
Abstract
Bone loss during advancing age is the net result of reduced modeling-based bone formation upon the outer (periosteal) envelope and unbalanced remodeling by basic multicellular units (BMUs) upon the three (intracortical, endocortical, and trabecular) components of the inner (endosteal) bone envelope. Each BMU deposits less bone than resorbed, reducing total bone volume and deteriorating the microstructure of the diminished residual bone volume.
Antiresorptive agents like bisphosphonates reduce, but do not abolish, the rate of bone remodeling – fewer BMUs remodel, “turn over,” the volume of bone. Residual unbalanced remodeling continues to slowly reduce total bone volume and deteriorate bone microstructure. By contrast, denosumab virtually abolishes remodeling so the decrease in bone volume and the deterioration in microstructure cease. The less remodeled matrix remains, leaving more time to complete the slow process of secondary mineralization which reduces the heterogeneity of matrix mineralization and allows it to become glycosylated, changes that may make the smaller and microstructurally deteriorated bone volume more brittle. Neither class of antiresorptive restores bone volume or its microstructure, despite increases in bone mineral density misleadingly suggesting otherwise. Nevertheless, these agents reduce vertebral and hip fractures by 50–60% but only reduce nonvertebral fractures by 20–30%.
Restoring bone volume, microstructure, and material composition, “curing” bone fragility, may be partly achieved using anabolic therapy. Teriparatide, and probably abaloparatide, produce mainly remodeling-based bone formation by acting on BMUs existing in their resorption, reversal, or formation phase at the time of treatment and by promoting bone formation in newly initiated BMUs. Romosozumab produces modeling-based bone formation almost exclusively and decreases the surface extent of bone resorption. All three anabolic agents reduce vertebral fracture risk relative to untreated controls; parathyroid hormone 1-34 and romosozumab reduce vertebral fracture risk more greatly than risedronate or alendronate, respectively. Evidence for nonvertebral or hip fracture risk reduction relative to untreated or antiresorptive-treated controls is lacking or inconsistent. Only one study suggests sequential romosozumab followed by alendronate reduces vertebral, nonvertebral, and hip fracture risk compared to continuous alendronate alone. Whether combined antiresorptive and anabolic therapy result in superior fracture risk reduction than monotherapy is untested.
We shall not cease from exploration
And the end of all our exploring
Will be to arrive where we started
And know the place for the first time.
T.S. Eliot, Little Gidding, The Four Quartets
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
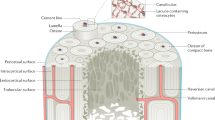
Bone remodeling is balanced during young adulthood; equal volumes of bone are resorbed and replaced by each basic multicellular unit (BMU) (Hattner et al. 1965). Around midlife, remodeling becomes unbalanced; each BMU resorbs a smaller volume of bone at discrete points upon the three (intracortical, endocortical, and trabecular) components of the endosteal (inner) surface of the bone, but the volume of new bone subsequently deposited is even less than the volume resorbed (Lips et al. 1978). In women, estrogen deficiency associated with menopause worsens age-related remodeling imbalance because the life span of osteoclasts increases, resulting in an increased volume of bone resorbed, while the life span of osteoblasts decreases further (Manolagas 2000). Less bone is deposited in a larger cavity worsening the focal net volume deficit. Bone loss and microstructural deterioration are amplified by the increased birth rate of BMUs associated with estrogen deficiency; more unbalanced remodeling events occur upon the three components of the endosteal surface, renewing, “turning over,” and eroding, a larger proportion of the ever-diminishing total bone volume (Parfitt 1984).
This remodeling imbalance is the necessary and sufficient morphological basis of bone loss (Parfitt 1984). Remodeling imbalance upon the intracortical surfaces of the Haversian canals enlarges them focally producing more surface upon which unbalanced remodeling can be initiated (Bjornerem et al. 2011). Canals traversing the inner cortex (adjacent to the medullary canal) coalesce, cavitating the cortex, a morphological effect that contributes more to cortical thinning than unbalanced endocortical remodeling (which enlarges the medullary canal) (Zebaze et al. 2010). Age-related resorption upon trabeculae causes them to thin. Trabeculae are lost after menopause as the larger resorption cavities perforate and remove them completely (Dempster et al. 1995). Trabeculae become less interconnected and more rod-like (Dempster et al. 1995). Loss of trabeculae with their surfaces results in slowing or cessation of trabecular bone remodeling because there is no longer a surface for remodeling to be initiated upon. Total bone surface may remain unchanged or increase, but it is now located in the cortical compartment (Bjornerem et al. 2011). Intracortical remodeling intensity increases, and bone loss becomes predominantly cortical (Zebaze et al. 2010). There is little trabecular bone remaining, and the more rapid bone loss erodes an ever-decreasing cortical bone volume predisposing to nonvertebral fractures.
Bone resorption by the BMU affects morphology at higher levels of resolution. For example, the radial extent of resorption defines the diameter of osteons in cortical bone and hemiosteons formed upon the endocortical and trabecular surfaces. The radial extent of resorption is delineated by the cement line which separates osteons from each other and the interosteonal (interstitial) bone matrix (Mohsin et al. 2006). The age-related reduction in the volume of bone resorbed by each BMU produces smaller osteons, whereas after menopause, the longer living osteoclasts excavate larger cavities producing larger osteons with a larger central canal (produced by the reduction in bone formation by each BMU) with fewer lamellae and a reduction in the population of osteocytes per osteon. The diminishing total cortical bone volume becomes more porous, there are more osteons of varying size, and the matrix mineral density of the diminishing total bone volume decreases due to rapid replacement of older more mineralized bone with younger less mineralized bone (Bjornerem et al. 2018). Advanced glycation end products increase collagen cross-linking compromising matrix ductility (Poundarik et al. 2015; Saito et al. 2008; Tang et al. 2007; Vashishth 2007). The resulting decline in total bone volume, microstructural deterioration, and altered material properties compromise bone strength predisposing to fractures (Seeman and Delmas 2006).
The loss of bone from the three components of the endosteal envelope proceeds with a concurrent decrease in periosteal modeling-based bone formation (Seeman 2003). Whether there is also modeling upon the endosteal envelope is uncertain and is difficult to establish in the face of rapid unbalanced remodeling which is likely to erode any concurrent modeling-based bone formation (Ominsky et al. 2015). Whatever the case, the cellular mechanisms determining the volumes of bone resorbed and formed by each BMU, the birth rate of the these BMUs, concurrent age-related reduction in periosteal and endosteal modeling-based bone formation, the compromised bone volume, microstructure, osteocyte population, and the material properties of bone are each rational targets for the prevention and reversal of bone fragility (Seeman and Martin 2015, 2019) (Fig. 1).

Drug therapy targets the cellular activity of the basic multicellular units (BMUs) and the surface extent of this activity upon the periosteal and three components of the endosteal surface. Antiresorptive agents reduce the volume of bone resorbed by each BMU and reduce the number of BMUs producing unbalanced remodeling and thereby slow bone loss. Anabolic agents may increase the volume of bone formed by each BMU and increase bone formation upon the periosteal and the endosteal surfaces
2 Antiresorptive Therapy
Antiresorptive agents are remodeling suppressants (Russell et al. 2008). These agents probably reduce the volume of bone resorbed by each BMU (Allen et al. 2010), but the most important benefit these agents confer is that they reduce the rate of unbalanced remodeling. The bisphosphonates, and weaker remodeling suppressants like selective estrogen receptor modulators (SERMs) and calcium supplements, slow unbalanced bone remodeling; they do not abolish it (Russell et al. 2008). Slowing remodeling reduces the rate of decrease in bone volume and microstructural deterioration, but the unsuppressed unbalanced remodeling continues to deteriorate the skeleton, albeit more slowly than without treatment. Denosumab, the most efficacious remodeling suppressant, virtually abolishes remodeling by inhibiting existing BMUs in their resorptive phase at the time of starting treatment and by preventing differentiation of precursors into mature bone-resorbing osteoclasts (McClung et al. 2006). Any further decrease in total bone volume and microstructural deterioration virtually stops.
Antiresorptive agents are not anabolic; they do not restore the reduced bone volume or microstructural deterioration present at the time of initiating treatment. This important limitation is not obvious because of the misleading effect antiresorptive agents have on bone mineral density (BMD). Slowing of the rate of remodeling by 50–60% using bisphosphonates or by 20–30% with SERMs or calcium supplements is expected to slow the decline in BMD. Stopping bone loss using denosumab should stop the decline in BMD.
Neither slowing nor stopping the decline in BMD is observed. On the contrary, there is a rapid initial increase in BMD during the first 6–12 months of therapy, a change erroneously interpreted as being an increase in bone matrix mass or volume (Fig. 2). This is a net increase, the result of four events produced by interrupting steady-state remodeling. At the onset of treatment surface-level remodeling is perturbed. There is a prompt reduction in the number of new BMUs excavating cavities upon the endosteal surface (which does not increase BMD). The cavities may be smaller because of a reduction in the volume of bone resorbed by bisphosphonates and, probably, denosumab (Allen et al. 2010). Concurrently, the many more cavities excavated upon the endosteal surface weeks before starting treatment enter their formation phase and refill (but still do so incompletely because remodeling imbalance is not corrected). The matrix of the many cavities incompletely refilling undergoes primary mineralization within days of being deposited, while matrix deposited weeks to months earlier (but no longer being remodeled) undergoes more complete secondary mineralization (a process taking many months if not years to complete) (Akkus et al. 2003).

(a) Before treatment. Remodeling is rapid and in steady state, similar numbers of BMUs resorb a volume of bone as BMUs refilling, but the filling is incomplete, so BMD decreases. (b) Early antiresorptive therapy. Remodeling is perturbed. Fewer BMUs excavate cavities, and each cavity is smaller. Concurrently, the many more BMUs excavating bone before treatment now refill, but incompletely (curved green arrows). Note that the number of BMUs refilling is similar in the treated and control groups (sloping white arrows). There is a rapid net increase in BMD (see text). (c) In the intermediate term, during the second and third year, remodeling returns to steady state but at a slower rate. The fewer and smaller cavities excavated during early antiresorptive treatment refill incompletely as similar numbers of new BMUs excavate smaller cavities. (d) In the longer term, (1) with denosumab treatment there is almost complete remodeling suppression, so BMD increases due to refilling of cavities excavated before treatment unopposed by the appearance of new cavities; secondary mineralization of the unremodeled matrix (depicted as whitening of orange matrix); and possible modeling-based bone formation (see text). (2) With bisphosphonates, secondary mineralization reaches completion, and BMD stabilizes. Cortical matrix volume may decrease, but this is not captured by the BMD measurement (see text). (3) Weak antiresorptives reduce remodeling by only 10–20%, so most unbalanced remodeling continues to reduce bone volume and cause microstructural deterioration
These four morphological features (newly excavated cavities, incompletely refilling cavities, incomplete primary and secondary mineralization) are part of the reversible remodeling space deficit in matrix and mineral produced by the normal delay in onset and slowness of the formation phase of remodeling and slowness of matrix mineralization (Parfitt 1980). Whether this early increase in BMD is mainly due to early bone matrix deposition or matrix mineralization is not known. It is unlikely to be matrix deposition because this is no different to the matrix deposition in controls at this stage. What differs from controls is the fewer cavities being excavated and the mineralization of the less remodeled bone matrix.
