
Abstract
Antibiotics are the bedrock of modern medicine but their efficacy is rapidly eroding due to the alarming emergence of multi-drug resistant bacteria. To begin to address this crisis, novel antibacterial agents that inhibit bacterial-specific cellular functions essential for growth, viability, and/or pathogenesis are urgently needed. Although the genomics era has contributed greatly to identifying novel antibacterial targets, it has failed to appropriately characterize, prioritize, and ultimately exploit such targets to significantly impact antibiotic discovery. Here we describe a contemporary view of new antibacterial target discovery; one which complements existing genomics strategies with a deeply rooted and fundamental understanding of target biology in the context of genetic networks and environmental conditions to rigorously identify high potential targets, and cognate inhibitors, for consideration as antibacterial leads.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Antibiotic
- Antibiotic resistance
- Conditional essential
- Drug target
- Outer membrane biogenesis
- Synthetic lethality
- Wall teichoic acid
1 Introduction: An Evolving View of New and Legitimate Antibacterial Targets
Antibiotics are extraordinarily valuable therapeutic agents whose widespread use has transformed human health since the early twentieth century, largely relegating historically uncontrollable and deadly bacterial infections to mild and conveniently treatable illnesses due to the high efficacy, wide availability, and relatively low cost of these antibiotics [1]. Their remarkable success is tempered by the increasing rise of multi-drug resistant bacteria that are recalcitrant to our existing repertoire of chemotherapies [2, 3], principally due to the lack of stewardship in health care and overuse in livestock for food production [4, 5]. Alarmingly, the rate at which drug-resistance is emerging is in stark contrast to the abrupt decline in the discovery of novel antimicrobials with which to treat them [3, 6]. Existing antibiotics in clinical use target a surprisingly small subset of essential processes [7], and the pipeline in recent years has been awash in “me-too” inhibitors of similar classes that are incremental modifications of existing compounds [8]. There is an obligation among the research community to identify inhibitors from compound collections that interdict novel targets in pathways essential for bacterial growth or infection for which resistance has not yet been widely disseminated. Despite this clarion call and the herculean efforts of many, success in the discovery of clinically relevant antimicrobials to novel targets has remained elusive in recent decades despite the dawn of the genomics era that has provided researchers detailed blueprints of promising targets in countless bacterial organisms. The causes for this failure are likely multi-faceted and overcoming stagnation may require (1) a paradigm shift that will integrate modern approaches with lessons from the past; (2) a broader definition of druggable targets to include those involved throughout the course of a bacterial infection in the host-pathogen context rather than relying on targets that disrupt growth in artificial environments in vitro; and (3) a shift away from the expectations of a novel broad-spectrum panacea to a more narrow spectrum-focused effort to find treatments for multi-drug resistant bacterial infections of high-priority.
In contrast to the scarcity of antibacterials with new mechanisms of action (MOA) that meet or exceed standard of care antibiotic treatments in recent years, there is no lack in the literature of the discovery of new and exciting antibacterial targets of potential utility [8, 9]. However, defining the quality of any particular drug target and its relative prioritization versus literally 1000s of other potential targets is difficult, and is often considered from an antiquated and subjective perspective rooted in the idea that any gene required for microbial growth and/or viability is considered a plausible drug target. In fact, the genomes of most bacterial pathogens typically comprise hundreds of essential genes (for example, E. coli contains ~300 essential genes) [10, 11] required to facilitate fundamental cellular functions; fungal genomes contain even more, typically approaching as many as ~1000 essential genes [12,13,14]. These numbers can be whittled down considerably by introducing additional sensible drug target prioritization criteria, such as conservation of the protein target amongst medically significant microbial pathogens (i.e., genetically predicted achievable “spectrum” for the activity and efficacy of the cognate drug to the selected target) and absence of the target in the human genome, hence mitigating the possibility of target-based cytotoxicity. This view, although seemingly necessary, is certainly not sufficient and the last 20 years of antibacterial discovery efforts only underscores the frailty of these simplistic considerations [15, 16]. Furthermore, such an approach neglects many valuable targets that are conserved in humans and yet are selectively inhibited by clinically successful antibiotics such as the ribosome, RNA polymerase, type II topoisomerase, dihydrofolate reductase, and the tRNA synthetases. Instead, antibiotic targets should be defined more rigorously and according to a continuum of validation criteria that describes their likelihood to deliver new therapeutics. Identifying and leveraging “high value” novel targets to discover new antibacterial leads requires a much greater level of biological insight and innovation to efficiently and unequivocally discover cognate small-molecule inhibitors. Here, we provide a contemporary perspective on the topic of new antibacterial targets; one streamlined to empirically identify and validate “druggable” targets and cognate inhibitors as antibiotic chemical starting points with demonstrated efficacy in a disease model of infection.
A central dogma driving the definition of a novel antibacterial target is that it is essential for the growth and/or viability of the pathogen(s) for which novel therapeutics are needed. Accordingly, cognate inhibitors of such targets are predicted to disrupt fundamental aspects of bacterial physiology and lead to cell death (i.e., bactericidal) or a growth arrest (bacteriostatic). Indeed, all successful antibiotics past and present meet this fundamental criterion. However, such successes whether pioneered by Fleming and Waxman or later by large pharmaceutical companies were almost entirely based on empiric screening of chemical collections (largely natural product extracts) displaying intrinsic antimicrobial activity [17] with target and MOA elucidation typically only achieved many years after their discovery and clinical use [7]. Decades later, success derived from the continued application of this strategy has fallen precipitously; whether resulting from (1) a diminishing return in discovering new leads versus the inefficient and time consuming rediscovery of known natural product compounds [18], (2) the perceived “undesirable” chemical space in which synthetic compound libraries tend to exist versus the physicochemical properties of natural products [16, 19, 20], and/or (3) the high therapeutic bar that clinically non-inferior new agents must achieve versus >70 years of standard of care antibiotics to which they are compared [21]. Consequently, a target-centric approach – fueled by the genomic era – has emerged where targets are first selected to screen and/or rationally design small-molecule inhibitors whose potency, spectrum, and safety can be later chemically optimized.
Defining robust validation criteria of a new antibiotic target spans three basic levels (Level-1, -2, and -3) in their broad continuum of characterization, where Level-3 targets are the most extensively substantiated. We propose defining Level-1 targets as having (1) genetic evidence under in vitro conditions that inactivation/inhibition of their function impairs growth and/or viability of the pathogen and (2) ideally, satisfy basic bioinformatics criteria pertaining to their spectrum and absence from man. In addition to these criteria, Level-2 targets also possess genetic verification that (1) abolishing target function impairs pathogenesis in a relevant animal model of disease and (2) that the target has been confirmed to be druggable by identifying whole-cell bioactive target-selective inhibitor(s) supported with (3) unambiguous MOA evidence (Table 1) [22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53]. Finally, Level-3 targets are those previously validated in a clinical setting by currently marketed antibacterial therapeutics. Such benchmarks clearly emphasize the enormous objectives sought within and between each level. Considering the sheer number of Level-1 targets that can easily be identified by surveying the scientific literature and perusing publically available databases, as well as the Level-3 targets which facilitate the developing of improved versions of existing agents (i.e., best in class agents [54]) rather than entirely new classes of antibiotics, our review will instead focus largely on new targets approaching or meeting Level-2 objectives.
2 Conditional Essentiality: Providing Novel Screens and Cognate Inhibitors to Validate New Druggable Targets
Although the importance of essential gene products serving as antibacterial drug targets is undisputed, the identification of novel antibacterial targets can be significantly expanded from this strict historical definition. Most important is to broaden the conditional context in which gene essentiality is defined. Routinely, gene essentiality is determined under rich nutritional conditions highly optimized for the growth of the pathogen in a laboratory setting but which does not reflect the more extreme conditions a pathogen must overcome during infection. To emphasize this point, large scale gene disruption experiments in E. coli identify ~300 genes required for growth on rich medium, whereas >100 additional genes are identified to be essential for growth strictly on minimal growth media [11]. Similar conclusions are drawn in yeast [55]. Indeed, anti-folates such as the early sulfa drugs and later, sulfamethoxazole and trimethoprim target the conditionally essential proteins folP and folA, respectively, and their activity is suppressed in vitro by exogenous addition of p-aminobenzoic acid (PABA) and thymidine [7, 56]. Therapeutic efficacy of these agents is nonetheless achieved because such metabolites are insufficiently low in an infectious setting to suppress the antibiotic effects of these agents. Conversely, exogenous fatty acids are present at sufficient concentrations in the host to support growth of type II fatty acid synthesis null mutant bacteria of the order Lactobacillales, including Streptococcus agalactiae and Streptococcus pneumoniae, illustrating that a precise understanding of the host environment is paramount when selecting metabolite-suppressed targets [57]. Considering the extent of additional biosynthetic pathways in which metabolite suppression is achieved, a robust chemical genetic strategy to identify new antibacterial inhibitors and empirically identify new druggable targets is certainly achievable [56]. One notable example of this approach relates to the discovery of ribocil, a synthetic mimic of the natural metabolite, flavin mononucleotide (FMN), which selectively targets a non-coding mRNA structural element (termed a FMN riboswitch) responsible for gene regulation within the riboflavin biosynthetic pathway [52, 53]. The structure of ribocil C (and other representative compounds discussed in this chapter) are given in Fig. 1. Here, ribocil and its cognate target, the FMN riboswitch, were identified by screening a bioactive compound collection for inhibitors whose bioactivity was specifically suppressed in the presence of exogenous riboflavin supplemented to the growth medium. Despite a conditional essentiality for de novo riboflavin biosynthesis by E. coli under in vitro conditions, genetic evidence demonstrates an absolute essential requirement for this metabolic pathway in a murine model of E. coli infection which is pharmacologically validated by demonstrating ribocil C provides dose-dependent efficacy in this model [52].


