
Abstract
Staudinger’s (1922) “macromolecular hypothesis” stating that most synthetic and natural polymers could be rationalized as extensive covalently linked linear macromolecules, followed by Crick and Watson’s (1953) revelation that life was actually based on poly(nucleotide), helical double-stranded variations of Staudinger’s linear architectures, launched two of the most significant technological revolutions of the twentieth century. After Staudinger, a total of four major polymer architectures were recognized and each architecture, namely, (I) linear, (II) crosslinked (bridged), (III) branched, and (IV) dendritic (hyperbranched), is highly valued for its intrinsic and unique macromolecular properties. Upon entering the twenty-first century, members of architectural class IV, dendritic polymers (i.e., dendrimers), have now been accepted by both chemists and physics as quantized nanoscale building blocks due to their atom mimicry features and are referred to as “soft superatoms.” Atom mimicry, manifested by both soft and hard superatoms (i.e., organic and inorganic nanoscale clusters), has provided the first steps towards a proposed new nano-periodic paradigm, based on first principles from traditional chemistry and physics, for unifying nanoscience.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Atom mimicry
- Dendrimers
- Dendritic effects
- Hard/soft nano-elements
- Nanocompounds/assemblies
- Nano-periodic system
- Superatoms
1 Introduction
1.1 Evolution from Basic Building Blocks to Higher Complexity
Understanding the hierarchical principles and parameters involved in the natural evolution of first matter to the present state of complexity has received substantial attention by all the major scientific disciplines. Advancement of the “Big Bang Theory” by physicists has provided a foundation for understanding the early evolution of subpicoscale particles to elemental atoms, presumably based on thermodynamic selection principles. On the other hand, biologists have defined an acceptable hypothesis for the evolution of micro- and macroscale matter to higher complexity, including life and organisms, based on certain environmental selection principles. Between these two extremes, however, resides the unresolved evolutionary domain of the chemist (see Fig. 1). Hierarchical matter in this domain is defined by dimensions between the subnanoscale and the micron level. Recently, J.M. Lehn [1] and others [2] have advanced certain molecular recognition, supramolecular/self-assembly principles as first steps toward qualitatively defining both the natural and synthetic evolution of matter in this size region. Contemporary chemists now view elemental atoms and small, molecular structures (i.e., monomers) as versatile, richly endowed building blocks with important surface chemistry that may be supramolecularly assembled or chemically bonded into an infinite number of combinatorial molecular libraries. These libraries consist of both precise well-defined subnanoscale molecular structures and perhaps less well-defined nanoscale structures that we now refer to as macromolecules or polymers.

Evolution of hierarchical building blocks, structural information transfer, and scientific disciplines leading to present material complexity as a function of time lapsed from “the big bang”
1.1.1 Atomic Elements→Small Molecules→Macromolecules→Megamolecules
The seminal “macromolecular hypothesis” proposed in 1922 [3, 4] by H. Staudinger initiated one of the most significant technological revolutions of the twentieth century, namely, the polymer (plastics) revolution [5]. Staudinger was not recognized for this monumental contribution by the Nobel committee until 1953. Coincidentally, in that same year Crick and Watson first reported the characterization and structure of DNA. Perhaps two of the most important chemistry discoveries in the twentieth century were Staudinger’s “macromolecular hypothesis” and elucidation of the linear polymer, double helix structure evolved by Nature, namely, DNA [6, 7]. Macromolecular DNA was found to be an elegant covalent biopolymer that was indeed consistent with and could be accounted for by Staudinger’s earlier macromolecular hypothesis. This work by Crick and Watson was later recognized by the Nobel Prize committee in 1962 and initiated an equally important scientific understanding of linear nucleotide biopolymers and their role in storing and transferring critical genetic information as the basis for life. In spite of that, during the first part of the twentieth century, there was an almost fanatical opposition to the notion of Staudinger that atoms or their compounds could be transformed into chemically bonded macromolecular structures. However, in an abstract way, Staudinger’s concept may now be viewed as an elaborate continuation of J. Dalton’s simple hypothesis (i.e., New System of Chemical Philosophy, published in 1808). In essence, the theme of chemically connecting (n′) multiples of atomic modules to produce small molecular structures (e.g., monomers) could simply be extended to include the chemical linking of monomers to produce covalent macromolecular structures (Fig. 2).

Historical overview of the major technology revolutions “traditional chemistry” and “polymer chemistry” and their associated pioneers
This earlier atom/molecular hypothesis by Dalton led to synthesis of an endless array of small molecules that are now recognized as our “traditional chemistry”. On the other hand, Staudinger’s macromolecular hypothesis led to vast libraries of macromolecular structures now referred to as “traditional polymer chemistry.” Although the intrinsic features of atoms or monomers as well as their rules for assembly [i.e., (n′) and (n)] are most assuredly different, the enormous role that each of these technologies has played in the improvement of the “human condition” and enhancement of the world economy is indisputable. These benefits were largely derived from unique and extraordinary new properties that emerged in each of these areas as the technologies advanced to higher levels of complexity.
1.2 The Role of Molecular Architecture in Producing New Properties
A pervasive pattern apparent in both small-molecule chemistry as well as macromolecular science is the significant role that architecture plays in the determination of new properties. As early as 1825, Swedish chemist Jacob Berzelius clearly demonstrated that small molecular structures possessing identical elemental compositions, but different spatial arrangements, invariably differed in one or more physico-chemical properties such as melting or boiling point, density, combustion behavior, etc. Referred to as “molecular isomerism,” these isomeric states have been widely recognized in traditional inorganic and organic chemistry as geometric/position isomerism, valence isomerism, optical stereoisomerism, tautomerism, etc. In the polymer world, these analogous structural issues are referred to collectively as “macromolecular or architectural isomerism” [8]. Such macromolecular structures derived from identical monomeric building blocks in the same stoichiometric proportions but in different architectural or spatial configurations may be expected to manifest substantially different properties and macroscopic behavior. Thus, it was not surprising that traditional polymer architectures such as crosslinked (bridged) and simple branched polymers (after Staudinger’s first linear architectures) clearly manifested uniquely different as well as complementary properties ideally suited for the emergence of a vast array of diverse commercial applications. Early commercial polymer development usually involved the manipulation of three key parameters: (1) architecture (i.e., thermoplastic versus thermoset configurations and gels); (2) elemental composition (i.e., monomer or copolymer); and (3) molecular weight and molecular weight distribution. Ultimately, all macroscopic properties were determined, including process ability and performance. The advent of a fourth new macromolecular architecture (i.e., dendritic) exhibiting totally unprecedented physico-chemical properties compared to the traditional architectural classes (i.e., linear, crosslinked, and simple branched; see Fig. 3) led in the 1990s to a fresh examination of macromolecular architecture categories [9] and their impact on new emerging properties [10, 11].

Nobel recognition, commercial applications, and emerging properties for the four major macromolecular architectures
Four major macromolecular architectural classes are now recognized based on their unequivocal importance in driving new and differentiated properties. These four major macromolecular architectural classes are: (I) linear, (II) crosslinked/bridged, (III) branched, and (IV) dendritic/hyperbranched (as illustrated in Fig. 4). The importance of macromolecular architecture has been amply recognized by a preponderance of Nobel awards associated with the discovery of such architectural features and their consequent properties. Since Staudinger’s seminal Nobel Prize in 1953, a total of ten individual scientists have now been recognized by the Nobel Committee for their contributions to polymer science (as shown in Fig. 3). These recognized contributions may be placed in the general categories noted below:

Atomic small molecule and macromolecular architectures, with the emergence of new properties as a function of higher complexity
Discovery or Pioneering Characterization of the First Two Major Architectural Classes
H. Staudinger (1953) – Discovered linear, class I architecture
P. Flory (1974) – Clarified and defined crosslinked, class II architecture
Pioneering Modification or Characterization of Linear Class I Architecture
G. Natta, K. Ziegler (1963) – Polymerization catalyst, stereochemistry, tacticity
B. Merrifield (1984) – Controlled polypeptide sequencing, monodispersity
A. Heeger, A. MacDiarmid, H. Shirakawa (2000) – Polymer backbone conductivity
R. Grubbs, R. Schrock (2005) – Polymerization catalyst, monodispersity
History has shown that each time a major new architecture has been discovered, it has been accompanied by the emergence of a plethora of new properties, concepts, applications, products, and activities, all of which have led to enhanced new commercial markets, quality of life, and prosperity. Since Staudinger’s original discovery, a total of four major macromolecular architectures have evolved: (I) linear, (II) crosslinked, (III) branched and now (IV) dendritic topologies, as illustrated in Fig. 4.
2 Traditional Polymer Chemistry
Over the past 90 years, Staudinger’s macromolecular synthesis strategy has evolved based on the catenation of reactive small molecular modules (monomers). Broadly speaking, these catenations involve the use of reactive (AB-type) monomers that may be engaged to produce large molecules with polydispersed masses. Such multiple bond formation may be driven by (1) chain growth, (2) ring opening, (3) step-growth condensation, or (4) enzyme-catalyzed processes. Staudinger first introduced this paradigm in the 1920s [4, 5, 12–14] by demonstrating that reactive monomers could be used to produce a statistical distribution of one-dimensional (linear) molecules with very high molecular weights (i.e., >106 Da). As many as 10,000 or more covalent bonds may be formed in a single chain reaction of monomers. Although these macro- or megamolecules may possess nanoscale dimensions, structure control of critical macromolecular design parameters, such as size, molecular shape, spatial positioning of atoms, or covalent connectivity – other than those affording linear or crosslinked topologies – is difficult. However, substantial progress has been made in controlling dispersity by using living polymerization techniques that afford dramatic control over molecular weight and certain structural elements, as described by Matyjaszewski, Grubbs, Schrock, and others [15–19].

Traditional polymerizations usually involve AB-type monomers based on substituted ethylenes or strained small ring compounds using chain reactions that may be initiated by free radical, anionic or cationic initiators [20]. Alternatively, AB-type monomers may be used in polycondensation reactions.
Multiple covalent bonds are formed to produce each macromolecule, generally giving statistical, polydispersed structures. In the case of controlled vinyl polymerizations, the average length of the macromolecule is determined by monomer to initiator ratios. If one visualizes these polymerizations as extraordinarily long sequences of individual reaction steps, the average number of covalent bonds formed per chain may be described as shown in Scheme 1.

Mathematical description of covalent bond formation as a function of AB monomer polymerization to produce linear polymers [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
The first traditional polymerization strategies generally produced linear architectures; however, it was soon found that branched topologies may be formed either by chain transfer processes or intentionally introduced by grafting techniques. In any case, the linear and branched architectural classes have traditionally defined the broad area of thermoplastics. Of equal importance is the major architectural class formed by the introduction of covalent (bridging) bonds between linear or branched polymeric topologies. These crosslinked (bridged) topologies were studied by Flory in the early 1940s and constitute the second major area of traditional polymer chemistry, namely, thermosets. These two broad areas of polymer science (i.e., thermoplastics and thermosets) account for billions of dollars of commerce and constitute a vast array of familiar macromolecular compositions and applications, as shown in Fig. 5.

Dates of discovery and production of commercial thermoplastic and thermoset polymers, organized according to their architectural class [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
Historically, even 50 years after Staudinger’s introduction of the macromolecular hypothesis, the entire field of polymer science was viewed to consist of only the two major architectural classes: (1) linear topologies as found in thermoplastics and (2) crosslinked architectures as found in thermosets. The major focus of polymer science during the time frame spanning the 1920s to the 1970s was on unique architecturally driven properties manifested by either linear or crosslinked topologies. Based on the unique properties exhibited by these synthetic topologies, it was possible to replace many natural polymers crucial to the World War II effort. This combination of availability and properties were of utmost strategic importance [21]. During the 1960s and 1970s, pioneering investigation into long chain branching (LCB) involving polyolefins and other related branched systems began to emerge [22, 23]. More recently, intense commercial interest has been focused on new polyolefin architectures based on random long branched and dendritic topologies [24, 25]. These architectures are reportedly produced by “metallocene” and “Brookhart-type” catalysts. By the end of the 1970s, there were three major architectural polymer classes and commercial commodities associated with these topologies, as described chronologically in Fig. 6.

Traditional macromolecular architectures, organized chronologically according to their commercial introduction
2.1 Comparison of Traditional Polymer Science with Dendritic Macromolecular Science
Covalent synthesis in traditional polymer science has evolved around the use of reactive modules (AB-type monomer) or ABR-type branch reagents that may be engaged in multiple covalent bond formation to produce large one-dimensional molecules of various lengths. Such multiple bond formation may be driven either by chain reactions, ring-opening reactions, or polycondensation schemes. These propagation schemes and products are recognized as class I, linear or class III, branched architectures. Alternatively, using combinations and permutations of divalent AB-type monomers and/or AB n , A n B polyvalent, branch cell-type monomers produces class II, crosslinked (bridged) architectures.
A comparison of the covalent connectivity associated with each of these architecture classes (Fig. 7) reveals that the number of covalent bonds formed per step for linear and branched topology is a multiple (n = degree of polymerization) related to the monomer-to-initiator ratios. In contrast, ideal dendritic (class IV) propagation involves the formation of an exponential number of covalent bonds per reaction step (also termed G, for generation), as well as amplification of both mass (i.e., number of branch cells) and number of terminal groups per generation.

Examples of architectural polymer classes (I–IV), polymer type, repeat units, and covalent connectivity associated with architectural class [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
Mathematically, the number of covalent bonds formed per generation (reaction step) in the synthesis of an ideal dendron or dendrimer varies according to a power function of the reaction steps, as shown in Scheme 2. It is clear that covalent bond amplification occurs in all dendritic synthesis strategies. In addition to new architectural consequences, this feature clearly differentiates dendritic growth processes from linear covalent bond synthesis as found in traditional polymer chemistry [26].

Mathematical description of covalent bond formation as a function of AB2 monomer polymerization to produce dendritic polymers [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
It should be apparent that, although all major architectural polymer classes are derived from common or related repeat units, the covalent connectivity is truly discrete and different. Furthermore, mathematical analysis of the respective propagation strategies clearly illustrates the dramatic differences in structure development as a function of covalent bond formation. It should be noted that linear, branched, and dendritic topologies differ substantially both in their covalent connectivity as well as in the terminal group to initiator site ratios. In spite of these differences, these open, unlooped macromolecular assemblies clearly manifest thermoplastic polymer-type behavior in contrast to the looped, bridged connectivity associated with crosslinked, thermoset systems. In fact, it is now apparent that these three “open assembly” topologies (i.e., linear, branched, and dendritic) represent a graduated continuum of architectural intermediacy between thermoplastic and thermoset behavior, as will be described later (Sect. 3.2).
In summary, classical polymer science has provided facile access to a vast variety of polydispersed nanoscale structures, with some control over topology, composition, and flexibility or rigidity. More recent advances, however, involving “living polymerization” strategies [18, 19, 27] have produced substantially enhanced control over macromolecular size distribution and dispersity. That withstanding, dendritic macromolecular chemistry still remains the major strategy and route to unparalleled control over topology, composition, size, mass, shape, and functional group placement. These features and properties truly distinguish the many successful nanostructures found in nature [28] and as such are of keen interest as synthetic nanomaterials and for many applications in nanomedicine.
3 The Dendritic State
3.1 History
The origins of the present three-dimensional (3D), dendritic branching concepts can be traced back to the initial introduction of infinite network theory by Flory [29–32] and Stockmayer [33, 34]. In 1943, Flory introduced the term “network cell,” which he defined as the most fundamental unit in a molecular network structure [35]. To paraphrase the original definition, it is the recurring branch juncture in a network system as well as the excluded volume associated with this branch juncture. Graessley [36] took the notion one step further by describing ensembles of these network cells as micronetworks. Extending the concept of Flory’s statistical treatment of Gaussian-coil networks, analogous species that are part of an open, branched or dendritic organization are known as “branch cells” and “dendritic assemblies.”
Statistical modeling by Gordon et al. [37, 38], Dusek [39], Burchard [40] and others reduced such branched species to graph theory designed to mimic the morphological branching of trees. These dendritic models were combined with cascade theory [41, 42] mathematics to give a reasonable statistical treatment for network-forming events at that time.
The growth of branched and dendritic macromolecules in the sol phase of a traditional crosslinking process may be thought of as geometric aggregations of various branch cells or dendritic (network) assemblies, as described above. Beginning as molecular species, they advance through the dimensional complexity hierarchy to oligomeric, macromolecular, megamolecular, and ultimately to infinite network macroscale systems. The intermediacy of dendritic architecture in this continuum will be discussed later (Sect. 3.2). Traditional network-forming systems (e.g., epoxy resins, urethanes, polyesters) progress through this growth process in a statistical, random fashion. The resulting infinite networks may be visualized as a collection of unequally segmented Gaussian chains between f-functional branch junctures, crosslinks (loops), and dangling terminal groups.
More recently, non-traditional polymerization strategies have evolved to produce a fourth new major polymer architectural class, now referred to as “dendritic polymers” [43]. This new architectural polymer class consists of four major subsets: (1) random hyperbranched, (2) dendrigrafts, (3) dendrons and (4) dendrimers. Dendrimers, the most extensively studied subset were discovered by the Tomalia group while in The Dow Chemical Company laboratories (1979) and represent the first example of synthetic, macromolecular dendritic architecture [43, 44]. First use of the term “dendrimer” appeared in preprints for the first SPSJ International Polymer Conference, held in Kyoto, Japan in 1984 [45]. The following year, a full article in Polymer Journal [46] (Fig. 8) described the first preparation of a complete family of Tomalia-type poly(amidoamine) (PAMAM) dendrimers (G = 1–7) and their use as precise, fundamental building blocks to form poly(dendrimers) or so-called “starburst” polymers. These poly(dendrimers) are now referred to as “megamers” [47, 48] and are described in more detail later in Sect. 6.4.3. Other pioneers in the dendritic polymer field include Vogtle, Newkome, Frechet, Majoral, and others. These historical contributions have been reviewed recently [52] .