The increase in BMD may correlate with fracture risk reduction (Bouxsein et al. 2019), but that does not necessarily mean this is a causal relationship. Indeed, the morphological basis of the fracture risk reduction relative to controls is incompletely understood but may include (1) the appearance of fewer new stress risers; (2) less stress imposed by cavities excavated before treatment because they partly refill; (3) less decrease in bone volume and microstructural deterioration relative to controls; and (4) initial increases in matrix mineral content which correct the reduced matrix mineral content produced by rapid matrix turnover. However, in the longer term, complete matrix mineralization may produce brittleness – loss of ductility (Akkus et al. 2003; Lloyd et al. 2017).
None of the changes restore microstructural deterioration, the consequence of remodeling imbalance. Restoring bone volume and microstructure can only be achieved using bone-forming agents. The initial rise in BMD is less with weaker antiresorptives because 70–80% of the pre-treatment remodeling rate remains unsuppressed and continues to produce bone loss and renew bone matrix so that secondary mineralization does not increase or does so minimally (again, at this early stage, the number of cavities refilling incompletely is no different in treated subjects and untreated controls) (Reid et al. 2008; Silverman et al. 2012) (Fig. 2b).
After about 6–12 months, perturbed remodeling (with more BMUs incompletely refilling cavities than BMUs excavating cavities) returns to steady state (Fig. 2c). Now, approximately equal numbers of cavities are incompletely refilling and being excavated, as was the case before treatment (except now remodeling proceeds more slowly and at a rate determined by the efficacy of the antiresorptive agent being administered). The incomplete refilling reduces total bone volume and causes microstructural deterioration, but this proceeds at a lower rate than in controls. The deterioration is undetectable using bisphosphonates because secondary mineralization of the declining total bone matrix volume is likely to obscure it.
Indeed, BMD continues to increase, more slowly than it did during the first 6–12 months of treatment, but eventually plateaus after about 3–4 years (Fig. 2d). The continued bone loss and microstructural deterioration is mainly cortical because bisphosphonates bind avidly to mineral in superficial subperiosteal and subendosteal bone and fail to penetrate and distribute in high concentration in deeper cortical bone; osteoclasts engulfing matrix free of bisphosphonate or having low concentration of the drug continue to resorb bone. The loss of total bone matrix volume is greater using weak antiresorptives. The decrease in BMD after about 12 months of treatment, reflecting a decrease in bone volume, is detectable because 70–80% of pre-treatment unbalanced remodeling continues when weaker antiresorptives are used (Reid et al. 2008; Silverman et al. 2012) (Fig. 2d). There is no, or little, increase in matrix mineralization to obscure the decrease in bone volume because the bone volume is not resident long enough to undergo complete mineralization.
When the antiresorptive agent used is denosumab, the changes are different. This drug virtually abolishes remodeling, so the reduction in bone matrix volume and worsening of microstructural deterioration cease. Instead of newly excavated cavities appearing as with bisphosphonates or weak antiresorptives, incomplete refilling of cavities excavated before treatment do so without being offset by the concurrent appearance of new cavities so total bone matrix volume may increase because the reduction of the reversible remodeling space deficit in matrix and mineral is more complete. This increase is not anabolism, the formation of new bone upon the periosteal surface enlarging the external perimeter of the bone; upon the intracortical surface of Haversian canals, narrowing them; upon the endocortical and trabecular surfaces thickening the cortices and trabeculae, respectively.
BMD continues to increase using denosumab beyond 5 years of treatment. While this might be the result of more complete secondary mineralization, there is evidence suggesting matrix mineral density does not increase beyond 5 years (Dempster et al. 2018). There is another reason BMD may continue to increase. Periosteal and perhaps endosteal bone modeling continue throughout adult life (Ominsky et al. 2015). Bone formation by endosteal bone modeling is either obscured or lost after menopause perhaps because the rapid unbalanced remodeling removes it. Studies in nonhuman primates suggest that modest endosteal modeling-based bone formation during adulthood is not removed because remodeling is virtually completely suppressed during denosumab therapy (Ominsky et al. 2015). Under these circumstances, total bone matrix volume might increase.
Modeling-based bone formation upon the endosteal surface is held to explain the continued increase in BMD beyond 5 years using denosumab despite the absence of comparable data in human subjects. It is also held to account for the low fracture rates during prolonged therapy up to 10 years. However, this interpretation is also not well founded for several reasons. The study was not placebo controlled beyond 3 years, and there was significant loss of the inception cohort so it is plausible that the low fracture rates may be the result of healthy user bias (Bone et al. 2017). There is no published evidence of a modeling effect upon the endosteal envelope in human subjects, and whatever the effect, evidence is needed that it is responsible for the low fracture incidence.
3 Anabolic Therapy
Reconstruction of the skeleton, “curing” bone fragility, requires anabolic therapy. There are three obvious targets for anabolic therapy – reduced remodeling-based bone formation by each BMU and reduced modeling-based bone formation upon the periosteal and each of the three (intracortical, endocortical, trabecular) components of the endosteal surface.
If anabolic agents increase remodeling-based bone formation, the increased volume of bone formed by each BMU might produce a positive BMU balance. If so, it is advantageous to increase the birth rate of BMUs. Widespread positively balanced remodeling events at many points upon the three components of the endosteal surface should focally increase bone volume of remaining trabeculae; thicken the cortex focally, thereby increasing compressive strength; and reduce intracortical porosity. Teriparatide and probably abaloparatide address some of these mechanisms.
Modeling-based bone formation upon the periosteal surface is a biomechanically advantageous location because resistance to bending is a fourth-power function of the radial distance of a unit bone volume from the neutral axis of a bone (Ruff and Hayes 1988). Modeling-based bone formation upon the endocortical surface may thicken the cortices increasing compressive strength, modeling-based bone formation upon trabecular surfaces will thicken trabeculae and may improve connectivity of trabecular still interconnected. Whether trabecular number can be increased has been proposed by splitting of trabeculae (Jiang et al. 2003), but otherwise, a mechanism for this is yet to be identified. Modeling upon surfaces of the intracortical canals may reduce cortical porosity. Romosozumab addresses some of these mechanisms (Chavassieux et al. 2019).
3.1 Primarily Remodeling-Based Anabolic Agents
3.1.1 Teriparatide (PTH 1-34)
PTH 1-34 increases bone matrix volume primarily by a remodeling-based anabolic action on cells of existing BMUs and new BMUs generated by its administration. This accounts for 70–80% of the anabolic action. Modeling-based anabolic effects initiated upon quiescent bone surfaces appear to contribute only 20–30% of the total anabolic action (Ma et al. 2006).
At any time, existing BMUs are more likely to be at various stages of their formation than resorption phases of their remodeling cycle because bone formation proceeds for ~3 months, whereas the resorption phase is ~3 weeks, and reversal phase is ~1 week (Hattner et al. 1965; Parfitt 2008; Tran Van et al. 1982). PTH 1-34 acting on BMUs in their reversal phase may promote differentiation of osteoblast lineage cells into mature osteoid-producing forms. Concurrent increases in matrix production occur as PTH 1-34 inhibits apoptosis of mature osteoblasts which synthesize osteoid overfilling resorption cavities upon trabecular and endocortical surfaces with spillover of newly formed osteoid onto adjacent quiescent surfaces.
Thus, the early response to PTH 1-34 is likely to be modeling-based bone formation upon quiescent bone surfaces with a concurrent increase in remodeling-based bone formation by existing BMUs, both increasing circulating P1NP. Presumably, the higher the baseline pre-treatment remodeling rate, the greater the numbers of BMUs in their formation phase facilitating this anabolic action (Chen et al. 2005). In addition, and again, concurrently, or soon after starting treatment, PTH 1-34 initiates new remodeling events by promoting osteocyte and osteoblast precursor production of RANKL, osteoclast formation, and bone resorption with a later rise in circulating CTX (Seeman and Martin 2015).
What determines the number and activity of new BMUs initiated by PTH 1-34 is not known, but they first enter their resorptive phase resulting in removal of mineralized bone, increased intracortical porosity, and excavated cavities upon endocortical and trabecular surfaces with later deposition of osteoid. These changes are likely to be accompanied by an increase in serum CTX, an event that does not necessarily signal the end of bone formation and closure of the so-called anabolic window, a period of time of maximal anabolic effect of PTH 1-34 (Bilezikian 2008).
This “anabolic window” is an ambiguous notion. It suggests that the limited anabolic effect is due to bone resorption caused by newly generated BMUs and so a “window” of opportunity that can be left “open” by antiresorptive therapy (Bilezikian 2008). The ambiguity is the failure to distinguish between the absolute and net volume of bone formed. The net volume of bone deposited or resorbed is the sum of (1) the volume of bone formed by modeling initiated by PTH 1-34; (2) the volume of bone formed by BMUs existing in their resorption, reversal, or formation phase at the time PTH 1-34 is started; and (3) the volume of bone formed by new BMUs initiated by PTH 1-34 when they enter their reversal and formation phases minus (4) the volume of bone resorbed by new BMUs initiated by PTH 1-34 during their resorption phase (before entering their formation phase).
The notion of offsetting the absolute increase in bone formation using antiresorptive agents is also problematic. Preventing the birth of new BMUs (using denosumab) or aborting their progression (using bisphosphonates) is likely to deprive the skeleton of the bone formation phase of these BMUs for PTH 1-34 to act upon.
Perhaps most importantly, the notion of the anabolic window diverts attention away from deliberations concerning the potential major mechanisms responsible for the limited anabolic effect of PTH peptides. Is the limited anabolic effect the result of a reduced osteoblast precursor pool and reduced proliferation, differentiation, and lifespan of osteoblasts (Kim et al. 2012)? Is the opportunity for remodeling-based bone formation greater initially, when remodeling is rapid, providing many BMUs in their formation phase at the time of starting PTH 1-34? What determines the number of new BMUs generated by PTH 1-34, and is their bone forming potential less than that of the existing BMUs? If anabolic activity is less in the newly initiated BMUs, this may partly account for the waning of the anabolic effect. What limits the modeling-based anabolic effect?