Structures of representative compounds cited in this review
Temperature sensitive (TS) growth phenotypes of gene depletion mutants also offer a simple strategy to consider both a wider array of drug targets and rapidly identify cognate inhibitors. For example, ltaS encodes a lipoteichoic acid (LTA) synthase responsible for the biogenesis of this basic cell-wall polymer common to Gram-positive pathogens [58, 59]. Genetic studies in S. aureus reveal that ltaS is dispensable at 30°C, albeit resulting in severe cell division and morphological defects. However, at elevated temperatures such as 37°C (the physiologically relevant temperature of infection), ltaS depletion mutants are not viable [58]. Taking advantage of this TS phenotype, Richter et al. screened for compounds that phenocopy the ltaS phenotype and thus inhibit S. aureus growth only at the elevated temperature. One such compound resulting from this screen, compound 1771, is proposed to inhibit LtaS by structurally mimicking the phosphatidylglycerol substrate of the synthase [24]. Whereas the above examples of conditional essentiality are straightforward, more innovative strategies to exploit this phenomenon are also possible. One particularly intriguing opportunity relates to bacterial gene essentiality in the context of host innate immunity. For over 50 years, it has been known that the extracellular capsule and diverse O-antigen types that coat the surface of Gram-negative bacteria protect these pathogens from the lethal effects of human serum [60,61,62,63,64]. Recently, the Schembri lab has revisited this biology. Using a genome-wide transposon mutagenesis strategy in clinical isolates of E. coli they uncovered multiple non-essential genes involved in O-antigen biosynthesis and in outer membrane (OM) biogenesis which when genetically inactivated, profoundly sensitize the bacterium to the killing effects of serum [65,66,67]. The clever exploitation of these (and other) phenotypes that are relevant to the infectious disease setting offers the design of robust cellular screens to identify cognate inhibitors, and so to expand the diversity of new antibacterial targets.
Beyond any particular growth condition and/or environmental context in which gene function is essential, conditional essentiality may also manifest in a unique genetic context. Synthetic lethality (SL) describes such a context in which a gene is dispensable in a wild-type genetic background, but not in a particular mutant background in which another gene has been inactivated [68]. Typically, this phenomenon applies to genes either involved in a common biological process or distinct but interdependent biological processes which partially compensate or “buffer” the loss of the other [54]. The most extensive demonstration of the myriad of intrinsic synthetic lethal genetic interactions within a microbial genome undoubtedly has been characterized in the bakers’ yeast, Saccharomyces cerevisiae [68,69,70]. However, SL is also emerging as an important approach to identifying new antibacterial targets [45, 71,72,73,74] as well as mapping genetic interaction networks between a known target (for example, a clinically validated antibiotic drug target) and new targets that if inactivated, enhance the activity of the clinically used antibiotic. Such chemical genetic interaction networks are highly analogous to SL and provide a powerful means to rationally identify cognate inhibitors that are chemically synergistic with the clinical antibiotic, thus offering a compelling combination agent strategy to improve existing antibiotics [54, 75]. An elegant implementation of this strategy has been applied rigorously to methicillin resistant S. aureus (MRSA) and methicillin resistant S. epidermidis (MRSE) as a means of restoring potent β-lactam efficacy against otherwise β-lactam resistant Staphylococci [47, 75, 76]. Here, a β-lactam genetic interaction network was first identified using antisense interference methodology [77, 78] to genetically deplete gene expression of ~250 possible targets and identify 24 distinct genes, which if partially inactivated render MRSA and MRSE specifically susceptible to β-lactam antibiotics [76]. Interestingly, many of these β-lactam potentiation targets contribute to various aspects of cell-wall peptidoglycan (PG) and wall teichoic acid (WTA) biosynthesis, offering a clear mechanistic basis for their SL when genetically knocked down in expression specifically in the context of sub MIC levels of β-lactams. Additionally, targets involved in other biologically significant processes, most notably cell division (e.g., FtsA, FtsZ, and FtsW), secretion (SpsB), and PG lipid II amidation were also revealed [76, 79]. Finally, the genetic prediction of β-lactam potentiation provided by this genetic interaction network was robustly verified by evaluating the effects of PC190723, a potent and highly selective inhibitor of FtsZ [46] in combination with diverse β-lactam antibiotics and demonstrating striking chemical synergy between these agents in vitro as well as in a murine deep thigh infection model of MRSA [47]. Consequently, the therapeutic context of PC190723 as a single-agent antibacterial lead targeting FtsZ [46] could be expanded into a role as a validated adjuvant, with analogy to β-lactamase inhibitors [80], as a result of its ability to restore the efficacy of β-lactams against methicillin resistant Staphylococci albeit through an entirely novel mechanism [47]. Subsequent examples reinforce this view, as demonstrated by the identification of target-specific inhibitors of MurG and MurJ-mediated PG biosynthesis [28, 29] and WTA-mediated biogenesis (see below).
Perhaps the most remarkable genetic context in which conditional essentiality was exploited to identify new antibacterial targets and screening opportunities for cognate inhibitors relates to the phenomenon of an “essential gene paradox.” First identified by Eric Brown and colleagues in both S. aureus and Bacillus subtilis, inactivation of genes involved in WTA biogenesis displays paradoxical growth phenotypes [81, 82]. Whereas early genes in WTA polymer synthesis are dispensable for growth in vitro, later stage enzymes in the pathway are indispensable for growth and result in a bacteriostatic terminal phenotype [83]. Remarkably, double deletion mutant analysis revealed that genetic inactivation of early stage WTA enzymes suppressed the essentiality of disrupting late stage enzymes in the pathway, perhaps by preventing sequestration of the essential bactoprenyl phosphate lipid carrier which otherwise accumulates in late stage mutants and which is also a shared lipid carrier essential for PG synthesis [50]. Regardless, the unique gene dispensability pattern within WTA biogenesis offers powerful whole-cell based phenotypic screens to identify early and late stage inhibitors of discrete biochemical enzymes within the pathway and their corresponding druggable targets [84]. Whole-cell screens designed to phenocopy the conditional essentiality of late stage lesions in WTA synthesis led to the discovery of targocil [33], targeting the membrane-associated subunit (TarG) of the WTA “flippase” responsible for transporting newly synthesized cytosolic WTA polymer to the cell surface [85]. Accordingly, targocil is bioactive against wild-type S. aureus (including MRSA) but its bioactivity is dramatically suppressed when assayed against S. aureus strains deleted of early stage enzymes, such as TarO. Underscoring the robustness of TarG as a druggable target, similar screens have identified multiple new chemotypes with broader Gram-positive bacterial spectrum targeting the WTA transporter [34].
Recently, we have described a chemical suppression-based screen that similarly relies on the opposing gene dispensability pattern of WTA genes to identity inhibitors of early stage WTA enzymes [32]. Here, the entire Merck corporate library was screened for compounds that restored growth of S. aureus bacteria that were growth arrested due to the bacteriostatic effect of a TarG inhibitor. Compounds that enable bacterial growth in this context phenocopy the restored growth of WTA double mutants defective in both early and late polymer synthesis and are predicted to target one of the early non-essential WTA biosynthetic enzymes. Two structurally distinct, synthetically derived chemicals named tarocin A and B were identified [32] and demonstrated to inhibit TarO, a glucosyltransferase responsible for the initial step in WTA polymer synthesis and previously demonstrated to be inhibited by the natural product, tunicamycin [27]. Thus TarO is uniquely druggable by both synthetic chemistry and natural products. As TarO is not essential for growth in a wild-type strain background, tarocins are non-bioactive (MIC values >256 μg/mL). Moreover, tarocins resensitize MRSA and MRSE to a broad diversity of β-lactams in vitro below the clinical breakpoint drug concentration defining β-lactam resistance and provide synergistic efficacy when paired with β-lactams in a murine infection model of MRSA infection [32]. Consequently, tarocins serve as novel and extensively validated non-bioactive adjuvants to pair with such antibiotics that are conceptually highly analogous to β-lactamase inhibitors used to restore β-lactam efficacy against Gram-negative pathogens [80].
3 Alternative Approaches to New Target Discovery
Historically, phenotypic screens have been enormously successful in identifying new classes of antibiotics. Best illustrative of this success is the discovery of thienamycin (the progenitor of imipenem and the entire carbapenem class of β-lactams), which was identified over 40 years ago from a natural product screen using a fluorescence-based readout of cell lysis indicative of cell-wall inhibitors [86]. Our reliance on phenotypic screens remains today. Recently, AstraZeneca researchers employed a high-throughput phenotypic screen utilizing the Citrobacter freundii AmpC β-lactamase, which when induced in E. coli serves as a sensor for inhibition of cell-wall biosynthesis. Screening over 1.2 million compounds against this reporter assay ultimately yielded specific whole-cell active inhibitors targeting LpxH (catalyzing the fourth step in lipopolysaccharide (LPS) biosynthesis) and the lipoprotein outer membrane localization (Lol) complex, LolCDE [25]. Both LPS and bacterial lipoproteins contribute greatly to the composition of the Gram-negative OM, and the discovery by this approach underscores the functional interrelationship (think synthetic lethality!) between the OM and PG synthesis. Importantly, this work also provides the first reported inhibitors of these essential enzymes [25, 26]. Additional examples of phenotypic screening campaigns discussed above and which similarly identify novel druggable targets and cognate inhibitors emphasize the continued success of this approach [32, 33, 52].