Abstract of the first full article describing the synthesis of a complete family of dendrimers [55]
This article will overview the dendritic architectural state, its unique architecturally driven properties, its role relative to traditional polymer science, and describe the many enabling features that dendrimers are expected to offer to the emerging nanotechnology revolution.
3.2 A Fourth Major New Architectural Polymer Class
Dendritic topology has now been recognized as a fourth major class of macromolecular architecture [49–51]. The signature for such a distinction is the unique repertoire of new properties manifested by this class of polymers [9, 26, 52–56]. Numerous synthetic strategies have been reported for the preparation of these materials, and have led to a broad range of dendritic structures. Currently, this architectural class namely, Dendritic (IV) consists of three dendritic subclasses: (IVa) random hyperbranched polymers, (IVb) dendrigraft polymers, and (IVc) dendrons/dendrimers (Fig. 9). The order of this subset, from IVa to IVc, reflects the relative degree of structural control present in each of these dendritic architectures.

Branch cell structural parameters: a branching angle, b rotation angle, l repeat unit length, Z terminal group, I molecular reference marker or core. Dendritic subclasses derived from branches: IVa random hyperbranched, IVb dendrigrafts, and IVc dendrons/dendrimers [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
All dendritic polymers are open covalent assemblies of branch cells. They may be organized as very symmetrical, monodispersed arrays, as is the case for dendrimers, or as irregular polydispersed assemblies that typically define random hyperbranched polymers. As such, the respective subclasses and the level of structure control are defined by the propagation methodology used to produce these assemblies, as well as by the branch cell construction parameters. The branch cell parameters are determined by the composition of the branch cell monomers, as well as by the nature of the “excluded volume” defined by the branch cell. The excluded volume of the branch cell is determined by the length of the arms, the symmetry, rigidity/flexibility, as well as the branching and rotation angles involved within each of the branch cell domains. As shown in Fig. 9, these dendritic arrays of branch cells usually manifest covalent connectivity relative to some molecular reference marker (I) or core. As such, these branch cell arrays may be very non-ideal and polydispersed (e.g. M w/M n ≅ 2–10), as observed for random hyperbranched polymers (IVa), or very ideally organized into highly controlled core–shell type structures, as noted for dendrons/dendrimers (IVc) (M w/M n ≅ 1.01–1.0001 and less). Dendrigraft (arborescent) polymers reside between these two extremes of structure control, frequently manifesting rather narrow polydispersities of M w/M n ≅ 1.1–1.5, depending on their mode of preparation.
3.3 Dendritic Polymer Subclasses
3.3.1 Random Hyperbranched Polymers
Flory first hypothesized dendritic polymer concepts [32, 30], which are now recognized to apply to statistical or random hyperbranched polymers. However, the first experimental confirmation of dendritic topologies did not produce random hyperbranched polymers but rather the more precise, structure-controlled, dendrimer architecture [43, 44, 46, 55]. This work was initiated nearly a decade before the first examples of random hyperbranched polymers were confirmed independently by Gunatillake, Odian et al. [57], as well as by and by Kim and Webster [58, 59] in 1988. At that time, Kim and Webster coined the popular term “hyperbranched polymers” that has been widely used to describe this subclass of dendritic macromolecules. Hyperbranched polymers are typically prepared by polymerization of AB x monomers. When x is 2 or more, polymerization gives highly branched random polymers, as long as A reacts only with B from another molecule. Reactions between A and B from the same molecule result in termination of polymerization by cyclization. This approach produces hyperbranched polymers with a degree of polymerization n, possessing one unreacted A functional group and [(x – 1) n + 1] unreacted B terminal groups. In a similar fashion, copolymerization of A2 and B3 or other such polyvalent monomers can give hyperbranched polymers [60, 61] if the polymerization is maintained below the gel point by manipulating monomer stoichiometry or limiting polymer conversion. Random hyperbranched polymers are generally produced by the one-pot polymerization of AB x -type monomers or macromonomers involving polycondensation, ring opening, or polyaddition reactions. Hence, the products usually have broad, statistical molecular weight distributions, much as observed for traditional polymers. Over the past decade, literally dozens of new AB2-type monomers have been reported, leading to an enormously diverse array of hyperbranched structures. Some general types include poly(phenylenes) obtained by the Suzuki coupling [58, 59]; poly(phenylacetylenes) prepared by the Heck reaction [62]; polycarbosilanes, polycarbosiloxanes [63], and poly(siloxysilanes) by hydrosilylation [64]; poly(ether ketones) by nucleophilic aromatic substitution [65]; and polyesters [66] or polyethers [67] by polycondensations or by ring-opening polymerization [68].
New advances beyond the traditional AB2 Flory-type, branch cell monomers have been reported by Fréchet and coworkers [69, 70]. They introduced the concept of latent AB2 monomers, referred to as self-condensing vinyl polymerizations (SCVP). These monomers, which possess both initiation and propagation properties, may follow two modes of polymerization: polymerization of the double bond (i.e., chain growth) and condensation of the initiating group with the double bond (i.e., step growth). Recent progress involving the derivative process of self-condensing, ring-opening polymerizations (SCROP) has been reviewed by Sunder et al. [71]. In addition, the use of enhanced processing techniques such as pseudo chain growth by slow monomer addition [72], allow somewhat better control of hyperbranched structures [71].
3.3.2 Dendrigraft Polymers
Dendrigraft polymers are the most recently discovered and currently the least understood subset of dendritic polymers. The first examples were reported in 1991 independently by Tomalia et al. [73] and Gauthier et al. [74]. Whereas traditional monomers are generally employed in constructing dendrimers, reactive oligomers or polymers are used in protect–deprotect or activation schemes to produce dendrigrafts. Consequently, dendrigraft polymers are generally larger structures than dendrimers, grow much faster, and amplify surface groups more dramatically as a function of generational development. Both hydrophilic [e.g., poly(oxazolines) and poly(ethyleneimines)] and hydrophobic (e.g., polystyrenes) dendrigrafts were reported in these early works. These first methodologies involved the iterative grafting of oligomeric reagents derived from living polymerization processes in various iterative “graft-on-graft” strategies. By analogy to dendrimers, each iterative grafting step is referred to as a generation. An important feature of this approach is that branch densities, as well as the size of the grafted branches, can be varied independently for each generation. Furthermore, by initiating these iterative grafting steps from either a point-like core or a linear core it is possible to produce spheroidal and cylindrical dendrigrafts, respectively. Depending on the graft densities and molecular weights of the grafted branches, ultrahigh molecular weight dendrigrafts (e.g., M w > 104 kDa) can be obtained at very low generation levels (e.g., G = 3). Dramatic molecular weight enhancements vis-à-vis other dendrimer propagation methodologies are possible using dendrigraft techniques [75]. Further elaboration of these dendrigraft principles allowed the synthesis of a variety of core–shell-type dendrigrafts, in which elemental composition as well as the hydrophobic or hydrophilic character of the core were controlled independently [74].
In general, the above methodologies have involved convergent-type grafting principles whereby preformed, reactive oligomers are grafted onto successive branched precursors to produce semicontrolled structures. Compared to dendrimers, dendrigraft structures are less controlled since grafting may occur along the entire length of each generational branch, and the exact branching densities are somewhat arbitrary and difficult to control. More recently, both Gnanou [76, 77] and Hedrick [78, 79] have developed approaches to dendrigrafts that mimic dendrimer topologies by confining the graft sites to the branch termini for each generation. These methods involve so-called “graft from” techniques and allow better control of branching topologies and densities as a function of generation. Topologies produced by these methods are reminiscent of the dendrimer architecture. Since the branch-cell arms are derived from oligomeric segments, the products are referred to as polymeric dendrimers [22, 78, 79]. These more flexible and extended structures exhibit unique and different properties compared to the more compact traditional dendrimers. Fréchet, Hawker, and coworkers [80] have utilized the techniques of living polymerization and a staged polymerization process (in which latent polymerization sites are incorporated within growing chains) to produce dendrigrafts of mixed composition and narrow polydispersity.
Another exciting development has been the emerging role that dendritic architecture is playing in the production of commodity polymers. A recent report by Guan et al. [24] has shown that ethylene polymerizes to dendrigraft polyethylene (dendri-polyethylene) at low pressures, in contrast to high-pressure conditions which produce only simple branched topologies. This occurs when using late-transition metal or Brookhart catalysts. Furthermore, these authors also state that small amounts of dendri-poly(ethylene) architecture may be expected from analogous early-transition-metal metallocene catalysts.
3.3.3 Dendrons and Dendrimers
Dendrons and dendrimers are the most intensely investigated subset of dendritic polymers. In the past decade, over 6,000 literature references have appeared dealing with this unique class of structure-controlled polymers. The word “dendrimer” is derived from the Greek words dendri- (tree-branch-like) and meros (part of), and was coined by Tomalia, et al. about 20 years ago in the first full paper on PAMAM dendrimers [45, 46]. Since this early disclosure, over 125 dendrimer compositions (families) and 1,100 dendrimer surface modifications have been reported. The two most widely studied dendrimer families are the Fréchet-type polyether compositions and the Tomalia-type PAMAM dendrimers. PAMAM dendrimers constitute the first dendrimer family to be commercialized, and represent the most extensively characterized and best-understood series at this time [55].
3.3.3.1 Dendrimer Synthesis: Divergent and Convergent Methods
In contrast to traditional polymers, dendrimers are unique core–shell structures possessing three basic architectural components (Fig. 10): a core, an interior of shells (generations) consisting of repeating branch-cell units, and terminal functional groups (the outer shell or periphery). In general, dendrimer synthesis involves divergent or convergent hierarchical assembly strategies that require the construction components shown in Scheme 3. Within each of these major approaches there may be variations in methodology for branch-cell construction or dendron construction. Many of these issues, together with experimental laboratory procedures, have been reviewed elsewhere [81–83].

Three-dimensional projection of dendrimer core–shell architecture for G = 4.5 poly(amidoamine) (PAMAM) dendrimer showing principal architectural components: (I) core, (II) interior, and (III) surface [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission

Strategies for dendrimer synthesis [52]. Copyright: Cambridge University Press
PAMAM dendrimers are synthesized by the divergent approach. This methodology involves in situ branch-cell construction in stepwise, iterative stages around a desired core to produce mathematically defined core–shell structures. Typically, ethylenediamine (core multiplicity N c = 4), ammonia (N c = 3), or cystamine (N c = 4) may be used as cores and allowed to undergo reiterative, two-step reaction sequences. These sequences consist of: (1) an exhaustive alkylation of primary amines (Michael addition) with methyl acrylate, and (2) amidation of amplified ester groups with a large excess of ethylenediamine to produce primary amine terminal groups (Fig. 10). This first reaction sequence on the exposed core creates G = 0 (i.e., the core branch cell), wherein the number of arms (i.e., dendrons) anchored to the core is determined by N c. Iteration of the alkylation–amidation sequence produces an amplification of terminal groups from one to two with the in situ creation of a branch cell at the anchoring site of the dendron that constitutes G = 1. Repeating these iterative sequences (Fig. 10) produces additional shells (generations) of branch cells that amplify mass and terminal groups according to the mathematical expressions shown in the box in Fig. 11). It is apparent that both the core multiplicity (N c) and branch cell multiplicity (N b) determine the precise number of terminal groups and mass amplification as a function of generation. One may view those generation sequences as quantized polymerization events. The assembly of reactive monomers [44, 84], branch cells [9, 55, 56], or dendrons [55, 85, 86] around atomic or molecular cores, to produce dendrimers according to divergent or convergent dendritic branching principles, has been well demonstrated. Such systematic filling of molecular space around cores with branch cells as a function of generational growth stages (branch-cell shells) – to give discrete, quantized bundles of nanoscale mass – has been shown to be mathematically predictable [10, 11, 26]. Predicted molecular weights have been confirmed by mass spectrometry [87–90] and other analytical methods [9, 52, 91, 92]. Predicted number of branch cells, number of terminal groups, and molecular weight as a function of generation for an ethylenediamine-core (N c = 4) PAMAM dendrimer are shown in Fig. 11. It should be noted that the molecular weight approximately doubles as one progresses from one generation to the next. The number of surface groups and branch cells amplify mathematically according to a power function, thus producing discrete, monodispersed structures with precise molecular weights and a nanoscale diameter enhancement, as described in Fig. 11. These predicted values are routinely verified by mass spectrometry for the earlier generations (i.e., G = 4–5); however, with divergent dendrimers, minor mass defects are often observed for higher generations as congestion-induced de Gennes dense packing begins to take effect [9, 52, 93, 94].

Dendritic branching mathematics for predicting the number of dendrimer surface groups, number of branch cells, and molecular weight. Calculated values are for [ethylenediamine core] dendri-poly(amidoamine) series with nanoscale diameters
4 Dendrimer Features of Interest to Nanoscience
Dendrimers may be viewed as unique, information processing, nanoscale devices. Each architectural component (core, interior, and surface) manifests a specific function, while at the same time defining properties for these nanostructures as they are grown generation by generation. For example, the core may be thought of as the molecular information center from which size, shape, directionality, and multiplicity are expressed via the covalent connectivity to the outer shells. Within the interior, one finds the branch cell amplification region, which defines the type and amount of interior void space that may be enclosed by the terminal groups as the dendrimer is grown. Branch cell multiplicity (N b) determines the density and degree of amplification as an exponential function of generation. The interior composition and amount of solvent-filled void space determines the extent and nature of guest–host (endoreceptor) properties that are possible within a particular dendrimer family and generation. Finally, the surface consists of reactive or passive terminal groups that may perform several functions. With appropriate functionality, they serve as a template polymerization region as each generation is amplified and covalently attached to the precursor generation. The surface groups may also serve as passive or reactive gates controlling entry or departure of guest molecules from the dendrimer interior. These three architectural components determine the physical and chemical properties, as well as the overall size, shape and flexibility of the dendrimers. It is important to note that dendrimer diameters increase linearly as a function of the number of shells or generations added, whereas the terminal functional groups increase exponentially as a function of generation. This dilemma enhances “tethered congestion” of the anchored dendrons, as a function of generation, due to the steric crowding of the end groups. As a consequence, lower generations are generally open, floppy structures, whereas higher generations become robust, less-deformable spheroids, ellipsoids, or cylinders depending on the shape and directionality of the core.
Tomalia-type PAMAM dendrimers are synthesized by the divergent approach. This methodology involves in situ branch cell construction in stepwise, iterative stages (i.e., G = 1, 2, 3 …) around a desired core to produce mathematically defined nanoscale core–shell structures. Typically, ethylenediamine (N c = 4) or ammonia (N c = 3) are used as nucleophilic cores and are allowed to undergo reiterative two-step reaction sequences involving: (1) exhaustive alkylation of primary amines (Michael addition) with methyl acrylate and (2) amidation of amplified ester groups (Fig. 10) with a large excess ethylenediamine to produce primary amine terminal groups.
This first reaction sequence on the exposed dendron (Fig. 12) creates G = 0 (i.e., the core branch cell), wherein the number of arms (i.e., dendrons) anchored to the core is determined by N c. Iteration of the alkylation/amidation sequence produces an amplification of terminal groups from one to two, with the in situ creation of a branch cell at the anchoring site of the dendron that constitutes G = 1. Repeating these iterative sequences produces additional shells (generations) of branch cells that amplify mass and terminal groups according to the mathematical expressions described in Fig. 11.