The morphological changes documented using PTH 1-34 do not meet the theoretical potential envisaged. The resorption of mineralized bone by newly initiated BMUs, and deposition of newly synthesized osteoid during remodeling- and modeling-based bone formation, is likely to account for the decrease in BMD during early treatment, a transitory event because primary and secondary mineralization of the newly deposited bone follow (Seeman and Martin 2019). Whether newly excavated cavities temporarily increase bone fragility is not known. The best documented benefits of PTH 1-34 are thickening of existing trabeculae and endocortical bone formation. Another possible mechanism that needs investigation is “corticalization” of trabeculae abutting the cortex. As these trabeculae thicken, they may coalesce to become cortical in appearance, the opposite of “trabecularization” of the cortex during aging (Zebaze et al. 2010). There is evidence of periosteal apposition reported using histomorphometry (Lindsay et al. 2006, 2007), but whether this is sufficient to result in cortical thickening, increased periosteal perimeter, and increased bending strength is uncertain.
There are other potential benefits of remodeling-based bone formation, but these have not been rigorously studied and so remain speculative. For example, initiation of new BMUs is likely to result in the formation of new osteons formed by new osteoid and populated by newly formed osteocytes and younger less cross-linked and less fully mineralized matrix, improving the ductility of bone. Greater numbers of osteons may result in a greater proportion of the total bone volume being osteonal and hemiosteonal rather than interstitial, improving resistance to microcrack propagation. It has been suggested that glycosylation interferes with the effect of non-collagenous proteins like osteocalcin and osteopontin to defend mineral crystals against excessive loading (Fantner et al. 2005; Poundarik et al. 2015; Sroga and Vashishth 2012; Thomas et al. 2017). During loading, non-collagenous proteins uncoil, reducing the stress upon mineral platelets (the most brittle component of bone). Advanced glycation end products may interfere with uncoiling and contribute to diffuse damage within osteons, a form of damage that differs from microdamage in interosteonal bone (Saito and Marumo 2010; Seref-Ferlengez et al. 2014).
These bone qualities are relevant, especially when consideration is given to combining PTH 1-34 and antiresorptives because these agents influence the material composition and microstructure of bone differently, often in opposite directions. For example, antiresorptives slow or inhibit bone remodeling facilitating secondary mineralization which increases the homogeneity of the bone matrix mineralization density distribution (Fuchs et al. 2011). A homogeneous material offers less resistant to microcrack propagation and fracture than a heterogeneous material (Lloyd et al. 2017; Mohsin et al. 2006). By contrast PTH 1-34 removes mineralized bone, replacing it with younger less mineralized bone, increasing the heterogeneity of the material (Misof et al. 2003; Paschalis et al. 2005). Slowing or inhibition of bone remodeling may also lead to accumulation of advanced glycation end products, whereas treatment with PTH 1-34 reduces collagen cross-linking (Allen et al. 2008; Saito et al. 2011).
PTH 1-34 reduces vertebral fracture risk by 60–70% (Miller et al. 2016; Nakamura et al. 2012; Neer et al. 2001), with only one study reporting nonvertebral fracture risk reduction (Neer et al. 2001). Whether hip fracture risk is reduced is unknown because no properly designed studies have been done to assess this endpoint. For reasons that are not understood, cessation of PTH 1-34 is followed by bone loss, so that antiresorptive therapy is needed to prevent or minimize this bone loss (Cosman et al. 2001; Ejersted et al. 1998).
3.1.2 Abaloparatide
Abaloparatide shares many of its amino acid residues with PTH 1-34 and PTHrP. All three peptides mediate their effects through the PTHrPR1 receptor. Like PTH 1-34, this drug is likely to mediate its anabolic effects through both remodeling- and modeling-based mechanisms. There is no published histomorphometric evidence examining the contributions of these two mechanisms by quantifying the volumes of bone deposited upon crenated surfaces (reflecting remodeling-based bone formation) versus smooth surfaces (reflecting modeling-based bone formation) (Hattersley et al. 2016; Moreira et al. 2017; Varela et al. 2017).
Claims that the anabolic effect of abaloparatide is achieved with less resorptive activity are based on the study of aged rats that have little resorptive activity, as assessed using biochemical markers and histomorphometry (Varela et al. 2017). Anabolic effects of PTH 1-34 free of resorptive activity have frequently been reported using this model (Kimmel et al. 1993; Liu and Kalu 1990; Ma et al. 2011; Shen et al. 1992; Wronski et al. 1993). Abaloparatide has less effects on both circulating measurements, P1NP and CTX, than does PTH 1-34, with less increase in CTX relative to the increase in P1NP, an observation claimed to account for the 1–2% higher BMD achieved using abaloparatide (Eastell et al. 2019).
Making inferences about the net amount of bone formed relative to that resorbed based on circulating remodeling markers is problematic (Seeman and Nguyen 2016). The concentrations of these markers are determined by the rate of remodeling, by the volumes of bone resorbed and deposited by each BMU, and, in the case of P1NP, by modeling activity. A blood sample contains a concentration of CTX produced by BMUs currently in their resorption phase. That same sample contains a concentration of P1NP produced by different BMUs at different locations and at different stages of the formation phase as well as P1NP produced by concurrent modeling activity.
In a phase 3 clinical trial, 2,463 postmenopausal women were randomized to 18 months of abaloparatide (80 μg daily), placebo, or open-label PTH 1-34 (20 μg daily); 1901 completed the study (Miller et al. 2016). Compared to placebo, abaloparatide reduced morphometric vertebral fractures (relative risk 0.14; 95% CI 0.05–0.39, P < 0.001) and major osteoporotic fractures (hazard ratio (HR) 0.30; 95% CI 0.15–0.61, P < 0.001) but not nonvertebral fracture (after removal of study participants from two Czech sites) (EMA 2018) or hip fracture. Although vertebral antifracture efficacy was demonstrated, interpretation of these findings is challenging. The number of women who sustained fractures in both the placebo and PTH 1-34 groups was approximately double that of the abaloparatide group within the first few weeks of the study, events likely to be independent of the interventions yet influencing the number of fractures in the first 6 months and total event rates. These earlier fracture events may account for the overall perceived better antifracture efficacy of abaloparatide compared to PTH 1-34 as the number of women having fractures in the two groups in the last 12 months of the 18-month study was similar.
3.2 Modeling-Based Anabolic Therapy
3.2.1 Romosozumab
Sclerostin, the osteocyte-derived protein product of the sost gene, inhibits bone formation by inhibiting Wnt signaling. Loss-of-function sost mutations produce increased bone mass of sclerosteosis and van Buchem disease (Balemans et al. 2001, 2002). A single injection of sclerostin antibody stimulates bone formation in rats without changes in resorption parameters (Ke et al. 2012). A phase I study of a monoclonal anti-sclerostin antibody, AMG 785 (romosozumab), increased bone formation markers and decreased the resorption marker, serum CTX (Padhi et al. 2011). A 12-month phase II randomized, placebo-controlled, multi-dose study of romosozumab in 410 women confirmed the rapid increase in BMD (McClung et al. 2014). In a phase III placebo-controlled study of 7,180 women, risk reductions after 12 months of treatment with romosozumab were 73% for vertebral fractures (risk ratio 0.27; 95% CI 0.16–0.47, P < 0.001) and 40% for major osteoporotic fracture (HR 0.60; 95% CI 0.40–0.90, P = 0.012) (Cosman et al. 2016). Fracture risk reduction with romosozumab was numerically lower for nonvertebral (HR 0.75; 95% CI 0.53–1.05, P = 0.10) and hip (HR 0.54; 95% CI 0.22–1.35, P = 0.18) fracture (Cosman et al. 2016).
In another study, 4,093 postmenopausal women with osteoporosis and a fragility fracture were assigned to monthly subcutaneous romosozumab (210 mg) or weekly oral alendronate (70 mg) for 12 months then open-label alendronate in both groups for a further 12 months (Saag et al. 2017). At 12 months, romosozumab reduced vertebral fracture risk by 37% (risk ratio 0.63; 95% CI 0.47–0.85, P = 0.003), but there were no statistically significant reductions in nonvertebral, major osteoporotic, or hip fracture. However, romosozumab followed by alendronate reduced nonvertebral fracture by 19% (HR 0.81; 95% CI 0.66–0.99, P = 0.04), clinical fracture by 27% (HR 0.73; 95% CI 0.61–0.88, P < 0.001), and hip fracture by 38% (HR 0.62; 95% CI 0.42–0.92, P = 0.02) compared to continuous alendronate. The trial demonstrated a higher incidence of cardiovascular events with romosozumab (50/2040) compared to alendronate (38/2014) at the end of 12 months, not at 24 months (Saag et al. 2018). These findings were not observed in the larger placebo-controlled FRAME study (Cosman et al. 2016).
Romosozumab is associated with a transitory increase in P1NP within 1 week and a rapid but sustained reduction in CTX for 12 months, perhaps because of a reduction in cells stimulating RANKL production and so reduced osteoclastogenesis (Ominsky et al. 2015). In a study of the transcriptional effects of anti-sclerostin treatment in mature ovariectomized rats, no effect on osteoprotegerin was noted (Nioi et al. 2015). Bone formation is modeling-based and is limited in duration, probably due to self-regulation in the Wnt pathway restricting proliferative drive in the osteoblast lineage. Increases in mRNA levels of Wnt signaling antagonists Sost and DKK-1 in osteoblastic cells, tibiae, and vertebrae have been reported (Taylor et al. 2016). Blocking both sclerostin and DKK-1 resulted in a more robust anabolic effect using a bispecific antibody with dual inhibition of sclerostin and DKK-1 to treat rodents and nonhuman primates (Florio et al. 2016; Holdsworth et al. 2018; Maeda et al. 2015). These anabolic effects are lost after stopping treatment. Bone mineral density decreases, associated with a decrease in bone formation and increase in bone resorption markers (Ominsky et al. 2017).
4 First-Line Therapy: Antiresorptive, Anabolic, Combined, or Sequential?
Antiresorptive agents are the first-line approach to therapy. They produce a relative fracture risk reduction. Bone strength stabilizes, or decreases, albeit more slowly than without treatment; it is not restored. Vertebral and hip fracture risk is reduced by ~50% relative to untreated controls, i.e., 50% of the women having fractures still do so despite treatment, while nonvertebral fracture risk is reduced by ~20%, i.e., 80% of the women having fractures still do so despite treatment, a particular concern given 80% of all fractures are nonvertebral (Reid 2015).