Repurposing existing antibacterial leads in a new therapeutic context breathes new life into old compounds (and targets) but requires novel biological insights to either enhance the activity of the agent or circumvent previously perceived limitations of the antibiotic. The discovery of PC190723 as a β-lactam potentiation adjuvant that restores β-lactam efficacy against MRSA is one example [47]. Another clever example is the repurposing of ClpP inhibitors in the context of chronic S. aureus infections mediated by persister cells. Persister cells reflect a small minority of planktonic cells in a bacterial community which are metabolically inactive or dormant [87,88,89] and consequently resistant to antibiotics whose mode of action is dependent on cell growth. In an elegant series of experiments, Lewis and colleagues demonstrated that the semi-synthetic acyldepsipeptide, ADEP4, which was previously shown to activate ClpP-mediated proteolysis by the bacterial proteasome [43, 90], effectively kills S. aureus persister cells within planktonic communities as well as biofilms [42]. To circumvent the unacceptably high frequency of resistance ADEP4 exhibits as a single agent (clpP null mutants are highly resistant) it was paired with rifampicin. Remarkably, the ADEP4-rifampicin combination demonstrated complete sterilization of both planktonic and persister cells in a murine chronic infection model of S. aureus [42]. Conceptually, activating – rather than inhibiting – the proteasome (or other proteases) provides a compelling new target and general strategy to treat chronic infections refractory to standard antibiotics.
Innovative strategies to optimize chemical libraries for antibacterial activity have also demonstrated significant success. Starting with the host defense antimicrobial peptide protegrin I, researchers at Polyphor have performed iterative synthesis of this starting point for the design and optimization of a library of peptidomimetics with improved antibacterial potency and reduced hemolytic activity [22]. One optimized macrocyclic compound, POL7080, was demonstrated in Pseudomonas aeruginosa to inhibit LPS biogenesis by targeting LptD, which functions in the final step of LPS transport to the outer leaflet of the OM [91]. Based on drug resistant mutant mapping studies to the target and significant protein sequence differences between P. aeruginosa LptD and orthologs across other Gram-negative pathogens, POL7080 is predicted to display a narrow antibiotic spectrum. However, a clear unmet clinical need for novel and effective narrow spectrum anti-Pseudomonas agents undoubtedly exists. Moreover, POL7080 displays impressive nanomolar anti-Pseudomonas activity in vitro and robust efficacy against P. aeruginosa in a lethal septicemia model of infection, achieving a median effective dose in the range of 0.25–0.55 μg/mL [22].
Revisiting natural product libraries as a source of new antibacterial leads involves clever methods to growing previously “unculturable” microorganisms, thereby potentially overcoming the asymptotic inefficiencies of natural product rediscovery currently faced by conventional means [18, 92]. Domesticating such microbes has recently been achieved utilizing a multichannel device (named an iChip) where soil microorganisms are diluted into separate channels and enclosed in a semi-permeable membrane to support diffusion of nutrients and growth when incubated in a soil environment [35]. Screening ~10,000 natural products using this method led to the discovery of teixobactin, an unusual depsipeptide demonstrated to target both lipid II and lipid III precursors of PG and WTA biosynthesis, respectively. Teixobactin displays potent Gram-positive activity, dramatic efficacy in multiple murine infection models, and a highly favorable resistance profile achieved by its dual targeting mechanism. Consistent with teixobactin’s unique mechanism, it is structurally distinct from vancomycin and other glycopeptides, lantibiotics, and defensins which solely target lipid II [93]. Unlike proteins encoding an essential enzyme activity, however, lipid II, lipid III, and the FMN riboswitch [52, 53] constitute non-conventional antibiotic targets. Whereas lipid II and III are essentially immutable lipid substrates, substantial mutation-based plasticity likely exists in non-coding RNA structural elements. Other successful antibiotics that interdict non-conventional targets include daptomycin and colistin, which disrupt membrane lipids, and bacitracin, which binds and sequesters the undecaprenylpyrophosphate lipid carrier from which PG and WTA are synthesized and translocated to the cell surface. One cannot help but think other classes of non-conventional antibiotic targets remain to be discovered.
In parallel to these efforts exploiting previously “unculturable” microbes, in silico methods have been developed to facilitate the design of new natural products for use as antimicrobials [94]. This approach utilizes bioinformatics to predict natural product structures from primary genomic sequence data and chemical synthesis to create these synthetic-bioinformatic natural products (syn-BNPs), which can then be assayed for antibacterial activity. A major problem with natural product drug discovery is the inability to access all biologically relevant chemical diversity through typical laboratory growth conditions, and this recently discovered method provides one potential solution to this issue. As a test case, Chu et al. show that, using sequence data from human commensals and pathogens, they are able to predict and synthesize a novel class of molecules, dubbed the humimycins, that inhibit the S. aureus lipid II flippase [94].
Notwithstanding the current view that in vitro-based biochemical high through-put screening (HTS) and downstream optimization of such synthetic chemistry hits has been largely unsuccessful in the search for new antibacterial leads with whole-cell potency [15, 16], there are quite compelling exceptions to this general rule, particularly as it applies to new Gram-positive targets with cognate inhibitors. In vitro HTS efforts against multiple isoforms of the CoaD enzyme, involved in the synthesis of the essential cofactor, coenzyme A (CoA), combined with structure-based optimization efforts has recently led to the discovery of a highly potent series of antibacterials with broad Gram-positive spectrum, in vivo efficacy across multiple models of infection, and an acceptably low frequency of resistance [41]. Although the further drug optimization of these compounds addressing solubility and tissue penetration was not achieved, both CoaD and the CoA biosynthetic pathway were rigorously validated and offer the potential for the discovery of new series with superior physicochemical properties.
4 Structural Biology Advances Driving Target Discovery
Targeting OM biogenesis factors of Gram-negative bacteria remains a highly attractive, yet underexploited approach in novel antibacterial drug discovery. The OM is an asymmetric bilayer composed of phospholipids in the inner leaflet, LPS in the outer leaflet, OM beta-barrel proteins (OMPs) integrated within the bilayer, and lipoproteins anchored to the inner leaflet [95]. Because the OM is essential, inhibiting its assembly by intervening in lipoprotein, β-barrel protein, or LPS biogenesis will compromise the viability of the cell. Recent structural data for proteins involved in OM biogenesis helps to prioritize such targets. A recently solved P. aeruginosa co-crystal structure of LspA, the signal II peptidase responsible for processing of lipoproteins, with its cognate inhibitor globomycin [40] serves as a significant starting point for rational drug design against this target. The β-barrel assembly machine (BamABCDE) and the LPS-transport subcomplex located at the OM (LptDE) are particularly attractive targets because they are not only druggable enzymes [22, 23] but also contain surface-accessible essential proteins. The principal difficulty with discovering Gram-negative antibacterial leads is identifying compounds that can cross the robust barrier created by the LPS layer and avoid efflux once inside the cell; targeting a surface-exposed protein would circumvent these issues. Additionally, targeting the LPS assembly machine (LptDE) would not only kill the cell, but also permeabilize the OM to other agents that normally have a difficult time traversing the membrane [22].
Bam complex structural data now provide significant insight into the mechanics behind β-barrel protein assembly into the OM in Gram-negative bacteria. The recently solved BamACDE crystal structure overlaid with a previously solved BamAB subcomplex crystal structure permitted the first structural model of a fully assembled Bam complex from E. coli [96]. This BamABCDE structure confirmed previously reported interactions amongst the Bam components as well as revealed new interactions and Bam protein conformations to allow for speculation of a mechanism of β-barrel assembly. Of note, Bakelar et al. found that when the lipoprotein subcomplex BamCDE binds, the essential β-barrel component BamA undergoes a conformational change opening the exit pore and lateral gate in the barrel. This opening may serve to destabilize the membrane locally (near the lateral gate) to allow for OMP insertion through reduction of the kinetic barrier, rather than the threading of nascent OMPs through the lumen of the barrel and out the lateral gate [97], since the BamA N-terminal soluble POTRA domains occlude the lumen of the barrel when the exit pore and lateral gate are open in this crystal structure. Recent genetic studies demonstrating that periplasmic components of the assembly process interact with substrate after much of the β-barrel has formed also support this mechanism of OMP insertion [98]. Another recent study has demonstrated that the only essential lipoprotein in the Bam complex, BamD, can be targeted with a peptide that mimics a substrate protein to which BamD normally binds in the assembly process, validating the druggability of this complex [23]. Understanding the critical points of interaction amongst the Bam components and movement of the Bam machine should enable the discovery of additional inhibitors of OM β-barrel protein assembly.
Like recent advances in the Bam complex structure dataset, the first crystal structures of the LPS-assembly subcomplex LptDE have shed light on a mechanism of LPS insertion into the outer leaflet of the OM [99, 100]. The crystal structures revealed a β-jellyroll N-terminal domain of LptD and an enormous 26-β-stranded C-terminal barrel domain, the largest β-barrel discovered to date. The barrel contains two lobes, one adjacent to the N-terminal domain and one occupied by the essential lipoprotein LptE. LptE not only acts as a plug in the barrel, but also plays a role in LptD assembly as well as LPS assembly [101, 102]. Based on crystallographic and genetic data, the authors speculate that the hydrophilic portion of LPS (O-antigen and core sugars) traverse the lumen of the open lobe of the barrel, while that the lipid component is shielded from the aqueous periplasm by the N-terminal domain of LptD and shuttled through a lateral gate opening between the first and last β-strands of LptD, ensuring specific insertion into the outer leaflet of the OM [99]. Blocking this lateral gate with a peptide or small molecule may be one way to disrupt the function of this essential LptDE translocon. Validation of this hypothesis would highlight how these structural advances can facilitate design of novel antimicrobials.