Comparison of molecular shape change, two-dimensional branch cell amplification, number of surface branch cells, number of surface Z groups, and molecular weight as function of generation for G = 0–6 [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
As early as 2001, Nobel Laureate Prof. B. Sharpless popularized a modular approach to organic synthesis that he referred to as “click chemistry” [95, 96]. This strategy was defined in the context of four major organic reaction categories:
-
1.
Addition of nucleophiles to activated double bonds (i.e., Michael addition chemistry)
-
2.
“Non-aldol”-type carbonyl chemistry (i.e., formation of amides, hydrazones, etc.)
-
3.
Nucleophilic ring opening of strained heterocyclic electrophiles (i.e., aziridines, epoxides, etc.)
-
4.
Huisgen-type 1,3-dipolar cycloaddition of azides to alkynes
It should be noted that the first three reaction categories of click chemistry, as described above, were used by Tomalia [9, 46] and Vögtle [97] as preferred iterative synthetic routes to the first reported examples of dendrimers and low molecular weight cascade molecules, respectively.
In 1968, Huisgen [98] reported the facile, high yield, chemoselective cycloaddition of organic azides with alkynes to form covalent 1,4-disubstituted 1,2,3-triazole linkages. More recently, Sharples and colleagues [96, 99] have shown that terminal alkynes may be catalyzed by Cu1+ salts in an orthogonal fashion to form the corresponding triazoles in very high yields. Because of the high chemoselectivity of these reactions, they may be selectively performed in the presence of a wide variety of competing or parallel reactions and/or functionalities without interference. These features make this approach very attractive for dendrimer syntheses. Click chemistry based on these copper-catalyzed Huisgen reactions has been used recently to synthesize dendrimers [99–101], dendronized linear polymers [102], and other dendritic architectures [103].
It is apparent that both the core multiplicity (N c) and branch cell multiplicity (N b) determine the precise number of terminal groups and mass amplification as a function of generation. One may view those generation sequences as quantized polymerization events. The assembly of reactive monomers [9, 44], branch cells [9, 55, 56], or dendrons [55, 85, 86] around atomic or molecular cores to produce dendrimers according to divergent or convergent dendritic branching principles has been well demonstrated. Such systematic filling of space around cores with branch cells, as a function of generational growth stages (branch cell shells), to give discrete, quantized bundles of mass has been shown to be mathematically predictable (Fig. 11) [10, 11, 26]. Predicted molecular weights have been confirmed by mass spectroscopy [87–89] and other analytical methods [9, 85, 91, 92, 104]. Predicted numbers of branch cells, numbers of terminal groups, and molecular weights as a function of generation for an ethylenediamine-core (N c = 4) PAMAM dendrimer are shown in Fig. 12. It should be noted that the molecular weights approximately double as one progresses to the next generation. The number of surface groups and branch cells amplify mathematically according to a power function, thus producing discrete, monodispersed structures with precise molecular weights and nanoscale diameter enhancement, as described in Fig. 11. These predicted values are routinely verified by mass spectroscopy for the earlier generations (i.e., G = 4–5); however, with divergent dendrimers, minor mass defects are often observed for higher generations as congestion-induced de Gennes dense packing begins to take affect (Fig. 12).
4.1 Dendrimer Shape Change: A Nanoscale Molecular Morphogenesis
As illustrated in Fig. 12, dendrimers undergo congestion-induced molecular shape changes from flat, floppy conformations to robust spheroids, as first predicted by Goddard and coworkers [84]. Shape change transitions were subsequently confirmed by extensive photo-physical measurements, pioneered by Turro and coworkers [105–108] and solvatochromic measurements by Hawker et al. [109]. Depending upon the accumulative core and branch cell multiplicities of the dendrimer family under consideration, these transitions were found to occur between G = 3 and G = 5. Ammonia-core, PAMAM dendrimers (N c = 3, N b = 2) exhibited a molecular morphogenesis break at G = 4.5, whereas the ethylenediamine-core PAMAM dendrimer family (N c = 4, N b = 2) manifested a shape change break at around G = 3–4 [84] and the Fréchet-type convergent dendrons (N b = 2) at around G = 4 [109]. It is readily apparent that increasing the core multiplicity from N c = 3 to N c = 4 accelerates congestion and forces a shape change at least one generation earlier. Beyond these generational transitions, one can visualize these dendrimeric shapes as nearly spheroidal or slightly ellipsoidal core–shell architectures. Studies by Tomalia and colleagues [110] as well as Schluter and colleagues [111] have shown that the cylindrical or rod-shaped dendrimers are routinely formed by dendronizing traditional linear polymers. These new constructs derived from linear polymer backbones are pendant dendrons and are referred to as “architectural copolymers” [52].
4.2 de Gennes Dense Packing: A Nanoscale Steric Phenomenon Not Observed in Traditional Polymers
As a consequence of excluded volume associated with the core, interior, and surface branch cells, steric congestion is expected to result due to tethered core connectivity. Furthermore, the number of dendrimer surface groups, Z, amplifies with each subsequent generation. This occurs according to geometric branching laws, which are related to core multiplicity (N c) and branch cell multiplicity (N b). These values are defined by the following equation:
Since the radii of the dendrimers increase in a linear manner as a function of generation number G, whereas the surface cells amplify according to N c N b G, it is implicit from this equation that generational reiteration of branch cells ultimately will lead to a so-called dense-packed state.
As early as 1983, de Gennes and Hervet [43, 112] proposed a simple equation, derived from fundamental principles, to predict dense-packed generation for PAMAM dendrimers. It was predicted that at this generation, ideal branching can no longer occur because available surface space becomes too limited for the mathematically predicted number of surface cells to occupy. This produces a “closed geometric structure.” The surface is “crowded” with exterior groups that, although potentially chemically reactive, are sterically prohibited from participating in ideal dendrimer growth.
This “critical packing state” does not preclude further dendrimer growth beyond this point in the genealogical history of the dendrimer preparation. On the contrary, although continuation of dendrimer step-growth beyond the dense-packed state cannot yield structurally ideal, next generation dendrimers, it can nevertheless occur, as indicated by further increases in the molecular weight of the resulting products. Predictions by de Gennes [112] suggested that the PAMAM dendrimer series should reach a critical packing state at G = 9–10. Experimentally, we observed a moderate molecular weight deviation from predicted ideal values beginning at G = 4–7 (Fig. 13). This digression became very significant at G = 7–8 and as dendrimer growth was continued to generation 12 [94]. The products thus obtained are of “imperfect” structure because of the inability of all surface groups to undergo further reaction. Presumably, some of these surface groups remain trapped or are sterically encumbered under the surface of the newly formed dendrimer shell, yielding a unique architecture possessing two types of terminal groups. This new surface group population will consist of both those groups that are accessible to subsequent reiteration reagents and those that will be sterically screened. The total number of these groups will not, however, correspond to the predictions of the mathematical branching law, but will fall between the value that was mathematically predicted for the next generations (i.e., G + 1) and that expected for the precursor generation. Thus, a mass-defective dendrimer “generation” is formed.

(a) Comparison of theoretical and observed molecular weights and percentage shell filling for ethylenediamine-core poly(amidoamine) (PAMAM) dendrimers as a function of generation for G = 1–10. (b) Comparison of theoretical and observed molecular weights and percentage shell filling for NH3-core PAMAM dendrimers as a function of generation for G = 1–12 [93]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
Dendrimer surface congestion can be appraised mathematically as a function of generation, from the following simple relationship:
where A z is the surface area per terminal group Z, A D the dendrimer surface area, N z the number of surface groups Z per generation, and r the dendrimer radius. This relationship predicts that at higher generations, the surface area per Z group becomes increasingly smaller and experimentally approaches the cross-sectional area or van der Waals dimension of the surface groups Z. The generation G thus reached is referred to as the “de Gennes dense-packed generation” [9, 26, 55]. Ideal dendritic growth without branch defects is possible only for those generations preceding this dense-packed state. This critical dendrimer property gives rise to self-limiting dendrimer dimensions, which are a function of the branch cell segment length (l), the core multiplicity N c, the branch cell juncture multiplicity N b, and the steric dimensions of the terminal group Z (Fig. 10). Whereas the dendrimer radius r in the above expression is dependent on the branch cell segment lengths l, large l values delay this congestion. On the other hand, larger N c and N b values and larger Z dimensions dramatically hasten it.
Additional physical evidence supporting the development of congestion as a function of generation is shown in the composite comparison of dendrimer nano-periodic property patterns as illustrated in Sect. 6.5.2. Plots of intrinsic viscosity [η] [9, 113], density z, surface area per Z group (A z), and refractive index n as a function of generation clearly show maxima or minima at G = 3–5, paralleling computer-assisted molecular-simulation predictions [84, 114], as well as extensive photochemical probe experiments reported by Turro and coworkers [105–108].
Clearly, this de Gennes dense-packed congestion would be expected to contribute to (1) sterically inhibited reaction rates and (2) sterically-induced stoichiometry [9]. Each of these effects was observed experimentally at higher generations. The latter would be expected to induce dendrimer mass defects at higher generations, which we have used as a diagnostic signature for appraising the de Gennes dense packing effect.
Theoretical dendrimer mass values were compared to experimental values by performing electrospray and MALDI-TOF mass spectrometry analysis on the respective PAMAM families (i.e., N c = 3 and 4) [88]. Note that there is essentially complete shell filling for the first five generations of the NH3-core PAMAM series (N c = 3, N b = 2) (Fig. 13b). A gradual digression from theoretical masses occurs for G = 5–8, followed by a substantial break (i.e., Δ = 23%) between G = 8 and 9. This discontinuity in shell saturation is interpreted as a signature for de Gennes dense packing. It should be noted that shell saturation values continue to decline monotonically beyond this breakpoint to a value of 35.7% of theoretical at G = 12. A similar trend is noted for the ethylenediamine-core PAMAM series (N c = 4, N b = 2); however, the shell saturation inflection point occurs at least one generation earlier (i.e., G = 4–7, see Fig. 13a). This suggests that the onset of de Gennes dense packing may be occurring between G = 7 and 8. Recent work by Halperin, Schluter and coworkers [111] describes a simple yet elegant strategy for detecting the onset of de Gennes dense packing by UV labeling dendrimer surfaces with the Sanger reagent, as a function of generation, and monitoring signal regression as an indication of congestion and dense packing. This protocol provides a photolabeling technique that corroborates mass spectrometry data, as shown in Fig. 13.
Unique features offered by the “dendritic state” that have no equivalency in classical polymer topologies are found almost exclusively in the dendron/dendrimer subset and to a slightly lesser degree in the dendrigrafts. They include:
-
1.
Nearly complete nanoscale size and mass monodispersity
-
2.
The ability to control congestion, shape, and nanocontainer/scaffolding properties as function of generation
-
3.
Mathematically defined exponential amplification and functionalization of dendrimer surface chemistry
-
4.
Nanoscale dimensions and shape mimicry of proteins
-
5.
Dendrimer interior guest–host encapsulation properties for both inorganic and organic guests
These features are captured to some degree with dendrigraft polymers; however, they are either absent or present to a vanishing small extent for random hyperbranched polymers.
5 Unique Quantized Dendrimer Properties
5.1 Critical Nanoscale Design Parameters
The structure-controlled features manifested by dendrons/dendrimers, such as: size, shape, surface chemistry, flexibility/rigidity, elemental composition, and architecture, have provided a unique window to a new systematic concept for unifying nanoscience and will be described later in Section 6. These nanolevel structure-controlled features are referred to as “critical nanoscale design parameters” (CNDPs).
5.1.1 Controlled Nanoscale Monodispersity
The monodispersed nature of dendrimers has been verified extensively by mass spectroscopy, size exclusion chromatography, gel electrophoresis, and transmission electron microscopy (TEM) [55, 115]. As is always the case, the level of monodispersity is determined by the skill of the synthetic chemist, as well as the isolation or purification methods utilized. In general, convergent methods produce the most nearly isomolecular dendrimers. This is because the convergent growth process allows purification at each step of the synthesis and eliminates cumulative effects due to failed couplings [85, 116]. Appropriately purified, convergent dendrimers are probably the most precise synthetic macromolecules that exist today.
As discussed earlier, mass spectroscopy has shown that PAMAM dendrimers produced by the divergent method are very monodisperse and have masses consistent with predicted values for the earlier generations (i.e., G = 0–5) (Fig. 13). Even at higher generations, as one enters the de Gennes dense packed region, the molecular weight distributions remain very narrow (i.e., 1.05) and consistent, in spite of the fact that experimental masses deviate substantially from predicted theoretical values. Presumably, de Gennes dense packing produces a very regular and dependable effect that is manifested by the observed narrow molecular weight distribution.
5.1.2 Controlled Nanoscale Shapes and Container or Scaffolding Properties
Systematic shape and unimolecular container or scaffolding behavior appears to be a nano-periodic property that is specific to each dendrimer family or series. These properties are determined by the size, shape, and multiplicity of the construction components used for the core, interior, and surface of the dendrimer (Fig. 12). Higher multiplicity components and those that contribute to “tethered congestion” will hasten the development of more rigid shapes, container properties, and less flexible surface scaffolding as a function of generation.
5.2 Amplification and Functionalization of Dendrimer Surface Groups
Dendrimers within a generational series can be expected to present their terminal groups in at least three different modes, namely, flexible, semi-flexible, or rigid functionalized scaffolding. Based on mathematically defined dendritic branching rules (i.e., Z = N c N b G), the various surface presentations become more congested and rigid as a function of increasing generation level. It is implicit that this surface amplification can be designed to control gating properties associated with unimolecular container development. Furthermore, dendrimers may be viewed as versatile nanosized objects that can be readily surface-functionalized with a vast array of chemical and application features. Presently, well over 1,000 diverse surface functionalities have been attached to dendrimer surfaces [52]. The ability to control and engineer these parameters provides an endless list of possibilities for utilizing dendrimers as modules for nanodevice design [11, 48, 50, 117]. Recent reviews have begun to focus on this area [118–122].
5.3 Nanoscale Dimensions and Shapes Mimic Those of Proteins
In view of the extraordinary structure control and nanoscale dimensions observed for dendrimers, it is not surprising to find extensive interest in the use of dendrimers as globular protein mimics. Based on their systematic, dimensional length scaling properties and electrophoretic/hydrodynamic [91, 92] behavior, they are widely recognized as artificial proteins [48, 123]. Substantial effort has been focused recently on the use of dendrimers for “site isolation” mimicry of proteins [9], enzyme-like catalysis [124], viral capsid mimicry [125] and other biomimetic applications [48, 126], drug delivery [119, 123, 127, 128], surface engineering [129], and light harvesting [130, 131]. These fundamental properties have in fact led to their commercial use as globular protein replacements for gene therapy, immunodiagnostics [132, 133], and a variety of other biological applications [52].
6 Dendrimers: Window to a New Nano-periodic System for Defining and Unifying Nanoscience
“Science will continue to advance regardless of disputes over priorities. However, confusion and disagreement over common scientific language and standards can plunge a discipline into chaos. Such was the case for 19th century traditional chemistry before the emergence of Mendeleyev’s Periodic Table of the Elements (1869).” From Mendeleyev’s Dream – The Quest for the Elements by P. Strathern [134].
Clearly the need for a unifying system and framework that provides a central dogma with predictive capabilities for a priori design assessment as well as for defining risk/benefit boundaries remains an urgent challenge for nanotechnology [135]. Historically, a similar challenge existed for traditional chemistry in the early nineteenth century. Prior to the emergence of a central dogma and a common scientific language, traditional chemistry was viewed as an empirical discipline, which was transformed into a precise, predictive science only after the advent of atomic/molecular theory, established stoichiometries, and the emergence of well-defined periodic property patterns as first described by Mendeleev in 1869 [134].
It is from this perspective that the National Science Foundation (NSF) sponsored a workshop entitled “Periodic patterns, relationships and categories of well-defined nanoscale building blocks” in 2007 [136]. This seminal workshop evolved an embryonic consensus that subsequently led to a proposed concept for defining and unifying nanoscience based on the integration of traditional chemistry “first principles” with certain critical hierarchical design parameters (CHDPs) [137, 138]. These CHDPs include size, shape, surface chemistry, flexibility/rigidity, composition, and architecture and appear to be conserved and transferred as a function of complexity (illustrated in Fig. 14).

Structural control of critical hierarchical design parameters (CHDPs), namely, size, shape, surface chemistry, flexibility/rigidity, composition, and architecture, required for bottom-up synthesis of higher nanostructural complexity manifesting atom mimicry
These highly conserved CHDP transformations were first reported for a wide range of divergent, structure-controlled dendrimer syntheses as early as 1990 [9]. These syntheses provided a remarkable window for observing CHDP-dependent structure control related to divergent dendrimer synthesis. This structure control and information transfer was observed from the atomic scale (critical atomic design parameters, CADP), i.e., 10−11 m → molecular/subnanoscale (critical molecular design parameters, CMDP), i.e., 10−10 m → nanoscale level (critical nanoscale design parameters, CNDP), i.e., 10−9 m, as shown in Fig. 15. Furthermore, it became readily apparent that these CHDPs defined discrete, reproducible hierarchical periodic property patterns. These patterns were uniquely different at each of these hierarchical levels. In essence, the predictions of Nobel Laureate physicist, P.W. Anderson in 1972 were observed to be fulfilled [139]. Simply stated, as one breaks hierarchical symmetry by advancement with well-defined building blocks to higher structural complexity, the whole becomes not only more than, but very different from the sum of its parts. As a consequence, one should expect to observe totally new emerging nanomaterial properties and patterns that are unprecedented and uncharacteristic compared to the less complex hierarchical precursors and building blocks involved in their construction.

Front cover of Angew Chem Int Ed Engl (1990), 29:138–175 first describing structural control of critical hierarchical design parameters (CHDP) from atoms to macroscopic matter observed during the divergent syntheses of all dendrimers [9]. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission
6.1 Elemental Picoscale Periodicity Derived from CADPs
It is generally accepted that very specific amounts and arrangements of quantized subatomic building block constituents (i.e., particles such as electrons, protons, and neutrons) are involved in the production of all known atomic elements. The unique quantities and ratios of these self-assembling subatomic building blocks, by definition, determine the discrete and unique physico-chemical properties of each atomic element. As a consequence, each atomic element possesses a unique list of CADPs that allows them to be reproducibly defined and structure-controlled as a function of CADPs such as size, shape, surface chemistry, flexibility (i.e., polarizability), elemental composition (i.e., number of protons, neutrons, and electrons), and architecture. As such, these CADP-derived picoscale building blocks are observed to manifest discrete and unique intrinsic properties individually, as well as very familiar periodic property relationships when compared to each other. These elemental periodic property trends or patterns based on CADPs provide the invaluable predictive value and are the very essence of Mendeleev’s Periodic Table, as illustrated in Fig. 16.