The most obvious explanation for these modest relative risk reductions and the continuing fracture risk is that antiresorptive agents only reduce the reversible component of the deficit in matrix and its mineral content by slowing the rate of remodeling, so that there are fewer excavated cavities and cavities with osteoid in various stages of incompleteness of mineralization (Seeman and Martin 2015, 2019). Antiresorptives do not restore bone volume or repair microstructural deterioration, irreversible deficits caused by rapid unbalanced remodeling.
The question is, do anabolic agents restore bone volume and its microstructure? Do they reduce fracture risk more effectively than antiresorptives? The underlying assumption justifying the use of anabolic therapy over antiresorptives as first-line therapy is that even partial restoration of bone volume and microstructural deterioration, with deposition of new bone of normal material composition, will reduce fracture risk more greatly than contraction of the remodeling space deficit in a bone of reduced and architecturally compromised matrix volume which eventually becomes homogeneously and fully mineralized (Parfitt 1980; Seeman and Martin 2019).
There are only two clinical trials that compare the antifracture efficacy of anabolic versus antiresorptive therapy. In the study reported by Saag et al., at 12 months, romosozumab reduced new vertebral (risk ratio 0.63; 95% CI 0.47–0.85, P = 0.003) and clinical fractures (HR 0.72; 95% CI 0.54–0.96, P = 0.03) compared to alendronate. Nonvertebral fracture risk was numerically lower in the romosozumab treated group (HR 0.74; 95% CI 0.54–1.01, P = 0.06) (Saag et al. 2017).
In the second study, Kendler et al. randomized 1,360 postmenopausal women with osteoporosis to PTH 1-34 (20 μg daily) or risedronate (35 mg daily) for 2 years (Kendler et al. 2018). Overall, 72% of participants had previously received bone targeted treatment. At 2 years, treatment with PTH 1-34 resulted in a 56% reduction in vertebral fracture risk (risk ratio 0.44; 95% CI 0.29–0.68, P < 0.0001). Nonvertebral fracture risk was numerically lower in the PTH 1-34 treated group (HR 0.66; 95% CI 0.39–1.10, P = 0.1). These changes were consistent across a range of participant characteristics (Geusens et al. 2018). Both studies demonstrate greater vertebral fracture risk reduction using anabolic than antiresorptive therapy; more evidence is needed to establish the superiority of anabolic agents in reducing nonvertebral and hip fracture risk.
4.1 Combining Antiresorptive and Anabolic Agents
There are no studies comparing the antifracture efficacy of combined antiresorptive and anabolic therapy versus either alone. Inferences regarding the potential advantage of combined therapy over single therapy are entirely based on studies in animal models and human subjects using non-invasive imaging methods and biochemical measurements of bone remodeling. In some animal models, ex vivo strength testing of bone samples has been examined.
It is widely held that antiresorptive therapy suppresses, “blunts,” remodeling-based bone formation by PTH. This is largely based on the use of alendronate in two papers published in the New England Journal of Medicine with an accompanying editorial (Black et al. 2003; Finkelstein et al. 2003; Khosla 2003). However, the data has not been confirmed in the majority of subsequent studies using other antiresorptives (Cosman et al. 2011; Kostenuik et al. 2001; Samadfam et al. 2007; Tsai et al. 2013). The notion of blunting was based on the assumption that a higher BMD or higher P1NP means more bone formation and a lack of response means less bone formation.
BMD is a problematic endpoint because it cannot differentiate changes in matrix mineralization from changes in matrix volume and structure. Remodeling-based anabolic therapy increases bone matrix volume by replacing more fully mineralized bone with young less fully mineralized bone. Modeling-based anabolic therapy adds young less fully mineralized bone to existing older bone. Imaging using radiation transmission often results in a net reduction in BMD because young less mineralized bone transmits, rather than attenuates, photons leading to the inference that bone “loss” and fragility have occurred when in fact bone volume has increased. Antiresorptives slow remodeling. Matrix no longer “turned over” undergoes more complete mineralization increasing BMD leading to the inference that bone “volume” or “mass” has increased and that bone strength has increased even though bone matrix volume is unchanged or decreased and the matrix has become less ductile (Lloyd et al. 2017).
Even if an increase or lack of an increase in BMD is accepted on face value, Figs. 1, 2, and 3 of the 12-month study by Black et al., which compared the effects on BMD of PTH 1-84 (100 mcg daily) and alendronate (10 mg daily) either alone or in combination, do not support the notion of blunting (Black et al. 2003) (Fig. 3). Relative to PTH 1-84 alone, combined therapy (1) did not produce a smaller increment in BMD, (2) did produce a greater increase in total hip BMD, (3) did reduce the decline in distal radius BMD, and (4) did prevent the reduction in total hip and femoral neck cortical vBMD produced by PTH 1-84 alone. Curiously, the increase in total hip and femoral neck cortical volume by PTH 1-84, a modeling effect, was prevented by combined therapy. Moreover, combined therapy increased trabecular vBMD less than PTH 1-84 monotherapy, but this may not be blunting. The antiresorptive might prevent PTH 1-84-mediated increase in intracortical remodeling, cortical porosity, and prevent the increase in cortical fragments making it seem that the rise in “trabecular” BMD is blunted (Zebaze and Seeman 2015). Blunting of the rise in P1NP and CTX is likely to be the result of suppressed remodeling rate rather than a reduction in the net volumes of bone deposited or resorbed by BMUs, respectively (Seeman and Nguyen 2016).

Mean percent changes in (a) areal bone mineral density and (b) volumetric bone density in postmenopausal women after 12 months of treatment with PTH 1-84 100-μg daily (green bars) or a combination of PTH 1-84 100-μg daily and alendronate 10-mg daily (orange bars). Data sourced from Black DM, Greenspan SL, Ensrud KE, et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med. 2003;349:1207–15
If blunting of the BMD response was due to fewer BMUs, then blunting should be more severe with co-administration of PTH with more effective remodeling suppressants like zoledronate, denosumab, or osteoprotegerin (OPG, an endogenous inhibitor of RANKL) than with alendronate. The opposite is reported, and many studies report additive effects (Cosman et al. 2011; Kostenuik et al. 2001; Samadfam et al. 2007; Tsai et al. 2013). Blunting is not greater with denosumab/PTH 1-34 than alendronate/PTH 1-34 even though denosumab suppresses remodeling more greatly than alendronate. Additive effects on BMD are reported with PTH 1-34/denosumab relative to PTH 1-34 alone (Tsai et al. 2013) and comparing PTH 1-34/OPG to PTH 1-34 alone (Samadfam et al. 2007).
The difficulties in using BMD as a phenotype are also present using high-resolution peripheral computed tomography. Tsai et al. report that combined PTH 1-34 and denosumab increased cortical vBMD, the net result of a reduction by PTH 1-34 and an increase using denosumab (Tsai et al. 2015). Combined therapy increased cortical matrix mineral density, yet PTH 1-34 decreased it (by replacing more mineralized bone with less mineralized bone), and denosumab had no effect. Combined therapy had no effect on porosity, yet PTH 1-34 increased it, while denosumab had no effect (Fig. 4). These findings do not entirely add up, probably because of methodological challenges in separating cortical from trabecular compartments and quantifying cortical porosity and trabecular density. Low image resolution and changes in matrix mineral density influence quantification of microstructure (Zebaze et al. 2010, 2013). Moreover, both bisphosphonates and PTH 1-34 reduce marrow fat composition (Fan et al. 2017; Veldhuis-Vlug and Rosen 2018). Replacing bone marrow fat with water or cells increases photon attenuation and may exaggerate the rise in BMD.

Mean percent changes in cortical volumetric bone mineral density (vBMD), matrix mineral density, and porosity at the distal tibia, as assessed by HR-pQCT, in postmenopausal women after 12 months of treatment with denosumab 60-mg (yellow), PTH 1-34 20-μg (red), or both (green). ∗p < 0.05, within group difference from baseline to 12 months. Data sourced from Tsai JN, Uihlein AV, Burnett-Bowie S-AM, et al. Comparative effects of teriparatide, denosumab, and combination therapy on peripheral compartmental bone density, microarchitecture, and estimated strength: the DATA-HRpQCT Study. J Bone Miner Res. 2015;30(1):39–45
A lower level of evidence is preclinical studies demonstrating that combined therapy increases the breaking strength of bone ex vivo more greatly than either drug alone. Only one study in rodents comparing PTH 1-34/alendronate and PTH 1-34/OPG versus PTH 1-34 alone has been reported. While trabecular bone volume increased more greatly than PTH 1-34 alone, bone strength assessed ex vivo was similar to PTH 1-34 monotherapy (Samadfam et al. 2007). These concerns suggest that caution is needed before making inferences about the comparative efficacy of these agents and that eventually, well designed and executed studies using fracture outcomes will be needed.
4.2 Sequential Therapy
4.2.1 Anabolic to Antiresorptive
Cessation of anabolic treatment results in loss of the benefits. Antiresorptives maintain or increase BMD, particularly denosumab because it is the most efficacious in suppressing remodeling (Cosman et al. 2016; Saag et al. 2017). In the DATA-Switch study, 2 years of PTH 1-34 followed by 2 years of denosumab resulted in further increases in BMD (Leder et al. 2015). At 48 months, women treated with combined PTH 1-34/denosumab for 2 years followed by denosumab alone had greater gains in BMD than those treated with PTH 1-34 followed by denosumab (Leder et al. 2015).
Bone et al. administered 24 months of alendronate after 18 months of abaloparatide or placebo (Bone et al. 2018). Treatment with alendronate was claimed to maintain the purported vertebral, nonvertebral, and major osteoporotic fracture risk reduction with abaloparatide relative to the placebo group. However, comparisons of the two groups are difficult to interpret as the placebo group may have lost bone and suffered microstructural deterioration during 18 months without active treatment. Although it is likely that stopping abaloparatide results in bone loss, as reported with PTH (Adami et al. 2008; Eastell et al. 2009), an abaloparatide group given placebo shown to be losing bone is needed to establish this and that administration of alendronate prevents this loss. Similar issues arise in the interpretation of antifracture efficacy at 24 months of romosozumab-denosumab compared to placebo-denosumab (Cosman et al. 2016).
At this time, the only study comparing fracture outcomes with sequential therapy against continuous monotherapy is the study by Saag et al., who reported greater vertebral, nonvertebral, and hip fracture reduction with romosozumab-alendronate compared to alendronate alone (Saag et al. 2017).
4.2.2 Antiresorptive to Anabolic
As discussed above, most (Boonen et al. 2008; Cosman et al. 2011; Kostenuik et al. 2001; Samadfam et al. 2007; Tsai et al. 2013), but not all (Delmas et al. 1995), studies suggest that concurrent administration of anabolic and antiresorptive agents does not blunt the effect on BMD or microstructure. The presence of many remodeling sites in their formative phase pre-treatment allows PTH to initiate remodeling-based bone formation by these BMUs (as they are unaffected by the concurrently started antiresorptive therapy).