5 Considering Antibacterial Drug Resistance as Contextual
A general theme to most new targets and cognate antibacterial leads highlighted in this review is their propensity for target-based drug resistance (Table 1). Often, this resistance likely reflects their single-target mode of action [103, 104]. We are also mindful of the disastrous impact drug resistance can have on antibacterial clinical development [105]. The likelihood that acceptable resistance profiles for Level-2 targets described here are achievable, however, either by structure-based design to improve and/or change drug-target binding contacts and/or increase potency should be considered on a case by case basis. One such example is that of antibiotic 2, a non-β-lactam inhibitor which not only inhibits the classic targets of penicillin, PBP1, but also allosterically inhibits the target responsible for β-lactam resistance in MRSA and MRSE, PBP2a [31]. It is also appropriate to be mindful that in vitro-based resistance studies may not always reflect the prevalence of resistance in an infectious setting. The broad-spectrum β-lactam mecillinam serves as an important example of the potential paradoxical resistance profiles of an antibiotic observed by in vitro testing versus that encountered in a clinical setting. Multiple different mecillinam resistance mechanisms in E. coli are commonly identified in vitro, ranging from target-based (pbpB) mutations to other processes including cell-wall synthesis, cell division, tRNA synthetases, and the ppGpp stringent response pathway [106]. Conversely, mecillinam-resistant E. coli from patients treated for a urinary tract infection are very rarely identified and reflect a single type of mutation: inactivation of cysB, a gene involved in cysteine biosynthesis [106]. Whereas all mutant classes selected in vitro share similar fitness costs, the cysB mutations uniquely lack a fitness cost in a more relevant urine-rich growth condition. Thus amongst a broad set of mutations that can confer mecillinam resistance under standard in vitro growth conditions, only cysB mutants are sufficiently fit to potentially persist in the urinary tract. Moreover, considering the high exposure level of the drug in the urine, few of these mutants cause resistance to the antibiotic in a clinical setting, likely because high mecillinam levels sufficiently impact the fitness of the pathogen in an environment where robust growth of the pathogen is required to offset their natural expulsion from the urinary tract. Therefore, understanding drug resistance in a more therapeutically relevant context is critical to avoid the risk of potentially deprioritizing new targets and antimicrobial leads solely based on their in vitro resistance profile.
It is also interesting to consider how a fundamental understanding of the genetic interactions within a single biological process combined with the discovery of multiple inhibitors to distinct targets within such a process can be leveraged in a systems biology-based combination agent strategy to mitigate drug resistance. Consider the WTA biosynthetic pathway (Fig. 2) [34, 48, 107, 108]. Tarocins restore the efficacy of β-lactams against MRSA with target-based resistance mapping to tarO [32]. Addition of a TarG inhibitor as a third component to this combination substantially reduced tarO-mediated resistance [32]. However, mechanistically this is not achieved by simply adding another antibiotic since the growth inhibitory activity of the TarG inhibitor is robustly suppressed by TarO inhibitors (Fig. 2a). Instead, in this three-way combination context the TarG inhibitor is inactive against the bacterial population sensitive to a tarocin-dicloxacillin combination (where TarO is inhibited) and only bioactive against tarocin-resistant tarO mutants (i.e., target-based mutations) (Fig. 2b) in the population that maintain TarO functional activity [33, 34]. Conversely, pre-existing tarO loss-of-function mutations which would suppress the activity of the TarG inhibitor are broadly and highly sensitive to β-lactams [27, 34] as well as strikingly attenuated in virulence across diverse animal infection studies [34, 108,109,110] (Fig. 2c). Consequently, such a three component combination therapeutic elegantly exploits a circuitry of genetic interactions and antibiotic hypersensitivities within the cell-wall network as well as avirulent phenotypes of tarO mutants to provide an integrated and interdependent means of mitigating target-based resistance of the β-lactam potentiator.


Triple combination strategy provides an interdependent mechanism of mitigating target-based resistance. The illustration depicts multiple scenarios in which of a triple combination of TarO and TarG inhibitors paired with a β-lactam antibiotic overcomes various potential mechanisms of resistance. (a) Synergistic activity of tarocin and β-lactam re-sensitizes β-lactam-resistant Gram-positive bacterial pathogens to provide broad Gram-positive antibacterial coverage; simultaneously inactivating L-638 such that it is non-bioactive in this context. (b) In this scenario, acquisition of TarO target-mediated mutations will confer resistance to tarocin but consequently “activate” L-638 antibacterial activity to re-establish broad Gram-positive coverage by the combination cocktail. In addition, Sakoulas et al. have demonstrated that β-lactams (e.g., nafcillin) enhance innate-immune mediated killing of MRSA despite its elevated MIC to the antibiotic [107]. (c) In this scenario, acquisition of Pbp target-mediated mutations that confer resistance to the β-lactam (a very rare event*) may occur but still allows for tarocin to inhibit WTA synthesis, which has been demonstrated to reduce virulence and biofilm formation of methicillin-sensitive S. aureus (MSSA), MRSA, and MRSE during infection [34, 48, 108]. (d) In this final scenario, resistance that may arise due to mutations to both Pbp and TarO targets (an extremely rare event) also activates L-638 antibiotic activity MSSA, MRSA, and MRSE as well as potentially providing broader Gram-positive coverage
6 Target Discovery Parallels Between Gram-positive WTA and Gram-negative OM Biogenesis
Recent progress made in the discovery of Level-2 targets participating in Gram-positive WTA biosynthesis also serves as an instructive example for how new targets may similarly be discovered in other critical biological processes, particularly OM biogenesis amongst Gram-negative pathogens (Fig. 3) [111,112,113]. Central to this success is a deep functional understanding of WTA biogenesis and cell surface assembly from a genetic, biochemical, structural, and pathogenesis perspective [83, 108, 114] and from which an integrated systems biology mindset can be applied. Discovery of robust WTA Level-2 targets such as TarG and TarO, whether “essential” or “non-essential” are actually conditionally essential in the context of an unorthodox gene dispensability pattern and β-lactam exposure [115]. Exploiting WTA genetics provides elegant whole-cell target-based screens to efficiently identify target-specific inhibitors of the pathway [32,33,34] as well as other biochemical pathways impinging on WTA biogenesis (UppS) [50, 51] or by leveraging synthetic lethal interactions within the WTA genetic interaction network (DltB) [45]. Such targets and cognate inhibitors can also be considered as adjuvants for developing synergistic antibiotic combination agents from a rational biology-based perspective [32, 47] and mindful of virulence phenotypes that may augment efficacy. Finally, entirely new anti-infective approaches may be derived from such a fundamental understanding of WTA biology, as elegantly shown by Lehar et al. at Genentech who report an efficacious WTA antibody-antibiotic conjugate to target intracellular reservoirs of S. aureus associated with chronic infections [36].




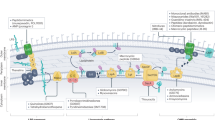
(a) Select Gram-positive cell-wall associated targets and cognate inhibitors. Representative diagram of a prototypical S. aureus bacterial cell surface displaying color-coded biologically relevant biosynthetic pathways: lipoteichoic acid (yellow); wall teichoic acid (salmon); peptidoglycan (blue); cell division (green). New antibacterial targets and cognate inhibitors described in the main text and Table 1 are highlighted. The potential antimicrobial spectrum for the reported inhibitors is designated: potential broad Gram-positive spectrum (red box); potential broad Gram-positive and Gram-negative spectrum (blue box). (b) Select Gram-negative OM-associated targets and cognate inhibitors. Representative diagram of Gram-negative bacteria and their relevant biosynthetic pathways: The Lpt pathway (in salmon), Lipoprotein processing and assembly (yellow), and OMP assembly (purple). Much of the machinery used in the biosynthesis of the peptidoglycan is conserved among Gram-negatives and Gram-positives (see (a) for potential targets). In Gram-negatives, tunicamycin has been demonstrated to additionally target WecA, an IM protein involved in the biosynthesis of O-antigen [111]. Potential Gram-negative spectrum targets (green boxes) and potential broad Gram-positive and Gram-negative spectrum (blue boxes) are indicated [112, 113]. OM outer membrane, IM inner membrane, PG peptidoglycan, PL phospholipid, LPS lipopolysaccharide, OMP OM β-barrel protein
Similarly remarkable advances in our understanding of Gram-negative OM biogenesis have also emerged over the last decade. Beyond the fundamental architecture and composition of the OM, we are gaining a deep functional understanding of the distinct biological assembly processes (i.e., Bam, Lpt, Lol, LPS, PG, capsule, and stress response signaling pathways) [116,117,118,119,120,121,122,123,124,125,126] contributing to its biogenesis and homeostasis as well as their functional interconnectivity [127, 128]. Synthetic lethal-based genetic strategies are also being employed to identify new OM targets such as LpoA and LpoB, two PBP accessory proteins central to PG biogenesis [71, 129], as well as to map genetic interactions within and between these biological processes [72, 102, 130, 131]. Such synthetic lethal interactions could be exploited to develop whole-cell pathway-based screens for novel OM biogenesis inhibitors. Parallels between the WTA essential gene paradox and analogous genetic dispensability patterns in O-antigen biogenesis also exist [132], suggesting similar whole-cell screening opportunities to identify inhibitors of O-antigen assembly are possible. Finally, recent work re-emphasizing the importance of O-antigen and other aspects of the OM in protecting E. coli from the lytic effects of human serum provide exciting new avenues of conditionally essential targets and screens to impair Gram-negative virulence [66, 67].