Critical atomic design parameters (CADPs): structure-controlled (a) size, (b) shape, (c) surface chemistry, (d) flexibility/polarizability, (e) architecture, and (f) elemental composition [94]
6.2 Chemists and Physicists Are Developing a Mutual Consensus on Nanoscale Atom Mimicry and Superatoms
Recent dialogue sparked by a plenary presentation to the American Physical Society in early 2012 [140] has led to the realization that both chemists and physicists have been thinking and working in parallel worlds concerning the general concept of nanoscale atom mimicry, nanoscale superatoms, and nanoclusters [141]. Although physicists have focused primarily on atom mimicry associated with hard particle, metal cluster-type electron orbital behavior, chemists have been more interested in heuristic nanoscale atom mimicry based on well-defined nanovalency, nanosterics, nanostoichiometries, and similar issues. Many of these features and properties have been associated with discrete soft nanoparticles such as dendrimers, proteins, viral capsids, DNA and RNA, nanolatexes, polymeric micelles, and monodispersed synthetic polymers.
It is now recognized and generally accepted, that more complex, large nanoscale collections (i.e., 103 times larger than atoms) of discretely organized atoms may manifest many physico-chemical and building block features that are reminiscent of individual atoms [142, 143]. These chemically bonded or supramolecularly assembled collections of atoms are generally homogeneous and monodisperse entities that exhibit well-defined size (i.e., mass), shape, surface chemistry (i.e., valency), flexibility/rigidity, atomic composition, and architecture. They are often referred to as nanoscale “superatoms,” [142–145] atom equivalents [146], or heuristic “atom mimics” [121, 137, 138, 147, 148].
A superatom is defined as any cluster of atoms that seems to exhibit the properties of elemental atoms. An early example of a hard superatom was the observed clustering of sodium atoms, when cooled from vapor, to preferentially form a magic number of cluster atoms (i.e., 2, 8, 20, 40, 58, etc.). The first two magic numbers (i.e., 2 and 8) are recognized as the number of electrons required to fill the first and second shells, respectively. Thus, superatom mimicry is related to the free electrons in the cluster that appear to occupy a new set of orbitals that are defined by the entire group of atoms involved in the cluster, rather than each individual atom separately. Superatoms appear to behave chemically in a way that will allow them to have a closed shell of electrons in this new cluster orbital counting scheme. Many examples of hard superatoms involving metal atom clusters have been reported by pioneering physicists such as Khanna, Castleman and coworkers [143, 144], and others [149].
This atom cluster behavior has also been observed and referred to by others as “nanoscale atom mimicry,” [137, 138], wherein certain heterogeneous, soft, non-metal atom clusters appear to exhibit combining patterns that produce well-defined stoichiometries and closed-shell-type behavior that is normally associated with naked, elemental atoms. More specifically, this nanoscale atom mimicry was noted in the 1990s [10, 11] for analogous soft superatoms such as dendrimers. For example, dendrimers possessing unfilled outer monomer shells were observed to be highly autoreactive, leading to dimer or oligomer formation. In contrast, ideal outer shell saturated dendrimers behaved like noble gas atomic elements and did not exhibit this autoreactivity. In fact this nanoscale atom mimicry constituted a primary hypothesis upon which a new nano-periodic system for unifying nanoscience was proposed [137]. More specifically, it provided a fundamental paradigm for explaining why many well-defined nanoscale building blocks (i.e., both soft and hard nano-elements) were observed to combine in well-defined stoichiometries. These soft and hard nano-elements have been observed to produce extensive libraries of literature-documented chemically bonded nanocompounds and supramolecularly derived nano-assemblies, as will be described later.
These superatoms or atom mimics appear to fulfill a pivotal role as nanoscale building blocks, much as elemental atoms function at the pico- or subnanoscale level. As such, these poly(atomic) structures or entities have been classified and referred to as “nano-element categories” [137, 138]. Furthermore, these nano-element categories have been shown to form stoichiometric nanocompounds or assemblies that exhibit well-defined intrinsic nano-periodic property patterns in much the same way as atomic elements and their compounds.
In the context of this perspective and using “traditional chemistry first principles” initiated by Lavoisier, Dalton, Mendeleev and others, a new systematic framework for unifying and defining nanoscience was proposed. Just as the nineteenth century first principles led to a central paradigm and a periodic system for traditional elemental atom and small molecule chemistry, it was proposed that a similar nano-periodic system might be defined for discrete, well-defined nanomodules at the nanolevel (Fig. 17).

Hierarchical dimensions influenced by the traditional elemental periodic system and the proposed nano-periodic system [138]
The initial nano-periodic framework of nano-elemental categories should be viewed as a “works in progress”. This framework is expected to be expanded and better articulated with time, just as Dalton’s original list of atomic elements has grown from 23 in 1808 to now over 117 known atomic elements [150]. The current system is based on 12 nano-element categories, which are differentiated equally into two main groups consisting of six categories each: (1) hard nano-element categories (i.e., inorganic modules) and (2) soft nano-element categories (i.e., organic modules). The inorganic-like, hard nano-element categories are arbitrarily designated as [H-1] metal nanoclusters, [H-2] metal chalcogenide nanocrystals, [H-3] metal oxide nanocrystals, [H-4] silica nanoparticles, [H-5] fullerenes, and [H-6] carbon nanotubes. The organic-like, soft nano-element categories include [S-1] dendrons/dendrimers, [S-2] nano-latexes, [S-3] polymeric micelles, [S-4] proteins, [S-5] viral capsids, and [S-6] RNA/DNA (Fig. 18). Single units of these various elements (i.e., chemically bonded or supramolecularly assembled modules) are 1–100 nm in at least one dimension, contain between 103 and 109 atoms with masses of 104–1010 Da. In order to be included as a nano-element category, each type of nanomaterial had to exhibit:

Concept overview: Using first principles and step logic that led to the “central dogma” for traditional chemistry, the criteria of nanoscale atom mimicry was applied to category I-type, well-defined nanoparticles. This produced 12 proposed nano-element categories, which were classified into six hard particle and six soft particle nano-element categories. Chemically bonding or assembling these hard and soft nano-elements leads to hard:hard, soft:hard or soft:soft types of nanocompound categories, many of which have been reported in the literature . Based on the discrete, quantized features associated with the proposed nano-elements and their compounds, an abundance of nano-periodic property patterns related to their intrinsic physico-chemical and functional/application properties have been observed and reported in the literature [137]. Copyright: Springer
-
1.
Discrete, well-defined monodispersity (i.e., >90% monodisperse as a function of size or mass)
-
2.
Exist as well-defined nanostructures, assemblies, or collections of units that mimic or behave like atoms
-
3.
Exhibit well-defined stoichiometries (i.e., quantitative constants) and mass-combining ratios when reacting or assembling with each other
-
4.
Exhibit discrete, nano-periodic property patterns as a function of one or more of their CNDPs (i.e., size, shape, surface chemistry, flexibility/rigidity, elemental composition, or architecture)
From this basic list of 12 nano-element categories, a nano-element road map leading to three combinatorial libraries of nanocompounds and nano-assemblies can be envisioned, namely, [hard-hard], [hard-soft], and [soft-soft] types as illustrated in Fig. 18. These nanocompounds and nano-assemblies can be characterized analytically by the proportion of each of these 12 basic nano-elements they contain, based on their discrete bonding/assembly capacities, valencies, stoichiometries, and mass-combining ratios. Many examples of these stoichiometric nanocompounds and assemblies are already documented in the literature and are described in more detail elsewhere [137, 138].
As described above, a fourth feature anticipated by this new nano-periodic system was the expectation that members of these hard and soft nano-elemental categories, as well as their nanocompounds and assemblies would be expected to manifest certain well-defined nano-periodic property patterns. These property patterns were expected to be dependent on one or more of their CNDPs. Just as atomic element periodic property patterns have been shown to be dependent on their intrinsic CADPs (Fig. 16) and are routinely utilized for predictive purposes in traditional small-molecule chemistry, it was hoped that similar relationships and behavior would be observed at the nanoscale level.
Recently, first steps toward the fulfillment of this expectation have been realized by publication of the first “Mendeleev-like nano-periodic tables” for predicting the self-assembly modes of soft nano-element modules. More specifically, the self-assembly properties of soft nano-elements such as amphiphilic dendrons (i.e., [S-1] nano-elements) were systematically investigated by Percec, Rosen and colleagues [151]. They reported a prediction accuracy for resulting self-assembled structures of 85–90% based on the a priori use of dendron CNDPs. These issues will be described later in Section 6.6.3.
6.3 Atom Mimicry: Nanoscale Superatoms and Atom Equivalents
6.3.1 Quantized Aufbau Components: Electrons, Atoms, and Monomer Units
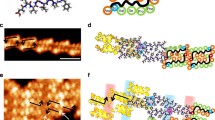
The selection process for various category I-type, hard and soft particle nano-elements (Fig. 18) was based on certain heuristic or experimentally demonstrated atom mimicry features. Earlier general atom mimicry comparisons were made based on the similarity of core–shell architecture exhibited by atoms and dendrimers [10, 11]; however, more detailed working examples that include (inorganic) hard metal nanoclusters are as illustrated in Fig. 19. In descending order, analogous (i.e., heuristic) aufbau components (i.e., electron, Au atoms, and β-alanine monomer units) leading to core–shell picoscale (atoms) and nanoscale hard matter (Au nanoclusters) and soft matter (dendrimers), respectively, are compared. This comparison illustrates aufbau component mimicry and quantized features required to produce core–shell-type structures at two diverse hierarchical dimensional levels. Well-defined sizes, atomic/molecular masses, and outer-shell saturation values (n) are inextricably connected to specific electron shell, atom shell, or monomer shell (generation) levels in each case. Such atom mimicry is clearly demonstrated for hard nanoparticle gold clusters and soft nanoparticle dendrimers. Similar architectural motif patterns may be observed to a lesser or greater degree in the pervasive core–shell taxonomy observed for all proposed nano-element categories, as described elsewhere [137, 138].

Comparison of atomic picoscale particles, hard nanoparticles, and soft nanoparticles. Center image Hard Matter. Reprinted from [155] with permission from Elsevier
Seminal work by Schmid [152, 153] and Rao [154] has shown that fundamental core–shell metal nanoclusters (i.e., Au and Pd) with magic numbers of metal atoms (i.e., 13, 55, 147, 309, 561, and 1,415) corresponding to closed atom shells 1, 2, 3, 4, 5, and 7, respectively, do indeed exist. As noted by Schmid, they are substantially more robust when ligand-stabilized [155]. Furthermore, they can be prepared routinely as monodisperse modules by chemical means [152, 156–159]. Wilcoxon et al. [160] have shown that these closed, metal nanocluster, core–shell assemblies can be isolated, analyzed and characterized using high pressure liquid chromatography (HPLC) methodologies. It is also noteworthy, that these basic hard particle nanomodules exhibit pervasive nano-periodic self-assembly features by organizing into giant, self-similar core–shell nanocrystals that are invariant to scaling [154]. Similar nano-periodic, self-assembly properties have also been noted for soft nanoparticles such as dendrimers [46, 161, 162] and are described later in Sect. 6.6.3.
6.3.2 Heuristic Comparison of Autoreactive Surface Chemistry Associated with Unsaturated Outer Shells in Atomic Elements and in Dendrimers
Without the benefit of quantum mechanics or electronic theory, nineteenth century chemists determined that an atom’s reactivity was associated with electron occupancy levels residing between the shell saturation levels that completed each period [134, 163, 164]. Furthermore, these elements combined with precise valencies and stoichiometries to give compounds with predictable combining mass ratios. As shown in Fig. 20, traditional chemistry recognizes that the noble gas configurations are associated with inertness due to their saturated outer electron shells. They do not exhibit any autoreactivity, unlike atomic elements penultimate to the noble gases that contain unsaturated outer electron shells. As such, halogen elements such as chlorine exhibit autoreactivity and exist as chlorine atom dimers. It should be noted in the far right column that ideal dendrimer structures (i.e., G = 1–5) possessing saturated outer monomer shells are compared heuristically to the respective atomic element noble gases.

Mendeleev periodic table, displaying horizontal autoreactive elements (i.e., chlorine dimer) in respective periods (1, 2, 3…) penultimate to the vertical column of non-autoreactive noble gases. Far right column displays ideal theoretical, shell-saturated PAMAM dendrimers (G = 1, 2, 3,…) as heuristic non-autoreactive nanoscale analogs of inert, noble gas elements. In the case where G = 2, shell-saturated dendrimer structure is equivalent to argon at the atomic level
In a similar fashion, as illustrated in Fig. 21, dendrimers possessing an unfilled outer monomer shell are found to be very autoreactive and combine to form dimers, etc. reminiscent of halogens (or more specifically chlorine). As such, it should be apparent that the G = 2 dendrimer possessing an unsaturated outer monomer shell behaves as a superatom analogue of chlorine. This dendrimer species, possessing an unsaturated monomer shell penultimate to the saturated ideal dendrimer structure, may proceed to form a dimeric nanocompound (i.e., megamer) by interdendrimer reactions or simply by combining intramolecularly to produce a macrocyclic site. In contrast, so-called ideal dendrimers in the far right column are heuristically analogous to atomic-level inert gas configurations. These ideal, outer-shell-saturated dendrimers possess saturated outer shell level monomer values commensurate with mathematically defined shell saturation values, as described earlier (Fig. 11).

Heuristic dendrimer-based periodic table based on monomer shell filling. Monomer aufbau stages (i.e., G = 0–4) mimic respective electron shell-filling stages in atoms. Autoreactive dendrimer species reside penultimate to the outer-shell-saturated stick configurations mimicking noble gas elements. These unfilled shell species are autoreactive, producing G = 2 dimers, wherein dendrimer species possessing 20 monomer units represent a “superatom” analog of elemental chlorine. The 21-monomer, shell-saturated analog is a G = 2 dendrimer mimicking argon. Molecular simulations of ideal, outer-shell-saturated dendrimer generations (i.e., G = 0–4) (far right column) are shown next to the core–shell (i.e. shell-saturated) stick configurations
These saturated outer monomer shell dendrimers are not autoreactive with each other or reagents possessing common surface functionality (i.e., either nucleophilic or electrophilic moieties, respectively). In summary, as illustrated in Fig. 22, this outer shell autoreactivity has been observed not only with atomic elements but also with dendrimers [10, 11], as well as with their related core–shell (tecto)dendrimers [48, 165]. In the case of atomic elements (i.e., far left column) outer electron shell saturation is fulfilled by autoreaction to produce an elemental dimer. In the middle column, the penultimate dendrimer species to the saturated ideal dendrimer structure presents an isolated functional group (i.e., amine or ester) in the outer monomer shell that may react with a co-reactive functional group (i.e., amine or ester) on the surface of a neighboring dendrimer to give dimer formation [10, 11]. Therefore, these two nanoscale dendrimer scaffoldings that combined to form dimer appear to be heuristically mimicking elemental atoms and, as such, are individually referred to as “soft superatoms” [141]. As illustrated in Fig. 22, core–shell tecto(dendimers) were also observed to follow an analogous autoreactivity pattern associated with unsaturated outer dendrimer shells [48, 165].

Quantized module reactivity patterns at the subnanoscale level (i.e., atoms), lower nanoscale level (i.e., dendrimers), and higher nanoscale level, i.e., core–shell tecto(dendrimers) involving outer unsaturated electron, monomer, or dendrimer principle valence shells [137] Copyright: Springer
6.3.3 Heuristic Comparison of Valency and Symmetry Features Shared by Atoms and Spheroidal Nanomodules
At the picoscale level, valence shell electron pair repulsion (VSEPR) theory is a widely recognized theoretical model that proposes the geometric arrangement of terminal atoms or groups of atoms surrounding a central atom in a covalent compound or charged ion. The concept is based solely on the repulsion of the electron pairs present in the valence shell of the central atom. The premise of VSEPR is that the valence electron pairs surrounding an atom mutually repel each other and therefore adopt an arrangement that minimizes this repulsion. In essence, the utilization of space by the valence electrons surrounding the central atom is defined by these charge repulsion events and ultimately determines the shape and molecular geometry of the resulting bonded structure. The number of electron pairs surrounding an atom, both bonding and non-bonding, is called its steric number. VSEPR theory mainly involves predicting the arrangement of electron pairs surrounding one or more central atoms in a molecule that are bonded to two or more other atoms. The geometry of these central atoms in turn determines the ultimate architecture or shape of the structure [166], as shown in Fig. 23a.