By contrast, remodeling is already suppressed in patients receiving long-term antiresorptive therapy before being switched to anabolic therapy. The early increase in BMD (due to a reduction in the reversible remodeling space deficit by antiresorptive therapy) has occurred (Parfitt 2008; Seeman and Martin 2015). Namely, the many BMUs present pre-treatment that have incompletely refilled, and secondary mineralization of the less remodeled matrix has occurred. When the antiresorptive is stopped, there are fewer BMUs available for PTH peptides or abaloparatide to initiate remodeling-based bone formation. This suggests that blunting of the effect of these peptides is plausible.
The question is whether previously administered bisphosphonates, which remain in the matrix for years (Russell et al. 2008), predispose to blunting of the effect of PTH peptides or abaloparatide when the antiresorptive is stopped and replaced by these anabolic agents. Namely, is there blunting of the effect of the anabolic agent on BMD, microstructure, material composition, and most important, blunting of any improvement in bone strength or antifracture efficacy.
These are challenging questions because of difficulties in developing the study design needed to address these issues. For example, in the setting of many years of treatment with bisphosphonates, BMD is likely to have increased, mainly due to near-complete secondary mineralization of the less remodeled bone matrix volume, not due to restoration of bone volume or microstructure (Seeman and Martin 2019). Indeed, the longer the duration of bisphosphonate therapy before switching to an anabolic agent, the more reduced and architecturally deteriorated the matrix volume is likely to be and the higher its matrix mineral density because of slow continued unbalanced remodeling (Seeman and Martin 2019). The deficit in bone volume and the microstructural deterioration are likely to be obscured by the increased matrix mineral density producing a higher BMD.
If a control group is chosen to receive PTH 1-34 or abaloparatide to define the “un-blunted” response, they must have had no prior treatment with bisphosphonates. How should this control group be chosen? If matched by age, the control group is likely to have higher baseline bone remodeling, lower BMD, and more deteriorated microstructure than the prior bisphosphonate-treated group (because the control group are untreated). If matched by BMD, the control group is likely to have lower baseline bone remodeling, higher bone volume, and better microstructure (because the prior bisphosphonate-treated group have had an increase in BMD mainly due to secondary mineralization). Assessment of blunting is problematic if comparisons are made using a percentage change when baselines differ. These issues need to be addressed in dealing with this subject as these methodological issues may be responsible for the contradictory findings in the literature (Cosman et al. 2017).
Several studies support blunting by antiresorptives with greater remodeling suppressant action. For example, Miller et al. report a lesser increase in remodeling markers and BMD responses to PTH 1-34 in patients previously treated with alendronate than risedronate, perhaps because baseline remodeling was lower in the alendronate group (Miller et al. 2008). There was a correlation between change in 3-month measures of P1NP and spine trabecular volumetric BMD but not areal BMD. In another study in which baseline remodeling was numerically higher in the prior raloxifene than prior bisphosphonate group, switching to PTH 1-34 resulted in similar numerical increases in remodeling markers, but spine (not hip) BMD and estimated strength increased more greatly in the prior raloxifene group (Cosman et al. 2009, 2013). (Comparisons were confined to “switch” versus “add” within a group, not the comparisons presented here.) These data support the notion of blunting of BMD responses when remodeling is more greatly suppressed at the time PTH 1-34 is started.
By contrast, several studies do not support the notion of blunting by previously administered antiresorptive therapy. For example, in a paired bone biopsy study, despite lower baseline remodeling and higher BMD in women treated with alendronate than untreated controls, the response to PTH 1-34 in women previously treated with alendronate was not blunted compared to the PTH 1-34 effect in untreated controls (Ma et al. 2014). Periosteal and endocortical bone formation increased, consistent with an anabolic response, and intracortical porosity also increased in both groups, consistent with unsuppressed stimulation of intracortical remodeling. Likewise, in another paired bone biopsy study, trabecular bone formation in response to PTH 1-34 was not blunted by prior alendronate therapy (Fahrleitner-Pammer et al. 2016).
If the antiresorptive used is denosumab, the question of blunting is less relevant because of a rapid offset of remodeling suppression when denosumab is stopped. Caution is needed because cessation of denosumab is associated with a rapid increase in bone remodeling markers, a decline in BMD within months of stopping therapy, and uncommonly, an increased risk of multiple vertebral fractures (Bone et al. 2011; Miller et al. 2011).
Indeed, this accelerated loss of bone may be exacerbated by switching to PTH 1-34. In the DATA-switch study, women treated with denosumab for 24 months switched to PTH 1-34 for a further 24 months had a reduction in spine and hip BMD in the first 12 months followed by a gradual increase in BMD (Leder et al. 2015). However, despite this increase in BMD, cortical and trabecular volumetric bone density decreased, and bone strength estimated by finite element analysis also reduced (Tsai et al. 2017).
Several factors may be responsible for these changes. The decline in BMD may be due to the replacement of bone with osteoid during remodelling- and/or modeling-dependent bone formation. The later increase in BMD may be the result of primary and secondary mineralization of this osteoid. The concurrent decrease in cortical and trabecular volumetric BMD may be the result of increased remodeling produced by cessation of denosumab and initiation of PTH 1-34, both of which increase cortical porosity and excavate cavities upon trabeculae. Whether bone fragility increases is uncertain but replacement of denosumab with PTH 1-34 needs to be done with caution.
5 Are We There Yet?
There is progress, but important needs remain unmet. Anti-vertebral fracture efficacy of antiresorptives and anabolic therapy is robustly established in controlled studies mostly of 1–3 years duration. There is now evidence of superior anti-vertebral fracture efficacy of anabolic than antiresorptive agents. This is consistent with the notion that antiresorptive agents only reduce the reversible remodeling space deficit, whereas anabolic agents partly restore bone volume, microstructure, and its material composition, changes that should restore bone strength more effectively than antiresorptive agents.
Should anabolic agents be first-line therapy? This notion may be premature for several reasons: (1) The superior anti-vertebral fracture efficacy compared to antiresorptives has only been shown in two studies (Kendler et al. 2018; Saag et al. 2017). (2) There is no evidence of hip fracture risk reduction using anabolic agents. (3) There is little evidence of nonvertebral fracture risk reduction using anabolic therapies. Only one (Neer et al. 2001) of several (Greenspan et al. 2007; Miller et al. 2016; Nakamura et al. 2012) studies using PTH regimens, and no critically evaluated study of abaloparatide (EMA 2018; Miller et al. 2016), supports anti-nonvertebral fracture efficacy relative to untreated controls. Nor is there evidence of superior nonvertebral antifracture efficacy of anabolic agents compared with antiresorptive agents, although the numerical reduction in nonvertebral fracture risk in both trials suggests this is likely. This uncertainty is of particular concern because nonvertebral fractures account for 80% of all fractures and 50% of all the morbidity and mortality (Tatangelo et al. 2019). However, there is evidence that sequential romosozumab followed by alendronate results in greater vertebral, nonvertebral, major osteoporotic, and hip fracture risk reduction than alendronate alone (Saag et al. 2017). Finally, whether combined antiresorptive and anabolic regimens result in greater vertebral, nonvertebral, and hip fracture risk reduction than either agent alone, and whether these risk reductions cost-effectively produce more quality-adjusted life years free of fracture, remains untested.
References
Adami S, San Martin J, Munoz-Torres M, Econs MJ, Xie L, Dalsky GP, McClung M, Felsenberg D, Brown JP, Brandi ML, Sipos A (2008) Effect of raloxifene after recombinant teriparatide [hPTH(1-34)] treatment in postmenopausal women with osteoporosis. Osteoporos Int 19:87–94. https://doi.org/10.1007/s00198-007-0485-y
Akkus O, Polyakova-Akkus A, Adar F, Schaffler MB (2003) Aging of microstructural compartments in human compact bone. J Bone Miner Res 18:1012–1019. https://doi.org/10.1359/jbmr.2003.18.6.1012
Allen MR, Gineyts E, Leeming DJ, Burr DB, Delmas PD (2008) Bisphosphonates alter trabecular bone collagen cross-linking and isomerization in beagle dog vertebra. Osteoporos Int 19:329–337. https://doi.org/10.1007/s00198-007-0533-7
Allen MR, Erickson AM, Wang X, Burr DB, Martin RB, Hazelwood SJ (2010) Morphological assessment of basic multicellular unit resorption parameters in dogs shows additional mechanisms of bisphosphonate effects on bone. Calcif Tissue Int 86:67–71. https://doi.org/10.1007/s00223-009-9315-x
