much limited due to its side effects, particularly nephrotoxicity [5]. The fungal infections have emerged into prominence after the onset of AIDS and HIV infections where these infections may prove to be fatal to the host [6]. The number of antifungal agents is limited as compared to antibacterial drugs because of the fact that the fungus is an eukaryotic organism that parasitizes an eukaryotic host where the narrow range of physiologic difference between them cause difficulties in developing safe and broad spectrum antifungal agents. There are limited number of classes of antifungal agents to combat fungal infections with limitations of toxicity and development of drug resistance [8, 9].
2 Challenges in Antifungal Therapy
The major challenge in the treatment of mycoses is the timely and correct diagnosis of the disease. This is the first very important step which is mainly dependent on the clinical symptoms, which are very peculiar in case of superficial infections like raised erythematous margins of the lesions with prominent scaling and many times present with itching. However, in case of systemic infections the symptoms are very often common to those caused by other bacterial infections particularly in case of the infections of the lung. Then comes the step of obtaining the sample from the site of infection which may be achieved through scraping from the active sites of the infection in case of the involvement of the skin (margins), hair, nail, and sputum in case of lung infection, blood in case of systemic infection, etc. The samples thus collected are subjected to direct microscopic examination using wet mount, KOH preparations or fungal specific stains such as lactophenol cotton blue. In case of deep seated infections biopsy is often required for establishing the correct diagnosis. From the obtained clinical specimen cultures are made generally in Sabouraud’s dextrose agar at 30–53°C. Very often the fungi take longer periods to grow and thus result in the delay in diagnosis of mucoses.
Advances in biological techniques particularly the molecular one have opened avenues for diagnostic methods that are not dependent on culture of the organisms. Specific metabolites and molecular probes are often used for the detection and identification of fungal infections [10,11,12,13]. PCR (polymerase chain reaction) has exhibited its utility in the diagnosis of microbial infections inclusive of mycoses [14,15,16,17,18]. In the genome the most conserved region is the ribosomal DNA having capability of phylogenetic divergence [19]. The rRNA gene has a large subunit (LSU) 28S rRNA and small subunit (SSU) 18S rRNA and 5.8S rRNA. The internal transcribed spacer (ITS) region I (ITSI) and ITSII are found between SSU rRNA and 5.8S rRNA and between 5.8S rRNA and LSU rRNA respectively and are more variable than the rest of the ribosomal gene subunits. In addition the intergenicspacer (IGS) region I (IGSI) and IGSII occur between the LSU and SSU sequence [20]. Further the single-stranded conformation polymorphism (SSCP) technique to identify sequence variations in a single strand of DNA due to its adoption to a unique conformation under non-denaturing conditions [18] has been used by various researchers [21,22,23,24]. Such molecular approaches have the advantage of detecting fungi directly in the clinical specimen and provide much faster and more sensitive fungal detection than the conventional culture-based methods.
The next important step in the direction of therapy is in vitro sensitivity tests for the isolated fungal strain against the available antifungal agents. This is achieved by exposing the test fungus against the known concentrations of various antifungal agents and determining the minimal inhibitory concentration values. This may be achieved by either disc diffusion method or more precisely by the twofold serial dilution method as per guidelines of the CLSI (Earlier NCCLS). There are a number of antifungal agents available for the treatment of mycoses. However their usefulness has been limited either by their selective activity or more recently this situation is further complicated because of the development of resistance in the fungal pathogens against the existing antifungal agents.
3 Available Antifungal Drugs, Spectrum of Activity, and Development of Resistance
The availability of antifungal agents is limited for therapy and the use of these drugs is further restricted by the issue of safety, resistance, and their efficacy profiles. Understanding the mode of action of different antifungal agents is an important prerequisite to explore drug resistance mechanisms. The emergence of resistance against drugs is an evolutionary process based on natural selection of organisms that enhances their ability to survive and multiply in presence of drug. Investment of a considerable amount of energy is required by competitive microbial communities for the production and elaboration of antimicrobial agents [25]. The evolution of resistance against antimicrobial agent is ubiquitous in nature and microbes evolve various strategies to combat the action of drugs. The development of new antibiotics is outpaced by the evolution of drug resistance due to which progressing our knowledge towards understanding evolutionary mechanisms gains utmost importance. The present antifungal arsenal has been discussed below.
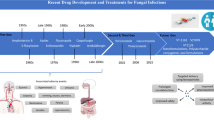
4 Polyenes
The Polyene antibiotics discovered in late 1950s have a broad spectrum fungicidal activity and were isolated from different species of Streptomyces which are soil born [26] (Fig. 1). Chemically the polyenes are the molecules that contain polyhydroxylic lactone ring of 20–40 carbon atoms with 4–7 conjugated double bonds, that’s why they are hydrophobic in nature. These are known to bind to the main component of fungal cell membrane, the ergosterol and result in the formation of transmembrane channels that allow the leakage of cell contents along with K+ and Na+ ions leading to the damage and death of the fungal cells [27]. The affinity of polyenes for ergosterol in fungal cell wall is higher than the affinity for cholesterol in mammalian cell; therefore they are less toxic to the latter. Yet this non-negligible toxicity cannot be ignored and explains the high toxicity associated with several side effects. Of the several polyene antibiotics only three, nystatin, natamycin, and amphotericin B, are in clinical use despite their side effects.

Structure of polyene antibiotics
4.1 Nystatin
Nystatin (1) is the first antifungal agent introduced for the clinical use which was discovered by E.L. Hazen and R.F. Brown in 1944 while doing their research in the division of Laboratories and Research, New York State Department of Health which was published in 1950 [28, 29]. It was isolated from an actinomycete Streptomyces noursei which is commercially described as mycostatin and is active against many moulds and yeast infections [29, 30]. Nystatin is insoluble in water and sparingly soluble in organic solvents. It is unstable under moist conditions, heat, and light sensitive and therefore stored in cold and dark places [31]. Nystatin structure has been resolved by chemical degradation and X-ray crystallography [32]. It consists of a 38-membered macrolide lactone ring containing single tetraene and diene moieties separated by two methylene groups [33].
This drug is not absorbed through oral route but is effective topically for oropharyngealcandidosis. Nystatin was licensed for use in 1951 and due to its greater potential activity that caused toxicity in the system its use has been restricted to topical administration for superficial (mucosal) Candida infections of the oropharynx, esophagus, and intestinal tract.
Later on a liposomal preparation of nystatin was prepared that enhanced survival and reduced the tissue burden of Aspergillus in experimental neutropenic rabbits with invasive pulmonary aspergillosis and mice with disseminated aspergillosis [34, 35].
4.2 Natamycin
Natamycin (2) also known as pimaricin has been reported to be produced during fermentation process by a soil inhabiting microorganism Streptomyces natalensis [36]. It is sparingly soluble in water and has been found to exhibit antifungal activity at low concentrations. Natamycin is being used in the treatment of mycotic keratitis an infection of the cornea especially the cases caused by the species of Aspergillus and Fusarium [37]. It is normally used as topical antifungal agent in the form of cream or drops where it exhibits absorption in very low quantities in the body. This antibiotic is very little absorbed from the GI tract and therefore not recommended for use against systemic fungal infections [38].
4.3 Amphotericin B
Amphotericin B (3) is a polyene antifungal agent which is produced by Streptomycin nodosus [39]. According to the modern pharmacological standards, it is notified that amphotericin B, an antifungal agent, is a very old drug and since long times it was the only therapeutic option for the treatment of invasive mycoses. This compound is amphoteric in nature with a primary amino group attached to the mycosamine ring and a carboxyl group on the macrocycle [40]. Amphotericin B forms deep yellow crystals that are sparingly soluble in organic solvents but insoluble in water [41].
Though it is not well absorbed after oral administration, it exhibits a wide spectrum of activity that is fungicidal in nature [42]. This drug can be used as an oral/topical formulation for the treatment of mucosal candidosis and intravenous amphotericin B for invasive fungal infections as a successful therapy [43]. It is proposed by most clinical medical mycologists as the drug of choice for all forms of invasive aspergillosis and cutaneous mucormycosis, blastomycosis, paracoccidioidomycosis, histoplasmosis, fusariosis, severe and moderate cryptococcal meningitis, coccidioidomycosis, candidosis, and Candida infections of the central nervous system [9]. The side effect of amphotericin B therapy causes serious nephrotoxicity where almost each patient contracts some defect in renal function [44].
The amphotericin B molecule is largely lipophilic and forms pore in the fungal membranes but does not cause pore formation in the mammalian cell membrane because its partition coefficient is lower for cholesterol which form the main constituent of mammalian cell membrane instead of ergosterol, which is found in fungal membrane. The drug gets saturated in fungal cell and leads to its lysis due to its higher partition coefficient for ergosterol. The fungicidal activity of amphotericin B is mediated by its binding with ergosterol that is supplemented by the secondary mechanism of membrane permeabilization through channel formation. In a recent study the cytocidal activity of amphotericin B has been attributed due to its ability to extract ergosterol from lipid bilayers by forming large, extramembranous aggregates [45,46,47,48,49]. Use of amphotericin B has certain limitations as its intravenous administration is associated with side effects such as fever, chills, headache, nausea, vomiting and nephrotoxicity. To overcome this problem different commercial lipid-based formulations of amphotericin B are available that cause less toxicity.
The clinically useful and established novel formulations are lipid combinations with amphotericin B, encapsulated in liposomes or in ribbon-like and disc-like lipid complexes while the others studied are amphotericin B–cochleate preparation and an arabinogalactan complex. To overcome the nephrotoxiciy of standard amphotericin B lipid formulation of amphotericin B can be used. The lipid formulation is very expensive as compared to the native formulation [25, 50]. Occurrence of resistance to polyenes in C. albicans is a rare event but recently increasing cases of resistance have been reported [51]. Filamentous fungi exhibit greater resistance to polyenes than yeasts. Aspergillus terreus is generally amphotericin B resistant whereas A. fumigates and A. flavus are becoming gradually more resistant [52]. Polyene resistance could be developed by reducing the substrate to which it binds, i.e., ergosterol content in plasma membrane. Mutation in ERG3 gene lowers the ergosterol content of plasma membrane leading to accumulation of alternative sterols, causing amphotericin B resistance. The polyene resistance is also associated with increased catalase activity, which increases its oxidative tolerance [53].
A biochemical hypothesis for amphotericin B resistance has been given by Hamilton Miller that the altered sterol content of the resistant cells should bind to smaller amounts of polyene than do susceptible cells, hence may become resistant.
5 Azoles
Azoles were first introduced in 1960s as derivatives of N-substituted imidazole such as econazole, ketoconazole, miconazole, and clotrimazole, and is the most widely used class of antifungal agents [54] (Fig. 2). The azoles form a group of fungistatic agents with broad spectrum activity and are classified into two groups: the imidazoles and the triazoles. These antifungals inhibit the cytochrome P450 dependent enzyme lanosterol 14-alpha-demethylase that converts the lanosterol (the main sterol found in fungal cell wall) to ergosterol and thus results in the depleted ergosterol in the cell membrane causing cell death [55]. Azole antifungals are widely used in the treatment of systemic and topical (athletes foot, ringworm etc.) fungal infections. However azoles being fungistatic have a disadvantage due to recurrence of fungal infections.

Structure of azole class of antifungals
5.1 Econazole
Econazole nitrate chemically 1-[2-[(4-Chlorophenyl) methoxy]-2-(2,4-dichlorophenyl)-ethyl]-1H-imidazole (4) is a white crystalline nitric acid salt of econazole. It is slightly soluble in water, ether, and alcohol, sparingly soluble in chloroform, and soluble in methanol [56]. This antifungal is commonly used as the nitrate salt for antifungal therapy [57] mainly in the form of a cream to treat tinea corporis, tinea pedis, athlete's foot, tinea cruris, tinea versicolor and cutaneous candidiasis. However about 3% of treated patients have been reported to exhibit side effects like burning, itching, erthema, and pruritic rash [58].
5.2 Clotrimazole
Clotrimazole (5) was first described in 1969 from At Bayer Research Laboratories. 1-(o-Chloro-alpha,alpha-diphenyl benzyl)imidazole (clotrimazole) is a white crystalline solid that is sparingly soluble in water but soluble in alcohol and most organic solvents [59]. This antifungal is also known as Canesten or Lotrimin and is the first imidazole derivative which was developed for the treatment of human mycoses. It played an important role in the treatment of fungal infections such as vaginal yeast infections, oral thrush, ringworm, athlete foot, and jock itch. It is a vital medicine in the list of WHO [60].
Clotrimazole kills fungal cell by altering the permeability of fungal cell wall and binds to phospholipids in the cell membrane that inhibit the biosynthesis of ergosterol and sterols for the cell membrane production which results in loss of intracellular elements and cellular death [61]. Clotrimazole is a broad spectrum antifungal agent used in the treatment of infections caused by dermatophytes, yeasts, and Malassezia furfur.
5.3 Miconazole
Miconazole (6) is a synthetic imidazole antifungal agent that is poorly soluble in water and most of the organic solvents [62]. It also has some antibacterial action and antiparasitic properties. The mode of action of miconazole is inhibiting the synthesis of ergosterol [63]. It is used for the treatment of topical fungal infections including vaginal candidiasis [64], onychomycosis [65], tineapedis [66], and pityriasis versicolor [67]. It has also been moderately successful in the treatment of systemic mycoses [68].