7 Conclusions
Table 1 summarizes multiple new antibacterial targets discovered in recent years that approach or satisfy Level-2 criteria of (1) bioinformatics-based pathogen spectrum and target-based cytotoxicity predictions; (2) druggable with cognate inhibitor(s) identified with compelling MOA validation; and (3) pharmacological and/or genetic demonstration that target inactivation provides efficacy in a relevant animal model of infection. Surveying this list illustrates a number of emerging trends. For example, many of the druggable targets are multi-spanning membrane proteins localized to the cytoplasmic membrane in Gram-positive bacteria, or resident in the periplasm or OM of Gram-negatives where they functionally serve as biosynthetic enzymes or transporters involved in cell-surface biogenesis. In part, their druggable nature likely reflects their cell-surface location and ability of small molecules to engage such targets without confronting cell permeability and/or efflux issues. The highly hydrophobic nature of such druggable targets does however “select” for cognate inhibitors with high cLogP values and physicochemical properties incompatible with high solubility and drug-like properties [20]. Multi-spanning membrane proteins are also highly challenging from the perspective of target X-ray crystal structure determination, compound co-crystallization, and hence structure-based design and compound optimization. In this way, bacterial druggable targets resemble the majority of known therapeutic targets in human disease (e.g., G-protein coupled receptors, ion channels, and other cell-surface targets), emphasizing the need for technical improvements and greater focus towards X-ray crystallography of complex bacterial membrane proteins. It is also evident that the level of small molecule MOA validation in many of these studies can vary considerably and mechanistic evidence in a whole-cell context is often overly weighted by phenomenological evidence rather than direct target engagement within the cellular milieu. We suggest that in addition to in vitro-based biochemical studies and structural biology evidence, isolation and characterization of causal drug resistant mutations are critically needed to unambiguously validate the MOA of cognate inhibitors of such privileged antibacterial drug targets. Evident also in Table 1 are Level-2 targets with often an unattractive frequency of resistance observed by cognate inhibitors. However, with few exceptions [22] these are lead candidate molecules, not pre-clinical or clinical candidate therapeutics. Considerable medicinal chemistry optimization is required and substantial attrition is certain. Identifying new series with more favorable resistance profiles is also possible and often warranted considering the importance of such targets. Identifying more dual-target opportunities would also likely mitigate resistance development [35, 44, 103, 104]. A greater understanding of drug resistance in a relevant infectious setting as well as potential genetic interaction circuits that can be pharmacologically interdicted to mitigate drug resistance also deserves greater consideration. Finally, a survey of Table 1 emphasizes a strong bias towards Gram-positive targets meeting Level-2 criteria despite the urgent need for new Gram-negative antibacterials. Perhaps in small part this reflects a lag time required to catch up to a growing government, industry and (and importantly) clinical perspective collectively shifting focus to addressing Gram-negative pathogens in recent years. In large part, however, this asymmetry is based on the OM barrier and extensive efflux pumps shared by Gram-negative bacteria that thwart the entry and concentration of potent and selective inhibitors; hence compounding the difficulty to identify and validate druggable targets [20, 133]. The recent commitments made by the Pew Charitable Trust, Welcome Trust, BARDA, NIAID, and most recently CARB-X to fund research centered around OM biogenesis, small molecule permeability, and drug efflux is timely and much needed to address this fundamental issue.
References
Piddock L (2012) The crisis of no new antibiotics – what is the way forward? Lancet Infect Dis 12(3):249–253. doi:10.1016/S1473-3099(11)70316-4
Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J (2009) Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis 48:1–12. doi:10.1086/595011
Brown ED, Wright GD (2016) Antibacterial drug discovery in the resistance era. Nature 529(7586):336–343. doi:10.1038/nature17042
Ventola CL (2015) The antibiotic resistance crisis: part 1: causes and threats. P T 40(4):277–283
Woolhouse M, Ward M, van Bunnik B, Farrar J (2015) Antimicrobial resistance in humans, livestock and the wider environment. Philos Trans R Soc B Biol Sci 370(1670):20140083. doi:10.1098/rstb.2014.0083
Kinch MS, Patridge E, Plummer M, Hoyer D (2014) An analysis of FDA-approved drugs for infectious disease: antibacterial agents. Drug Discov Today 19(9):1283–1287. doi:10.1016/j.drudis.2014.07.005
Walsh C, Wencewicz T (2016) Antibiotics: challenges, mechanisms, opportunities. ASM Press, Washington, DC
Butler MS, Blaskovich MA, Cooper MA (2015) Antibiotics in the clinical pipeline at the end of 2015. J Antibiot (Tokyo). DOI: 10.1038/ja.2016.72
Klahn P, Brönstrup M (2016) New structural templates for clinically validated and novel targets in antimicrobial drug research and development. Curr Top Microbiol Immunol. DOI: 10.1007/82_2016_501
Gerdes SY, Scholle MD, Campbell JW, Balázsi G, Ravasz E, Daugherty MD, Somera AL, Kyrpides NC, Anderson I, Gelfand MS, Bhattacharya A, Kapatral V, D’Souza M, Baev MV, Grechkin Y, Mseeh F, Fonstein MY, Overbeek R, Barabási AL, Oltvai ZN, Osterman AL (2003) Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol 185(19):5673–5684. doi:10.1128/JB.185.19.5673-5684.2003
Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H (2006) Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi:10.1038/msb4100050
Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, Chu AM, Connelly C, Davis K, Dietrich F, Dow SW, El Bakkoury M, Foury F, Friend SH, Gentalen E, Giaever G, Hegemann JH, Jones T, Laub M, Liao H, Liebundguth N, Lockhart DJ, Lucau-Danila A, Lussier M, M’Rabet N, Menard P, Mittmann M, Pai C, Rebischung C, Revuelta JL, Riles L, Roberts CJ, Ross-MacDonald P, Scherens B, Snyder M, Sookhai-Mahadeo S, Storms RK, Véronneau S, Voet M, Volckaert G, Ward TR, Wysocki R, Yen GS, Zimmermann K, Philippsen P, Johnston M, Davis RW (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285(5429):901–906. doi:10.1126/science.285.5429.901
Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418(6896):387–391. doi:10.1038/nature00935
Roemer T, Jiang B, Davison J, Ketela T, Veillette K, Breton A, Tandia F, Linteau A, Sillaots S, Marta C, Martel N, Veronneau S, Lemieux S, Kauffman S, Becker J, Storms R, Boone C, Bussey H (2003) Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol 50(1):167–181. doi:10.1046/j.1365-2958.2003.03697.x
Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat Rev Drug Discov 6(1):29–40. doi:10.1038/nrd2201
Tommasi R, Brown DG, Walkup GK, Manchester JI, Miller AA (2015) ESKAPEing the labyrinth of antibacterial discovery. Nat Rev Drug Discov 14(8):529–542. doi:10.1038/nrd4572
Lewis K (2012) Antibiotics: recover the lost art of drug discovery. Nature 485(7399):439–440. doi:10.1038/485439a
Baltz RH (2006) Marcel Faber roundtable: is our antibiotic pipeline unproductive because of starvation, constipation or lack of inspiration? J Ind Microbiol Biotechnol 33(7):507–513. doi:10.1007/s10295-005-0077-9
O’Shea R, Moser HE (2008) Physicochemical properties of antibacterial compounds: implications for drug discovery. J Med Chem 51(10):2871–2878. doi:10.1021/jm700967e
Brown DG, May-Dracka TL, Gagnon MM, Tommasi R (2014) Trends and exceptions of physical properties on antibacterial activity for Gram-positive and Gram-negative pathogens. J Med Chem 57(23):10144–10161. doi:10.1021/jm501552x
Projan SJ (2008) Whither antibacterial drug discovery? Drug Discov Today 13(7–8):279–280. doi:10.1016/j.drudis.2008.03.010
Srinivas N, Jetter P, Ueberbacher BJ, Werneburg M, Zerbe K, Steinmann J, Van der Meijden B, Bernardini F, Lederer A, Dias RL, Misson PE, Henze H, Zumbrunn J, Gombert FO, Obrecht D, Hunziker P, Schauer S, Ziegler U, Käch A, Eberl L, Riedel K, DeMarco SJ, Robinson JA (2010) Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 327(5968):1010–1013. doi:10.1126/science.1182749
Hagan CL, Wzorek JS, Kahne D (2015) Inhibition of the β-barrel assembly machine by a peptide that binds BamD. Proc Natl Acad Sci U S A 112(7):2011–2016. doi:10.1073/pnas.1415955112
Richter SG, Elli D, Kim HK, Hendrickx AP, Sorg JA, Schneewind O, Missiakas D (2013) Small molecule inhibitor of lipoteichoic acid synthesis is an antibiotic for Gram-positive bacteria. Proc Natl Acad Sci U S A 110(9):3531–3536. doi:10.1073/pnas.1217337110
Nayar AS, Dougherty TJ, Ferguson KE, Granger BA, McWilliams L, Stacey C, Leach LJ, Narita S, Tokuda H, Miller AA, Brown DG, McLeod SM (2015) Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay. J Bacteriol 197(10):1726–1734. doi:10.1128/JB.02552-14
McLeod SM, Fleming PR, MacCormack K, McLaughlin RE, Whiteaker JD, Narita S, Mori M, Tokuda H, Miller AA (2015) Small-molecule inhibitors of gram-negative lipoprotein trafficking discovered by phenotypic screening. J Bacteriol 197(6):1075–1082. doi:10.1128/JB.02352-14
Campbell J, Singh AK, Santa Maria Jr JP, Kim Y, Brown S, Swoboda JG, Mylonakis E, Wilkinson BJ, Walker S (2011) Synthetic lethal compound combinations reveal a fundamental connection between wall teichoic acid and peptidoglycan biosyntheses in Staphylococcus aureus. ACS Chem Biol 6(1):106–116. doi:10.1021/cb100269f
Mann PA, Müller A, Xiao L, Pereira PM, Yang C, Ho Lee S, Wang H, Trzeciak J, Schneeweis J, Dos Santos MM, Murgolo N, She X, Gill C, Balibar CJ, Labroli M, Su J, Flattery A, Sherborne B, Maier R, Tan CM, Black T, Onder K, Kargman S, Monsma Jr FJ, Pinho MG, Schneider T, Roemer T (2013) Murgocil is a highly bioactive staphylococcal-specific inhibitor of the peptidoglycan glycosyltransferase enzyme MurG. ACS Chem Biol 8(11):2442–2451. doi:10.1021/cb400487f
Huber J, Donald RG, Lee SH, Jarantow LW, Salvatore MJ, Meng X, Painter R, Onishi RH, Occi J, Dorso K, Young K, Park YW, Skwish S, Szymonifka MJ, Waddell TS, Miesel L, Phillips JW, Roemer T (2009) Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem Biol 16(8):837–848. doi:10.1016/j.chembiol.2009.05.012
Mott JE, Shaw BA, Smith JF, Bonin PD, Romero DL, Marotti KR, Miller AA (2008) Resistance mapping and mode of action of a novel class of antibacterial anthranilic acids: evidence for disruption of cell wall biosynthesis. J Antimicrob Chemother 62(4):720–729. doi:10.1093/jac/dkn261
Bouley R, Kumarasiri M, Peng Z, Otero LH, Song W, Suckow MA, Schroeder VA, Wolter WR, Lastochkin E, Antunes NT, Pi H, Vakulenko S, Hermoso JA, Chang M, Mobashery S (2015) Discovery of antibiotic (E)-3-(3-Carboxyphenyl)-2-(4-cyanostyryl)quinazolin-4(3H)-one. J Am Chem Soc 137(5):1738–1741. doi:10.1021/jacs.5b00056
Lee SH, Wang H, Labroli M, Koseoglu S, Zuck P, Mayhood T, Gill C, Mann P, Sher X, Ha S, Yang SW, Mandal M, Yang C, Liang L, Tan Z, Tawa P, Hou Y, Kuvelkar R, DeVito K, Wen X, Xiao J, Batchlett M, Balibar CJ, Liu J, Xiao J, Murgolo N, Garlisi CG, Sheth PR, Flattery A, Su J, Tan C, Roemer T (2016) TarO-specific inhibitors of wall teichoic acid biosynthesis restore β-lactam efficacy against methicillin-resistant staphylococci. Sci Transl Med 8(329):329ra32. DOI: 10.1126/scitranslmed.aad7364.