Balemans W, Ebeling M, Patel N, van Hul E, Olson P, Dioszegi M, Lacza C, Wuyts W, van den Ende J, Willems P, Paes-Alves AF, Hill S, Bueno M, Ramos FJ, Tacconi P, Dikkers FG, Stratakis C, Lindpaintner K, Vickery B, Foernzler D, van Hul W (2001) Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum Mol Genet 10:537–543. https://doi.org/10.1093/hmg/10.5.537
Balemans W, Patel N, Ebeling M, van Hul E, Wuyts W, Lacza C, Dioszegi M, Dikkers FG, Hildering P, Willems PJ, Verheij JB, Lindpaintner K, Vickery B, Foernzler D, van Hul W (2002) Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet 39:91–97. https://doi.org/10.1136/jmg.39.2.91
Bilezikian JP (2008) Combination anabolic and antiresorptive therapy for osteoporosis: opening the anabolic window. Curr Osteoporos Rep 6:24–30. https://doi.org/10.1007/s11914-008-0005-9
Bjornerem A, Ghasem-Zadeh A, Bui M, Wang X, Rantzau C, Nguyen TV, Hopper JL, Zebaze R, Seeman E (2011) Remodeling markers are associated with larger intracortical surface area but smaller trabecular surface area: a twin study. Bone 49:1125–1130. https://doi.org/10.1016/j.bone.2011.08.009
Bjornerem A, Wang X, Bui M, Ghasem-Zadeh A, Hopper JL, Zebaze R, Seeman E (2018) Menopause-related appendicular bone loss is mainly cortical and results in increased cortical porosity. J Bone Miner Res 33:598–605. https://doi.org/10.1002/jbmr.3333
Black DM, Greenspan SL, Ensrud KE, Palermo L, McGowan JA, Lang TF, Garnero P, Bouxsein ML, Bilezikian JP, Rosen CJ, Pa THSI (2003) The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med 349:1207–1215. https://doi.org/10.1056/NEJMoa031975
Bone HG, Bolognese MA, Yuen CK, Kendler DL, Miller PD, Yang YC, Grazette L, San Martin J, Gallagher JC (2011) Effects of denosumab treatment and discontinuation on bone mineral density and bone turnover markers in postmenopausal women with low bone mass. J Clin Endocrinol Metab 96:972–980. https://doi.org/10.1210/jc.2010-1502
Bone HG, Wagman RB, Brandi ML, Brown JP, Chapurlat R, Cummings SR, Czerwinski E, Fahrleitner-Pammer A, Kendler DL, Lippuner K, Reginster JY, Roux C, Malouf J, Bradley MN, Daizadeh NS, Wang A, Dakin P, Pannacciulli N, Dempster DW, Papapoulos S (2017) 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol 5:513–523. https://doi.org/10.1016/S2213-8587(17)30138-9
Bone HG, Cosman F, Miller PD, Williams GC, Hattersley G, Hu MY, Fitzpatrick LA, Mitlak B, Papapoulos S, Rizzoli R, Dore RK, Bilezikian JP, Saag KG (2018) ACTIVExtend: 24 months of alendronate after 18 months of abaloparatide or placebo for postmenopausal osteoporosis. J Clin Endocrinol Metab 103:2949–2957. https://doi.org/10.1210/jc.2018-00163
Boonen S, Marin F, Obermayer-Pietsch B, Simoes ME, Barker C, Glass EV, Hadji P, Lyritis G, Oertel H, Nickelsen T, McCloskey EV, Investigators E (2008) Effects of previous antiresorptive therapy on the bone mineral density response to two years of teriparatide treatment in postmenopausal women with osteoporosis. J Clin Endocrinol Metab 93:852–860. https://doi.org/10.1210/jc.2007-0711
Bouxsein ML, Eastell R, Lui LY, Wu LA, de Papp AE, Grauer A, Marin F, Cauley JA, Bauer DC, Black DM, Project FBQ (2019) Change in bone density and reduction in fracture risk: a meta-regression of published trials. J Bone Miner Res 34:632–642. https://doi.org/10.1002/jbmr.3641
Chavassieux P, Chapurlat R, Portero-Muzy N, Roux JP, Garcia P, Brown JP, Libanati C, Boyce RW, Wang A, Grauer A (2019) Bone-forming and antiresorptive effects of Romosozumab in postmenopausal women with osteoporosis: bone histomorphometry and microcomputed tomography analysis after 2 and 12 months of treatment. J Bone Miner Res 34:1597–1608. https://doi.org/10.1002/jbmr.3735
Chen P, Satterwhite JH, Licata AA, Lewiecki EM, Sipos AA, Misurski DM, Wagman RB (2005) Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res 20:962–970. https://doi.org/10.1359/JBMR.050105
Cosman F, Nieves J, Woelfert L, Formica C, Gordon S, Shen V, Lindsay R (2001) Parathyroid hormone added to established hormone therapy: effects on vertebral fracture and maintenance of bone mass after parathyroid hormone withdrawal. J Bone Miner Res 16:925–931. https://doi.org/10.1359/jbmr.2001.16.5.925
Cosman F, Wermers RA, Recknor C, Mauck KF, Xie L, Glass EV, Krege JH (2009) Effects of teriparatide in postmenopausal women with osteoporosis on prior alendronate or raloxifene: differences between stopping and continuing the antiresorptive agent. J Clin Endocrinol Metab 94:3772–3780. https://doi.org/10.1210/jc.2008-2719
Cosman F, Eriksen EF, Recknor C, Miller PD, Guanabens N, Kasperk C, Papanastasiou P, Readie A, Rao H, Gasser JA, Bucci-Rechtweg C, Boonen S (2011) Effects of intravenous zoledronic acid plus subcutaneous teriparatide [rhPTH(1-34)] in postmenopausal osteoporosis. J Bone Miner Res 26:503–511. https://doi.org/10.1002/jbmr.238
Cosman F, Keaveny TM, Kopperdahl D, Wermers RA, Wan X, Krohn KD, Krege JH (2013) Hip and spine strength effects of adding versus switching to teriparatide in postmenopausal women with osteoporosis treated with prior alendronate or raloxifene. J Bone Miner Res 28:1328–1336. https://doi.org/10.1002/jbmr.1853
Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, Hofbauer LC, Lau E, Lewiecki EM, Miyauchi A, Zerbini CA, Milmont CE, Chen L, Maddox J, Meisner PD, Libanati C, Grauer A (2016) Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med 375:1532–1543. https://doi.org/10.1056/NEJMoa1607948
Cosman F, Nieves JW, Dempster DW (2017) Treatment sequence matters: anabolic and antiresorptive therapy for osteoporosis. J Bone Miner Res 32:198–202. https://doi.org/10.1002/jbmr.3051
Delmas PD, Vergnaud P, Arlot ME, Pastoureau P, Meunier PJ, Nilssen MH (1995) The anabolic effect of human PTH (1-34) on bone formation is blunted when bone resorption is inhibited by the bisphosphonate tiludronate--is activated resorption a prerequisite for the in vivo effect of PTH on formation in a remodeling system? Bone 16:603–610. https://doi.org/10.1016/8756-3282(95)00113-r
Dempster DW, Birchman R, Xu R, Lindsay R, Shen V (1995) Temporal changes in cancellous bone structure of rats immediately after ovariectomy. Bone 16:157–161
Dempster DW, Brown JP, Fahrleitner-Pammer A, Kendler D, Rizzo S, Valter I, Wagman RB, Yin X, Yue SV, Boivin G (2018) Effects of long-term denosumab on bone histomorphometry and mineralization in women with postmenopausal osteoporosis. J Clin Endocrinol Metab 103:2498–2509. https://doi.org/10.1210/jc.2017-02669
Eastell R, Nickelsen T, Marin F, Barker C, Hadji P, Farrerons J, Audran M, Boonen S, Brixen K, Gomes JM, Obermayer-Pietsch B, Avramidis A, Sigurdsson G, Gluer CC (2009) Sequential treatment of severe postmenopausal osteoporosis after teriparatide: final results of the randomized, controlled European Study of Forsteo (EUROFORS). J Bone Miner Res 24:726–736. https://doi.org/10.1359/jbmr.081215
Eastell R, Mitlak BH, Wang Y, Hu M, Fitzpatrick LA, Black DM (2019) Bone turnover markers to explain changes in lumbar spine BMD with abaloparatide and teriparatide: results from ACTIVE. Osteoporos Int 30:667–673. https://doi.org/10.1007/s00198-018-04819-1
Ejersted C, Oxlund H, Eriksen EF, Andreassen TT (1998) Withdrawal of parathyroid hormone treatment causes rapid resorption of newly formed vertebral cancellous and endocortical bone in old rats. Bone 23:43–52. https://doi.org/10.1016/s8756-3282(98)00072-6
EMA (2018) Eladynos (Abaloparatide) Assessment Report – EMA/CHMP/581111/2018. https://www.ema.europa.eu/en/documents/assessment-report/eladynos-epar-refusal-public-assessment-report_en.pdf
Fahrleitner-Pammer A, Burr D, Dobnig H, Stepan JJ, Petto H, Li J, Krege JH, Pavo I (2016) Improvement of cancellous bone microstructure in patients on teriparatide following alendronate pretreatment. Bone 89:16–24. https://doi.org/10.1016/j.bone.2016.05.004
Fan Y, Hanai JI, Le PT, Bi R, Maridas D, DeMambro V, Figueroa CA, Kir S, Zhou X, Mannstadt M, Baron R, Bronson RT, Horowitz MC, Wu JY, Bilezikian JP, Dempster DW, Rosen CJ, Lanske B (2017) Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab 25:661–672. https://doi.org/10.1016/j.cmet.2017.01.001
Fantner GE, Hassenkam T, Kindt JH, Weaver JC, Birkedal H, Pechenik L, Cutroni JA, Cidade GA, Stucky GD, Morse DE, Hansma PK (2005) Sacrificial bonds and hidden length dissipate energy as mineralized fibrils separate during bone fracture. Nat Mater 4:612–616. https://doi.org/10.1038/nmat1428
Finkelstein JS, Hayes A, Hunzelman JL, Wyland JJ, Lee H, Neer RM (2003) The effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med 349:1216–1226. https://doi.org/10.1056/NEJMoa035725
Florio M, Gunasekaran K, Stolina M, Li X, Liu L, Tipton B, Salimi-Moosavi H, Asuncion FJ, Li C, Sun B, Tan HL, Zhang L, Han CY, Case R, Duguay AN, Grisanti M, Stevens J, Pretorius JK, Pacheco E, Jones H, Chen Q, Soriano BD, Wen J, Heron B, Jacobsen FW, Brisan E, Richards WG, Ke HZ, Ominsky MS (2016) A bispecific antibody targeting sclerostin and DKK-1 promotes bone mass accrual and fracture repair. Nat Commun 7:11505. https://doi.org/10.1038/ncomms11505
Fuchs RK, Faillace ME, Allen MR, Phipps RJ, Miller LM, Burr DB (2011) Bisphosphonates do not alter the rate of secondary mineralization. Bone 49:701–705. https://doi.org/10.1016/j.bone.2011.05.009
Geusens P, Marin F, Kendler DL, Russo LA, Zerbini CA, Minisola S, Body JJ, Lespessailles E, Greenspan SL, Bagur A, Stepan JJ, Lakatos P, Casado E, Moericke R, Lopez-Romero P, Fahrleitner-Pammer A (2018) Effects of teriparatide compared with risedronate on the risk of fractures in subgroups of postmenopausal women with severe osteoporosis: the VERO trial. J Bone Miner Res 33:783–794. https://doi.org/10.1002/jbmr.3384