For example, when two electron pairs surround the central atom, their mutual repulsion is minimal when they lie at opposite poles of the central sphere. Therefore, the central atom is predicted to adopt a linear geometry. If three electron pairs surround the central atom, their repulsion is minimized by placing them at the vertices of a triangle centered on the atom. Therefore, the predicted geometry is trigonal. Similarly, for four electron pairs, the optimal arrangement is tetrahedral, for five electron pairs it is trigonal bipyrimidal, for six electron pairs it is octahedral, etc., thus defining a wide range of defined symmetries and geometries, as illustrated in Fig. 23a. Essentially, all of these geometries are manifestations of core–shell (i.e. nucleus–electron) relationships, which yield reproducible geometries defining one of the important CADPs for atoms, namely shape. These features are in turn translated into shape-defining features, which are conserved in the resulting molecular structure. Now consider a similar analysis at the nanoscale level using the space-filling features of spheroids (Fig. 23b). At the nanoscale level, similar heuristic core–shell relationships have been analyzed mathematically using spheroids. More importantly, these relationships have also been demonstrated experimentally using spherical dendrimers to produce core–shell tecto(dendrimers) and are described later (see Sects. 6.3.3 and 6.4.3).
Mathematically [167], these core–shell relationships have been analyzed as a function of the ratio of the core spheroid (r 1) and shell spheroid (r 2) radii [167], wherein the core spheroid size is systematic increased relative to the shell spheroid. Quite remarkably, this treatment produces many important symmetries and geometries that appear to mimic those observed for atoms at the picoscale level in the context of the VSEPR theory. For example, at an r 1/r 2 value of 0.155, a valence of 3 shell spheroids and a trigonal geometry (D 3h) is observed. At values for r 1/r 2 = 0.255–0.414, one observes a valency of 4 with tetrahedral (T h) symmetry, and at r 1/r 2 = 0.255 one observes a valency of 8 with octahedral (Oh) symmetry (see also Sect. 6.4.3). In essence, these valencies and geometries represent space-saturated values around core atoms or core spheroids, respectively. These space saturation values around a core may be engineered by simply tuning the relative core and shell radii. This provides a powerful and useful strategy for defining valency for all surface-reactive spheroidal nano-objects. It can be seen that when the core reagent is small and the shell reagent is large, only a very limited number of shell-type reagents can be attached to saturate the space surrounding the core (i.e., r 1/r 2 = 0.155–1.20). Quite remarkably, when r 1/r 2 = 1, as would be the case for metal nanoclusters, a valency of 12 and an icosahedral (I h) symmetry is observed (see Fig. 21 and Sect. 6.4.3). This is consistent for core–shell-type metal nanoclusters (i.e., gold nanoclusters), as reported by Schmidt et al. [152, 153] (Fig. 19). However, when r 1/r 2 ≥ 1.20 more space surrounding the core allows the attachment of more spheroidal shell reagents p to discrete saturation values (N max). This saturation value (N max) is discrete and can be determined from the general expression described by the Mansfield–Tomalia–Rakesh equation [167] (described later in Sect. 6.4.3).
6.4 Combining Soft and Hard Nano-element Categories to Create Combinatorial Libraries of Nanocompounds and Nano-assemblies
6.4.1 Recent Literature Examples Fulfilling and Verifying Atom Mimicry and Superatom Behavior by Forming 3D Nanoscale Lattices, Nanocompounds, and Nano-assemblies Reminiscent of Atomic Elements
Very recently, important examples describing the chemical combination and assembly of these proposed hard and soft nano-element categories (i.e., superatoms) as described in Fig. 24 have now appeared in the literature and are referred to as “nanoscale atom mimicry” at the nanoscale. In each case, our early concept has been fulfilled and validated by these authors, who have referred to these nanoscale building blocks as “atom equivalents” (i.e., Mirkin and coworkers [146]) or “nanoscale atoms” (i.e., Roy, Brus and coworkers [168]). In the first case, Mirkin and coworkers [146] have reported the assembly of metal nanoclusters [H-1], metal chalcogenide nanocrystals (quantum dots) [H-2], and metal oxide nanocrystals [H-3] using complementary DNA [S-6] to give [H-1:(S-6)n], [H-2:(S-6)n], or [H-3:(S-6)n] type 3D nanoscale unit cell lattices. Quite remarkably, these nanoscale unit cell lattices mimic inorganic salt lattices formed from atomic elements. In the second case, Roy et al. [168] have shown that by combining fullerene (C60) [H-5] (i.e., 0.71 nm) with various metal chalcogenide nanocrystals [H-2] (i.e., 0.85–0.92 nm), a solid-state material is formed that they described as a “super atomic relative" of the cadmium iodide (CdI2) structure type. Furthermore, they stated that the constituent clusters (i.e., [H-5] and [H-2]) interacted electronically to produce a magnetically ordered phase at low temperature, akin to atoms in a solid-state compound.

Both soft matter (organic) and hard matter (inorganic) categories of these quantized nanomodules have been proposed and referred to as soft and hard nano-element categories, respectively. These nano-element categories (see Figs. 18 and 24) were proposed on the basis of selection criteria and assumptions described elsewhere [137, 138]. Furthermore, these first 12 soft and hard nano-element categories, designated [S-n] and [H-n], respectively, have been reported to form a wide range of soft particle and soft-hard particle nanocompounds and assemblies. Both the nano-elements and their nanocompounds are widely recognized to exhibit new emerging properties and nano-periodic property patterns [137, 138]. Leading references to these literature examples (designated by X in the combinatorial nanocompound library in Fig. 24) are described in greater detail elsewhere [137]. This account will focus only on several selected examples of nanocompound formation (designated by an asterisk in Fig. 24 ) that involve either chemical reactions or supramolecular, self-assembly interactions between dendrons/dendrimers and/or other nano-element categories. For example, self-assembling certain [S-1]-type amphiphilic dendrons, according to Percec and colleagues. [169], produces vast libraries of stoichiometric spherical or cylindrical supramolecular dendrimers [S-1] n .
These assemblies may be viewed as nanocompounds/assemblies of the [S-1]-type nano-element category, much as S8 is viewed to be a molecular compound of the atomic element sulfur. Combining dendrimers with other dendrimers has produced core–shell tecto(dendrimers), i.e., [S-1:(S-1)n]-type core–shell nanocompounds with well-defined stoichiometries. Similarly, covalent grafting of linear poly(ethyleneglycol)s produces discrete [S-1:(S-3)n]-type core–shell compounds . On the other hand, covalent attachment of fullerenes produced precise [S-1:(H-4)n]-type core–shell structures. Combining dendrimers with metal nanoclusters has produced a variety of unique, i.e., [(H-1)n:(S-1)] and [(S-1):(H-1)n], core–shell-type nanocompounds, as designated in Fig. 24. Specific literature examples of these proposed nanocompounds/assemblies will be described in the remaining sections of this review.
6.4.2 (Dendrons) n [S-1] n : Self-Assembly into Supramolecular Spherical or Cylindrical Dendrimer-Type Nanocompounds and Nano-assemblies
Perhaps some of the most compelling examples of precise stoichiometric [S-1]-type nano-assemblies are the enormous libraries of spherical and cylindrical supramolecular dendrimers (i.e., supramolecular megamers) reported by Percec and colleagues [151, 169, 170]. Percec’s amphiphilic dendrons have been shown to exhibit heuristic atom mimicry features reminiscent of atomic elements, namely, precise mass-combining ratios and unique emerging properties. Just as atomic elements such as phosphorous and sulfur aggregate into discrete P4 and S8 clusters, respectively [171], so do appropriately functionalized Percec dendrons (Fig. 25). Whereas earlier Zimmerman-type dendron self-assemblies [172, 173] have generally involved small, single-digit aggregation numbers, many of Percec’s dendrons self-assemble into supramolecular dendrimers requiring large double-digit aggregation numbers. For example, the number of dendrons leading to hollow, spherical supramolecular dendrimers involved aggregation numbers of 72–155 [170]. Recently, a remarkably large supramolecular dendrimer derived from (770)-dendrons (i.e., 1.73 × 106 g/mol) has been reported [151]. This giant supramolecular dendrimer completes a continuum that has been defined between small filled and large hollow dendrimers, all of which appear to be defined by the primary structure of the precursor dendrons.

Self-assembly of Percec-type amphiphilic dendrons (i.e., [S-1]-type nano-elements) into spherical supramolecular dendrimers (i.e., [S-1] n , where n = discrete, stoichiometric aggregation number that ranges between 72 and 155 for various [S-1] n -type stoichiometric nanocompounds and nano-assemblies) [170]. Copyright: 2008 American Chemical Society
6.4.3 Dendrimer(G)-(Dendrimer(G)) n [S-1(G):(S-1(G)) n ] Core–Shell-Type Nanocompounds
Covalent, saturated-shell, nanocompounds (Fig. 26) can be prepared by a two-step approach involving, firstly, self-assembly of an excess of carboxylic acid-terminated dendrimers (i.e., shell reagent) around a limited amount of amine-terminated dendrimer (i.e., core reagent) in the presence of LiCl to form a charge-neutralized dendriplex. This was followed by covalent amide bond formation between the core and dendrimer shell reagents using a carbodiimide reagent [165, 174, 175]. The resulting nanocompounds are outer shell saturated, core–shell tecto(dendrimers). They have also been referred to as “covalent megamers” and are prime examples of precise polydendrimer cluster structures that are reminiscent of metal nanoclusters (Fig. 19). These structures may be mathematically predicted by the Mansfield–Tomalia–Rakesh equation [121, 167] (see Sect. 6.5.3) and have been unequivocally verified by experimental mass spectrometry, gel electrophoresis, and atomic force field microscopy (AFM) [121, 174–176].

The saturated-shell architecture approach to covalent megamer synthesis. All surface dendrimers are terminated with carboxylic acid [165]
6.4.4 Dendrimer-(Fullerene) n [S-1:(H-5) n ] Core–Shell-Type Nanocompounds
Covalent, stoichiometric [dendrimer core:fullerene shell] nanocompounds were readily formed by allowing a [core:1,2-diaminoethane];(4→2); {dendri-poly(amidoamine)-(NH2)64} (G = 4) PAMAM dendrimer to react with an excess of buckminsterfullerene (C60) [177]. In the presence of an excess of C60, only 30 C60 moieties bonded to the dendrimer surface to produce a well-defined, stoichiometric [dendrimer (core):fullerene (shell) n ] nanocompound, i.e., [S-1:(H-5)30] core–shell-type as shown in Fig. 27. These structures were characterized extensively by MALDI-TOF, thermogravimetric analysis (TGA), UV–vis spectroscopy, and Fourier transform infrared (FTIR) spectroscopy. Such nanocompounds exhibited new fullerene-like solubility and photo-properties by readily generating singlet 1O2 in either aqueous or organic solvents. However, they offered other unique features such as larger size and nanocontainer-type properties that would normally be associated with the dendrimer core interior.

Core–shell architecture of the PAMAM core:fullerene shell [S-1:(H-5)30] type of nanocompound. Z indicates terminal –NH2 or –NH– groups on the PAMAM dendrimer core component of the core–shell nanocompound [177]
6.5 Nano-periodic Physico-Chemical Property Patterns
6.5.1 Historical Picoscale, Atomic Element Periodic Patterns Contributing to Emergence of Mendeleev’s Periodic Table
The emergence of Mendeleev’s Periodic Table (1869) classifying the fundamental elemental building blocks of the universe, provided a central idea or dogma for a new science. Much like the axioms for geometry, Newtonian physics, and Darwinian biology, the area of traditional chemistry now had a central idea (dogma) upon which this discipline could be systematically defined, unified, and grown. However, history shows that many minor, yet important, documented periodic property patterns were required for the elements that ultimately contributed to the final consolidation and framework for Mendeelev’s Periodic Table [178]. A small sampling of these well-known minor periodic element property patterns is given below:
-
Elemental chemical and physical properties repeated in a series of periodic intervals as a function of atomic weight both horizontally and vertically [166]
-
Valency in the early elements appeared to increase as a function of atomic weight
-
De Chancourtois’ “telluric screw,” which demonstrated periodic property patterns that appeared to repeat or become similar after every 16 atomic weight units
In a similar fashion, analogous nano-periodic property patterns are accumulating. Many have been documented in the literature and are described briefly in Sect. 6.4.2. There is no doubt that collectively these nano-periodic property patterns will eventually evolve into a grand, encompassing framework that should be expected to define an ultimate version of a Mendeelev-like nano-periodic system. A small sampling of examples is presented in the following section.
6.5.2 Intrinsic Dendrimer-Based Periodic Patterns of Chemical Reactivity and Physical Size
Intrinsic viscosity [η] is a physical property (expressed in dL/g), which in essence is the ratio of volume to mass. As the generation number increases and transition occurs to a spherical shape, the volume of a spherical dendrimer increases in cubic fashion while its mass increases exponentially; hence, the value of [η] must decrease once a certain generation is reached. This prediction has now been confirmed for many different dendrimer families [9, 116, 179]. Because of this feature, the soft particle dendron/dendrimer-based, [S-1]-type nano-elements are unique macromolecules that exhibit completely different physico-chemical properties (i.e., nano-periodic property patterns) compared to compositionally isomeric traditional linear, crosslinked, or branched polymers. This is largely due to the dendritic architecture that induces congestion properties. These properties emerge as a function of generational growth (Figs. 28 and 29) to produce unprecedented nano-periodic property patterns that are intrinsic and uniquely characteristic of dendrons and dendrimers.


Molecular volume of PAMAM dendrimers as a function of generation and pH. Dendrimer samples were deposited on mica from solutions of pH = 1 (diamonds) and pH = 6 (triangles). The squares depict the theoretical volumes for generations 5-9 based on known molecular weights and estimated densities [174]. Copyright: 2002 American Chemical Society
Dendrimer-based intrinsic viscosities [η] initially increase in a classical fashion as a function of molar mass (i.e., generation), but dramatically decline beyond a critical generation due to a congestion-induced shape change. A dendrimer shape change occurs from an extended, compressible, floppy configuration in the early generations (i.e., G = 0–3) to more rigid globular shapes in the later generations (i.e., G = 4–10) (Fig. 28). In effect, for the Tomalia-type PAMAM series at critical generations (i.e., G = 3–4 and higher) the dendrimer acts more like an Einstein spheroid [9, 84, 114].
The dendrimer density z (atomic mass units per unit volume) clearly minimizes between generations 4 and 5. It then begins to increase as a function of generation due to the increasingly larger, exponential accumulation of surface groups. Since refractive indices are directly related to density parameters, their values minimize and parallel the above density relationship.
Plots of intrinsic viscosity [η], density (d), surface area per Z group (A z) and refractive index as a function of generation clearly show intrinsic maxima or minima at G = 3–5 for this Tomalia-type PAMAM dendrimer series. These data corroborate computer-assisted molecular-simulation predictions [9, 180], as well as extensive photochemical probe experiments reported by Turro et al, and others [55, 105–108, 181].
Atomic force microscopy studies by Betley et al. [174] clearly demonstrated that dendrimers exhibit well-defined, monodispersed molecular volumes as a function of generation and pH, as shown in Fig. 29.
The dendrimer radius (r) is dependent on the branch cell segment length l, such that large l values delay congestion. On the other hand, larger N c and N b values and larger Z dimensions dramatically enhance congestion. These congestion properties are unique for each dendrimer family; wherein, N c and N b determine the generation levels within a family that will exhibit nano-encapsulation properties. Higher N c and N b values predict that lower generation levels will produce appropriate surface congestion properties, to manifest encapsulation features as shown in Fig. 30.

Congestion-induced dendrimer shape changes (I, II, III) with development of nanocontainer properties for a family of [core:1,2-diaminoethane];(4→2); dendri-poly(amidoamine)–(NH2)Z} (G = 0–10) PAMAM dendrimers with core multiplicity N c = 4 and branch cell multiplicity N b = 2. Distances between Z surface groups are shown as a function of generation [138]
These congestion issues are consistently observed universally as periodic patterns characteristic of all dendrimer families including so-called giant redox active metallo-dendrimers recently reported by Astruc and coworkers [182].
6.5.3 Spheroidal Valency Defined by Nanosterics
Clearly, these fundamental dendrimer properties illustrate the unique and intrinsic nano-periodic property patterns manifested by this soft matter, [S-1]-type nano-element category. Many other nano-periodic property patterns have been documented for the behavior, assembly, and reactions of dendrimers with other dendrimers, as well as with other well-defined nano-element categories. For example, work on this soft matter, [S-1]-type nano-element category [121, 167, 175] has demonstrated that mathematically defined, periodic size properties of spheroidal dendrimers can determine the chemical reactivity patterns with other dendrimers. These reactivity patterns, based on the relative sizes of a targeted dendrimer cores and dendrimer shell components, strongly influence the assembly of precise dendrimer clusters (i.e., core–shell (tecto)dendrimers). Mathematical relationships (i.e. the Mansfield–Tomalia–Rakesh equation) predict dendrimer cluster saturation levels (i.e., magic numbers for dendrimer shells) as a function of the core dendrimer size relative to the size of the shell dendrimers that are being used to construct the dendrimer cluster (Fig. 31) [167, 183]. These periodic property patterns and magic shell relationships are reminiscent of those observed for the self-assembly of [H-1]-type metal nanocrystals; wherein, the predicted number of touching spheroids for the first shell surrounding a central core metal atom is 12 when r 1/r 2 = 1.00. This is a well-known value (i.e., 12 atoms) for the first shell of all core–shell metal atom self-assemblies [152, 154, 156] (see Fig. 19).