Swoboda JG, Meredith TC, Campbell J, Brown S, Suzuki T, Bollenbach T, Malhowski AJ, Kishony R, Gilmore MS, Walker S (2009) Discovery of a small molecule that blocks wall teichoic acid biosynthesis in Staphylococcus aureus. ACS Chem Biol 4(10):875–883. doi:10.1021/cb900151k
Wang H, Gill CJ, Lee SH, Mann P, Zuck P, Meredith TC, Murgolo N, She X, Kales S, Liang L, Liu J, Wu J, Santa Maria J, Su J, Pan J, Hailey J, Mcguinness D, Tan CM, Flattery A, Walker S, Black T, Roemer T (2013) Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem Biol 20(2):272–284. doi:10.1016/j.chembiol.2012.11.013
Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schäberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, Lewis K (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517(7535):455–459. doi:10.1038/nature14098
Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Morisaki JH, Kim J, Park S, Darwish M, Lee BC, Hernandez H, Loyet KM, Lupardus P, Fong R, Yan D, Chalouni C, Luis E, Khalfin Y, Plise E, Cheong J, Lyssikatos JP, Strandh M, Koefoed K, Andersen PS, Flygare JA, Wah Tan M, Brown EJ, Mariathasan S (2015) Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 527(7578):323–328. doi:10.1038/nature16057
Tokunaga M, Loranger JM, Wu HC (1983) Isolation and characterization of an Escherichia coli clone overproducing prolipoprotein signal peptidase. J Biol Chem 258(20):12102–12105
Dev IK, Harvey RJ, Ray PH (1985) Inhibition of prolipoprotein signal peptidase by globomycin. J Biol Chem 260(10):5891–5894
Xiao Y, Gerth K, Müller R, Wall D (2012) Myxobacterium-produced antibiotic TA (myxovirescin) inhibits type II signal peptidase. Antimicrob Agents Chemother 56(4):2014–2021. doi:10.1128/AAC.06148-11
Vogeley L, El Arnaout T, Bailey J, Stansfeld PJ, Boland C, Caffrey M (2016) Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 351(6275):876–880. doi:10.1126/science.aad3747
de Jonge BL, Walkup GK, Lahiri SD, Huynh H, Neckermann G, Utley L, Nash TJ, Brock J, San Martin M, Kutschke A, Johnstone M, Laganas V, Hajec L, Gu RF, Ni H, Chen B, Hutchings K, Holt E, McKinney D, Gao N, Livchak S, Thresher J (2013) Discovery of inhibitors of 4′-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob Agents Chemother 57(12):6005–6015. DOI: 10.1128/AAC.01661-13
Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K (2013) Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503(7476):365–370. doi:10.1038/nature12790
Brötz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H (2005) Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11(10):1082–1087. doi:10.1038/nm1306
Tomašić T, Šink R, Zidar N, Fic A, Contreras-Martel C, Dessen A, Patin D, Blanot D, Müller-Premru M, Gobec S, Zega A, Kikelj D, Peterlin Mašič L (2012) Dual inhibitor of MurD and MurE ligases from Escherichia coli and Staphylococcus aureus. ACS Med Chem Lett 3(8):626–630. doi:10.1021/ml300047h
Pasquina L, Santa Maria Jr JP, McKay Wood B, Moussa SH, Matano LM, Santiago M, Martin SE, Lee W, Meredith TC, Walker S (2016) A synthetic lethal approach for compound and target identification in Staphylococcus aureus. Nat Chem Biol 12(1):40–45. doi:10.1038/nchembio.1967
Haydon DJ, Stokes NR, Ure R, Galbraith G, Bennett JM, Brown DR, Baker PJ, Barynin VV, Rice DW, Sedelnikova SE, Heal JR, Sheridan JM, Aiwale ST, Chauhan PK, Srivastava A, Taneja A, Collins I, Errington J, Czaplewski LG (2008) An inhibitor of FtsZ with potent and selective anti-staphylococcal activity. Science 321(5896):1673–1675. doi:10.1126/science.1159961
Tan CM, Therien AG, Lu J, Lee SH, Caron A, Gill CJ, Lebeau-Jacob C, Benton-Perdomo L, Elsen N, Wu J, Deschamps K, Petcu M, Wong S, Daigneault E, Kramer S, Liang L, Maxwell E, Claveau D, Vaillencourt J, Skorey K, Tam J, Wang H, Meredith TC, Sillaots S, Wang-Jarantow L, Reid JC, Parthasarathy G, Sharma S, Baryshnikova A, Lumb KJ, Soisson SM, Roemer T (2012) Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci Transl Med 4(126):126ra35. doi:10.1126/scitranslmed.3003592
Mann PA, Müller A, Wolff KA, Fischmann T, Wang H, Reed P, Hou Y, Li W, Müller CE, Xiao J, Murgolo N, Sher X, Mayhood T, Sheth PR, Mirza A, Labroli M, Xiao L, McCoy M, Gill CJ, Pinho MG, Schneider T, Roemer T (2016) Chemical genetic analysis and functional characterization of staphylococcal wall teichoic acid 2-epimerases reveals unconventional antibiotic drug targets. PLoS Pathog 12(5):e1005585. doi:10.1371/journal.ppat.1005585
Zhu W, Zhang Y, Sinko W, Hensler ME, Olson J, Molohon KJ, Lindert S, Cao R, Li K, Wang K, Wang Y, Liu YL, Sankovsky A, de Oliveira CA, Mitchell DA, Nizet V, McCammon JA, Oldfield E (2013) Antibacterial drug leads targeting isoprenoid biosynthesis. Proc Natl Acad Sci U S A 110(1):123–128. doi:10.1073/pnas.1219899110
Farha MA, Czarny TL, Myers CL, Worrall LJ, French S, Conrady DG, Wang Y, Oldfield E, Strynadka NC, Brown ED (2015) Antagonism screen for inhibitors of bacterial cell wall biogenesis uncovers an inhibitor of undecaprenyl diphosphate synthase. Proc Natl Acad Sci U S A 112(35):11048–11053. doi:10.1073/pnas.1511751112
Czarny TL, Brown ED (2016) A small-molecule screening platform for the discovery of inhibitors of undecaprenyl diphosphate synthase. ACS Infect Dis 2(7):489–499. doi:10.1021/acsinfecdis.6b00044
Howe JA, Wang H, Fischmann TO, Balibar CJ, Xiao L, Galgoci AM, Malinverni JC, Mayhood T, Villafania A, Nahvi A, Murgolo N, Barbieri CM, Mann PA, Carr D, Xia E, Zuck P, Riley D, Painter RE, Walker SS, Sherborne B, de Jesus R, Pan W, Plotkin MA, Wu J, Rindgen D, Cummings J, Garlisi CG, Zhang R, Sheth PR, Gill CJ, Tang H, Roemer T (2015) Selective small-molecule inhibition of an RNA structural element. Nature 526(7575):672–677. doi:10.1038/nature15542
Howe JA, Xiao L, Fischmann TO, Wang H, Tang H, Villafania A, Zhang R, Barbieri CM, Roemer T (2016) Atomic resolution mechanistic studies of ribocil: a highly selective unnatural ligand mimic of the E. coli FMN riboswitch. RNA Biol 13(10):946–954. doi:10.1080/15476286.2016.1216304
Roemer T, Boone C (2013) Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol 9(4):222–231. doi:10.1038/nchembio.1205
Hillenmeyer ME, Fung E, WildenhainJ PSE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, Altman RB, Davis RW, Nislow C, Giaever G (2008) The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320(5874):362–365. doi:10.1126/science.1150021
Zlitni S, Ferruccio LF, Brown ED (2013) Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat Chem Biol 9(12):796–804. doi:10.1038/nchembio.1361
Yao J, Rock CO (2015) How bacterial pathogens eat host lipids: implications for the development of fatty acid synthesis therapeutics. J Biol Chem 290(10):5940–5946. doi:10.1074/jbc.R114.636241
Gründling A, Schneewind O (2007) Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc Natl Acad Sci U S A 104(20):8478–8483. doi:10.1073/pnas.0701821104
Oku Y, Kurokawa K, Matsuo M, Yamada S, Lee BL, Sekimizu K (2009) Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J Bacteriol 191(1):141–151. doi:10.1128/JB.01221-08
Beckmann I, Subbaiah TV, Stocker BAD (1964) Rough mutants of Salmonella typhimurium. II. Serological and chemical investigations. Nature 201:1299–1301
Nelson BW, Roantree RJ (1967) Analyses of lipopolysaccharides extracted from penicillin-resistant, serum-sensitive Salmonella mutants. J Gen Microbiol 48(2):179–188. doi:10.1099/00221287-48-2-179
Joiner KA, Schmetz MA, Sanders ME, Murray TG, Hammer CH, Dourmashkin R, Frank MM (1985) Multimeric complement component C9 is necessary for killing of Escherichia coli J5 by terminal attack complex C5b-9. Proc Natl Acad Sci U S A 82(14):4808–4812
Schiller NL (1988) Characterization of the susceptibility of Pseudomonas aeruginosa to complement-mediated killing: role of antibodies to the rough lipopolysaccharide on serum-sensitive strains. Infect Immun 56(3):632–639