5.4 Ketoconazole
Ketoconazole (7), discovered in 1976, is a member of imidazole synthetic compounds series which has a broad spectrum antifungal profile. 1-Acetyl-4-[4-[[2-(2,4-dichlorophenyl)-2(1H-imidazole-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazine is a weak basic compound that occurs as a white crystalline solid [69]. Ketoconazole is a racemic compound, consisting of the cis-2S,4R and cis-2R,4S isomers and it has seen that the 2S,4R isomer was more active than its 2R,4S enantiomer [70].
Ketoconazole was the first available compound for the oral treatment of systemic fungal infections in the early 1980s [71]. Ketoconazole is less soluble in water and administered orally [72] and in a range of formulations for topical administration such as creams (in treatment of cutaneous candidasis, pityriasis versicolor, candidal paronychia) and shampoos [73]. It shows toxic effects against yeast and interferes with other membrane lipids or enzymes. Ketoconazole inhibits the enzyme cytochrome P450 14-alpha-demethylase (P45014DM) which plays an important role in sterol biosynthesis pathway that leads from lanosterol to ergosterol [74].
High oral dose of ketoconazole may cause hepatotoxicity. Higher therapeutic doses may also produce endocrine abnormalities by reduction in circulating testosterone levels and blocks both testicular and adrenal androgen biosynthesis [75]. Ketoconazole is highly protein bound, has poor CNS penetration, and is not suitable for treating CNS infections. There is no intravenous preparation [76]. Oral ketoconazole is effective in patients with candidosis, coccidioidomycosis, blastomycosis, histoplasmosis, paracoccidioidomycosis, and cutaneous dermatophyte infections [77].
Later on the first generation triazoles such as fluconazole and itraconazole were introduced which are the imidazoles having five membrane ring atoms with one, two, and three nitrogen molecules. Fluconazole and itraconazole exhibited a broader antifungal activity spectrum as compared to the imidazoles and had a significant improved safety profile in comparison of amphotericin B and ketoconazole. Despite their prevalent use they face certain clinical limitations such as suboptimal level of activity spectrum, development of resistance and toxicity. In order to rectify these problems, several analogues have been derived. The second generation triazoles such as voriconazole, ravuconazole, and posaconazole possess higher potency and have increased efficacy against the emerging pathogens. Azoles perform their activity on cell membrane by inhibiting the ergosterol biosynthesis [78]. The major targets of most azoles are gene ERG11 encoded cytochrome P450 lanosterol 14α-demethylase, it leads to generation of faulty intermediate namely 14-methylergosta-8, 24(28)-dien-3,6-diol, which is toxic and is responsible for inhibition of fungus [79]. Increase in azole resistance is mainly due to its fungistatic nature instead of fungicidal. Resistance against fluconazole among HIV patients with OPC is the direct consequence of excess use of itraconazole and fluconazole [80]. About one-third population of patients with AIDS has azole resistant C. albicans in their oral tract [81]. Candida species employs various mechanisms to develop resistance against azoles as follows:
-
Over expression of efflux pumps: C. albicans overexpresses the efflux pumps in response to drug which results in efflux of drugs from cells thus reducing the drug concentration at action site. Two gene families namely MDR (Multi-Drug Resistance) genes of the major facilitator class and CDR genes belonging to the ATP-binding super cassette family. Up-regulation of CDR genes is responsible for resistance against most azoles while MDR encoded pumps exhibit narrow fluconazole specific spectrum [82].
-
Modification of target: Mutations in the ERG11 gene, which encodes lanosterol 14α-demethylase, decrease azole affinity to the target site. Fluconazole has been used against a variety of mycotic infections and resistance to this antifungal has been documented. The two yeasts Candida glabrata and C. krusei with inherent low susceptibilities to fluconazole have been reported at a greater frequency from patients [83].
-
Up-regulation of target enzyme: Candida isolates overexpress the ERG11 gene which results in reduced azole susceptibility [84]. The overexpression of gene results in accumulation of target molecules.
-
Development of alternative pathways: Organisms express alternative genes in order to bypass the pathway. Azole exposure results in ergosterol depletion from the membrane and leads to accumulation of toxic metabolite namely 14α-methyl 3, 6-diol. Additive mutation in ERG3 gene prevents the formation of this toxic product from 14α-methyl fecosterol and leads to accumulation of nontoxic sterols [85].
5.5 Fluconazole
Fluconazole (8) was formulated in 1981 and marketed in 1990. It is a novel bi-striazole which is metabolically stable, water soluble, low lipophilicity, and plasma protein binding antifungal agent. Fluconazole acts by inhibiting ergosterol enzyme biosynthesis in fungal cells through inhibition of a cytochrome P450 enzyme dependent 14 alpha-sterol demethylase [86]. This leads to the accumulation of methylated sterols which break fungal membrane structure resulting in growth arrest. Fluconazole antifungal is administered orally, intravenously, or both and is used to treat broad spectrum of fungal infections and has a very low incidence of side effects. It is used to treat Candida infections of the vagina (“yeast infections”), mouth, throat, and bloodstream [87]. It is also used to prevent infections in people with weak immune systems, including those due to cancer chemotherapy, bone marrow transplantation patients, premature babies, and oropharyngeal candidosis, neutropenia, sporotrichosis infections [87, 88].
5.6 Itraconazole
Itraconazole (9) discovered in 1984 is another triazole antifungal agent with broad spectrum antifungal activity [89]. It contains a weakly basic 1,2,4-triazole and a non-basic 1,2,4-triazol-3-one moieties in its structure and requires an acidic environment for optimum solubilization and oral absorption [90].
It is insoluble in water and available in oral form. It is active against Aspergillus, Candida, and Cryptococcus species [91]. Itraconazole has been useful in the treatment of chronic cavitary pulmonary disease, extrapulmonary blastomycosis, disseminated non-meningeal histoplasmosis, osseous/articular and lymphocutaneoussporotrichosis in non-immunosuppressed patients [92]. Itraconazole has recently been repositioned as anticancer agent [93]. Traconazole is the only inhibitor in this class that has been exposed to reduce both hedgehog signaling pathway and angiogenesis. These different actions are unrelated to inhibition of the cytochrome P450 lenosterol 14 alpha demethylase. The anti-angiogenic action of itraconazole is associated with inhibition of glycosylation VEGFR2, phosphorylation, trafficking, and cholesterol biosynthesis pathways.
5.7 Voriconazole
Voriconazole (10) is a low molecular weight, water soluble broad spectrum triazole effective against the treatment of invasive aspergillosis and esophageal candidiasis [94, 95]. It shows activity against Aspergillus spp., Fusarium spp., Candida spp., Cryptococcus neoformans, Fusarium, and Scedosporium infections including the fluconazole resistant or less susceptible spp. of C. glabrata and C. krusei [96, 97]. It showed serious drug–drug interactions and side effects like skin rash and transaminase elevation and hallucinations [98,99,100,101,102].
5.8 Posaconazole
Posaconazole (11), a triazole antifungal drug, was approved by the US FDA in September 2006 for the prophylaxis of invasive Aspergillus and Candida infections in severely immune-compromised patients [103]. It shows in vitro activity against Aspergillus, Candida spp., Cryptococcus spp., and Histoplasma spp. and also effective against infections caused by the zygomycetes than voriconazole [8, 104]. The most common side effects of posaconazole are gastrointestinal complaints, nausea, vomiting, abdominal pain, headache, elevation of liver enzymes, and skin rash [105,106,107].
5.9 Ravuconazole
Ravuconazole (12), a triazole, is a broad spectrum antifungal agent. It shows activity against Candida spp. even isolates that are resistant to fluconazole, Aspergillus, Cryptococcus, and many dermatophytic fungi [107,108,109]. Ravuconazole shows long elimination half-life and high protein binding [110, 111].
5.10 Other Azoles
5.10.1 Imidazoles
Azoles being popular as antifungal agents have been considered for various modifications (Fig. 3). Among the imidazoles, a series of N-[(1,1′-biphenyl)-4-ylmethyl]-1H-imidazol-1-amine derivatives (13) reported by Setzu et al. [112] showed better antifungal activity with substitutions at 2-position (R1) of the phenyl ring compared to substitution at the 4- position (R2) when tested in Candida neoformans. However the most potent compound in the series with chloro substitutions at both 2 and 4 positions (R1 and R2) of the phenyl ring had a MIC value of 0.8 μg/mL against Trichophyton rubrum compared to miconazole (0.4 μg/mL). Imidazole modifications were also made by introducing nitro group at 5-position resulting in potent antifungal compounds. The analogs of 14 having R1 substituted by morpholineor piperidine, R2 and X substituted by H showed good activity against Sclerophoma pityophila [113]. Effective antifungal activity was also observed in another series of 5-nitro imidazoles having phenyl piperidine substitution at R separated by 2-hydroxypropyl methanedithioatespacer (15) [114]. The compound had MIC = 3 μg/mL against Trichophyton tonsurans. However the compound was less effective than miconazole (MIC = 0.2 μg/mL) or ornidazole (MIC = 0.8 μg/mL). In another imidazole containing series 2-(1H-imidazol-1-yl)-1-phenylethanone-O-2-(1H-imidazol-1-yl)-1-phenyl-ethyl oxime derivatives (16) were synthesized by inverting the oxime group present in oxiconazole [115]. The most active compound in the series having substitutions at R=Ethoxy morpholine, R1=H, R2=Me, and X=Cl is an effective antifungal compound against C. glabrata (MIC = 0.06 μg/mL), C. parapsilosis (MIC = 0.004 μg/mL), and C. albicans (MIC = 1 μg/mL). Another compound in the series where X=F also showed good antifungal activity in the above fungal species [C. glabrata (MIC = 0.25 μg/mL), C. parapsilosis (MIC = 0.03 μg/mL), and C. albicans (MIC = 8 μg/mL)]. In the same series modification of the R2 to imidazole and substitution at R=F resulted in less active compound (17) with MIC values of 4, 8, 2 μg/mL in C. glabrata, C. parapsilosis, and C. albicans, respectively. In another series of imidazole derivatives having a pyrrole ring (18) it was observed that compounds having R1=Cl, R2=R3=R4=R5=H, and R3=CH3 (MIC = 0.062 μg/mL), C3H7 (MIC = 0.016 μg/mL), CH2–c–C3H5 (MIC = 0.016 μg/mL), CH2=CH2 (MIC = 0.032 μg/mL), CH2–CH=CH2 (MIC = 0.016 μg/mL), CH2–CH=C(CH3)2 (MIC = 0.065 μg/mL) had comparable activity with miconazole (MIC = 0.062 μg/mL) and itraconazole (MIC = 0.062 μg/mL) and better than fluconazole (MIC = 0.25 μg/mL) in C. albicans [116]. In a series of 2,4,5- trisubstituted imidazoles (19), the best compounds had an indole moiety at the 2-position of the imidazole ring while the 4 and 5 positions were having substituted phenyl moiety . Three compounds [1: (R1=R2=F), 2: (R1=Cl, R2=H), 3:(R1=Br, R2=H)] in the series showed MIC = 8 μg/mL in C. albicans [117, 118].