(a) Symmetry properties of core–shell tecto(dendrimer) structures when r 1/r 2 < 1.20. (b) Sterically induced stoichiometry (SIS) defined shell capacities (N max), based on the respective core and shell radii, when r 1/r 2 < 1.20. (c) Mansfield–Tomalia–Rakesh equation for calculating the maximum shell-filling value (capacity) (N max ), when r 1/r 2 > 1.20 [121, 138, 167]
6.6 First Steps Towards a “Central Dogma” for Synthetic Nanochemistry: Dendrimer-Based Nanochemistry
One of the highest priority challenges and barriers hindering continued progress of the international nanoscience technology movement is the absence of a “central paradigm and a Mendeleev-like periodic system” for unifying and defining nanoscience.
Historically, the development of such a central paradigm and systematic framework was absolutely critical for the seminal transformation in the early nineteenth century of an empirical alchemy movement to a systematic, highly predictable scientific discipline recognized as traditional small-molecule chemistry [134].
As described in this chapter and elsewhere, substantial progress has been made toward resolving this challenge by the introduction of a systematic, unifying framework based on the first principles of traditional chemistry [137, 138]. In review, this concept was inspired by the pervasive heuristic “atom mimicry” behavior observed for a broad range of monodisperse, well-defined nanoparticle categories [137, 138]. Ample evidence has now emerged that supports the premise that CADPs such as size, shape, surface chemistry, flexibility/rigidity, composition, and architecture may be conserved and translated hierarchically from the picoscale to the nanoscale level if suitable structure-controlled, bottom-up synthesis strategies are employed [137]. These conserved features were first observed with well-defined bottom-up structure-controlled, soft nanoparticles such as dendrons, dendrimers, and dendronized polymers [138, 151, 169]. An abundance of literature data has now shown that at least 12 categories of both soft and hard nano-elements (i.e., SNE, HNE) exhibit atom mimicry features and pervasive nano-periodic property patterns or trends related to their CNDPs. Hard and soft nanomodule categories (i.e., atom collections of 103–109 atoms) have been shown to behave heuristically like “nanosized superatoms” by exhibiting remarkably well-defined stoichiometries and mass-combining ratios to form covalent nanocompounds and non-bonding nano-assemblies. Furthermore, as predicted in the original concept paper [137] and described briefly in this chapter, both the hard and soft nano-element categories (designated [HNE-n] and [SNE-n]), as well as their resulting nanocompounds and assemblies appear to manifest both physico-chemical and functional/ application property trends reminiscent of Mendeleev-like property patterns normally associated with the atomic elements (Fig. 32).

The first examples of Mendeleev-like nano-periodic tables have recently fulfilled the predictions for expected nanoscale property patterns and trends [137, 138]. Percec and Rosen [151] have reported the first three nano-periodic tables for predicting the self-assembly patterns for [S-1]-type amphiphilic dendrons, with predictive accuracies of 85 to >90%, based on knowledge of the primary dendron CNDPs, namely, size, shape, surface/apex chemistry, and flexibility/rigidity [94].
We now examine recent progress reported by Percec, Rosen and colleagues [151] that has clearly demonstrated the first working examples of predictive, Mendeleev-like nano-periodic tables. These Percec nano-periodic tables clearly demonstrate a priori predictions for the mode of [S-1]-type amphiphilic dendron self-assembly into supramolecular dendrimers with 85–90% accuracy. Quite remarkably, as proposed in the original concept [137], these self-assembly modes may be accurately predicted based on simply knowing the CNDPs (size, shape, surface chemistry, and flexibility) for the amphiphilic dendron primary structure, as will be described in the next section.
6.6.1 Percec’s Quest for Synthetic Mimicry of Biological Quasi-equivalence with [S-1]-Type Amphiphilic Dendrons
As early as 1992, Percec et al. [184] compared the similarity of supramolecular nanocylinders obtained from his amphiphilic dendrons with the supramolecular assembly of protein subunits to produce the cylindrical viral capsids that surround RNA in the tobacco mosaic virus (TMV). More recently, Percec [185] reviewed the historical inspiration provided by Klug’s seminal Nobel work on the structure of TMV [186, 187]. Percec was able to show unequivocally that dendrons behave much like protein subunits to produce a rich variety of cylindrical and spherical supramolecular dendrimers that exhibit quasi-equivalency, much as noted in many viral capsids. Based on accelerated design strategies involving synthetic amphiphilic dendrons, Percec et al. [188–191] were able to demonstrate the quasi-equivalent mimicry of biological systems by using retrostructural analysis [191] of their periodic and quasi-periodic supramolecular dendrimer assemblies, as outlined in Fig. 33. This remarkable comparison corroborates and documents many dendron libraries and other examples of dendron/dendrimer-based “protein mimicry” [192–194].

Dependency of self-assembly patterns leading to tertiary and quaternary dendron assemblies on primary structure-controlled dendron CNDPs such as size, shape, surface/apex chemistry, and flexibility [151]. Copyright: 2009 American Chemical Society
6.6.2 Tobacco Mosaic Virus as a Compelling Example of a Supramolecular Core–Shell Nanocompound [S-6:(S-4)2130] Exhibiting Well-Defined Stoichiometry: Self-Assembly of an [S-4]-Type Protein Subunit Shell Around an [S-6]-Type ss-RNA Core
More than three decades ago, important stoichiometric, self-assembly relationships were noted by Klug [186, 187, 195] between the single-stranded (ss)-RNA core and the self-assembling protein subunits in the formation of tobacco mosaic viruses. The stoichiometric relationship between the viral core and the viral capsid was carefully documented by X-ray studies. This work rigorously demonstrated that exactly 2,130 protein subunits assembled to form a viral capsid shell around an ss-RNA core to produce tobacco mosaic virus of 18 nm diameter, 300 nm length, and helical symmetry. Elucidation of this self-assembly process together with the unprecedented characterization of this viral assembly by X-ray analysis garnered the Nobel Prize for A. Klug in 1982. In the context of the systematic nano-periodic concept [137], this viral construct may be viewed as a supramolecular, stoichiometric core–shell [S-6:(S-4)2130]-type nano-assembly as described in Fig. 34.

Tobacco mosaic virus (TMV): an example of a well-defined nanocompound [S-6:(S-4)2130] consisting of an ss-RNA (core) and protein subunits (shell), with nanoscale dimensions of 18 nm diameter and 300 nm length, and a helical symmetry [195, 206]. Reproduced with permission from the Society for General Microbiology
6.6.3 A Library of Amphiphilic Dendron Self-Assembly Directed by the CNDPs
Inspired by Klug’s work on TMV, the Percec group synthesized and analyzed innumerable libraries of self-assembling amphiphilic dendrons [169]. For each library, the dendron primary structures were compared to the tertiary structures of the self-assembled supramolecular dendrimers and the quaternary structure of the crystal lattices. A sampling of these libraries reveals primary dendron structures derived from AB2; 3,4-dendrons, AB2; 3,5-dendrons, and AB3; 3,4,5-dendrons, to mention a few [151]. A typical library for an AB2; 3,4-disubstituted biphenyl dendron family is characterized as a function of dendron CNDPs such as generation (size), surface or apex chemistry, shape, and flexibility (as shown in Fig. 35). These analyses clearly showed that important dendron parameters such as (1) the molecular solid angle (α′) of the dendron, (2) the morphology (shape) of the supramolecular dendrimer, and (3) the aggregation number (μ) (i.e. supramolecular dendrimer stoichiometry) varied in a predictive manner to reveal important self-assembly patterns as a function of dendron generation. It should be noted that very precise reproducible stoichiometries were observed for these dendron self-assemblies, as evidenced by their discrete aggregation numbers, namely, [S-1] n (Fig. 35).

For example, these library analyses revealed interesting patterns such as an increase in the generation number causes a change in molecular solid angle (α′) and typically a transition from lamellar to columnar and spherical assemblies. Increasing the generation number does not necessarily increase the diameter of the supramolecular dendrimer, but generally reduces the aggregation number (μ) or number of dendrons required to form a supramolecular sphere or the cross-section of a supramolecular column. Deviations from these patterns usually indicate the formation of hollow core supramolecular dendrimers or other novel mechanisms of self-assembly. Generally, AB3; 3,4,5-trisubstituted libraries exhibit more spherical structures as compared to AB2; 3,4-disubstituted dendron libraries.
Furthermore, it was shown by Percec and coworkers [151] that simply by knowing the four CNDPs (size, shape, surface chemistry, and flexibility) of the primary dendron structure, one could predict self-assembly patterns leading to tertiary and quaternary structures with greater than 85–93% accuracy, as shown in Fig. 36.

Nano-periodic table I: Primary dendron structures [S-1] versus 3D supramolecular dendrimer structures [S-1]μ for all libraries of AB3 supramolecular dendrimers. Bn benzyl ether, Pr phenylpropylether, Bp biphenyl-4-methyl ether, BpPr biphenylpropyl ether [151]. Copyright: 2009 American Chemical Society
6.6.4 First Nano-periodic Tables for Predicting Amphiphilic Dendron Self-Assembly to Supramolecular Dendrimers Based on the CNDPs
Like proteins, the primary structures of the amphiphilic dendrons determine their tertiary structure. As such, Percec has compared dozens of his AB2- and AB3-derived dendron libraries in an effort to determine trends or “nano-periodic self-assembly patterns” as proposed by others [137]. Percec’s seminal comparison produced the first three Mendeleev-like, predictive nano-periodic tables for the self-assembly of aryl ether dendrons [151]. The first of these nano-periodic tables is shown in Fig. 36. The three nano-periodic tables summarize the tertiary and quaternary structures that are formed for similar primary dendron structures, but using different dendron building blocks. They provide predictive nano-periodic tables that describe general trends in the sequence–structure relationship (i.e., primary → secondary → tertiary → quaternary structures). Furthermore, they identify clustered regions where specific structures will be found. The supramolecular dendrimer structures formed may be classified into lamellar, columnar, or spherical morphologies by analogy to β-sheets, helical structures of fibrillar proteins, and the pseudo-spherical structure of globular proteins. In all three nano-periodic tables, G = 1 dendrons behave similarly and exhibit a high proportion of lamellar and columnar structures, including hollow columnar structures.
6.6.5 Aufbau Intermediates Involved in the Dimensional Enhancement of Soft Nano-element [S-1] Category Complexity
As stated earlier, the “central dogma” for traditional soft and hard matter chemistry emerged from the first initiatives of Lavoisier, Dalton, and others in the early nineteenth century. It was initially focused on the simple combinatorial bonding of atoms to form small molecules (i.e., monomers, branch cell monomers), much as illustrated in Fig. 37. Synthetic soft matter chemistry, initiated by Wöhler, witnessed steady progress throughout the nineteenth and twentieth century toward more complex molecular structures and architectures, including dendrons and dendrimers. The aufbau process for bottom-up construction of such well-defined soft matter, nano-element category [S-1]-type structures (i.e., dendrons and dendrimers) by covalent bonding and non-bonding supramolecular strategies is outlined in this section, as illustrated earlier in Scheme 3.

Mathematically defined, bottom-up aufbau roadmap for constructing and transferring CADP → CMDPs to produce CNDP-conserved nanoscale [S-1]-type nano-element category complexity [94]
Essentially, all other proposed hard-soft nano-element categories (Fig. 18) evolve from aufbau strategies that allow the control and conservation of critical hierarchical design parameters (CHDPs) from the atomic to the nanoscale level (i.e., CADP → CMDP → CNDP). Nature has already evolved very exquisite aufbau strategies for synthesizing other important soft matter nano-element categories such as proteins [S-4], viral capsids [S-5], and DNA/RNA [S-6].
It is noteworthy, that an “aufbau roadmap” leading to the dendron/dendrimer soft nano-element category [S-1] can be mathematically defined from the atomic and small molecule dimensional levels. It is apparent that that this aufbau strategy is dependent on conserved CADPs and CMDPs to produce precise mathematically defined covalent structures such as linear and branch cell monomers (Fig. 37). When assembled according to well-defined divergent or convergent dendritic amplification principles, they produce precise mathematically defined covalent dendron, dendrimer, or core–shell tecto(dendrimer) structures (Fig. 26). Presumably, analogous mathematical relationships exist for Percec-type self-assembling dendrons to produce supramolecular dendrimers (as described in Fig. 25).
7 Conclusions
In summary, polymer science has progressed and advanced dramatically in the 60 years that have lapsed since Herman Staudinger was recognized for his revolutionary macromolecular hypothesis in 1953. Most notable, has been the enormous impact that Staudinger’s paradigm has had on international commerce and enhancement of the human condition. This influence has been so substantial that the twentieth century has been referred to as the “plastic's century” [196]. The explosive activity during the twentieth century in the field of polymer science has been directly connected to the many important new emerging properties these materials have presented to society in such diverse areas as transportation, shelter, clothing, food, and healthcare, to mention a few. There is no doubt that these new properties were driven by emergence of the four major architecture classes, namely, (I) linear, (II) crosslinked, (III) branched, and (IV) dendritic polymers. Based on their macromolecular physico-chemical properties and low cost of production, the first three major macromolecular architectures (I–III) have constituted the bulk of all commercial polymer products used by society. Since feedstocks for these three early macromolecular architectures have been based primarily on non-renewable petroleum and fossil fuels, the impact of these materials has not been totally positive for society or the environment. As such, many new commercial polymer platforms have turned to renewable or biodegradable feedstocks and polymer compositions.
In contrast, the fourth major architectural class, namely, dendrimers/dendritic polymers have been found to be more suited for very important, but smaller volume, markets such as catalysis, electronics, diagnostics, protein mimics, and nanomedicine to mention a few. In that regard, using strictly abiotic methods, it has been widely demonstrated over the past decade that dendrimers [52, 55] can be routinely constructed with a control that rivals the structural regulation found in biological systems. The close scaling of size [123, 197], shape, and quasi-equivalency of surfaces [188, 189, 198] observed between nanoscale biostructures and various dendrimer families/generational levels are both striking and provocative [54, 123, 188, 189, 197–201]. These remarkable similarities suggest a broad strategy based on rational biomimicry as a means for creating a repertoire of structure-controlled, size- and shape-variable dendrimer assemblies. Successful demonstrations of such a biomimetic approach has proved it to be a versatile and powerful synthetic strategy for systematically accessing virtually any desired combination of size, shape, and surface chemistry in the nanoscale region. Future extensions will involve combinational variation of dendrimer module parameters such as families (interior compositions), surfaces, generational levels, or architectural shapes (i.e., spheroids, rods, etc.).
In conclusion, it is hoped that the remarkable features described for the dendritic state throughout this account will provide fresh new perspectives and positive expectations for continued growth in the field. There is enormous optimism for the emergence of entirely new, unprecedented properties and applications based on the hybridization of these quantized dendrimer nanosized building blocks with other similar quantized soft and hard nano-building blocks. Quite remarkably, convergence of the dendritic state with the world of nanoscience has already inspired a unique perspective and scientific window to a new concept and systematic framework for unifying and defining nanoscience [136–138]. Recent reports by Percec, Rosen and colleagues [151, 169] have provided the first steps toward fulfillment of this nano-periodic concept by predicting a priori nano-periodic self-assembly property patterns for dozens of amphiphilic dendrons. These Percec–Rosen tables are Mendeleev-like in that they have accurately predicted nano-periodic property patterns for dendron self-assembly by simply using nano-periodic CNDP concept criteria. More recent work by chemists such as Mirkin and colleagues [146], Roy, Brus and colleagues [168] and physicists such as Khanna, Castleman and colleagues [143, 144, 202, 203], and others [204, 205] are fulfilling and validating the nano-periodic concept based on atom mimicry and nanoscale superatoms by documenting very sophisticated examples of hard/hard, hard/soft and soft/soft nanocompounds and nano-assemblies and their new properties. Continued progress in this area will undoubtedly lead to a deeper understanding of this proposed nano-periodic paradigm, which unifies both soft and hard nanomatter, as well as providing a more scientifically grounded basis for the emergence and future growth of two important scientific disciplines: stoichiometric synthetic organic nanochemistry and synthetic inorganic nanochemistry (Fig. 38).