Greenspan SL, Bone HG, Ettinger MP, Hanley DA, Lindsay R, Zanchetta JR, Blosch CM, Mathisen AL, Morris SA, Marriott TB, Treatment of Osteoporosis with Parathyroid Hormone Study G (2007) Effect of recombinant human parathyroid hormone (1-84) on vertebral fracture and bone mineral density in postmenopausal women with osteoporosis: a randomized trial. Ann Intern Med 146:326–339. https://doi.org/10.7326/0003-4819-146-5-200703060-00005
Hattersley G, Dean T, Corbin BA, Bahar H, Gardella TJ (2016) Binding selectivity of abaloparatide for PTH-type-1-receptor conformations and effects on downstream signaling. Endocrinology 157:141–149. https://doi.org/10.1210/en.2015-1726
Hattner R, Epker BN, Frost HM (1965) Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature 206:489–490
Holdsworth G, Greenslade K, Jose J, Stencel Z, Kirby H, Moore A, Ke HZ, Robinson MK (2018) Dampening of the bone formation response following repeat dosing with sclerostin antibody in mice is associated with up-regulation of Wnt antagonists. Bone 107:93–103. https://doi.org/10.1016/j.bone.2017.11.003
Jiang Y, Zhao JJ, Mitlak BH, Wang O, Genant HK, Eriksen EF (2003) Recombinant human parathyroid hormone (1-34) [teriparatide] improves both cortical and cancellous bone structure. J Bone Miner Res 18:1932–1941. https://doi.org/10.1359/jbmr.2003.18.11.1932
Ke HZ, Richards WG, Li X, Ominsky MS (2012) Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases. Endocr Rev 33:747–783. https://doi.org/10.1210/er.2011-1060
Kendler DL, Marin F, Zerbini CAF, Russo LA, Greenspan SL, Zikan V, Bagur A, Malouf-Sierra J, Lakatos P, Fahrleitner-Pammer A, Lespessailles E, Minisola S, Body JJ, Geusens P, Möricke R, López-Romero P (2018) Effects of teriparatide and risedronate on new fractures in post-menopausal women with severe osteoporosis (VERO): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet 391:230–240. https://doi.org/10.1016/s0140-6736(17)32137-2
Khosla S (2003) Parathyroid hormone plus alendronate--a combination that does not add up. N Engl J Med 349:1277–1279. https://doi.org/10.1056/NEJMe038143
Kim SW, Pajevic PD, Selig M, Barry KJ, Yang JY, Shin CS, Baek WY, Kim JE, Kronenberg HM (2012) Intermittent parathyroid hormone administration converts quiescent lining cells to active osteoblasts. J Bone Miner Res 27:2075–2084. https://doi.org/10.1002/jbmr.1665
Kimmel DB, Bozzato RP, Kronis KA, Coble T, Sindrey D, Kwong P, Recker RR (1993) The effect of recombinant human (1-84) or synthetic human (1-34) parathyroid hormone on the skeleton of adult osteopenic ovariectomized rats. Endocrinology 132:1577–1584. https://doi.org/10.1210/endo.132.4.8462456
Kostenuik PJ, Capparelli C, Morony S, Adamu S, Shimamoto G, Shen V, Lacey DL, Dunstan CR (2001) OPG and PTH-(1-34) have additive effects on bone density and mechanical strength in osteopenic ovariectomized rats. Endocrinology 142:4295–4304. https://doi.org/10.1210/endo.142.10.8437
Leder BZ, Tsai JN, Uihlein AV, Wallace PM, Lee H, Neer RM, Burnett-Bowie SA (2015) Denosumab and teriparatide transitions in postmenopausal osteoporosis (the DATA-switch study): extension of a randomised controlled trial. Lancet 386:1147–1155. https://doi.org/10.1016/S0140-6736(15)61120-5
Lindsay R, Cosman F, Zhou H, Bostrom MP, Shen VW, Cruz JD, Nieves JW, Dempster DW (2006) A novel tetracycline labeling schedule for longitudinal evaluation of the short-term effects of anabolic therapy with a single iliac crest bone biopsy: early actions of teriparatide. J Bone Miner Res 21:366–373. https://doi.org/10.1359/JBMR.051109
Lindsay R, Zhou H, Cosman F, Nieves J, Dempster DW, Hodsman AB (2007) Effects of a one-month treatment with PTH(1-34) on bone formation on cancellous, endocortical, and periosteal surfaces of the human ilium. J Bone Miner Res 22:495–502. https://doi.org/10.1359/jbmr.070104
Lips P, Courpron P, Meunier PJ (1978) Mean wall thickness of trabecular bone packets in the human iliac crest: changes with age. Calcif Tissue Res 26:13–17
Liu CC, Kalu DN (1990) Human parathyroid hormone-(1-34) prevents bone loss and augments bone formation in sexually mature ovariectomized rats. J Bone Miner Res 5:973–982. https://doi.org/10.1002/jbmr.5650050911
Lloyd AA, Gludovatz B, Riedel C, Luengo EA, Saiyed R, Marty E, Lorich DG, Lane JM, Ritchie RO, Busse B, Donnelly E (2017) Atypical fracture with long-term bisphosphonate therapy is associated with altered cortical composition and reduced fracture resistance. Proc Natl Acad Sci U S A 114:8722–8727. https://doi.org/10.1073/pnas.1704460114
Ma YL, Zeng Q, Donley DW, Ste-Marie LG, Gallagher JC, Dalsky GP, Marcus R, Eriksen EF (2006) Teriparatide increases bone formation in modeling and remodeling osteons and enhances IGF-II immunoreactivity in postmenopausal women with osteoporosis. J Bone Miner Res 21:855–864. https://doi.org/10.1359/jbmr.060314
Ma YL, Zeng QQ, Porras LL, Harvey A, Moore TL, Shelbourn TL, Dalsky GP, Wronski TJ, Aguirre JI, Bryant HU, Sato M (2011) Teriparatide [rhPTH (1-34)], but not strontium ranelate, demonstrated bone anabolic efficacy in mature, osteopenic, ovariectomized rats. Endocrinology 152:1767–1778. https://doi.org/10.1210/en.2010-1112
Ma YL, Zeng QQ, Chiang AY, Burr D, Li J, Dobnig H, Fahrleitner-Pammer A, Michalska D, Marin F, Pavo I, Stepan JJ (2014) Effects of teriparatide on cortical histomorphometric variables in postmenopausal women with or without prior alendronate treatment. Bone 59:139–147. https://doi.org/10.1016/j.bone.2013.11.011
Maeda A, Ono M, Holmbeck K, Li L, Kilts TM, Kram V, Noonan ML, Yoshioka Y, McNerny EM, Tantillo MA, Kohn DH, Lyons KM, Robey PG, Young MF (2015) WNT1-induced secreted protein-1 (WISP1), a novel regulator of bone turnover and WNT signaling. J Biol Chem 290:14004–14018. https://doi.org/10.1074/jbc.M114.628818
Manolagas SC (2000) Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev 21:115–137. https://doi.org/10.1210/edrv.21.2.0395
McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, Peacock M, Miller PD, Lederman SN, Chesnut CH, Lain D, Kivitz AJ, Holloway DL, Zhang C, Peterson MC, Bekker PJ, Group AMGBLS (2006) Denosumab in postmenopausal women with low bone mineral density. N Engl J Med 354:821–831. https://doi.org/10.1056/NEJMoa044459
McClung MR, Grauer A, Boonen S, Bolognese MA, Brown JP, Diez-Perez A, Langdahl BL, Reginster JY, Zanchetta JR, Wasserman SM, Katz L, Maddox J, Yang YC, Libanati C, Bone HG (2014) Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med 370:412–420. https://doi.org/10.1056/NEJMoa1305224
Miller PD, Delmas PD, Lindsay R, Watts NB, Luckey M, Adachi J, Saag K, Greenspan SL, Seeman E, Boonen S, Meeves S, Lang TF, Bilezikian JP (2008) Open-label study to determine how prior therapy with alendronate or risedronate in postmenopausal women with osteoporosis influences the clinical effectiveness of teriparatide I. early responsiveness of women with osteoporosis to teriparatide after therapy with alendronate or risedronate. J Clin Endocrinol Metab 93:3785–3793. https://doi.org/10.1210/jc.2008-0353
Miller PD, Wagman RB, Peacock M, Lewiecki EM, Bolognese MA, Weinstein RL, Ding B, San Martin J, McClung MR (2011) Effect of denosumab on bone mineral density and biochemical markers of bone turnover: six-year results of a phase 2 clinical trial. J Clin Endocrinol Metab 96:394–402. https://doi.org/10.1210/jc.2010-1805
Miller PD, Hattersley G, Riis BJ, Williams GC, Lau E, Russo LA, Alexandersen P, Zerbini CA, Hu MY, Harris AG, Fitzpatrick LA, Cosman F, Christiansen C, Investigators AS (2016) Effect of Abaloparatide vs placebo on new vertebral fractures in postmenopausal women with osteoporosis: a randomized clinical trial. JAMA 316:722–733. https://doi.org/10.1001/jama.2016.11136
Misof BM, Roschger P, Cosman F, Kurland ES, Tesch W, Messmer P, Dempster DW, Nieves J, Shane E, Fratzl P, Klaushofer K, Bilezikian J, Lindsay R (2003) Effects of intermittent parathyroid hormone administration on bone mineralization density in iliac crest biopsies from patients with osteoporosis: a paired study before and after treatment. J Clin Endocrinol Metab 88:1150–1156. https://doi.org/10.1210/jc.2002-021988
Mohsin S, O'Brien FJ, Lee TC (2006) Osteonal crack barriers in ovine compact bone. J Anat 208:81–89. https://doi.org/10.1111/j.1469-7580.2006.00509.x
Moreira CA, Fitzpatrick LA, Wang Y, Recker RR (2017) Effects of abaloparatide-SC (BA058) on bone histology and histomorphometry: the ACTIVE phase 3 trial. Bone 97:314–319. https://doi.org/10.1016/j.bone.2016.11.004
Nakamura T, Sugimoto T, Nakano T, Kishimoto H, Ito M, Fukunaga M, Hagino H, Sone T, Yoshikawa H, Nishizawa Y, Fujita T, Shiraki M (2012) Randomized teriparatide [human parathyroid hormone (PTH) 1-34] once-weekly efficacy research (TOWER) trial for examining the reduction in new vertebral fractures in subjects with primary osteoporosis and high fracture risk. J Clin Endocrinol Metab 97:3097–3106. https://doi.org/10.1210/jc.2011-3479
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH (2001) Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441. https://doi.org/10.1056/NEJM200105103441904
Nioi P, Taylor S, Hu R, Pacheco E, He YD, Hamadeh H, Paszty C, Pyrah I, Ominsky MS, Boyce RW (2015) Transcriptional profiling of laser capture microdissected subpopulations of the osteoblast lineage provides insight into the early response to sclerostin antibody in rats. J Bone Miner Res 30:1457–1467. https://doi.org/10.1002/jbmr.2482
Ominsky MS, Libanati C, Niu QT, Boyce RW, Kostenuik PJ, Wagman RB, Baron R, Dempster DW (2015) Sustained modeling-based bone formation during adulthood in cynomolgus monkeys may contribute to continuous BMD gains with denosumab. J Bone Miner Res 30:1280–1289. https://doi.org/10.1002/jbmr.2480