Structure of imidazole containing molecules as antifungal agents
5.10.2 Triazoles
Bile conjugates of fluconazole (20) (Fig. 4) have shown good antifungal activity when the R position of the steroid moiety is substituted by H or OH, the activity was in between 2.18 and 25 μg/mL when evaluated in different fungal species (S. schenckii, C. albicans, C. parapsilosis) [119]. The triazole derivatives of 1-(1H-1,2,4-triazole-1-yl)-2-(2,4-difluorophenyl)-3-(N-cyclopropyl-N-substituted-amino)-2-propanol (21) were effective antifungal agents, most of them had broad antifungal activity with MIC80 less than 0.125 μg/mL [120]. The compounds having R=CH3, CH2CH3, CH2CHCH2, (CH2)3CH3, (CH2)4CH3, (CH2)6CH3, (CH2)7CH3 were the most potent ones having MIC80 in the range of 0.125–8 μg/mL against C. albicans, C. parapsilosis, C. neoformans, C. tropicalis, T. rubrum, A. fumigatus, M. canis, and F. compacta. Fluconazole at similar bioassay condition showed MIC80 range of 0.5–32 μg/mL in the fungal species mentioned above. In this series retention of antifungal activity was observed when the R group is substituted by benzyl group (22) having different substituents at the phenyl ring [X=H, 3F, 3Cl, 3CH3, 4-NO2, 2NO2, 2CN, 4CN, (2,4-Cl), 2CH3, 4CH3, 4F]. All these compounds had MIC80 value less than 0.125 μg/mL in C. albicans. Heterocyclic derivatives of fluconazole having N1-Indazole, indole, indoline, benzimidazole, azaindole, and benztriazole (23) were also synthesized where the R=N1-indazole and X=Cl, Cl substitution was the most potent candidate (MIC80 = 0.0007 μg/mL) than fluconazole (MIC80 = 0.020 μg/mL) against C. albicans [121]. In this series (23) better antifungal activity was observed by the replacement of N1-indazole by azaindole moiety having X=Cl, Cl (MIC80 = 0.0031 μg/mL) and X=F, F (MIC80 = 0.007 μg/mL). Another compound where R=3-ethoxycarbonylmethyl-1H-indole and X=Cl, Cl also showed good antifungal activity (MIC80 = 0.006 μg/mL). The syntheses of triazole derivatives with varying olefinic chain length for two series have been reported where in first case the optimum chain length of n = 2 having the structure 24 and varying olefinic chain length (n = 0–3) in structure 25 has shown excellent in vitro activity against Candida, Cryptococcus, and Aspergillus spp. with antifungal activity MIC ranging 0.016–0.125 μg/mL [122]. This is better than fluconazole that is having the MIC range 0.5–4 μg/mL in the above-mentioned species. A series of 1-[((hetero)aryl- or piperidinylmethyl) amino]-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols evaluated against C. albicans and A. fumigatus showed compound 26 having X=F, F, R=R3=H, and R=N-Boc to be the most potent one (MIC80 = 3 ng/mL) and better than fluconazole (MIC80 = 190 ng/mL). In this series methyl substitution of the nitrogen atom in the linker reduces the activity 20 times (MIC80 = 60 ng/mL) when compared to 26 [123]. A series of triazole derivatives targeting lanosterol 14α-demethylase (CYP51) with a general structure 1-(1H-1,2,4-triazole-1-yl)-2-(2,4-difluorophenyl)-3-(N-isoproyl-N-substituted-amino)-2-propanol depicted good antifungal activity when R=4-H3COC6H4, 4-H5C2OC6H4 with MIC80 ranging 0.0156–1 μg/mL in 27. In the same series different esters at 4-position of the phenyl ring having R=CH3, CH2CH3, CH2CH2CH3 in 28 had MIC80 in the range of 0.0156–64 μg/mL [124]. A series of fluconazole derivatives (29) with benzothiazinone substituent depicted slightly better antifungal activity. The compound 29 (X=S, R1=H, R2=R3=F) (0.25 μg/mL) showed improved activity than the benzoxazinone replacement [X=O, R1=H, R2=R3=F (0.5 μg/mL)] but both were found to be better than fluconazole (1 μg/mL) [125]. Another synthesized triazole containing compound (30) based on QSAR study with R=4-FC6H4, 4-CONH2C6H4, 4-C5H4N was having comparable activity (0.0625–0.5 μg/mL) with itraconazole (1–2 μg/mL) when tested in A. fumigatus, C. parapsilosis, C. tropicalis, C. neoformans, M. lauosum, and T. rubrum with best activity in M. lauosum [126]. A series of triazole compounds having hydrophobic substitution or CN group with the general structure 31 (R=3,4- (CH3)2, 4-tBu, CN) was having comparable potency (0.125–64 μg/mL) with fluconazole (1–64 μg/mL) and itraconazole (0.125–1 μg/mL) [127]. A series of carboxylic acid esters of fluconazole showed higher activity than fluconazole against C. albicans (ATCC 14053) in SDB medium. The carboxylic acid esters of fluconazole having R=O-2-bromooctanoyl and O-11-bromoundecanoyl (32) (Fig. 5) have MIC values of 111 μg/mL and 198 μg/mL as compared to fluconazole that is having an MIC value greater than 4,444 μg/mL under similar bioassay conditions. Another series of fatty alcohol phosphate triester derivatives 33 has also been synthesized where compounds having R1=CNCH2CH2: R2=n-C11H23, R1=CNCH2CH2: R2=n-CH2=CH–C9H18, R1=CH3: R2=n-C11H23, R1=CH3: R2=n-CH2=CH–C9H18, R1=CH3: R2=n-C8H17 have MIC values ranging from 12 to 1,658 μg/mL [128]. A series of triazole derivatives having 5-substituted tetrazole ring and having Ar =2-nBuOC6H6 attached to piperazine (34) is the most active with MIC values of 1.0–8.0 μg/mL, against Candida sp. [129]. In another series involving d-glucose derivatives of 1,2,3-Triazoles (35), chain length is important for antifungal activity with n = 8 having 14 times better activity than fluconazole with no activity when the chain length was increased to n = 12 [130]. Substituted 1,2,4-triazole and benzotriazole derivatives having phenoxypropyl piperazine side chains showed the linker length of three carbon atoms (n = 3) between piperazine and the phenyl ring to be crucial for antifungal activity (36). Compounds with R=H was having an MIC of 0.0156 μg/mL; however substitution at R by CH3 (2,3,4 positions), 4-C(CH3)3, 4-Cl, 3-NO2, 4-Br has good antifungal activity against C. albicans with MIC values ranging from 0.0156 to 0.25 μg/mL [131]. Benztriazole having no substitution (R1=R2=H) at 5, 6 positions was found to have an MIC value of 0.8 μg/mL while substitution at R1=R2=CH3 and R1=R2=NO2 was found to have same MIC value of 1.6 μg/mL in C. albicans (37) [129]. In the triazole series following the structural requirements in fluconazole a halogenated phenyl ring and tertiary alcoholic oxygen is preserved (38). In this series compounds having a phenyl ring with one halogen or trifluoro substituent were found to be active in Candida spp., Aspergillus spp., and C. neoformans with MIC ranging from 0.015 to 8 μg/mL. The most active compound in the series had an MIC ≤ 0.015 μg/mL in C. parapsilopsis while having good activity for C. krusei (MIC = 0.25 μg/mL) and C. glabrata (MIC = 1 μg/mL). A series of triazole molecules were synthesized where imidazole ring (A) was connected with variable spacer (X) to a substituted phenyl ring (39). The active compounds in the series were found to have X=C–C, C=C, C≡C, imidazolidine-2-one, 1H-imidazol-2(3H)-one, and R=4-Cl, 4-F with MIC80 ranging from 0.015 to 4 μg/mL in the Candida sp. (C. albicans, C. glabrata, C. krusei, C. tropicalis, C. parapsilosis, C. neoformans). All the compounds have better activity in C. albicans with an MIC80 of ≤0.015 μg/mL as compared to fluconazole (MIC80 = 4 μg/mL) [132, 133]. A series of triazoles were synthesized where one triazole ring of fluconazole was modified into benzoxazinone (X=O, n = 1), benzothiazinone (X=S, n = 1), and benzoxazolinone (X=O; n = 0) moiety, with the most active compounds 40 having R=H, Cl and MIC = 0.06 μg/mL in C. glabrata [134].

Structure of molecules having triazole structure and fluconazole modifications

Molecular structures having triazole moiety and fluconazole modifications
6 Pyrimidine Analogue
5-Fluorocytosine or flucytosine (5-FC) (41) (Fig. 6), an antimetabolite, was first synthesized in 1957 and its antifungal property discovered in 1964 [135]. It is used for the treatment of invasive mycoses where it is effective against yeasts [136]. 5-FC is a fluorine analogue which inhibits nucleotide biosynthesis as it enters inside the fungal cells via cytosine permease and get deaminated to 5-fluorouracil (5-FU) by cytosine deaminase. 5-FU is a specific inhibitor of an enzyme essential for DNA synthesis namely thymidylate synthetase. This antifungal is selectively toxic to fungi as there is little or no cytosinedeaminase in mammalian cells [137]. The drug application is limited by the high prevalence of resistance in fungal species. Surveys conducted by Defever et al. and Stiller et al. [85, 137] on C. albicans estimated that 50–60% of the Candida isolates were susceptible, 30–40% were partially resistant along with 4–6% were highly resistant. 5-FC is administered in combination with other drugs such as fluconazole and amphotericin Bat present and rarely used as a sole agent. Resistance against 5-FC is developed due to mutational loss of permease activity. The resistance caused due to decreased uptake of 5-FC is prevalent in C. glabrata and S. cerevisiae, but this phenomenon is of least importance in case of C. albicans or C. neoformans. The mutational loss of the pyrimidine salvage enzymes forms the basis of resistance in laboratory or clinical strains of C. neoformans and C. albicans [138,139,140].

Structure of pyrimidine and allylamine antifungal agents
7 Allylamines
Allylamines form the newly developed class of ergosterol synthesis inhibitors. They are functionally and chemically very distinct from other classes of ergosterol binding antifungal agents [141] (Fig. 6). Allylamines inhibits the early steps of ergosterol biosynthesis leading to accumulation of squalene and absence of other sterol derivatives [142, 143]. Although clinical failures have been reported in treatment cases of allylamines yet human pathogenic fungi do not exhibit any associated resistance. Its resistance mechanism is poorly understood and further researches are required in this area. Important members of this group include naftifine and terbinafine.
7.1 Terbinafine
In Europe terbinafine (42) became available in 1991 whereas it got approval in the USA in 1996 [144]. Its hydrochloride salt is crystalline hydrophobic in nature but soluble in methanol, dichloromethane, and ethanol. This antifungal is mainly effective for dermatophytic fungi and used for superficial infections [145]. Terbinafineis recognized as inhibitor of fungal ergosterol biosynthesis by inhibiting squaleneepoxidase, an essential component of fungal cell. Fungal cell death is due to accumulation of squalene, which may increase permeability leading to disruption of cellular organization. Terbinafine hydrochloride may induce subacute cutaneous erythematous and people with this have been advised to know the possible risks with their physicians before the start of therapy [146].
A number of adverse drug reactions and side effects have been reported with oral terbinafine hydrochloride which may possibly due to longer duration of treatment and due to its extensive distribution in the body [144, 147].
7.2 Naftifine
Naftifine (43) is a synthetic, broad spectrum, allylamine antifungal agent which is used as a topical medication for the treatment of fungal infections. Naftifine hydrochloride is a white crystalline powder that is soluble in polar solvents such as ethanol and methylene chloride [148]. Naftifine hydrochloride, with potent in vitro antifungal activity against dermatophytes, was found to be effective against tinea cruris, tinea corporis, and tinea pedis as a topical agent [149]. It has shown very good activity against Trichophyton rubrum, Trichophyton mentagrophytes, Trichophyton tonsurans, Epidermophyton floccosum, and Microsporum canis, Microsporum audouini, and Microsporum gypseum; and fungistatic activity against Candida species including Candida albicans [150, 151]. The mode of action of naftifine is not so clear but it seems to block the sterol biosynthesis via inhibition of the squalene 2,3-epoxidase enzyme [152]. This inhibition results in the accumulation of squalene, which is known to be toxic to fungi.
7.3 Amorolfin
Amorolfine (44) is a new topical water soluble antifungal drug of the morpholine derivatives. It inhibits D14 reductase and D 7-D8 isomerase which reduce ergosterol and accumulates in the fungal cytoplasmic membrane. This antifungal is used for the treatment of infections caused by dermatophyitic fungi and has been very effective in the treatment of onychomycosis [153, 154].
7.4 Butenafine
Butenafine (45) is a new synthetic benzylamine which has a broad spectrum of antifungal activity and used for the topical treatment of dermatophytoses caused fungi such as Trichophyton mentagrophytes, Microsporum canis, and Trichophyton rubrum. Its structure and mode of action are similar to allylamines as it inhibits sterol synthesis by blocking squalene epoxidation resulting in depletion of egosterol which is an essential lipid component of fungal cell membrane [147, 155]. The dermatophytes isolated from Tinea cruris have been found to be susceptible to both terbinafine and butenafine. The butenafine 1% cream has been found to exhibit supremacy over 1% terbinafine cream with statistically significant difference [156].
8 Indoles
Several compounds incorporating the indole moiety have also been reported as antifungal agents (Fig. 7). A series of 1H-Indole-4,7-diones derivatives have been synthesized by masking the indole nitrogen atom with CH3 or with substituted phenyl groups (46, 47). The compounds having substituted phenyl ring were active for C. krusei, C. neoformans, and A. niger with the most active compound having R2=Cl (MIC = 0.8 μg/mL; Candida krusei) and a methyl ester attached to 3-position of the indole ring in 47. A series of 5,6-bis(arylthio)-1H-indole-4,7-diones (48) showed moderate activity with an MIC range of 1.6–100 μg/mL with the most active compound (MIC = 1.6 μg/mL) having R1=Cl, R2=H for Candida tropicalis. The other substitutions such as R1=CH3, H and R2=H, Cl, Br, I, OCH3, CH3; R1=H, CH3, F, Cl and R2=H, Cl, Br, F, OH in all the 1H-Indole-4,7-dione series had potent antifungal activity with MICs ranging from 0.8 to 100 μg/mL [157]. The aminoguanidine derivatives of N-arylsulfonyl-3-acylindoles indicated that incorporation of electron donating groups at R1 and R2 improve antifungal activity. Variations were also made regarding the length of alkyl chain at R3 (methyl, ethyl, propyl) (49). The compounds with R1=4-Me, R2=H, R3=Me (P. oryzae = 79.64%, A. alternata = 79.15%, B. sorokinianum = 82.28%) and R1=R2=4-Me, R3=Me (P. oryzae = 84.84%, A. alternata = 82.98%, B. sorokinianum = 80.58%) had good antifungal activity [158].

Structure of indole antifungals
A series of compounds having indole fused with benzoquinone moiety having substitutions R1=H, OH, F; R2=CH3O, H, CH3, Br, Cl, I, F, OH, R3=C2H5, CH3, n-Pr (50) had potent antifungal activity with MIC 6.3–100 μg/mL in the Candida and Aspergillus sp. [159]. In another series of indole (51) substitution at R1=CH3CH2S, H; R2=C2H5, CH3, n-Pr resulted in compounds with MIC of 1.6–100 μg/mL [159]. A series of 1-benzyl-3-(imidazol-1-ylmethyl)indole derivatives (52) showed that compound having Z=H, R1=H, R2=CH, and X=4-Cl to be the most potent in the series with an MIC of 1 μg/mL against C. albicans (CA980001). Compounds having Z=H/H/H/H/H (substitution for five compounds C1/C2/C3/C4/C5 at position Z), R1=CH3/H/i-propyl/H/n-butyl, R2=H/H/H/H/H and X=4-Cl/4-F/4-Cl/2,4-diCl/4-Cl have MIC values of 3, 4,5,5,3.5 μg/mL respectively for the C. albicans. However none of these compounds are better than fluconazole (MIC = 0.02 μg/mL). Most of these compounds were less potent for A. fumigatus (AF980003) with the best compound (MIC = 8 μg/mL) having Z=Br, R1=H, R2=H, and X=2-Cl and 16 times less active than itraconazole [160]. Compounds having substituted-10-methyl-1,2,3,4-tetrahydro-pyrazino[1,2-a]indoles structure (53) with R=4-ClC6H4 was the most potent in the series having MIC values of 31.25, 15.62, and 31.25 μg/mL against A. niger, A. fumigatus, and A. flavus, respectively [161].