Traditional scientific disciplines and the expected new nano-periodic system or framework and new scientific disciplines (i.e., synthetic organic and inorganic nanochemistry) as a function of the hierarchical building block [52]. Copyright: Cambridge University Press
Abbreviations
- BC:
-
Branch cell
- CADP:
-
Critical atomic design parameters
- CHDP:
-
Critical hierarchical design parameters
- CMDP:
-
Critical molecular design parameters
- CMicDP:
-
Critical micron design parameters
- CNDP:
-
Critical nanoscale design parmeters
- FTIR:
-
Fourier transform infrared
- G:
-
Generation
- HNE:
-
Hard nano-element
- LCB:
-
Long chain branching
- MALDI-TOF:
-
Matrix-assisted laser desorption ionization–time of flight
- M n :
-
Number-average molecular weight
- M w :
-
Weight-average molecular weight
- N b :
-
Branch cell multiplicity
- N c :
-
Core multiplicity
- NSF:
-
National Science Foundation
- PAMAM:
-
Poly(amidoamine)
- ROMP:
-
Ring-opening metathesis polymerization
- SCROP:
-
Self-condensing ring-opening polymerization
- SCVP:
-
Self-condensing vinyl polymerization
- SIS:
-
Sterically induced stoichiometry
- SNE:
-
Soft nano-element
- TEM:
-
Transmission electron microscopy
- TMV:
-
Tobacco mosaic virus
- UV:
-
Ultraviolet
- VESPR:
-
Valence shell electron pair repulsion
References
Lehn J-M (2012) Perspectives in chemistry–steps towards complex matter. Angew Chem Int Ed 52:2836–2850
Prigogine I (1972) Phys Today 25(12):38–44
Staudinger H (1926) Die Chemie der hochmolekularen organischen Stoffe im Sinne der Kekuleschen Strukturlehre. Ber Deut Chem Ges 59:3019–3043
Staudinger H (1961) From organic chemistry to macromolecules, a scientific autobiography. Wiley, New York, p 79
Staudinger H (1970) From organic chemistry to macromolecules. Wiley, New York
Watson JD (1976) Molecular biology of the gene. W.A. Benjamin, Menlo Park
Watson JD, Crick FHC (1953) Molecular structure of nucleic acids. Nature 171:4356
Tomalia DA, Dvornic PR, Uppuluri S, Swanson DR, Balogh L (1997) Skeletal macromolecular isomerism: a comparison of dendritic polymer properties to those of classical macromolecular architectures. Polym Mater Sci Eng 77:95–96
Tomalia DA, Naylor AM, Goddard WA III (1990) Starburst dendrimers: molecular level control of size, shape, surface chemistry, topology and flexibility from atoms to macroscopic matter. Angew Chem Int Ed Engl 29(2):138–175
Tomalia DA (1993) Starburst/cascade dendrimers: fundamental building blocks for a new nanoscopic chemistry set. Aldrichimica Acta 26(4):91–101
Tomalia DA (1994) Starburst/cascade dendrimers: fundamental building blocks for a new nanoscopic chemistry set. Adv Mater 6:529–539
Staudinger H (1919) Schweiz Chem Z 105(28–33):60–64
Staudinger H (1920) Ber 53:1073
James LK (1994) Hermann Staudinger 1881–1965. In: James LK (ed) Nobel laureates in chemistry 1901–1992. History of modern chemistry science series. American Chemical Society, Washington, DC, pp 359–367
Boydston AJ, Holcombe TW, Unruh DA, Frechet JMJ, Grubbs RH (2009) A direct route to cyclic organic nanostructures via ring-expansion metathesis polymerization of a dendronized macromonomers. J Am Chem Soc 131:5388–5389
Matyjaszewski K (1996) Cationic polymerizations: mechanisms, synthesis and applications. Marcel Dekker, New York
Matyjaszewski K (ed) (1997) Controlled radical polymerization. ACS symposium series, vol 685. ACS, Washington DC
Goethals EJ, Verdonck B (2005) Living and controlled cationic polymerization. In: Jagur-Grodzinski J (ed) Living and controlled polymerization. Nova Sciences, New York, pp 131–172
Aoshima S, Kanaoka S (2009) A renaissance in living cationic polymerization. Chem Rev 109(11):5245–5287
Elias H-G (1987) Mega molecules. Springer, Berlin
Morawetz H (1985) Polymers. The origin and growth of a science. Wiley, New York
Roovers J (ed) (1999) Branched polymers I. Advances in polymer science, vol. 142. Springer, Berlin
Roovers J (ed) (2000) Branched polymers II. Advances in polymer science, vol 143. Springer, Berlin
Guan Z, Cotts PM, McCord EF, McLain SJ (1999) Chain walking: a new strategy to control polymer topology. Science 283:2059–2062
Scheirs J, Kaminsky W (eds) (2000) Metallocene-based polyolefins, vols 1 and 2. Wiley, Brisbane
Lothian-Tomalia MK, Hedstrand DM, Tomalia DA (1997) A contemporary survey of covalent connectivity and complexity. The divergent synthesis of poly(thioether) dendrimers. Amplified, genealogical directed synthesis leading to the de gennes dense packed state. Tetrahedron 53:15495–15513
Muller HE, Matyjaszewski K (2009) Controlled and living polymerizations: methods and materials. Wiley-VCH, Weinheim
Goodsell DS (2000) Biomolecules and nanotechnology. Am Scientist 88:230–237
Flory PJ (1941) Molecular size distribution in three dimensional polymers. I. Gelation. J Am Chem Soc 63:3083–3090
Flory PJ (1952) Molecular size distributiion in three-dimensional polymers; IV. branched polymers containing A-R-Bf-1 type units. J Am Chem Soc 74:2718–2723
Flory PJ(1953) Ann N Y Acad Sci 57(4):327
Flory PJ (1953) Principles of polymer chemistry. Cornell University Press, Ithaca
Stockmayer WH (1944) J Chem Phys 11:45
Zimm B, Stockmayer WH (1949) J Chem Phys 17:1301
Flory PJ, Rehner J (1943) J Chem Phys 11:512
Graessley WW (1975) Macromolecules 8:865
Gordon M, Dobson GR (1975) J Chem Phys 43:35
Gordon M, Malcolm GN (1966) Proc R Soc (Lond) A295:29
Dusek K (1979) Makromol Chem Suppl 2:35
Burchard W (1988) Adv Polym Sci 48:1
Good IJ (1948) Proc Cambridge Philos Soc 45:360
Good IJ (1963) Proc R Soc (Lond) A263:54
Tomalia DA, Fréchet JMJ (2002) Discovery of dendrimers and dendritic polymers: a brief historical perspective. J Polym Sci A Polym Chem 40(16):2719–2728
Tomalia DA (1995) Dendrimer molecules. Sci Am 272(5):42–46
Tomalia DA, Dewald JR, Hall MJ, Martin SJ, Smith PB (1984) In: Preprints of the 1st SPSJ International polymer conference. Society of Polymer Science, Kyoto, p 65
Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P (1985) A new class of polymers: starburst dendritic macromolecules. Polym J (Tokyo) 17:117–132
Tomalia DA, Swanson DR (2001) Laboratory synthesis and characterization of megamers: core-shell tecto(dendrimers). In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 617–629
Tomalia DA, Brothers HM II, Piehler LT, Durst HD, Swanson DR (2002) Partial shell-filled core-shell tecto(dendrimers): a strategy to surface differentiated nano-clefts and cusps. Proc Natl Acad Sci USA 99(8):5081–5087
Tomalia DA, Brothers HM II, Piehler LT, Hsu Y (1995) Polym Mater Sci Eng 73:75
Tomalia DA (1996) Starburst dendrimers – nanoscopic supermolecules according to dendritic rules and principles. Macromol Symp 101:243–255
Naj AK (1996) Persistent inventor markets a molecule. Wall Street J Feb 26(1996):B1
Tomalia DA, Christensen JB, Boas U (2012) Dendrimers, dendrons, and dendritic polymers: discovery applications and the future. Cambridge University Press, New York
Campagna S, Ceroni P, Puntoriero F (2012) Designing dendrimers. Wiley, Hoboken
Hecht S, Fréchet JMJ (2001) Dendritic encapsulation of function: applying nature’ site isolation principle from biomimetics to materials science. Angew Chem Int Ed 40(1):74–91
Fréchet JMJ, Tomalia DA (2001) Dendrimers and other dendritic polymers. Wiley, Chichester
Newkome GR, Moorfield CN, Vögtle F (1996) Dendritic molecules. Wiley-VCH, Weinheim
Gunatillake PA, Odian G, Tomalia DA (1988) Thermal polymerization of a 2-(carboxyalkyl)-2-oxazoline. Macromolecules 21:1556–1562
Kim YH, Webster OW (1988) Polym Prepr 29:310
Kim YH, Webster OW (1990) Water-soluble hyperbranched polyphenylene: a unimolecular micelle. J Am Chem Soc 112:4592–4593
Emrick T, Chang HT, Frèchet JMJ (2000) The preparation of hyperbranched aromatic and aliphatic polyether epoxies by chloride-catalyzed proton transfer polymerization from ABn and A2 + B3 monomers. J Poly Sci A 38:4850–4869
Emrick T, Fréchet JMJ (1999) Self-assembly of dendritic structures. Curr Opin Coll Interface Sci 4:15–23
Bharati P, Moore JS (1997) Solid-supported hyperbranched polymerization: evidence for self-limited growth. J Am Chem Soc 119:3391
Muzafarov AM, Rebrov EA, Gorbatsevich OB, Golly M, Gankema H, Moller M (1996) Degradable dendritic polymers — a template for functional pores and nanocavities. Macromol Symp 102:35
Miravet JF, Frèchet JMJ (1998) New hyperbranched poly(siloxysilanes): variation of the branching pattern and end-functionalization. Macromolecules 31:3461–3468
Chu F, Hawker CJ (1993) A versatile synthesis of isomeric hyperbranched polyetherketones. Polym Bull 30:265–272
Hawker CJ, Lee R, Frechet JMJ (1991) One-step synthesis of hyperbranched dendritic polyesters. J Am Chem Soc 113:4583–4588
Uhrich KE, Hawker CJ, Frèchet JMJ, Turner SR (1992) One-pot synthesis of hyperbranched polyethers. Macromolecules 25:4583–4587
Liu M, Vladimirov N, Frèchet JMJ (1999) A new approach to hyperbranched polymers by ring-opening polymerization of an AB monomer: 4-(−2-hydroxyethyl)-e-caprolactone. Macromolecules 32:6881–6884
Fréchet JMJ, Henmi M, Gitsov I, Aoshima S, Leduc MR, Grubbs RB (1995) Self-condensing vinyl polymerization: an approach to dendritic materials. Science 269:1080–1083
Hawker CJ, Farrington PJ, Mackay ME, Wooley KL, Frèchet JMJ (1995) Molecular ball bearings: the unusual melt viscosity behavior of dendritic macromolecules. J Am Chem Soc 117:4409–4410
Sunder A, Heinemann J, Frey H (2000) Controlling the growth of polymer trees: concepts and perspectives for hyperbranched polymers. Chem Eur J 6(14):2499–2506
Gong C, Miravet J, Frèchet JMJ (2000) Intramolecular cyclization in the polymerization of ABx monomers: approaches to the control of molecular weight and polydispersity in hyperbranched poly(siloxysilane). J Polym Sci A 37:3193–3201
Tomalia DA, Hedstrand DM, Ferrito MS (1991) COMBBURST™ dendrimers – a new macromolecular architecture. Macromolecules 24:1435–1438
Gauthier M, Li J, Dockendorff J (2003) Arborescent polystyrene-graft-poly(2-vinylpyridine) copolymers as unimolecular micelles. Synthesis from acetylated substrates. Macromolecules 36:2642–2648
Kee RA, Gauthier M, Tomalia DA (2001) Semi-controlled dendritic structure synthesis. In: Frechet JMJ, Tomalia, DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 209–235
Six J-L, Gnanou Y (1995) From star-shaped to dendritic poly(ethylene oxide)s: toward increasingly branched architectures by anionic polymerization. Macromol Symp 95:137–150
Taton D, Cloutet E, Gnanou Y (1998) Novel amphiphilic branched copolymers based on polystyrene and poly(ethylene oxide). Macromol Chem Phys 199:2501–2510
Trollsas M, Hedrick JL (1998) Dendrimer-like star polymers. J Am Chem Soc 120:4644–4651
Trollsas M, Hedrick JL (1998) Hyperbranched poly(e-caprolactone) derived from intrinsically branched AB2 macromonomers. Macromolecules 31:4390–4395
Grubbs RB, Hawker CJ, Dao J, Frèchet JMJ (1997) A tandem approach to graft and dendritic graft copolymers based on “living” free radical polymerizations. Angew Chem Int Ed Engl 36:270–272
Kleij AW, Ford A, Jastrzebski JTBH, Van Koten G (2001) Dendritic polymers applications: catalysts. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 485–513
Fréchet JMJ, Ihre H, Davey M (2001) Preparation of Frechet-type polyether dendrons and aliphatic polyester dendrimers by convergent growth: an experimental primer. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 569–586
Van Genderen MHP, Mak MHA, Berg DB-VD, Meijer EW (2001) Synthesis and characterization of poly(propylene imine) dendrimers. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 605–616
Naylor AM, Goddard WA III, Keifer GE, Tomalia DA (1989) Starburst dendrimers 5. Molecular shape control. J Am Chem Soc 111:2339–2341
Hawker CJ, Fréchet JMJ (1990) Preparation of polymers with controlled molecular architecture. a new convergent approach to dendritic macromolecules. J Am Chem Soc 112:7638–7647
Zeng F, Zimmerman SC (1997) Dendrimers in supramolecular chemistry: from molecular recognition to self-assembly. Chem Rev 97(5):1681–1712
Kallos GJ, Tomalia DA, Hedstrand DM, Lewis S, Zhou J (1991) Molecular weight determination of a polyamidoamine starburst polymer by electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom 5(9):383–386
Dvornic PR, Tomalia DA (1995) Genealogically directed syntheses (polymerizations): direct evidence by electrospray mass spectroscopy. Macromol Symp 98:403–428
Hummelen JC, van Dongen JLJ, Meijer EW (1997) Electrospray mass spectrometry of poly(propylene imine) dendrimers–the issue of dendritic purity of polydispersity. Chem Eur J 3(9):1489–1493
Peterson J, Allikmaa V, Subbi J, Pehk T, Lopp M (2003) Structural deviations in poly(amidoamine) dendrimers: a MALDI-TOF MS analysis. Eur Polym J 39:33–42
Brothers HM II, Piehler LT, Tomalia DA (1998) Slab-gel and capillary electrophoretic characterization of polyamidoamine dendrimers. J Chromatogr A 814:233–246
Zhang C, Tomalia DA (2001) Gel electrophoresis characterization of dendritic polymers. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 239–252
Tomalia DA, Fréchet JMJ (2001) Introduction to the dendritic state. In: Frechet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 3–44
Tomalia DA (2012) Dendritic effects: dependency of dendritic nano-periodic property patterns on critical nanoscale design parameters (CNDPs). New J Chem 36:264–281
Kolb HC, Finn MG, Sharpless KB (2001) Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed 40:2004–2021
Kolb HC, Sharpless KB (2003) The growing impact of click chemistry on drug discovery. DDT 8(24):1128–1137
Buhleier E, Wehner W, Vögtle F (1978) Cascade – and nonskid-chain-like syntheses of molecular cavity topologies. Synthesis 405:155–158
Huisgen R (1968) Cycloadditions - definition, classification, and characterization. Angew Chem Int Ed 7(5):321–328
Wu P, Feldman AK, Nugent AK, Hawker CJ, Scheel A, Voit B, Pyun J, Frechet JM, Sharpless KB, Fokin VV (2004) Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(I)-catalyzed ligation of azides and alkynes. Angew Chem Int Ed 43:3928–3932
Joralemon MJ, O'’eilly RK, Matson JB, Nugent AK, Hawker CJ, Wooley KL (2005) Dendrimers clicked together divergently. Macromolecules 38:5436–5443
Wu P, Malkoch M, Hunt JN, Vestberg R, Kaltgrad E, Finn MG, Fokin VV, Sharpless KB, Hawker CJ (2005) Multivalent, bifunctional dendrimers prepared by click chemistry. Chem Commun 2005:5775–5777
Helms B, Mynar JL, Hawker CJ, Frechet JM (2004) Dendronized linear polymers via “Click chemistry”. J Am Chem Soc 126:15020–15021
Joralemon MJ, O'’eilly RK, Hawker CJ, Wooley KL (2005) Shell click-crosslinked (SCC) nanoparticles: a new methodology for synthesis and orthogonal functionalization. J Am Chem Soc 127:16892–16899
Sharma A, Desai A, Ali AR, Tomalia DA (2005) Polyacrylamide gel electrophoresis separation and detection of polyamidoamine dendrimers possessing various cores and temrinal groups. J Chromatogr A 1081:238–244
Turro NJ, Barton JK, Tomalia DA (1991) Molecular recognition and chemistry in restricted reaction spaces. Acc Chem Res 24(11):332–340
Gopidas KR, Leheny AR, Caminati G, Turro NJ, Tomalia DA (1991) Photophysical investigation of similarities between starburst dendrimer and anionic micelles. J Am Chem Soc 113:7335–7342
Ottaviani MF, Turro NJ, Jockusch S, Tomalia DA (1996) Characterization of starburst dendrimers by EPR. 3. Aggregational processes of a postively charged nitroxide surfactant. J Phys Chem 100:13675–13686