Ominsky MS, Boyd SK, Varela A, Jolette J, Felx M, Doyle N, Mellal N, Smith SY, Locher K, Buntich S, Pyrah I, Boyce RW (2017) Romosozumab improves bone mass and strength while maintaining bone quality in ovariectomized cynomolgus monkeys. J Bone Miner Res 32:788–801. https://doi.org/10.1002/jbmr.3036
Padhi D, Jang G, Stouch B, Fang L, Posvar E (2011) Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res 26:19–26. https://doi.org/10.1002/jbmr.173
Parfitt AM (1980) Morphological basis of bone mineral measurements: transient and steady state effects of treatment in osteoporosis. Miner Electrolyte Metab 4:273–287
Parfitt AM (1984) Age-related structural changes in trabecular and cortical bone: cellular mechanisms and biomechanical consequences. Calcif Tissue Int 36(Suppl 1):S123–S128
Parfitt AM (2008) Skeletal heterogeneity and the purposes of bone remodelling: implications for the understanding of osteoporosis. Academic Press, Cambridge
Paschalis EP, Glass EV, Donley DW, Eriksen EF (2005) Bone mineral and collagen quality in iliac crest biopsies of patients given teriparatide: new results from the fracture prevention trial. J Clin Endocrinol Metab 90:4644–4649. https://doi.org/10.1210/jc.2004-2489
Poundarik AA, Wu PC, Evis Z, Sroga GE, Ural A, Rubin M, Vashishth D (2015) A direct role of collagen glycation in bone fracture. J Mech Behav Biomed Mater 52:120–130. https://doi.org/10.1016/j.jmbbm.2015.08.012
Reid IR (2015) Short-term and long-term effects of osteoporosis therapies. Nat Rev Endocrinol 11:418–428. https://doi.org/10.1038/nrendo.2015.71
Reid IR, Ames R, Mason B, Reid HE, Bacon CJ, Bolland MJ, Gamble GD, Grey A, Horne A (2008) Randomized controlled trial of calcium supplementation in healthy, nonosteoporotic, older men. Arch Intern Med 168:2276–2282. https://doi.org/10.1001/archinte.168.20.2276
Ruff CB, Hayes WC (1988) Sex differences in age-related remodeling of the femur and tibia. J Orthop Res 6:886–896. https://doi.org/10.1002/jor.1100060613
Russell RG, Watts NB, Ebetino FH, Rogers MJ (2008) Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 19:733–759. https://doi.org/10.1007/s00198-007-0540-8
Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, Maddox J, Fan M, Meisner PD, Grauer A (2017) Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med 377:1417–1427. https://doi.org/10.1056/NEJMoa1708322
Saag KG, Petersen J, Grauer A (2018) Romosozumab versus alendronate and fracture risk in women with osteoporosis. N Engl J Med 378:195–196. https://doi.org/10.1056/NEJMc1714810
Saito M, Marumo K (2010) Collagen cross-links as a determinant of bone quality: a possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos Int 21:195–214. https://doi.org/10.1007/s00198-009-1066-z
Saito M, Mori S, Mashiba T, Komatsubara S, Marumo K (2008) Collagen maturity, glycation induced-pentosidine, and mineralization are increased following 3-year treatment with incadronate in dogs. Osteoporos Int 19:1343–1354. https://doi.org/10.1007/s00198-008-0585-3
Saito M, Marumo K, Kida Y, Ushiku C, Kato S, Takao-Kawabata R, Kuroda T (2011) Changes in the contents of enzymatic immature, mature, and non-enzymatic senescent cross-links of collagen after once-weekly treatment with human parathyroid hormone (1-34) for 18 months contribute to improvement of bone strength in ovariectomized monkeys. Osteoporos Int 22:2373–2383. https://doi.org/10.1007/s00198-010-1454-4
Samadfam R, Xia Q, Goltzman D (2007) Co-treatment of PTH with osteoprotegerin or alendronate increases its anabolic effect on the skeleton of oophorectomized mice. J Bone Miner Res 22:55–63. https://doi.org/10.1359/jbmr.060915
Seeman E (2003) Periosteal bone formation--a neglected determinant of bone strength. N Engl J Med 349:320–323. https://doi.org/10.1056/NEJMp038101
Seeman E, Delmas PD (2006) Bone quality--the material and structural basis of bone strength and fragility. N Engl J Med 354:2250–2261. https://doi.org/10.1056/NEJMra053077
Seeman E, Martin TJ (2015) Co-administration of antiresorptive and anabolic agents: a missed opportunity. J Bone Miner Res 30:753–764. https://doi.org/10.1002/jbmr.2496
Seeman E, Martin TJ (2019) Antiresorptive and anabolic agents in the prevention and reversal of bone fragility. Nat Rev Rheumatol 15:225–236. https://doi.org/10.1038/s41584-019-0172-3
Seeman E, Nguyen TV (2016) Bone remodeling markers: so easy to measure, so difficult to interpret. Osteoporos Int 27:33–35. https://doi.org/10.1007/s00198-015-3374-9
Seref-Ferlengez Z, Basta-Pljakic J, Kennedy OD, Philemon CJ, Schaffler MB (2014) Structural and mechanical repair of diffuse damage in cortical bone in vivo. J Bone Miner Res 29:2537–2544. https://doi.org/10.1002/jbmr.2309
Shen V, Dempster DW, Mellish RW, Birchman R, Horbert W, Lindsay R (1992) Effects of combined and separate intermittent administration of low-dose human parathyroid hormone fragment (1-34) and 17 beta-estradiol on bone histomorphometry in ovariectomized rats with established osteopenia. Calcif Tissue Int 50:214–220
Silverman SL, Chines AA, Kendler DL, Kung AW, Teglbjaerg CS, Felsenberg D, Mairon N, Constantine GD, Adachi JD, Bazedoxifene Study Group (2012) Sustained efficacy and safety of bazedoxifene in preventing fractures in postmenopausal women with osteoporosis: results of a 5-year, randomized, placebo-controlled study. Osteoporos Int 23:351–363. https://doi.org/10.1007/s00198-011-1691-1
Sroga GE, Vashishth D (2012) Effects of bone matrix proteins on fracture and fragility in osteoporosis. Curr Osteoporos Rep 10:141–150. https://doi.org/10.1007/s11914-012-0103-6
Tang SY, Zeenath U, Vashishth D (2007) Effects of non-enzymatic glycation on cancellous bone fragility. Bone 40:1144–1151. https://doi.org/10.1016/j.bone.2006.12.056
Tatangelo G, Watts J, Lim K, Connaughton C, Abimanyi-Ochom J, Borgstrom F, Nicholson GC, Shore-Lorenti C, Stuart AL, Iuliano-Burns S, Seeman E, Prince R, March L, Cross M, Winzenberg T, Laslett LL, Duque G, Ebeling PR, Sanders KM (2019) The cost of osteoporosis, osteopenia, and associated fractures in Australia in 2017. J Bone Miner Res 34:616–625. https://doi.org/10.1002/jbmr.3640
Taylor S, Ominsky MS, Hu R, Pacheco E, He YD, Brown DL, Aguirre JI, Wronski TJ, Buntich S, Afshari CA, Pyrah I, Nioi P, Boyce RW (2016) Time-dependent cellular and transcriptional changes in the osteoblast lineage associated with sclerostin antibody treatment in ovariectomized rats. Bone 84:148–159. https://doi.org/10.1016/j.bone.2015.12.013
Thomas CJ, Cleland TP, Zhang S, Gundberg CM, Vashishth D (2017) Identification and characterization of glycation adducts on osteocalcin. Anal Biochem 525:46–53. https://doi.org/10.1016/j.ab.2017.02.011
Tran Van PT, Vignery A, Baron R (1982) Cellular kinetics of the bone remodeling sequence in the rat. Anat Rec 202:445–451. https://doi.org/10.1002/ar.1092020403
Tsai JN, Uihlein AV, Lee H, Kumbhani R, Siwila-Sackman E, McKay EA, Burnett-Bowie SA, Neer RM, Leder BZ (2013) Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet 382:50–56. https://doi.org/10.1016/S0140-6736(13)60856-9
Tsai JN, Uihlein AV, Burnett-Bowie SA, Neer RM, Zhu Y, Derrico N, Lee H, Bouxsein ML, Leder BZ (2015) Comparative effects of teriparatide, denosumab, and combination therapy on peripheral compartmental bone density, microarchitecture, and estimated strength: the DATA-HRpQCT study. J Bone Miner Res 30:39–45. https://doi.org/10.1002/jbmr.2315
Tsai JN, Nishiyama KK, Lin D, Yuan A, Lee H, Bouxsein ML, Leder BZ (2017) Effects of denosumab and teriparatide transitions on bone microarchitecture and estimated strength: the DATA-Switch HR-pQCT study. J Bone Miner Res 32:2001–2009. https://doi.org/10.1002/jbmr.3198
Varela A, Chouinard L, Lesage E, Smith SY, Hattersley G (2017) One year of abaloparatide, a selective activator of the PTH1 receptor, increased bone formation and bone mass in osteopenic ovariectomized rats without increasing bone resorption. J Bone Miner Res 32:24–33. https://doi.org/10.1002/jbmr.3003
Vashishth D (2007) The role of the collagen matrix in skeletal fragility. Curr Osteoporos Rep 5:62–66. https://doi.org/10.1007/s11914-007-0004-2
Veldhuis-Vlug AG, Rosen CJ (2018) Clinical implications of bone marrow adiposity. J Intern Med 283:121–139. https://doi.org/10.1111/joim.12718
Wronski TJ, Yen CF, Qi H, Dann LM (1993) Parathyroid hormone is more effective than estrogen or bisphosphonates for restoration of lost bone mass in ovariectomized rats. Endocrinology 132:823–831. https://doi.org/10.1210/endo.132.2.8425497
Zebaze R, Seeman E (2015) Cortical bone: a challenging geography. J Bone Miner Res 30:24–29. https://doi.org/10.1002/jbmr.2419
Zebaze RM, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, Mackie EJ, Seeman E (2010) Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet 375:1729–1736. https://doi.org/10.1016/S0140-6736(10)60320-0
Zebaze R, Ghasem-Zadeh A, Mbala A, Seeman E (2013) A new method of segmentation of compact-appearing, transitional and trabecular compartments and quantification of cortical porosity from high resolution peripheral quantitative computed tomographic images. Bone 54:8–20. https://doi.org/10.1016/j.bone.2013.01.007
Disclosures
SKR has no disclosures. ES has received research support from Amgen and MSD and has received lecture fees from Amgen, Eli Lilly, and Allergan. He is a director and board member of StraxCorp.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Ramchand, S.K., Seeman, E. (2020). Reduced Bone Modeling and Unbalanced Bone Remodeling: Targets for Antiresorptive and Anabolic Therapy. In: Stern, P.H. (eds) Bone Regulators and Osteoporosis Therapy. Handbook of Experimental Pharmacology, vol 262. Springer, Cham. https://doi.org/10.1007/164_2020_354
Download citation
DOI: https://doi.org/10.1007/164_2020_354
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-57377-5
Online ISBN: 978-3-030-57378-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)