9 Quinolines
In a quinoline series (Fig. 8) compounds 54 having nitro substitutions at 5 and 7 positions of the quinoline ring and hydroxyl group at the 8 position had less antifungal activity (MIC80 = 1.95 μmol/L) compared to fluconazole (MIC80 = 0.06 μmol/L) against C. albicans. Two other compounds 55 and 56 had similar activity (MIC80 = 1.95 μmol/L) in C. albicans, the former had the quinoline ring substituted at position 8 by OH group and at position 2 by N-phenylethanimine moiety having 4′-OH substituent at the phenyl ring and the latter had same substitution at the quinoline ring (8-OH group) but a saturated linker with a methoxy group attached to the carbon next to the amine group with a phenyl ring having 2,5 diCl and 4-NO2 substitution [162]. In another series of quinoline derivatives compounds having substitution at the 2-position by γ-pyridyl ring and at the C4 (R1) and/or C8 (R2) by methyl or isopropyl groups were found to be active. Substitutions at the same position by α-Furyl or α-thienyl group yielded inactive compounds (57). However some compounds having the γ-pyridyl ring were devoid of antifungal activity that indicated the importance of substituents at the C4/C8 position to be important for antifungal activity. The most active compounds in the series had C4=methyl and C8=methyl, isopropyl with an MIC value of 12.5 μg/mL [163]. The derivatives of norfloxacin (1-Ethyl-6-fluoro-1,4-dihydro-4-oxo-7(1-piperazinyl) quinoline-3-carboxylic acid) having R=3-(2,4-dichlorophenyl)propyl-2-en-1-one and 2-(2-methoxyphenoxy)ethyl-1-one (58) were found to inhibit the growth of R. solani by 83% and 94% at a concentration of 200 mg/L that is comparable to carbendazim (59) (100% inhibition under similar bioassay conditions) [164]. A series of 5-methyl benzothieno[3,2-b]quinolinium compounds were synthesized where two compounds having R=3-OMe, 4-Cl and for both Y=OTf (60) [C. neoformans (IC50 = 6 μg/mL), C. albicans (IC50 = 1.5 μg/mL), A. fumigatus (IC50 = 0.4 μg/mL)] and R=4-Cl, Y=OTf [C. neoformans (IC50 = 4 μg/mL), C. albicans (not determined), A. fumigatus (IC50 = 6 μg/mL)] were observed to be active [165]. The seco analog (61) of the benzothienoquinoline (60) resulted in N-methyl-3-phenylthio-quinolinium salt. In this series the most active compound having R=H and R1=5-cyclohexylpentyl group was found to be active in C. neoformans (IC50 = 0.5 μg/mL), C. albicans (IC50 = 2.7 μg/mL), A. fumigatus (IC50 = 8.6 μg/mL), C. krusei (IC50 = 0.7 μg/mL) [165]. In the isoquinoline analog hexahydro-4H-pyrazino[2,1-a]isoquinolin-4-one series the most potent compound had better activity than fluconazole (2–64 μg/mL) with R1=H, R2=F, R3=(CH2)8CH3 in 62 and an MIC range of 4–16 μg/mL against different fungal species such as T. rubrum, C. neoformans, M. gypseum, and A. fumigatus [166].

Structure of quinoline, quinazoline, quinazolinone, and isoquinoline antifungals
10 Quinazolines
In the quinazoline class (Fig. 8) the most potent compound (63) had R=m-ClC6H4 and Ar=p-CH3C6H4 group having MIC values of 13.70, 17.07, 16.62 μg/mL against A. nigers, C. albicans, and F. oxysporum, respectively. At the same bioassay condition clotrimazole had slightly better activity (A. nigers = 12.98 μg/mL, C. albicans = 6.21 μg/mL, and F. oxysporum = 10.78 μg/mL) than the most potent compound in the series [167]. In the quinazoline class of compounds, the compound 64 (Ar=p-FC6H4) and 65 (Ar=p-FC6H4, Ar′=p-ClC6H4) showed less antifungal activity than Ticonazole (trosyd) [168].
11 Napthalenes
In a series of naphthalene derivatives (Fig. 9) compounds having R=7 or 8-NO2 group at the naphthalene ring of 66 with X=S, Se showed better or comparable activity (MIC = 0.53–25 μg/mL) than fluconazole (MIC = 25 μg/mL) on S. cerevisiae. Better antifungal activity was also observed in S. cerevisiae (MIC = 3.12 μg/mL) when the NO2 group was replaced by R=7-SO2NH2 and X=S, Se. One of the analogs of 66 having X=S and 7-SO2NH2 substitution was also active (MIC = 0.53 μg/mL) towards C. neoformans like fluconazole (MIC = 0.53 μg/mL) [169]. The butenafine derivative (67) with R=CH3 (MIC = 0.125 μg/mL) had comparable activity to butenafine (MIC = 0.125 μg/mL) in C. neoformans. The terbinafine derivative (68) with R=CH3 retained antifungal activity (MIC = 0.5 μg/mL) towards C. neoformans comparable to Butenafine (MIC = 0.25 μg/mL); however R=CH2F, CHF2, CF3, and CN resulted in less active compounds [170].

Structure of naphthalene and thiazole antifungals
12 Thiazoles
In a series of thiazole derivatives (Fig. 9) compound having the structure 69 had an MIC of 8 μg/mL in C. tropicalis and in A. niger. The compound was also active in S. cerevisiae with MIC of 16 μg/mL. Another compound with structure (70) was also active in C. tropicalis. Compounds having the general structure of 71 with R=4-OH–C6H4 was active (MIC = 16 μg/mL) in C. tropicalis while R=2,3-di-ClC6H5 was active in S. cerevisiae with MIC of 16 μg/mL. Compounds with general structure 72 having R=C6H5, 3,4,5 -(OCH3)3–C6H2, 4-OH–C6H4, 2,3-diCl–C6H3 showed good activity with MIC ranging from 16 to 31.25 μg/mL in S. cerevisiae, C. tropicalis, and A. niger [171]. A series incorporating thiazole, thiazolidinone, and adamantine structures were synthesized where all the compounds were more potent than ketoconazole and bifonazole (73) under same biological assay condition. The various substituents at R=2-Cl, 3-Cl, 4-Cl, 2-NO2, 3-NO2, 4-NO2, 4-OH, (4-OH and 3-OCH3), (4-OH and 3,5-OCH3) and 4-OCH3 of 73 were having MIC in the range of 0.52–2.38 μg/mL in different fungal species (P. funiculosum, P. ochrochloron, T. viride, A. fumigatus, A. niger, A. flavus, A. versicolor, F. fulvum) [172, 173]. A series of [4-(4′-substituted-phenyl)thiazol-2-yl]hydrazine derivatives (74) showed better activity in C. glabrata and C. albicans with MIC values within 0.125–16 μg/mL. Under same assay conditions clotrimazole was found to have MIC values in the range of 2–8 μg/mL in both C. glabrata and C. albicans while fluconazole antifungal activity (MIC) varied from 4 to 16 μg/mL in C. glabrata and 4–64 μg/mL in C. albicans. The most active compounds for C. albicans (MIC = 0.125 μg/mL) had Het=Thiophen-2-yl, Pyridin-3-yl, Pyridin-4-yl, Benzodioxol-5-yl, Indol-3-yl, Coumarin-3-yl, R=H, CH3 and R1=CH3, OCH3 [174].
13 Echinocandins
Echinocandins (75) (Fig. 10) are the most recent antifungals available for use. Echinocandins are water soluble, large hetrodimeric amphipathic polypeptides. This antifungal drug inhibit 1,3- β-d-glucansynthetase, resulting in damage of the cell wall of fungi, cell lysis, and cell death and are also called as “penicillin of antifungals” [175, 176]. Echinocandins are poorly absorbed through oral route; therefore they are administered intravenously to cure the localized and systemic fungal infections. It has a broad range of activity against all Candida species, also used in empirically in febrile neutropenia and stem cell transplant. At present medically used echinocandins like caspofungin, micafungin, and anidulafungin are semisynthetic derivatives with clinical use due to their solubility, antifungal spectrum, and pharmacokinetic properties [177].

Structure of echinocandin antifungals
13.1 Anidulafungin
Anidulafungin (76) is a semisynthetic lipopeptide antifungal approved by Food and Drug Administration. It was buildup by Eli Lily under clinical development at Vicuron Pharmaceuticals. It is the fermented product of the mold Aspergillus nidulans. Anidulafungin is used for the treatment of the persons who have high risk for serious fungal infections include patients with organ transplantation or hematopoietic stem cell transplantation, HIV infection/AIDS, malignancies, high-dose steroid therapy, and invasive Aspergillus infections [178]. It inhibits β-1,3-d-glucan synthase as glucan is a major structural component of the cell wall of pathogenic fungi, resulting in cell death.
13.2 Caspofungin
Caspofungin (77) is a semi-synthetic water soluble lipopeptide antifungal drug which belongs to member of echinocandins. Caspofunginis is a fermented product of the fungus Glareal-ozoyensis. Caspofungin is administered intravenously and it inhibits the synthesis of component beta-(1,3)-d-glucan of fungal cell wall [179]. It is used for the treatment of fungal infections such as Candida infection (intra-abdominal abscesses, pleural cavity, perotonotis infections and esophagitis) and invasive aspergillosis [180].
13.3 Micafungin
Micafungin (78) is an echinocandin antifungal agent which was approved by FDA in March 2008. Micafungin is administered through intravenous route. Beta-(1,3)-d-glucanan is an essential component of fungal cell wall and the production of which is inhibited by micafungin. This drug is used in the treatment of infections caused by Candida sp. [181].
14 Miscellaneous
Diverse structural classes of compounds have been evaluated for antifungal activity. A series of benzoxazole derivatives (79) (Fig. 11) with fluorine substitution at different position of the phenyl ring were synthesized. All these compounds were synthesized as isosteric analogues of benzoheterocyclic-N-myristoyltransferase inhibitors. The most potent compound against C. tropicalis (MIC80 = 0.0625 μg/mL) had R=2-F substitution on the phenyl ring with better antifungal activity than fluconazole (C. tropicalis: MIC80 = 4 μg/mL) while another compound having R=2,3,4-trifluoro substitution in the phenyl ring had equipotent activity (MIC80 = 0.25 μg/mL) in C. albicans, C. parasilosis, and C. tropicalis. The compound (R=2,3,4-trifluoro substituted phenyl ring) had equivalent activity like fluconazole (MIC80 = 0.25 μg/mL) against C. albicans and better activity than fluconazole in C. parasilosis (MIC80 = 4 μg/mL) and C. tropicalis (MIC80 = 4 μg/mL) [182]. In a series of 2-Acylhydrazino-5-arylpyrroles (80) the most active compound with X=CN, Ar=4-OMePh and R=Et as substituent had an MIC of 0.39 μg/mL in C. albicans that is equipotent to amphotericin B (MIC = 0.39 μg/mL) and better than fluconazole (MIC = 0.78 μg/mL) under similar bioassay condition. The compound also showed good activity in other fungal species [C. glabrata (MIC = 0.78 μg/mL), C. parapsilosis (MIC = 0.78 μg/mL), C. krusei (MIC = 0.78 μg/mL)]. Substitution with R=iPr, 4-OMeBz when X is −COOC2H5 decreases activity drastically (MIC >100 μg/mL); however with X=CN fungal activity for R=iPr improved to great extent (MIC = 3.12 μg/mL) as observed against C. albicans. Hence the CN group is vital for antifungal activity [183]. A series of antifungal compounds having spiro[cyclopropane-1,4′-pyrazol-3-one] as the basic structural moiety (81) with R1=H, CH3 and R2=CO2Me, CO2Et, CO2iPr, CO2tBt, CN, CONEt2 had weak antifungal activity (MIC = 25 μg/mL) in C. albicans as compared to miconazole and itraconazole (MIC = 2 μg/mL) [184]. In a series of N-alkyl substituted urea derivatives two compounds having R1=F, R2=H and R1=H, R2=F had MIC values of 3.1 and 3.5 μg/mL against T. rubrumas compared to ketoconazole (MIC = 3.9 μg/mL) on the same species (82). None of the compounds in the series are better than ketoconazole for A. niger (MIC = 7.8 μg/mL) except an analogue having the structure 83 had an MIC = 12.5 μg/mL [185]. A series of 5-Arylamino- and 6-arylthio-4,7-dioxobenzoselenazoles were synthesized where the best compound with R1=R3=Cl, R2=H (84) had an MIC = 1.6 μg/mL better than 5-Fluorocytosine (MIC = 12.5 μg/mL) in C. albicans. The compound had MIC value of 3.2 μg/mL in C. tropicalis (5-Fluorocytosine : MIC = 12.5 μg/mL). The other active compounds in the series (85) had MIC values of 3.2 μg/mL (R1=R3=H, R2=NO2), 6.3 μg/mL (R1=R2=R3=H), 6.3 μg/mL (R1=R3=H, R2=F), 6.3 μg/mL (R1=R3=H, R2=CH3), 6.3 μg/mL (R1=R2=R3=H) against C. albicans. The activity of other compounds in the series varied from 6.3 to 50 μg/mL in C. tropicalis, C. krusei, A. niger, and A. flavus [186]. A series of benzofuran compounds (86–90) (Figs. 11 and 12) with different substitutions R1=H, CH3, C2H5, CN and R2=H, CH3, Cl on the phenyl ring have good antifungal activity in C. albicans, C. tropicalis, C. Krusei, A. niger, A. flavus, and C. neoformans (MIC = 1.6–50 μg/mL). The two best compounds against C. albicans with R1=CH3, R2=H (89) (Fig. 12) and R1=C2H5, R2=H (89) were equipotent (MIC = 1.6 μg/mL) and was better than 5-Fluorocytosine (MIC = 6.3 μg/mL) and fluconazole (MIC = 50 μg/mL). These two compounds were also active in C. tropicalis and A. niger with both having MIC of 3.2 μg/mL in the two fungal species [187]. In the benzotriazine series the most active compound having R1=H, R2=H (91) was more potent than hymexazol [188]. In the chalcones (92) compounds with R1=H, 4-Br and R2=H, 4-NO2, 2-NO2 had good antifungal activity with the potent compounds having electron withdrawing substituents at the para position of the phenyl ring [189].