Schiller NL, Joiner KA (1986) Interaction of complement with serum-sensitive and serum-resistant strains of Pseudomonas aeruginosa. Infect Immun 54(3):689–694
Greenfield LK, Whitfield C (2012) Synthesis of lipopolysaccharide O-antigens by ABC transporter-dependent pathways. Carbohydr Res 356:12–24. doi:10.1016/j.carres.2012.02.027
Phan MD, Peters KM, Sarkar S, Lukowski SW, Allsopp LP, Gomes Moriel D, Achard ME, Totsika M, Marshall VM, Upton M, Beatson SA, Schembri MA (2013) The serum resistome of a globally disseminated multidrug resistant uropathogenic Escherichia coli clone. PLoS Genet 9(10):e1003834. doi:10.1371/journal.pgen.1003834
Sarkar S, Ulett GC, Totsika M, Phan MD, Schembri MA (2014) Role of capsule and O-antigen in the virulence of uropathogenic Escherichia coli. PLoS One 9(4):e94786. doi:10.1371/journal.pone.0094786
Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C (2009) Systematic mapping of genetic interaction networks. Annu Rev Genet 43:601–625. doi:10.1146/annurev.genet.39.073003.114751
Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Pagé N, Robinson M, Raghibizadeh S, Hogue CW, Bussey H, Andrews B, Tyers M, Boone C (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294(5550):2364–2368. doi:10.1126/science.1065810
Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, Prinz J, St Onge RP, VanderSluis B, Makhnevych T, Vizeacoumar FJ, Alizadeh S, Bahr S, Brost RL, Chen Y, Cokol M, Deshpande R, Li Z, Lin ZY, Liang W, Marback M, Paw J, San Luis BJ, Shuteriqi E, Tong AH, van Dyk N, Wallace IM, Whitney JA, Weirauch MT, Zhong G, Zhu H, Houry WA, Brudno M, Ragibizadeh S, Papp B, Pál C, Roth FP, Giaever G, Nislow C, Troyanskaya OG, Bussey H, Bader GD, Gingras AC, Morris QD, Kim PM, Kaiser CA, Myers CL, Andrews BJ, Boone C (2010) The genetic landscape of the cell. Science 327(5964):425–431. doi:10.1126/science.1180823
Paradis-Bleau C, Markovski M, Uehara T, Lupoli TJ, Walker S, Kahne DE, Bernhardt TG (2010) Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143(7):1110–1120. doi:10.1016/j.cell.2010.11.037
Babu M, Díaz-Mejía JJ, Vlasblom J, Gagarinova A, Phanse S, Graham C, Yousif F, Ding H, Xiong X, Nazarians-Armavil A, Alamgir M, Ali M, Pogoutse O, Pe’er A, Arnold R, Michaut M, Parkinson J, Golshani A, Whitfield C, Wodak SJ, Moreno-Hagelsieb G, Greenblatt JF, Emili A (2011) Genetic interaction maps in Escherichia coli reveal functional crosstalk among cell envelope biogenesis pathways. PLoS Genet 7(11):e1002377. doi:10.1371/journal.pgen.1002377
Typas A, Nichols RJ, Siegele DA, Shales M, Collins SR, Lim B, Braberg H, Yamamoto N, Takeuchi R, Wanner BL, Mori H, Weissman JS, Krogan NJ, Gross CA (2008) High-throughput, quantitative analyses of genetic interactions in E. coli. Nat Methods 5(9):781–787
Peters JM, Colavin A, Shi H, Czarny TL, Larson MH, Wong S, Hawkins JS, Lu CH, Koo BM, Marta E, Shiver AL, Whitehead EH, Weissman JS, Brown ED, Qi LS, Huang KC, Gross CA (2016) A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 165(6):1493–1506. doi:10.1016/j.cell.2016.05.003
Roemer T, Schneider T, Pinho MG (2013) Auxiliary factors: a chink in the armor of MRSA resistance to β-lactam antibiotics. Curr Opin Microbiol 16(5):538–548. doi:10.1016/j.mib.2013.06.012
Lee SH, Jarantow-Wang L, Sillaots S, Cheng H, Meredith TC, Thompson J, Roemer T (2011) Antagonism of chemical genetic interaction networks resensitize MRSA to β-lactam antibiotics. Chem Biol 18(11):1379–1389. doi:10.1016/j.chembiol.2011.08.015
Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, C KG, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu Zy ZY, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW (2002) A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol 43(6):1387–1400. doi:10.1046/j.1365-2958.2002.02832.x
Xu HH, Trawick JD, Haselbeck RJ, Forsyth RA, Yamamoto RT, Archer R, Patterson J, Allen M, Froelich JM, Taylor I, Nakaji D, Maile R, Kedar GC, Pilcher M, Brown-Driver V, McCarthy M, Files A, Robbins D, King P, Sillaots S, Malone C, Zamudio CS, Roemer T, Wang L, Youngman PJ, Wall D (2010) Staphylococcus aureus TargetArray: comprehensive differential essential gene expression as a mechanistic tool to profile antibacterials. Antimicrob Agents Chemother 54(9):3659–3670. doi:10.1128/AAC.00308-10
Münch D, Roemer T, Lee SH, Engeser M, Sahl HG, Schneider T (2012) Identification and in vitro analysis of the GatD/MurT enzyme-complex catalyzing lipid II amidation in Staphylococcus aureus. PLoS Pathog 8(1):e1002509. doi:10.1371/journal.ppat.1002509
Drawz SM, Bonomo RA (2010) Three decades of β-lactamase inhibitors. Clin Microbiol Rev 23(1):160–201. doi:10.1128/CMR.00037-09
D’Elia MA, Pereira MP, Chung YS, Zhao W, Chau A, Kenney TJ, Sulavik MC, Black TA, Brown ED (2006) Lesions in teichoic acid biosynthesis in Staphylococcus aureus lead to a lethal gain of function in the otherwise dispensable pathway. J Bacteriol 188:4183–4189. doi:10.1128/JB.00197-06
D’Elia MA, Millar KE, Beveridge TJ, Brown ED (2006) Wall teichoic acid polymers are dispensable for cell viability in Bacillus subtilis. J Bacteriol 188:8313–8316. doi:10.1128/JB.01336-06
Sewell EW, Brown ED (2014) Taking aim at wall teichoic acid synthesis: new biology and new leads for antibiotics. J Antibiot (Tokyo) 67(1):43–51. doi:10.1038/ja.2013.100
Pasquina LW, Santa Maria JP, Walker S (2013) Teichoic acid biosynthesis as an antibiotic target. Curr Opin Microbiol 16(5):531–537. doi:10.1016/j.mib.2013.06.014
Schirner K, Stone LK, Walker S (2011) ABC transporters required for export of wall teichoic acids do not discriminate between different main chain polymers. ACS Chem Biol 6(5):407–412. doi:10.1021/cb100390w
Cordes EH (2014) Hallelujah moments: tales of drug discovery (chapter 9). Oxford University Press, New York
Bigger JW (1944) Treatment of staphylococcal infections with penicillin. Lancet 244:497–500
Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K (2004) Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett 230(1):13–18. doi:10.1016/S0378-1097(03)00856-5
Lewis K (2010) Persister cells. Annu Rev Microbiol 64:357–372. doi:10.1146/annurev.micro.112408.134306
Sass P, Josten M, Famulla K, Schiffer G, Sahl HG, Hamoen L, Brötz-Oesterhelt H (2011) Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc Natl Acad Sci U S A 108(42):17474–17479. doi:10.1073/pnas.1110385108
Wu T, McCandlish AC, Gronenberg LS, Chng SS, Silhavy TJ, Kahne D (2006) Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A 103(31):11754–11759. DOI: 10.1073/pnas.0604744103
Koehn FE, Carter GT (2005) Rediscovering natural products as a source of new drugs. Discov Med 5(26):159–164
Schneider T, Kruse T, Wimmer R, Wiedemann I, Sass V, Pag U, Jansen A, Nielsen AK, Mygind PH, Raventós DS, Neve S, Ravn B, Bonvin AM, De Maria L, Andersen AS, Gammelgaard LK, Sahl HG, Kristensen HH (2010) Plectasin, a fungal defensin, targets the bacterial cell wall precursor Lipid II. Science 328(5982):1168–1172. doi:10.1126/science.1185723
Chu J, Vila-Farres X, Inoyama D, Ternei M, Cohen LJ, Gordon EA, Reddy BV, Charlop-Powers Z, Zebroski HA, Gallardo-Macias R, Jaskowski M, Satish S, Park S, Perlin DS, Freundlich JS, Brady SF (2016) Discovery of MRSA active antibiotics using primary sequence from the human microbiome. Nat Chem Biol 12:1004–1006. doi:10.1038/nchembio.2207
Silhavy TJ, Kahne D, Walker S (2010) The bacterial cell envelope. Cold Spring Harb Perspect Biol 2(5):a000414. doi:10.1101/cshperspect.a000414
Bakelar J, Buchanan SK, Noinaj N (2016) The structure of the β-barrel assembly machinery complex. Science 351(6269):180–186. doi:10.1126/science.aad3460
Noinaj N, Kuszak AJ, Gumbart JC, Lukacik P, Chang H, Easley NC, Lithgow T, Buchanan SK (2013) Structural insight into the biogenesis of β-barrel membrane proteins. Nature 501(7467):385–390. doi:10.1038/nature12521