Structure of different miscellaneous antifungals

Structure of different chemical class of antifungals
In the pyrimidinone series three compounds having substitutions as R1=C6H5, 4-Me2NC6H5, 4-Me2NC6H5 and R2=C2H5 and X=S, S, O (93) had MIC = 0.35 μg/mL against A. niger. Another compound having R1=2-HOC6H4, R2=C2H5, X=O prevents the radical growth of T. koningii after 24 and 48 h completely (100%) [190]. The isoxazolidine derivatives (94) having R1=OCH3, F and R2=C6H5, COOC2H5, CH2OH had MIC values ranging from 2.5 to 3 mM in A. flavus that is comparable to nystatin (3 mM) [191]. In the carbazole series introduction of azole (imidazole or 1,2,4-triazole) ring increased activity with better antifungal activity (2–4 μg/mL) for R=C4H8, C2H4 in (95) [192]. In the 3,5-disubstituted furanones series compounds with structure (96) and (97) had equal MIC values of 0.49 μmol/L in C. albicans. Other compounds in the series having Z=4-OCH3, 4-I, 3-Br and 4-COOCH3 had MIC values of 0.97, 0.48, 0.97, and 0.48 μmol/L in C. albicans. Modification in the phenyl ring with Z=3-COOH, 4-COOH, 4-OH (98) also resulted in active compounds against C. albicans with MIC values of 0.48, 0.97, and 0.97 μmol/L, respectively. Amphotericin B and fluconazole had MIC values of 0.03 and 1 μmol/L in the same assay system for C. albicans [193]. In the 2-amino tetraline series compounds with R=(CH2)9CH3 had better antifungal activity with the two potent compounds having 5-OH and 5-OCH3 substitutions in the phenyl ring having equal MIC values of 0.3125 μmol/L against C. albicans (99). Another compound having R=(CH2)8CH3 and 6-OCH3 was active (MIC = 0.0625 μmol/L) in C. albicans strain resistant to fluconazole (MIC >64 μmol/L) (100) [194].
15 Cationic Peptides
The cationic peptides are small cationic and amphipathic molecules isolated from plants, mammals, and microorganisms with antifungal activity with great potential for development as new therapeutic agents [195]. Cecropins isolated from the hemolymph of the giant silk moth (Hyalophora cecropia) is constituted by 35–37 residues with a strongly basic N-terminal linked to a neutral C-terminal by a flexible glycine–proline link. Both Hyalophora and Drosophila Cecropin (Cecropin A and B) inhibited growth of S. cerevisiae, D. uninucleata, G. candidum, and M. anisopliae in MICs ranging from 0.4 to 4 mM [196]. The LD50 value of Cecropin was also evaluated on germinating and non-germinating A. flavus, A. fumigatus, A. niger, F. moniliforme, and F. oxysporum. Cecropin B had LD50 values of 3.0, 0.5, 2, 0.2, and 1 μM in A. flavus, A. fumigatus, A. niger, F. moniliforme, and F. oxysporum respectively while for non-germinating F. Moniliforme and F. oxysporum the LD50 value was 0.2 μM for both species. Dermaseptin peptides found in skin secretions of Phyllomedusinae frogs reported in the same study had LD50 values of 4, 0.05, 2, 0.3, and 0.8 μM in A. flavus, A. fumigatus, A. niger, F. moniliforme, and F. oxysporum [197]. Indolicin, the shortest linearly occurring peptide consisting of 39% tryptophan and 23% proline (ILPWKWPWWPWRR), is found in the cytoplasmic granules of bovine neutrophil. Indolicin disrupt the structure of cell membranes as examined on interaction with T. beigelii [198]. Histatins are histidine rich peptides isolated from human saliva and had strong antifungal activity in different Candida spp. (C. albicans, C. glabrata, C. guillermondii, C. krusei, C. lambica, C. parapsilosis, C. pseudo -tropicalis, C. stellatoidea, and C. tropicalis) with histatin 5 showing the strongest fungicidal activity against C. albicans (MIC = 100 μM) [199]. Magainins from Xenopuslaevis (the African frog) had antifungal activity against Candida spp., C. neoformans, and Saccharomyces cerevisiae. Magainin 2 acts as an antifungal against C. neoformans (MIC = 6.25 μg/mL), C. glabrata (MIC = 25.0 μg/ml), C. tropicalis (MIC, 12.5 μg/mL), and C. krusei (MIC = 12.5–25.0 μg/mL) with relatively low activity against C. albicans (MIC > 80 μg/mL) [200, 201]. Bombinin-H isolated from skin of Bombina genus are glycine rich peptides active against fungi, especially bombinin-like peptides-1 in C. albicans (MIC = 3–0.4 μM). Bombinins H2 and H4 also have antifungal activity against C. albicans, C. guillermondii, and C. tropicalis. Bombinin H2 had MIC values of 3.1, 1.3, 1.1 μM in C. albicans, C. guillermondii, and C. tropicalis respectively while Bombinin H4 had MIC values of 1.6, 0.7, and 0.6 μM for the above species [202, 203]. The antifungal activities of the amphibian cationic peptides have been reported elsewhere [204]. The cationic peptides bind to cholesterol and ergosterolin fungal cell membranes leading to fungal lysis [205]. Dolastatin 10, a synthetic cationic peptide, targeted at intracellular tubulin and inhibits microtubule assembly and tubulin-dependent GTP binding and have effective fungicidal activity against C. neoformans [206].
16 Monoclonal Antibodies
Since the fungi are eukaryotic organisms, a character shared with the host, it is difficult to develop a safe drug like antibacterials which are directed against prokaryotic organisms. In view of this an approach directed towards monoclonal antibodies against at least most common fungal pathogens like Candida albicans, Aspergillus fumigatus, and Cryptococcus neoformans is desirable. Identification and characterization of the proteins that are immunologically dominant and exhibit strong immune responses during mycoses could have vital repercussions for evolving new diagnostic, prophylactic, and therapeutic techniques for mycoses. Therefore efforts focused on the discovery of useful inhibitors of fungal specific, chitin, cell wall glucan and mannoprotein biosynthesis may play a very important role. In the absence of a safe and wide spectrum antimycotic agent, efforts may be directed for the development of monoclonal antibodies (MAbs). The MAbs have improved the specificity of immune procedures and have served as useful research methods and tools such as isolation, purification, and characterization of microbial antigens and development of assays methods for antibody and antigen detection [207,208,209]. In market there are many monoclonal antibodies available against a number of challenging diseases like cancer and many more diseases (Table 1). The antibodies are either developed in mouse which may be humanized, chimeric or in humans. The MAbs often exhibit adverse reactions like HAMA which is common for MAbs developed in mouse.
To overcome these types of side effects, an approach leading to the identification of active peptide sequences from the hypervariable regions of the hybridoma clone may be helpful. This way a library of peptide sequences may be synthesized and evaluated for antifungal activity which may have specific activity against fungi. The peptide sequences thus generated may not only have specific antifungal activity but may also result in specific diagnostic tolls.
17 Conclusions
Fungal diseases are global health problem with rising prevalence of infections in immunocompromised hosts related to cases of cancer, AIDS, diabetes, cystic fibrosis and in invasive surgical procedures. The three major fungal diseases in immunocompromised subjects are candidosis, aspergillosis, and cryptococcosis. Azoles, the most common clinically antifungals among the other candidates (polyenes, pyrimidines, allylamines, and echinocandins), suffer from developing resistance with drug–drug interactions and drug toxicity. This chapter presented the most common antifungals used for human health and also a brief update about the latest developments in antifungal agents.
CDRI Communication No:9207
References
Hawksworth DL (2004) Fungal diversity and its implications for genetic resource collections. Stud Mycol 50:9–18
Carris LM, Little CR, Stiles CM (2012) Introduction to fungi. Plant Health Instructor. doi:10.1094/PHI-I-2012-0426-01
Martin DS, Jones CP (1940) Further studies on the practical classification of the Monilias. J Bacteriol 39(5):609–630
Sobel JD, Vazquez J (1990) Candidemia and systemic candidiasis. Semin Respir Infect 5:123–137
Rippon JW (1982) Medical mycology: the pathogenic fungi and the pathogenic actinomycetes. Saunders, Philadelphia
Stein DK, Sugar AM (1989) Fungal infections in the immunocompromised host. Diagn Microbiol Infect Dis 12:221S–228S
Larriba G, Rubio Coque JJ, Ciudad A, Andaluz E (2000) Candida albicans molecular biology reaches its maturity. Int Microbiol 3:247–252
Carrillo-Munoz AJ, Giusiano G, Ezkurra PA, Quindos G (2006) Antifungal agents: mode of action in yeast cells. Rev Esp Quimioter 19:130–139
Andriole VT (1999) Current and future antifungal therapy: new targets for antifungal agents. J Antimicrob Chemother 44:151–162
Ahmad S, Khan Z, Mustafa AS, Khan ZU (2002) Seminested PCR for diagnosis of candidemia: comparison with culture, antigen detection, and biochemical methods for species identification. J Clin Microbiol 40:2483–2489
Fujita S, Lasker BA, Lott TJ, Reiss E, Morrison CJ (1995) Microtitration plate enzyme immunoassay to detect PCR amplified DNA from Candida species in blood. J Clin Microbiol 33:962–967
Iwastu TM, Miyaji M, Taguchi H, Okamoto S (1982) Evaluation of skin test for chromoblastomycosis using antigen prepared from cultural filtrates of Fonsecaea pedrosoi, Phlalophora verrucosa, Wangiella dermatitidis and Exophiala jeanselmei. Mycopathologia 77:59–64
Wu Z, Tsumura Y, Blomquist G, Wang X (2003) 18S rRNA gene variation among common airborne fungi, and development of specific oligonucleotide probes for the detection of fungal isolate. Appl Environ Microbiol 69:5389–5397
Ferrer C, Colom F, Frases S, Mulet E, Abad JL, Alio JL (2001) Detection and identification of fungal pathogens by PCR and by ITS2 and 5.8S ribosomal DNA typing in ocular infections. J Clin Microbiol 39:2873–2879
Ferrer C, Munoz G, Alio JL, Colom F (2002) Polymerase chain reaction diagnosis in fungal keratitis caused by Alternaria alternata. Am J Ophthalmol 133:398–399
Holmberd K, Feroze F (1996) Evaluation of an optimized system for random amplified polymorphic DNA (RAPD)-analysis for genotypic mapping of Candida albicans strains. J Clin Lab Anal 10:59–69
Hui M, Ip M, Chan PK, Chin ML, Cheng AF (2000) Rapid identification of medically important Candida to species level by polymerase chain reaction and single-strand conformational polymorphism. Diagn Microbiol Infect Dis 38:95–99
Humphreis SE, Gudnason V, Whittall R, Day INM (1997) Single stranded conformation polymorphism analysis with high throughput modifications and its use in mutation detection in familial hypercholesterolemia. Clin Chem 43:427–435
Iwen PC, Hinrichs SH, Rupp ME (2002) Utilization of the internal transcribed spacer region as molecular targets to detect and identify human fungal pathogens. Med Mycol 40:87–109
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic, New York, pp 315–322
Gillman LM, Gunton J, Turenne CY, Wolfe J, Kabani AM (2001) Identification of Mycobacterium species by multiple-fluorescence PCR-single-strand conformation polymorphism analysis of the 16S rRNA gene. J Clin Microbiol 39:3085–3091
Kumeda Y, Asao T (1996) Single-strand conformation polymorphism analysis of PCR-amplified ribosomal DNA internal transcribed spacers to differentiate species of Aspergillus section Flavi. Appl Environ Microbiol 62:2947–2952
Mora D, Ricci G, Gugliemetti S, Daffonchio D, Fortina MG (2003) 16S-23S rRNA intergenic spacer region sequence variation in Streptococcus thermophilus and related dairy streptococci and development of a multiplex ITS-SSSP analysis for their identification. Microbiology 149:807–813
Rath PM, Ansorg R (2000) Identification of medically important Aspergillus species by single stranded conformational polymorphism (SSCP) of the PCR-amplified intergenic spacer region. Mycoses 43:381–386
Pfaller MA, Messer SA, Boyken L, Tendolkar S, Hollis RJ, Diekema DJ (2004) Geographic variation in the susceptibilities of invasive isolates of Candida glabrata to seven systemically active antifungal agents: a global assessment from the ARTEMIS Antifungal Surveillance Program conducted in 2001 and 2002. J Clin Microbiol 42:3142–3146
Hazen EL (1960) Nystatin. Ann N Y Acad Sci 89:258–266
Mayers DL (2009) Antimicrobial drug resistance: mechanism of drug resistance vol. 1. Humana Press/Springer, Totowa/New York, p 299
Hazen EL, Brown R (1950) Two antifungal agents produced by a soil actinomycete. Science 112:423
Hazen EL, Brown R (1951) Fungicidin, an antibiotic produced by a soil actinomycete. Proc Soc Exp Biol Med 76:93
Harris EJ, Pritzker HG, Laski B, Eisen A, Steiner JW, Shack L (1958) The effect of nystatin (mycostatin) on neonatal candidiasis (thrush)- a method of eradicating thrush from hospital nurseries. Can Med Assoc J 79(11):891–896
Sklenář Z, Ščigel V, Horáčkova K, Slanař O (2013) Compounded preparations with nystatin for oral and oromucosal administration. Acta Pol Pharm Drug Res 70:759–762
Lencelin JM et al (1988) Tetrahedron Lett 29:2827
Pandey RC, Rinehart KL (1976) J Antibiot 29:1035
Groll AH, Gonzalez CE, Giri N et al (1999) Liposomal nystatin against experimental pulmonary aspergillosis in persistently neutropenic rabbits: efficacy, safety and non-compartmental pharmacokinetics. J Antimicrob Chemother 44(3):397–401
Wallace TL, Paetznick V, Cossum PA, Lopez-Berestein G, Rex JH, Anaissie E (1997) Activity of liposomal nystatin against disseminated Aspergillus fumigatus infection in neutropenic mice. Antimicrob Agents Chemother 41(10):2238–2243
Farid MA, El-Enshasy HA, El-Diwany AI, El-Sayed ESA (2000) Optimization of the cultivation medium for natamycin production by Streptomyces natalensis. J Basic Microbiol 40(3):157–166
Lalitha P, Kumar VR, Prajna NV, Fothergill AW (2008) In vitro natamycin susceptibility of ocular isolates of Fusarium and Aspergillus species: comparison of commercially formulated natamycin eye drops to pharmaceutical-grade powder. J Clin Microbiol 46(10):3477–3478
Vandeputte P, Ferrari S, Coste AT (2012) Antifungal resistance and new strategies to control fungal infections. Int J Microbiol 2012:1–27. doi:10.1155/2012/713687
Caffrey P, Lynch S, Flood E, Finnan S, Oliynyk M (2001) Amphotericin biosynthesis in Streptomyces nodosus: deductions from analysis of polyketide synthase and late genes. Chem Biol 8(7):713–723
Matsumori N, Sawada Y, Murata M (2005) Mycosamine orientation of amphotericin B controlling interaction with ergosterol: sterol-dependent activity of conformation-restricted derivatives with an amino-carbonyl bridge. J Am Chem Soc 127:10667–10675
Barratt G, Bretagne S (2007) Optimizing efficacy of amphotericin B through nanomodification. Int J Nanomedicine 2:301–313
Ogita A, Fujita KI, Tanaka T (2012) Enhancing effects on vacuole-targeting fungicidal activity of amphotericin B. Front Microbiol 3:100
Gallis H, Drew RH, Pickard WW (1990) Amphotericin B: 30 years of clinical experience. Rev Infect Dis 12(2):308–329
Laniado-Laborín R, Cabrales-Vargas MN (2009) Amphotericin B: side effects and toxicity. Rev Iberoam Micol 26(4):223–227
Czub J, Baginski M (2006) Modulation of amphotericin B membrane interaction by cholesterol and ergosterol--a molecular dynamics study. J Phys Chem B 110(33):16743–16753
Palacios DS, Dailey I, Siebert DM, Wilcock BC, Burke MD (2011) Synthesis-enabled functional group deletions reveal key underpinnings of amphotericin B ion channel and antifungal activities. Proc Natl Acad Sci U S A 108(17):6733–6738
Gray KC, Palacios DS, Dailey I et al (2012) Amphotericin primarily kills yeast by simply binding ergosterol. Proc Natl Acad Sci U S A 109(7):2234–2239
Wilcock BC, Endo MM, Uno BE, Burke MD (2013) C2-OH of amphotericin B plays an important role in binding the primary sterol of human cells but not yeast cells. J Am Chem Soc 135(23):8488–8491
Anderson TM, Clay MC, Cioffi AG et al (2014) Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat Chem Biol 10(5):400–406
Messer SA, Jones RN, Fritsche TR (2006) International surveillance of Candida spp. and Aspergillus spp.: report from the SENTRY Antimicrobial Surveillance Program (2003). J Clin Microbiol 44:1782–1787
Sokol-Anderson ML, Brajtburg J, Medoff G (1986) Amphotericin B-induced oxidative damage and killing of Candida albicans. J Infect Dis 154:76–83
Maertens JA (2004) History of the development of azole derivatives. Clin Microbiol Infect 10(Suppl 1):1–10
Odds FC, Brown AJ, Gow NA (2003) Antifungal agents: mechanisms of action. Trends Microbiol 11:272–279
Fromtling RA (1988) Overview of medically important antifungal azole derivatives. Clin Microbiol Rev 1:187–217
Sheehan DJ, Hitchcock CA, Sibley CM (1999) Current and emerging azole antifungal agents. Clin Microbiol Rev 12:40–79
Elkasabgy NA (2014) Ocular supersaturated self-nanoemulsifying drug delivery systems (S-SNEDDS) to enhance econazole nitrate bioavailability. Int J Pharm 460:33–44
Thienpont D, Van Cutsem J, Van Nueten JM, Niemegeers CJ, Marsboom R (1975) Bilogical and toxicological properties of econazole, a broad-spectrum antimycotic. Arzneimittelforschung 25:224–230
Heel RC, Brogden RN, Speight TM, Avery GS (1978) Econazole: a review of its antifungal activity and therapeutic efficacy. Drugs 16(3):177–201
Waitz JA, Moss EL, Weinstein MJ (1971) Chemotherapeutic evaluation of clotrimazole (Bay b 5097, 1 (o-chloro- - -diphenylbenzyl) imidazole). Appl Microbiol 22:891–898
World Health Organization (2013) WHO model list of essential medicines. World Health Organization. October 2013. Edition 18. http://www.who.int/medicines/publications/essentialmedicines/en/index.html. Retrieved 22 Apr 2014
Haller I (1985) Mode of action of clotrimazole: implications for therapy. Am J Obstet Gynecol 152(7 Pt 2):939–944
Rai VK, Dwivedi H, Yadav NP, Chanotiya CS, Saraf SA (2014) Solubility enhancement of miconazole nitrate: binary and ternary mixture approach. Drug Dev Ind Pharm 40:363–9045
Morita T, Nozawa Y (1985) Effects of antifungal agents on ergosterol biosynthesis in Candida albicans and Trichophyton mentagrophytes: differential inhibitory sites of naphthiomate and miconazole. J Invest Dermatol 85:434–437
Puolakka J, Tuimala R (1983) Comparison between oral ketoconazole and topical miconazole in the treatment of vaginal candidiasis. Acta Obstet Gynecol Scand 62:575–577
Rollman O (1982) Treatment of onychomycosis by partial nail avulsion and topical miconazole. Dermatologica 165:54–61
Brugmans JB, Van Cutsem JM, Thienpont DC (1970) Treatment of long-term tinea pedis with miconazole. Arch Dermatol 102:428–432
Van Cutsem J, Reyntjens A (1978) Miconazole treatment of pityriasis versicolor a review. Mykosen 21(3):87–91
Sung JP, Grendahl JG, Levine HB (1977) Intravenous and intrathecal miconazole therapy for systemic mycoses. West J Med 126:5–13
Balata G, Mahdi M, Bakera RA (2010) Improvement of solubility and dissolution properties of ketoconazole by solid dispersions and inclusion complexes. Asian J Pharm Sci 5:1–12
Rotstein DM, Kertesz DJ, Walker KAM et al (1992) J Med Chem 35:2818
Hume AL, Kerkering TM (1983) Ketoconazole. Drug Intell Clin Pharm 17:169–174
Terrell CL (1999) Antifungal agents. Part II. The azoles. Mayo Clin Proc 74:78–100
Gary G (2013) Optimizing treatment approaches in seborrheic dermatitis. J Clin Aesthet Dermatol 6:44–49
Venkateswarlu K, Kelly SL (1996) Biochemical characterisation of ketoconazole inhibitory action on Aspergillus fumigatus. FEMS Immunol Med Microbiol 16:11–20
Wood A (1994) Oral azole drugs as systemic antifungal therapy. N Engl J Med 330:263–272
Perfect JR, Durack DT (1985) Penetration of imidazoles and triazoles into cerebrospinal fluid of rabbits. J Antimicrob Chemother 16:81–86
Van Tyle JH (1984) Ketoconazole. Mechanism of action, spectrum of activity, pharmacokinetics, drug interactions, adverse reactions and therapeutic use. Pharmacotherapy 4:343–373
Akins RA (2005) An update on antifungal targets and mechanisms of resistance in Candida albicans. Med Mycol 43:285–318
Sanglard D, Odds FC (2002) Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2:73–85
Albertson GD, Niimi M, Cannon RD, Jenkinson HF (1996) Multiple efflux mechanisms are involved in Candida albicans fluconazole resistance. Antimicrob Agents Chemother 40:2835–2841
Orozco AS, Higginbotham LM, Hitchcock CA, Parkinson T, Falconer D, Ibrahim AS, Ghannoum MA, Filler SG (1998) Mechanism of fluconazole resistance in Candida krusei. Antimicrob Agents Chemother 42:2645–2649
Kelly SL, Lamb DC, Kelly DE, Manning NJ, Loeffler J, Hebart H, Schumacher U, Einsele H (1997) Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol delta 5,6-desaturation. FEBS Lett 400:80–82
Bossche HV, Marichal P, Odds FC (1994) Molecular mechanisms of drug resistance in fungi. Trends Microbiol 2:393–400
Romani L (2004) Immunity to fungal infections. Nat Rev Immunol 4:1–23
Stiller RL, Bennett JE, Scholer HJ, Wall M, Polak A, Stevens DA (1982) Susceptibility to 5-fluorocytosine and prevalence of serotype in 402 Candida albicans isolates from the United States. Antimicrob Agents Chemother 22:482–487
Zervos M, Meunier F (1993) Fluconazole (diflucan): a review. Int J Antimicrob Agents 3:147–170
Philpott-Howard JN, Wade JJ, Mufti GJ, Brammer KW, Ehninger G (1993) Randomized comparison of oral fluconazole versus oral polyenes for the prevention of fungal infection in patients at risk of neutropenia. Multicentre Study Group. J Antimicrob Chemother 31:973–984
Martin MV (1999) The use of fluconazole and itraconazole in the treatment of Candida albicans infections: a review. J Antimicrob Chemother 44:429–437
Willems L, Geest VD, De Beule K (2001) Itraconazole oral solution and intravenous formulations: a review of pharmacokinetics and pharmacodynamics. J Clin Pharm Ther 26:159–169
Jaruratanasirikul S, Kleepkaew A (1997) Influence of an acidic beverage (Coca-Cola) on the absorption of itraconazole. Eur J Clin Pharmacol 66:235–237
Odds FC, Oris M, Dorsselaer PV, Gerven FV (2000) Activities of an intravenous formulation of itraconazole in experimental disseminated Aspergillus, Candida, and Cryptococcus infections. Antimicrob Agents Chemother 44:3180–3183
Kauffman CA (1996) Role of azoles in antifungal therapy. Clin Infect Dis 22(2):S148–S153
Aftab BT, Dobromilskaya I, Liu JO, Rudin CM (2011) Itraconazole inhibits angiogenesis and tumor growth in non-small cell lung cancer. Cancer Res 71:6764–6772
Saravolatz LD, Johnson LB, Kauffman CA (2003) Voriconazole: a new triazole antifungal agent. Clin Infect Dis 36:630–637
Van Duin D, Cleare W, Zaragoza O, Nosanchuk JD, Casadevall A (2014) Effects of voriconazole on Cryptococcus neoformans. Antimicrob Agents Chemother 48:2014–2020