Lee J, Xue M, Wzorek JS, Wu T, Grabowicz M, Gronenberg L, Sutterlin HA, Davis RM, Ruiz N, Silhavy TJ, Kahne DE (2016) Characterization of a stalled complex on the β-barrel assembly machine. Proc Natl Acad Sci U S A 113(31):8717–8722. doi:10.1073/pnas.1604100113
Dong H, Xiang Q, Gu Y, Wang Z, Paterson NG, Stansfeld PJ, He C, Zhang Y, Wang W, Dong C (2014) Structural basis for outer membrane lipopolysaccharide insertion. Nature 511(7507):52–56. doi:10.1038/nature13464
Qiao S, Luo Q, Zhao Y, Zhang XC, Huang Y (2014) Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature 511(7507):108–111. doi:10.1038/nature13484
Freinkman E, Chng SS, Kahne D (2011) The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc Natl Acad Sci U S A 108(6):2486–2491. DOI: 10.1073/pnas.1015617108
Chimalakonda G, Ruiz N, Chng SS, Garner RA, Kahne D, Silhavy TJ (2011) Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A 108(6):2492–2497. doi:10.1073/pnas.1019089108
Silver LL (2011) Challenges of antibacterial discovery. Clin Microbiol Rev 24(1):71–109. doi:10.1128/CMR.00030-10
Silver LL (2016) Appropriate targets for antibacterial drugs. Cold Spring Harb Perspect Med. pii: a030239. DOI: 10.1101/cshperspect.a030239
O'Dwyer K, Spivak AT, Ingraham K, Min S, Holmes DJ, Jakielaszek C, Rittenhouse S, Kwan AL, Livi GP, Sathe G, Thomas E, Van Horn S, Miller LA, Twynholm M, Tomayko J, Dalessandro M, Caltabiano M, Scangarella-Oman NE, Brown JR (2015) Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrob Agents Chemother 59(1):289–298. doi:10.1128/AAC.03774-14
Thulin E, Sundqvist M, Andersson DI (2015) Amdinocillin (mecillinam) resistance mutations in clinical isolates and laboratory-selected mutants of Escherichia coli. Antimicrob Agents Chemother 59(3):1718–1727. doi:10.1128/AAC.04819-14
Sakoulas G, Okumura CY, Thienphrapa W, Olson J, Nonejuie P, Dam Q, Dhand A, Pogliano J, Yeaman MR, Hensler ME, Bayer AS, Nizet V (2014) Nafcillin enhances innate immune-mediated killing of methicillin-resistant Staphylococcus aureus. J Mol Med (Berl) 92(2):139–149. doi:10.1007/s00109-013-1100-7
Weidenmaier C, Peschel A (2008) Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol 6:276–287. doi:10.1038/nrmicro1861
Weidenmaier C, Kokai-Kun JF, Kristian SA, Chanturiya T, Kalbacher H, Gross M, Nicholson G, Neumeister B, Mond JJ, Peschel A (2004) Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat Med 10(3):243–245. doi:10.1038/nm991
Weidenmaier C, Peschel A, Xiong YQ, Kristian SA, Dietz K, Yeaman MR, Bayer AS (2005) Lack of wall teichoic acids in Staphylococcus aureus leads to reduced interactions with endothelial cells and to attenuated virulence in a rabbit model of endocarditis. J Infect Dis 191(10):1771–1777. doi:10.1086/429692
Alexander DC, Valvano MA (1994) Role of the rfe gene in the biosynthesis of the Escherichia coli O7-specific lipopolysaccharide and other O-specific polysaccharides containing N-acetylglucosamine. J Bacteriol 176(22):7079–7084
Walsh SI, Craney A, Romesberg FE (2016) Not just an antibiotic target: exploring the role of type I signal peptidase in bacterial virulence. Bioorg Med Chem 24:6370–6378. doi:http://dx.doi.org/10.1016/j.bmc.2016.09.048
Brown DG (2016) Drug discovery strategies to outer membrane targets in Gram-negative pathogens. Bioorg Med Chem. pii: S0968-0896(16)30325-X. DOI: 10.1016/j.bmc.2016.05.004
Brown S, Santa Maria JP, Walker S (2013) Wall teichoic acids of Gram-positive bacteria. Annu Rev Microbiol 67:313–336. doi:10.1146/annurev-micro-092412-155620
D’Elia MA, Pereira MP, Brown ED (2009) Are essential genes really essential? Trends Microbiol 17:433–438. doi:10.1016/j.tim.2009.08.005
O’Neil PK, Rollauer SE, Noinaj N, Buchanan SK (2015) Fitting the pieces of the β-barrel assembly machinery complex. Biochemistry 54(41):6303–6311. doi:10.1021/acs.biochem.5b00852
Plummer AM, Fleming KG (2016) From chaperones to the membrane with a BAM! Trends Biochem Sci 41(10):872–882. doi:10.1016/j.tibs.2016.06.005
Simpson BW, May JM, Sherman DJ, Kahne D, Ruiz N (2015) Lipopolysaccharide transport to the cell surface: biosynthesis and extraction from the inner membrane. Philos Trans R Soc Lond B Biol Sci. 370(1679). pii: 20150029. DOI: 10.1098/rstb.2015.0029
May JM, Sherman DJ, Simpson BW, Ruiz N, Kahne D (2015) Lipopolysaccharide transport to the cell surface: periplasmic transport and assembly into the outer membrane. Philos Trans R Soc Lond B Biol Sci. 370(1679). pii: 20150027. DOI: 10.1098/rstb.2015.0027
Konovalova A, Silhavy TJ (2015) Outer membrane lipoprotein biogenesis: Lol is not the end. Philos Trans R Soc Lond B Biol. 370(1679) pii: 20150030. DOI: 10.1098/rstb.2015.0030
Whitfield C, Trent MS (2014) Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem 83:99–128. doi:10.1146/annurev-biochem-060713-035600
Typas A, Banzhaf M, Gross CA, Vollmer W (2011) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10(2):123–136. doi:10.1038/nrmicro2677
Lovering AL, Safadi SS, Strynadka NC (2012) Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81:451–478. doi:10.1146/annurev-biochem-061809-112742
Woodward L, Naismith JH (2016) Bacterial polysaccharide synthesis and export. Curr Opin Struct Biol 40:81–88. doi:10.1016/j.sbi.2016.07.016
Rowley G, Spector M, Kormanec J, Roberts M (2006) Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol 4(5):383–394. doi:10.1038/nrmicro1394
Guest RL, Raivio TL (2016) Role of the Gram-negative envelope stress response in the presence of antimicrobial agents. Trends Microbiol 24(5):377–390. doi:10.1016/j.tim.2016.03.001
Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67(4):593–656. doi:10.1128/MMBR.67.4.593-656.2003
Gray AN, Egan AJ, Van't Veer IL, Verheul J, Colavin A, Koumoutsi A, Biboy J, Altelaar AF, Damen MJ, Huang KC, Simorre JP, Breukink E, den Blaauwen T, Typas A, Gross CA, Vollmer W (2015) Coordination of peptidoglycan synthesis and outer membrane constriction during Escherichia coli cell division. eLife 4. DOI: 10.7554/eLife.07118
Typas A, Banzhaf M, van den Berg van Saparoea B, Verheul J, Biboy J, Nichols RJ, Zietek M, Beilharz K, Kannenberg K, von Rechenberg M, Breukink E, den Blaauwen T, Gross CA, Vollmer W (2010) Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143(7):1097–1109. doi:10.1016/j.cell.2010.11.038
Rigel NW, Schwalm J, Ricci DP, Silhavy TJ (2012) BamE modulates the Escherichia coli β-barrel assembly machine component BamA. J Bacteriol 194(5):1002–1008. doi:10.1128/JB.06426-11
Rigel NW, Ricci DP, Silhavy TJ (2013) Conformation-specific labeling of BamA and suppressor analysis suggest a cyclic mechanism for β-barrel assembly in Escherichia coli. Proc Natl Acad Sci U S A 110(13):5151–5156. doi:10.1073/pnas.1302662110
Cuthbertson L, Powers J, Whitfield C (2005) The C-terminal domain of the nucleotide-binding domain protein Wzt determines substrate specificity in the ATP-binding cassette transporter for the lipopolysaccharide O-antigens in Escherichia coli serotypes O8 and O9a. J Biol Chem 280(34):30310–30319. doi:10.1074/jbc.M504371200
Silver LL (2016) A Gestalt approach to Gram-negative entry. Bioorg Med Chem 24:6379–6389. doi:http://dx.doi.org/10.1016/j.bmc.2016.06.044
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG, part of Springer Nature
About this chapter
Cite this chapter
Sutterlin, H.A., Malinverni, J.C., Lee, S.H., Balibar, C.J., Roemer, T. (2017). Antibacterial New Target Discovery: Sentinel Examples, Strategies, and Surveying Success. In: Fisher, J.F., Mobashery, S., Miller, M.J. (eds) Antibacterials. Topics in Medicinal Chemistry, vol 25. Springer, Cham. https://doi.org/10.1007/7355_2016_31
Download citation
DOI: https://doi.org/10.1007/7355_2016_31
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-68096-5
Online ISBN: 978-3-319-68097-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)