Rafael Z, Javier P (2008) Adv Sepsis 6:90
Ghannoum MA, Kuhn DM (2002) Eur J Med Res 7:242
Denning DW, Ribaud P, Milpied H, Raoul N, Eckhard T, Andrea H (2002) Clin Infect Dis 34:563
Pascual A, Calandra T, Bolay S et al (2008) Clin Infect Dis 46:201
Lewis RE (2008) Clin Infect Dis 46:212
Zonios DL, Gea-Banacloche J, Childs R (2008) Clin Infect Dis 47:e7–e10
Pasqualotto AC, Xavier MO, Andreolla HF, Linden R (2010) Voriconazole therapeutic drug monitoring: focus on safety. Expert Opin Drug Saf 9:125–137
Kauffman CA, Malani AN, Easley C, Kirkpatrick P (2007) Posaconazole. Nat Rev Drug Discov 6(3):183–184
Ullmann AJ, Lipton JH, Vesole DH (2007) N Engl J Med 356:335
Keating GM (2005) Drugs 65:1553
Torres HA, Hachem RY, Chemaly RF, Kantoyiannis DP, Raad I (2005) Lancet Infect Dis 5:775
Yamazumi T, Pfaller MA, Messer SA (2000) Antimicrob Agents Chemother 44:6
Mikamo H, Yin XH, Hayasaki Y et al (2002) Penetration of ravuconazole, a new triazole antifungal, into rat tissues. Chemotherapy 48:7–9
Pfaller MA, Messer SA, Hollis RJ (2002) Antimicrob Agents Chemother 46:1723
Pasqualotto AC, Denning DW (2008) New and emerging treatments for fungal infections. J Antimicrob Chemother 61(Suppl 1):19–30. doi:10.1093/jac/dkm428
Marino MR, Mummanei V, Norton J, et al (2001) Ravuconazole exposure-response relationship in HIV-patients with oropharyngeal candidiasis. In: Abstracts of the forty-first interscience conference on Antimicrobial Agents and Chemotherapy, Chicago. American Society for Microbiology, Washington, DC. Abstract J-1622
Giovanna Setzu M, Stefancich G, La Colla P, Castellano S (2002) Synthesis and antifungal properties of N-[(1,1′-biphenyl)-4-ylmethyl]-1H-imidazol-1-amine derivatives. Farmaco 57:1015–1018
Günay NS, Çapan G, Ulusoy N, Ergenç N, Ötük G, Kaya D (1999) 5-Nitroimidazole derivatives as possible antibacterial and antifungal agents. Farmaco 54:826–831
Olender D, Żwawiak J, Lukianchuk V, Lesyk R, Kropacz A, Fojutowski A, Zaprutko L (2009) Synthesis of some N-substituted nitroimidazole derivatives as potential antioxidant and antifungal agents. Eur J Med Chem 44:645–652
Rossello A, Bertini S, Lapucci A, Macchia M, Martinelli A, Rapposelli S, Herreros E, Macchia B (2002) Synthesis, antifungal activity, and molecular modeling studies of new inverted oxime ethers of oxiconazole. J Med Chem 45:4903–4912
Di Santo R, Tafi A, Costi R, Botta M, Artico M, Corelli F, Forte M, Caporuscio F, Angiolella L, Palamara AT (2005) Antifungal agents. 11. N-substituted derivatives of 1-[(aryl)(4-aryl-1H-pyrrol-3-yl)methyl]-1H-imidazole: synthesis, anti-Candida activity, and QSAR studies. J Med Chem 48:5140–5153
Lorus Therapeutic, Inc. (2011) 2,4,5-trisubstituted imidazoles and their use as anti-microbial agents. US7884120
Lorus Therapeutic, Inc. (2013) 2,4,5-trisubstituted imidazoles and their use as anti-microbial agents. US8394815
Pore VS, Aher NG, Kumar M, Shukla PK (2006) Design and synthesis of fluconazole/bile acid conjugate using click reaction. Tetrahedron 62:11178–11186
Zhao QJ, Song Y, Hu HG, Yu SC, Wu QY (2007) Design, synthesis and antifungal activity of novel triazole derivatives. Chin Chem Lett 18:670–672
Lebouvier N, Pagniez F, Duflos M, Le Pape P, Na YM, Le Baut G, Le Borgne M (2007) Synthesis and antifungal activities of new fluconazole analogues with azaheterocycle moiety. Bioorg Med Chem Lett 17:3686–3689
Uchida T, Somada A, Kagoshima Y, Konosu T, Oida S (2008) Carbon analogs of antifungal dioxane-triazole derivatives: synthesis and in vitro activities. Bioorg Med Chem Lett 18:6538–6541
Guillon R, Giraud F, Logé C, Le Borgne M, Picot C, Pagniez F, Le Pape P (2009) Design of new antifungal agents: synthesis and evaluation of 1-[(1H-indol-5-ylmethyl)amino]-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols. Bioorg Med Chem Lett 19:5833–5836
Dan ZG, Zhang J, Yu SC, Hu HG, Chai XY, Sun QY, Wu QY (2009) Design and synthesis of novel triazole antifungal derivatives based on the active site of fungal lanosterol 14a-demethylase (CYP51). Chin Chem Lett 20:935–938
Borate HB, Maujan SR, Sawargave SP, Chandavarkar MA, Vaiude SR, Joshi VA, Wakharkar RD, Iyer R, Kelkar RG, Chavan SP, Kunte SS (2010) Fluconazole analogues containing 2H-1,4-benzothiazin-3(4H)-one or 2H-1,4-benzoxazin-3(4H)-one moieties, a novel class of anti-Candida agents. Bioorg Med Chem Lett 20:722–725
He QQ, Liu CM, Li K, Cao YB (2007) Design, synthesis of novel antifungal triazole derivatives with high activities against Aspergillus fumigatus. Chin Chem Lett 18:421–423
He QQ, Li K, Cao YB, Dong HW, Zhao LH, Liu CM, Sheng CQ (2007) Design, synthesis and molecular docking studies of novel triazole antifungal compounds. Chin Chem Lett 18:663–666
Nam N-H, Sardari S, Selecky M, Parang K (2004) Carboxylic acid and phosphate ester derivatives of fluconazole: synthesis and antifungal activities. Bioorg Med Chem 12:6255–6269
Upadhayaya RS, Jain S, Sinha N, Kishore N, Chandra R, Arora SK (2004) Synthesis of novel substituted tetrazoles having antifungal activity. Eur J Med Chem 39:579–592
Wei JJ, Jin L, Wan K, Zhou CH (2011) Synthesis of novel D-glucose-derived benzyl and alkyl 1,2,3-triazoles as potential antifungal and antibacterial agents. Bull Korean Chem Soc 32:229–238
Che X, Sheng C, Wang W, Cao Y, Xu Y, Ji H, Dong G, Miao Z, Yao J, Zhang W (2009) New azoles with potent antifungal activity: design, synthesis and molecular docking. Eur J Med Chem 44:4218–4226
Daewoong Pharmaceutical Co. (2011) Antifungal triazole derivatives. US7968579
Daewoong Pharmaceutical Co. (2011) Antifungal triazole derivatives, method for the preparation thereof and pharmaceutical composition containing same. US8063229
Council of Scientific & Industrial Research and FDC Ltd. (2012) Antifungal compounds containing benzothiazinone, benzoxazinone, or benzoxazolinone and process thereof. US8129369
Loyse A, Dromer F, Day J, Lortholary O, Harrison TS (2013) Flucytosine and cryptococcosis: time to urgently address the world wide accessibility of a 50-year-old antifungal. J Antimicrob Chemother 68:2435–2444
Perumalla S, Pedireddi V, Sun C (2013) Design, synthesis, and characterization of new 5-flucytosine salts. Mol Pharm 10:2462–2466
Defever KS, Whelan WL, Rogers AL, Beneke ES, Veselenak JM, Soll DR (1982) Candida albicans resistance to 5-fluorocytosine: frequency of partially resistant strains among clinical isolates. Antimicrob Agents Chemother 22:810–815
Hector RF, Domer JE, Carrow EW (1982) Immune responses to Candida albicans in genetically distinct mice. Infect Immun 38:1020–1028
Polak A, Scholer HJ (1975) Mode of action of 5-fluorocytosine and mechanisms of resistance. Chemotherapy 21:113–130
Whelan WL, Kerridge D (1984) Decreased activity of UMP pyrophosphorylase associated with resistance to 5-fluorocytosine in Candida albicans. Antimicrob Agents Chemother 26:570–574
Hector RF (1993) Compounds active against cell walls of medically important fungi. Clin Microbiol Rev 6:1–21
Cassone A, Bernardis FD, Torososantucci A (2005) An outline of the role of anti-Candida antibodies within the context of passive immunization and protection from candidiasis. Curr Mol Med 5:377–382
Cassone A, Mason RE, Kerridge D (1981) Lysis of growing yeast-form cells of Candida albicans by echinocandin: a cytological study. Sabouraudia 19:97–110
Gupta AK, Shear NH (1997) Terbinafine: an update. J Am Acad Dermatol 37:979–988
Darkes MJM, Scott LJ, Goa KL (2003) Terbinafine: a review of its use in onychomycosis in adults. Am J Clin Dermatol 4:39–65
Callen JP, Hughes P, Kulp-Shorten C (2001) Subacute cutaneous lupus erythematosus induced or exacerbated by terbinafine: a report of 5 cases. Arch Dermatol 137L:1196–1198
Ryder NS (1992) Terbinafine: mode of action and properties of the squalene epoxidase inhibition. Br J Dermatol 126(Suppl 39):2–7
Georgopoulos A, Petranyi G, Mieth H, Drews J (1981) In vitro activity of naftifine, a new antifungal agent. Antimicrob Agents Chemother 19:386–389
Venugopal PV, Venugopal TV (1994) Antidermatophytic activity of allylamine derivatives. Indian J Pathol Microbiol 37:381–388
Gupta AK, Ryder JE, Cooper EA (2008) Naftifine: a review. J Cutan Med Surg 12:51–58
Ghannoum M et al (2013) In vitro antifungal activity of naftifine hydrochloride against dermatophytes. Antimicrob Agents Chemother 57:4369–4372
Ryder NS, Dupont MC (1985) Inhibition of squalene epoxidase by allylamine antimycotic compounds. A comparative study of the fungal and mammalian enzymes. Biochem J 230:765–770
Regli P, Ferrari H (1989) In vitro action spectrum of a new antifungal agent derived from morpholine: amorolfin. Pathol Biol 37:617–620
Hänel H, Smith-Kurtz E, Pastowsky S (1991) Therapy of seborrheic eczema with an antifungal agent with an antiphlogistic effect. Mycoses 34(Suppl 1):91–93
Singal A (2008) Butenafine and superficial mycoses: current status. Expert Opin Drug Metab Toxicol 4:999–1005
Das S, Barbhuniya JN, Biswas I, Bhattacharya S, Kundu PK (2010) Studies on comparison of the efficacy of terbinafine 1% cream and butenafine 1% cream for the treatment of Tinea cruris. Indian Dermatol Online J 1:8–9
Ryu C-K, Lee JY, Park R-E, Ma M-Y, Nho J-H (2007) Synthesis and antifungal activity of 1H-indole-4,7-diones. Bioorg Med Chem Lett 17:127–131
Xu H, Wang Y-Y (2010) Antifungal agents. Part 5: synthesis and antifungal activities of aminoguanidine derivatives of N-arylsulfonyl-3-acylindoles. Bioorg Med Chem Lett 20:7274–7277
Ryu C-K, Lee S-Y, Kim NY, Hong JA, Yoon JH, Kim A (2011) Synthesis and antifungal evaluation of 6-hydroxy-1H-carbazole-1,4(9H)-diones. Bioorg Med Chem Lett 21:427–430
Na Y-M, Borgne ML, Pagniez F, Baut GL, Pape PL (2003) Synthesis and antifungal activity of new 1-halogenobenzyl-3-imidazolylmethylindole derivatives. Eur J Med Chem 38:75–87
Tiwari RK, Verma AK, Chhillar AK, Singh D, Singh J, Kasi Sankar V et al (2006) Synthesis and antifungal activity of substituted-10-methyl-1,2,3,4-tetrahydropyrazino[1,2-a]indoles. Bioorg Med Chem 14:2747–2752
Musiol R, Jampilek J, Buchta V, Silva L, Niedbala H, Podeszwa B et al (2006) Antifungal properties of new series of quinoline derivatives. Bioorg Med Chem 14:3592–3598
Meléndez Gómez CM, Kouznetsov VV, Sortino MA, Álvarez SL, Zacchino SA (2008) In vitro antifungal activity of polyfunctionalized 2-(hetero)arylquinolines prepared through imino Diels–Alder reactions. Bioorg Med Chem 16:7908–7920
Yu Z, Shi G, Sun Q, Jin H, Teng Y, Tao K et al (2009) Design, synthesis and in vitro antibacterial/antifungal evaluation of novel 1-ethyl-6-fluoro-1,4-dihydro-4-oxo-7(1-piperazinyl)quinoline-3-carboxylic acid derivatives. Eur J Med Chem 44:4726–4733
Boateng CA, Eyunni SVK, Zhu XY, Etukala JR, Bricker BA, Ashfaq MK et al (2011) Benzothieno[3,2-b]quinolinium and 3-(phenylthio)quinolinium compounds: synthesis and evaluation against opportunistic fungal pathogens. Bioorg Med Chem 19:458–470
Tang H, Zheng C, Lv J, Wu J, Li Y, Yang H et al (2010) Synthesis and antifungal activities in vitro of novel pyrazino [2,1-a] isoquinolin derivatives. Bioorg Med Chem Lett 20:979–982
Jatav V, Kashaw S, Mishra P (2008) Synthesis, antibacterial and antifungal activity of some novel 3-[5-(4-substituted phenyl) 1,3,4-thiadiazole-2-yl]-2-styryl quinazoline-4(3H)-ones. Med Chem Res 17:169–181
Abdel-Gawad SM, El-Gaby MSA, Ghorab MM (2000) Synthesis and antifungal activity of novel pyrano[2′,3′:4,5]thiazolo[2,3-b]quinazolines, pyrido[2′,3′:4,5]thiazolo[2,3-b]quinazolines and pyrazolo[2′,3′:4,5]thiazolo[2,3-b]quinazolines. Farmaco 55:287–292
Jalilian AR, Sattari S, Bineshmarvasti M, Daneshtalab M, Shafiee A (2003) Synthesis and in vitro antifungal and cytotoxicity evaluation of substituted 4,5-dihydronaphtho[1,2-d][1,2,3]thia(or selena)diazoles. Farmaco 58:63–68
Fuglseth E, Otterholt E, Høgmoen H, Sundby E, Charnock C, Hoff BH (2009) Chiral derivatives of Butenafine and Terbinafine: synthesis and antifungal activity. Tetrahedron 65:9807–9813
Mallikarjuna BP, Sastry BS, Suresh Kumar GV, Rajendraprasad Y, Chandrashekar SM, Sathisha K (2009) Synthesis of new 4-isopropylthiazole hydrazide analogs and some derived clubbed triazole, oxadiazole ring systems – a novel class of potential antibacterial, antifungal and antitubercular agents. Eur J Med Chem 44:4739–4746
Omar K, Geronikaki A, Zoumpoulakis P, Camoutsis C, Soković M, Ćirić A et al (2010) Novel 4-thiazolidinone derivatives as potential antifungal and antibacterial drugs. Bioorg Med Chem 18:426–432
Pitta E, Tsolaki E, Geronikaki A, Petrovic J, Glamoclija J, Sokovic M et al (2015) 4-Thiazolidinone derivatives as potent antimicrobial agents: microwave-assisted synthesis, biological evaluation and docking studies. MedChemComm 6:319–326
Chimenti F, Bizzarri B, Bolasco A, Secci D, Chimenti P, Granese A et al (2011) Synthesis and biological evaluation of novel 2,4-disubstituted-1,3-thiazoles as anti-Candida spp. agents. Eur J Med Chem 46:378–382
Stan CD, Tuchiluş C, Stan CI (2002) Echinocandins--new antifungal agents. Rev Med Chir Soc Med Nat Iasi 118:528–536
Sucher AJ, Chahine EB, Balcer HE (2009) Echinocandins: the newest class of antifungals. Ann Pharmacother 43:1647–1657
Spampinato C, Leonardi D (2013) Candida infections, causes, targets, and resistance mechanisms: traditional and alternative antifungal agents. Biomed Res Int 2013:1–13
Vazquez J, Sobel JD (2006) Anidulafungin: a novel echinocandin. Clin Infect Dis 43:215–222
Denning DW (2003) New drug classes echinocandin antifungal drugs. Lancet 362:1142–1151
Letscher-Bru V, Herbrecht R (2003) Caspofungin: the first representative of a new antifungal class. J Antimicrob Chemother 51:513–521
Chandrasekar PH, Sobel JD (2006) Micafungin: a new echinocandin. Clin Infect Dis 42:1171–1178
Sheng C, Xu H, Wang W, Cao Y, Dong G, Wang S et al (2010) Design, synthesis and antifungal activity of isosteric analogues of benzoheterocyclic N-myristoyltransferase inhibitors. Eur J Med Chem 45:3531–3540
Onnis V, De Logu A, Cocco MT, Fadda R, Meleddu R, Congiu C (2009) 2-Acylhydrazino-5-arylpyrrole derivatives: synthesis and antifungal activity evaluation. Eur J Med Chem 44:1288–1295
Maruoka H, Kashige N, Eishima T, Okabe F, Fujioka T, Miake F et al (2008) Synthesis and antifungal activity of spiro[cyclopropane-1,4′-pyrazol-3-one] derivatives. J Heterocycl Chem 45:1883–1887
Zheng Q-Z, Cheng K, Zhang X-M, Liu K, Jiao Q-C, Zhu H-L (2010) Synthesis of some N-alkyl substituted urea derivatives as antibacterial and antifungal agents. Eur J Med Chem 45:3207–3212
Ryu C-K, Han J-Y, Jung O-J, Lee S-K, Lee JY, Jeong SH (2005) Synthesis and antifungal activity of noble 5-arylamino- and 6-arylthio-4,7-dioxobenzoselenazoles. Bioorg Med Chem Lett 15:679–682
Ryu C-K, Song AL, Lee JY, Hong JA, Yoon JH, Kim A (2010) Synthesis and antifungal activity of benzofuran-5-ols. Bioorg Med Chem Lett 20:6777–6780
Xu H, Fan L-L (2011) Antifungal agents. Part 4: synthesis and antifungal activities of novel indole[1,2-c]-1,2,4-benzotriazine derivatives against phytopathogenic fungi in vitro. Eur J Med Chem 46:364–369
López SN, Castelli MV, Zacchino SA, Domínguez JN, Lobo G, Charris-Charris J et al (2001) In vitro antifungal evaluation and structure–activity relationships of a new series of chalcone derivatives and synthetic analogues, with inhibitory properties against polymers of the fungal cell wall. Bioorg Med Chem 9:1999–2013
Singh OM, Singh SJ, Devi MB, Devi LN, Singh NI, Lee S-G (2008) Synthesis and in vitro evaluation of the antifungal activities of dihydropyrimidinones. Bioorg Med Chem Lett 18:6462–6467
Ravi Kumar KR, Mallesha H, Basappa, Rangappa KS (2003) Synthesis of novel isoxazolidine derivatives and studies for their antifungal properties. Eur J Med Chem 38:613–619
Zhang F-F, Gan L-L, Zhou C-H (2010) Synthesis, antibacterial and antifungal activities of some carbazole derivatives. Bioorg Med Chem Lett 20:1881–1884
Šenel P, Tichotová L, Votruba I, Buchta V, Špulák M, Kuneš J et al (2010) Antifungal 3,5-disubstituted furanones: from 5-acyloxymethyl to 5-alkylidene derivatives. Bioorg Med Chem 18:1988–2000
Yao B, Ji H, Cao Y, Zhou Y, Zhu J, Lü J et al (2007) Synthesis and antifungal activities of novel 2-aminotetralin derivatives. J Med Chem 50:5293–5300
Hilchie AL, Wuerth K, Hancock REW (2013) Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat Chem Biol 9:761–768
Rodrigues EG, Dobroff AS, Taborda CP, Travassos LR (2009) Antifungal and antitumor models of bioactive protective peptides. An Acad Bras Cienc 81:503–520
Ekengren S, Hultmark D (1999) Drosophila cecropin as an antifungal agent. Insect Biochem Mol Biol 29:965–972
De Lucca AJ, Bland JM, Jacks TJ, Grimm C, Walsh TJ (1998) Fungicidal and binding properties of the natural peptides cecropin B and dermaseptin. Med Mycol 36:291–298
Lee DG, Kim HK, Kim SA, Park Y, Park SC, Jang SH et al (2003) Fungicidal effect of indolicidin and its interaction with phospholipid membranes. Biochem Biophys Res Commun 305:305–310
Raj PA, Edgerton M, Levine MJ (1990) Salivary histatin 5: dependence of sequence, chain length, and helical conformation for candidacidal activity. J Biol Chem 265:3898–3905
Zasloff M (1987) Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci U S A A84:5449–5453
Giacometti A, Cirioni O, Barchiesi F, Del Prete MS, Scalise G (1999) Antimicrobial activity of polycationic peptides. Peptides 20:1265–1273
Mangoni ML, Grovale N, Giorgi A, Mignogna G, Simmaco M, Barra D (2000) Structure-function relationships in bombinins H, antimicrobial peptides from Bombina skin secretions. Peptides 21:1673–1679
Simmaco M et al (2003) Defense peptides in the amphibian immune system. In: Ascenzi P, Polticelli F, Visca P (eds) Bacterial, plant, and animal toxins. Research Signpost, Kerala
Hancock REW, Rozek A (2002) Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett 206:143–149
Pettit RK, Pettit GR, Hazen KC (1998) Specific activities of dolastatin 10 and peptide derivatives against Cryptococcus neoformans. Antimicrob Agents Chemother 42:2961–2965
Cassone A, Torosantucci A, Boccanera M, Pellengrini G, Palma C, Malavasi G (1988) Production and characterization of a monoclonal antibody to a cell surface, glucomannoprotein constituent of Candida albicans and other pathogenic Candida species. J Med Microbiol 27:233–238
De Wit MYL, Klaster PR (1988) Purification and characterization of a 36kDa antigen of Mycobacterium leprae. J Gen Microbiol 134:1541–1548
Chaturvedi AK, Kavishwar A, Shiva Keshava GB, Shukla PK (2005) Monoclonal immunoglobulin G1 directed against Aspergillus fumigatus cell wall glycoprotein protects against experimental murine aspergillosis. Clin Diagn Lab Immunol 12:1063–1068
Sgro C (1995) Side-effects of a monoclonal antibody, muromonab CD3/orthoclone OKT3: bibliographic review. Toxicology 105:23–29
Kettner SC et al (1999) Use of abciximab-modified thrombelastography in patients undergoing cardiac surgery. Anesth Analg 89:580–584
Zhang Y et al (2014) Daclizumab reduces CD25 levels on T cells through monocyte-mediated trogocytosis. Mult Scler 20:156–164
Borker A, Choudhary N (2011) Rituximab. Indian Pediatr 48:627–632
Boekhout AH, Beijnen JH, Schellens JHM (2011) Trastuzumab. Oncologist 16:800–810
Scott LJ, Lamb HM (1999) Palivizumab. Drugs 58:303–305
Valle E, Gross M, Bickston SJ (2001) Infliximab. Expert Opin Pharmacother 2:1015–1025
Onrust SV, Wiseman LR (1999) Basiliximab. Drugs 57:207–213, discussion 214
McGavin JK, Spencer CM (2001) Gemtuzumab ozogamicin. Drugs 61:1317–1324
Frampton JE, Wagstaff AJ (2003) Alemtuzumab. Drugs 63:1229–1243, discussion 1245–6
Savk E (2007) Efalizumab. Anti-inflamm Anti-Allergy Agents Med Chem 6:205–210
Mease PJ (2007) Adalimumab in the treatment of arthritis. Ther Clin Risk Manag 3:133–148
Witzig TE et al (2002) Randomized controlled trial of yttrium-90-labeled ibritumomab tiuxetan radioimmunotherapy versus rituximab immunotherapy for patients with relapsed or refractory low-grade, follicular, or transformed B-cell non-Hodgkin’s lymphoma. J Clin Oncol 20:2453–2463
Mukherji SK (2010) Bevacizumab (Avastin). AJNR Am J Neuroradiol 31:235–236
Graham J, Muhsin M, Kirkpatrick P (2004) Cetuximab. Nat Rev Drug Discov 3:549–550
Corren J et al (2009) Safety and tolerability of omalizumab. Clin Exp Allergy 39:788–797
Selewski DT, Shah GV, Segal BM, Rajdev PA, Mukherji SK (2010) Natalizumab (Tysabri). Am J Neuroradiol 31:1588–1590
Saltz L, Easley C, Kirkpatrick P (2006) Panitumumab. Nat Rev Drug Discov 5:987–988
Blick SK, Keating GM, Wagstaff AJ (2007) Ranibizumab. Drugs 67:1199–1206, discussion 1207–9
Davis J (2008) Eculizumab. Am J Health Syst Pharm 65:1609–1615
Goel N, Stephens S (2010) Certolizumab pegol. MAbs 2:137–147
Cingoz O (2009) Ustekinumab. MAbs 1:216–221
Mazumdar S, Greenwald D (2009) Golimumab. MAbs 1:422–431
Dhimolea E (2010) Canakinumab. MAbs 2:3–13
Keating MJ, Dritselis A, Yasothan U, Kirkpatrick P (2010) Ofatumumab. Nat Rev Drug Discov 9:101–102
Venkiteshwaran A (2009) Tocilizumab. MAbs 1:430–435
Cummings SR et al (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361:756–765
Sondak VK, Smalley KSM, Kudchadkar R, Grippon S, Kirkpatrick P (2011) Ipilimumab. Nat Rev Drug Discov 10:411–412
Sanz I, Yasothan U, Kirkpatrick P (2011) Belimumab. Nat Rev Drug Discov 10:335–336
Ansell SM (2014) Brentuximab vedotin. Blood 124:3197–3200
Zagouri F et al (2013) Pertuzumab in breast cancer: a systematic review. Clin Breast Cancer 13:315–324
Diéras V, Bachelot T (2014) The success story of trastuzumab emtansine, a targeted therapy in HER2-positive breast cancer. Target Oncol 9:111–122
Shah A (2014) Obinutuzumab: a novel anti-CD20 monoclonal antibody for previously untreated chronic lymphocytic leukemia. Ann Pharmacother 48:1356–1361
Rhee VF et al (2010) Siltuximab, a novel anti-interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol 28:3701–3708
Mosli MH, Feagan BG (2013) Vedolizumab for Crohn’s disease. Expert Opin Biol Ther 13:455–463
Javle M, Smyth EC, Chau I (2014) Ramucirumab: successfully targeting angiogenesis in gastric cancer. Clin Cancer Res 20:5875–5881
Sanford M, McKeage K (2015) Secukinumab: first global approval. Drugs 75:329–338
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Shukla, P.K., Singh, P., Yadav, R.K., Pandey, S., Bhunia, S.S. (2016). Past, Present, and Future of Antifungal Drug Development. In: Saxena, A. (eds) Communicable Diseases of the Developing World. Topics in Medicinal Chemistry, vol 29. Springer, Cham. https://doi.org/10.1007/7355_2016_4
Download citation
DOI: https://doi.org/10.1007/7355_2016_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-78252-2
Online ISBN: 978-3-319-78254-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)