Jockusch J, Ramirez J, Sanghvi K, Nociti R, Turro NJ, Tomalia DA (1999) Comparison of nitrogen core and ethylenediamine core starburst dendrimers through photochemical and spectroscopic probes. Macromolecules 32:4419–4423
Hawker CJ, Wooley KL, Frèchet JMJ (1993) Solvatochromism as a probe of the microenvironment in dendritic polyethers: transition from an extended to a globular structure. J Am Chem Soc 115(10):4375–4376
Yin R, Zhu Y, Tomalia DA (1998) Architectural copolymers: rod-shaped, cylindrical dendrimers. J Am Chem Soc 120:2678–2679
Zhang B, Yu H, Schluter AD, Halperin A, Kroger M (2013) Synthetic regimes due to packing constraints in dendritic molecules confirmed by labelling experiments. Nat Commun. doi:10.1038/ncomms2993
de Gennes PG, Hervet HJ (1983) Statistics of starburst polymers. J Phys Lett (Paris) 44:351–360
Mourey TH, Turner SR, Rubinstein M, Frechet JMJ, Hawer CJ, Wooley KL (1992) Unique behavior of dendritic macromolecules: intrinsic viscosity of polyether dendrimers. Macromolecules 25(9):2401–2406
Maiti PK, Cagin T, Wang G, Goddard WA III (2004) Structure of PAMAM dendrimers: generation 1 through 11. Macromolecules 37:6236–6254
Bauer BJ, Amis EJ (2001) Characterization of dendritically branched polymers by small angle neutron scattering (SANS), small angle X-ray scattering (SAXS), and transmission electron microscopy (TEM). In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 255–284
Fréchet JMJ (1994) Functional polymers and dendrimers: reactivity, molecular architectures, and interfacial energy. Science 263:1710–1715
Soler-Illia GJ de AA, Rozes L, Boggiano MK, Sanchez C, Turrin C-O, Caminade A-M, Majoral J-P (2000) New mesotextured hybrid materials made from assemblies of dendrimers and titanium (IV)-oxo-organo clusters. Angew Chem Int Ed 39(23):4250
Boas U, Christensen JB, Heegaard PMH (2006) Dendrimers in medicine and biotechnology. The Royal Society of Chemistry, Cambridge
Menjoge AR, Kannan RM, Tomalia DA (2010) Dendrimer-based drug and imaging conjugates: design considerations for nanomedical applications. Drug Discov Today 15(5/6):171–185
Mintzer MA, Grinstaff MW (2011) Biomedical applications of dendrimers: a tutorial. Chem Soc Rev 40:173–190
Tomalia DA (2005) Birth of a new macromolecular architecture: dendrimers as quantized building blocks for nanoscale synthetic polymer chemistry. Prog Polym Sci 30:294–324
Tomalia DA (2005) The dendritic state. Mater Today 2005(March):34–46
Esfand R, Tomalia DA (2001) Poly(amidoamine) (PAMAM) dendrimers: from biomimicry to drug delivery and biomedical applications. Drug Discov Today 6(8):427–436
Piotti ME, Rivera F, Bond R, Hawker CJ, Frèchet JMJ (1999) Synthesis and catalytic activity of unimolecular dendritic reverse micelles with “internal” functional groups. J Am Chem Soc 121(40):9471–9472
Lim J, Kostiainen M, Maly J, da Costa VCP, Annunziata O, Pavan GM, Simanek EE (2013) Synthesis of large dendrimers with the dimensions of small viruses. J Am Chem Soc 135:4660–4663
Bieniarz C (1998) Dendrimers: applications to pharmaceutical and medicinal chemistry. In: Encyclopedia of pharmaceutical technology, vol 18. Marcel Dekker, New York, pp 55–89
Svenson S, Tomalia DA (2005) Dendrimers in biomedical applications–reflections on the field. Adv Drug Deliv Rev 57:2106–2129
Cheng Y (2012) Dendrimer-based drug delivery systems. Wiley, Hoboken
Tully DC, Fréchet JMJ (2001) Dendrimers at surfaces and interfaces: chemistry and applications. Chem Commun 2001:1229–1239
Jiang D-L, Aida T (2001) Dendritic polymers: optical and photochemical properties. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 425–439
Adronov A, Fréchet JMJ (2000) Light-harvesting dendrimers. Chem Commun 2000:1701–1710
Singh P, Moll F III, Lin SH, Ferzli C (1996) Starburst dendrimers: a novel matrix for multifunctional reagents in immunoassays. Clin Chem 42(9):1567–1569
Singh P (2001) Dendrimer-based biological reagents: preparation and applications in diagnostics. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and dendritic polymers. Wiley, Chichester, pp 463–484
Strathern P (2000) Mendeleyev’s dream. The Berkley Publishing Group, New York
Bell TE (2007) Understanding risk assessment of nanotechnology. http://toxipedia.org/download/attachments/5998572/Understanding_nanoRisk_Assessment.pdf
Tomalia DA (2008) Periodic patterns, relationships and categories of well-defined nanoscale building blocks. National Science Foundation Final Workshop Report pp 1–156. http://www.nsf.gov/crssprgm/nano/GC_Charact08_Tomalia_nsf9_29_08.pdf
Tomalia DA (2009) In quest of a systematic framework for unifying and defining nanoscience. J Nanopart Res 11:1251–1310
Tomalia DA (2010) Dendrons/dendrimer: quantized, nano-element like building blocks for soft-soft and soft-hard nano-compound synthesis. Soft Matter 6:456–474
Anderson PW (1972) More is different. Science 177:393–396
Tomalia DA (2012) In quest of a systematic framework for unifying and defining nanoscience. In: Abstracts American Physical Society March meeting 2012, no W34.00001. http://meetings.aps.org/link/BAPS.2012.MAR.W34.1
Kemsley J (2013) Developing superatom science. Chem Eng News 2013:24–25
Hirsch A, Chen Z, Jiao H (2000) Spherical aromaticity in Ih symmetrical fullerenes: The 2 (N+1)2 rules. Angew Chem Int Ed 39(21):3915–3917
Bergeron DE, Castleman AW Jr, Morisato T, Khanna SN (2004) Formation of Al13I−: evidence for the superhalogen character of Al13. Science 304:84–87
Bergeron DE, Roach PJ, Castleman AW Jr, Jones NO, Khanna SN (2005) Al cluster superatoms as halogens in polyhalides and as alkaline earths in iodide salts. Science 307:231–235
Reveles JU, Khanna SN, Roach PJ, Castleman AW Jr (2006) Multiple valence superatoms. PNAS 103(49):18405–18410
Zhang C, Macfarlane RJ, Young KL, Choi CHJ, Hao L, Auyeung E, Liu G, Zhou X, Mirkin CA (2013) A general approach to DNA-programmable atom equivalents. Nat Mater. doi:10.1038/NMAT3647
Tabakovic I, Miller LL, Duan RG, Tully DC, Tomalia DA (1997) Dendrimers peripherally modified with anion radicals that form Pi-dimers and Pi-stacks. Chem Mater 9:736–745
Tomalia DA (1997) Nanoscopic modules for the construction of higher ordered complexity. In: Michl J (ed) Modular chemistry: proceedings of the NATO advanced research workshop on modular chemistry. Kluwer, The Netherlands, pp 183–191
Ball P (2005) A new kind of alchemy. New Scientist 2495:30
Heilbronner E, Dunitz JD (1993) Reflections on symmetry. Wiley-VCH, New York, p 40
Rosen BM, Wilson DA, Wilson CJ, Peterca M, Won BC, Huang C, Lipski LR, Zeng X, Ungar G, Heiney PA, Percec V (2009) Predicting the structure of supramolecular dendrimers via the analysis of libraries of AB3 and constitutional isomeric AB2 biphenylpropyl ether self-assembling dendrons. J Am Chem Soc 131:17500–17521
Schmid G, Meyer-Zaika W, Pugin R, Sawitowski T, Majoral J-P, Caminade A-M, Turrin C-O (2000) Naked Au 55 clusters: dramatic effect of a thiol-terminated dendrimer. Chem Eur J 6(9):1693–1697
Schmid G (2004) Nanoparticles. Wiley-VCH, Weinheim
Thomas PJ, Kulkarni GU, Rao CNR (2001) Magic nuclearity giant clusters of metal nanocrystals formed by mesoscale self-assembly. J Phys Chem B 105:2515–2517
Schmid G (1990) Clusters and colloids: bridges between molecular and condensed material. Endeavour 14(4):172–178
Rao CNR (1994) Chemical approaches to the synthesis of inorganic materials. Wiley Eastern, New Delhi
Vargaftik MN, Moiseev II, Kochubey DI, Zamaraev KI (1991) Giant palladium clusters: synthesis and characterization. Faraday Discuss 92:13–29
Teranishi T, Hori H, Miyake M (1997) ESR study on palladium nanoparticles. J Phys Chem B101:5774–5776
Schmid G, Harms M, Malm J-O, Bovin J-O, van Ruitenbeck J, Zanbergen HW, Fu WT (1993) Ligand-stabilized giant palladium clusters:promising candidates in heterogeneous catalysis. J Am Chem Soc 115:2046–2048
Wilcoxon JP, Martin JE, Provencio P (2000) Size distribution of gold nanoclusters studied by liquid chromatography. Langmuir 16(25):9912–9920
Jackson JL, Chanzy HD, Booy FP, Drake BJ, Tomalia DA, Bauer BJ, Amis EJ (1998) Visualization of dendrimer molecules by transmission electron microscopy (TEM): staining methods and cryo-TEM of vitrified solutions. Macromolecules 31:6259–6265
Tomalia DA, Baker H, Dewald J, Hall M, Kallos G, Martin S, Roeck J, Ryder J, Smith P (1986) Dendritic macromolecules: synthesis of starburst dendrimers. Macromolecules 19:2466–2468
Pullman B (1998) The atom in the history of human thought. Oxford University Press, New York
Weaver JH (1987) The world of physics, vol 2. Simon & Schuster, New York, pp 1–941
Uppuluri S, Piehler LT, Li J, Swanson DR, Hagnauer GL, Tomalia DA (2000) Core-shell tecto(dendrimers): I. Synthesis and characterization of saturated shell models. Adv Mater 12(11):796–800
Atkins PW (1991) Quanta: a handbook of concepts, 2nd edn. Oxford University Press, Oxford
Mansfield ML, Rakesh L, Tomalia DA (1996) The random parking of spheres on spheres. J Chem Phys 105(8):3245–3249
Roy X, Lee C-H, Crowther AC, Schenck CL, Besara T, Lalancette RA, Siegrist T, Stephens PW, Brus LE, Kim P, Steingerwald ML, Nuckolls C (2013) Nanoscale atoms in solid-state chemistry. Science 341:157–160
Rosen BM, Wilson CJ, Wilson DA, Peterca M, Imam MR, Percec V (2009) Dendron-mediated self-assembly, disassembly, and self-organization of complex systems. Chem Rev 109:6275–6540
Percec V, Peterca M, Dulcey AE, Imam MR, Hudson SD, Nummelin S, Adelman P, Heiney PA (2008) Hollow spherical supramolecular dendrimers. J Am Chem Soc 130:13079–13094
Zumdahl SS, Zumdahl SA (2007) Chemistry. Houghton Mifflin, Boston
Zimmerman SC, Zeng F, Reichert EC, Kolotuchin SV (1996) Self-assembling dendrimers. Science 271:1095–1098
Zimmerman SC, Lawless LJ (2001) Supramolecular chemistry of dendrimers. In: Vögtle F, Schalley CA (eds) Dendrimers IV, Topics in current chemistry, vol 217. Springer, Berlin, pp 95–120
Betley TA, Hessler JA, Mecke A, Banaszak Holl MM, Orr BG, Uppuluri S, Tomalia DA, Baker JR Jr (2002) Tapping mode atomic force microscopy investigation of poly(amidoamine) core-shell tecto(dendrimers) using carbon nanoprobes. Langmuir 18:3127–3133
Tomalia DA, Uppuluri S, Swanson DR, Li J (2000) Dendrimers as reactive modules for the synthesis of new structure controlled, higher complexity - megamers. Pure Appl Chem 72:2343–2358
Uppuluri S, Swanson DR, Brothers HM II, Piehler LT, Li J, Meier DJ, Hagnauer GL, Tomalia DA (1999) Tecto(dendrimer) core-shell molecules: macromolecular tectonics for the systematic synthesis of larger controlled structure molecules. Polym Mater Sci Eng (ACS) 80:55–56
Jensen AW, Maru BS, Zhang X, Mohanty D, Fahlman BD, Swanson DR, Tomalia DA (2005) Preparation of fullerene-shell dendrimer-core nanoconjugates. Nano Lett 5(6):1171–1173
Scerri ER (2007) The periodic table. Oxford University Press, New York
Caminade A-M, Laurent R, Majoral J-P (2005) Characterization of dendrimers. Adv Drug Deliv Rev 57:2130–2146
Tanis I, Tragoudaras D, Karatasos K, Anastasiadis SH (2009) Molelcular dynamics simulations of hyperbranched poly(ester amide):statics, dynamics, and hydrogen bonding. J Phys Chem B 113:5356–5368
Enoki O, Katoh H, Yamamoto K (2006) Synthesis and properties of a novel phenylazomethine dendrimer with a tetraphenylmethane core. Org Lett 8(4):569–571
Ornelas C, Ruiz J, Belin C, Astruc D (2009) Giant dendritic molecular electrochrome batteries with ferrocenyl and pentamethylferrocencyl termini. J Am Chem Soc 131:590–601
Tomalia DA, Frechet JM (2005) Introduction to dendrimers and dendritic polymers. Prog Polym Sci 30:217–219
Percec V, Heck J, Lee M, Ungar G, Alvarez-Castillo A (1992) Poly{2-vinyloxyethyl 3,4,5-tris[4-(n-dodecanyloxy)benzyloxy]benzoate}: a self-assembled supramolecular polymer similar to tobacco mosaic virus. J Mater Chem 2:1033–1039
Percec V (2006) Bioinsprired supramolecular liquid crystals. Philos Trans R Soc A 364:2709–2719
Klug A (1983) From macromolecules to biological assemblies (Nobel lecture). Angew Chem Int Ed 22:565–582
Klug A (1999) Tobacco mosiac virus particle structure and the initiation of disassembly. Philos Trans R Soc Lond B 354:531–535
Percec V, Johansson G, Ungar G, Zhou JP (1996) Fluorophobic effect induces the self-assembly of semifluorinated tapered monodendrons containing crown ethers into supramolecular columnar dendrimers which exhibit a homeotropic hexagonal columnar liquid crystalline phase. J Am Chem Soc 118(41):9855–9866
Percec V, Ahn C-H, Unger G, Yeardly DJP, Moller M (1998) Controlling polymer shape through the self-assembly of dendritic side-groups. Nature 391:161–164
Percec V, Ahn C-H, Cho W-D, Jamieson AM, Kim J, Leman T, Schmidt M, Gerle M, Moller M, Prokhorova SA, Sheiko SS, Cheng SZD, Zhang A, Ungar G, Yeardley DJP (1998) Visualizable cylindrical macromolecules with controlled stiffness from backbones containing libraries of self-assembling dendritic Side Groups. J Am Chem Soc 120:8619–8631
Percec V, Cho W-D, Ungar G, Yeardley DJP (2001) Synthesis and structural analysis of two constitutional isomeric libraries of AB2-based monodendrons and supramolecular dendrimers. J Am Chem Soc 123:1302–1315
Lockman JW, Paul NM, Parquette JR (2005) The role of dynamically correlated conformational equilibria in the folding of macromolecular structures. A model for the design of folded dendrimers. Prog Polym Sci 30:423–452
Huang B, Prantil MA, Gustafson TL, Parquette JR (2003) The effect of global compaction on the local secondary structure of folded dendrimers. J Am Chem Soc 125:14518–14530
Tomalia DA, Huang B, Swanson DR, Brothers HM II, Klimash JW (2003) Structure control within poly(amidoamine) dendrimers: size, shape and regio-chemical mimicry of globular proteins. Tetrahedron 59:3799–3813
Butler PG, Klug A (1978) The assembly of a virus. Sci Am 239(5):62–69
Hermes ME (1996) Enough for one lifetime: Wallace Carothers, inventor of nylon. American Chemical Society and Chemical Heritage Foundation, Philadelphia
Ottaviani MF, Sacchi B, Turro NJ, Chen W, Jockush S, Tomalia DA (1999) An EPR study of the interactions between starburst dendrimers and polynucleotides. Macromolecules 32:2275–2282
Hudson SD, Jung H-T, Percec V, Cho W-D, Johansson G, Ungar G, Balagurusamy VSK (1997) Direct visualization of individual cylindrical and spherical supramolecular dendrimers. Science 278:449–452
Goodson III T (2001) Optical effects manifested by PAMAM dendrimer metal nano-composites. In: Fréchet JMJ, Tomalia DA (eds) Dendrimers and other dendritic polymers. Wiley, Chichester, pp 515–541
Bosman AW, Janssen HM, Meijer EW (1999) About dendrimers: structure, physical properties, and applications. Chem Rev 99:1665–1688
Sayed-Sweet Y, Hedstrand DM, Spindler R, Tomalia DA (1997) Hydrophobically modified poly(amidoamine) (PAMAM) dendrimers: their properties at the air-water interface and use as nanoscopic container molecules. J Mater Chem 7(7):1199–1205
Castleman AW Jr, Khanna SN (2009) Clusters, superatoms and building blocks of new materials. J Phys Chem C 113:2664–2675
Castleman AW Jr, Khanna SN, Sen A, Reber AC, Qian M, Davis KM, Peppernick SJ, Ugrinov A, Merritt MD (2007) From designer clusters to synthetic crystalline nanoassemblies. Nano Lett 7(9):2734–2741
Kostiainen M, Hiekkataipale P, Laiho A, Lemieux V, Seitsonen J, Ruokolainen J, Ceci P (2013) Electrostatic assembly of binary nanoparticle superlattices using protein cages. Nat Nanotechnol 8:52–56
Qian M, Reber AC, Ugrinov A, Chaki NK, Mandal S, Saavedra HM, Khanna SN, Sen A, Weiss PS (2010) Cluster-assembled materials: toward nanomaterials with precise control over properties. ACS Nano 4(1):235–240
Butler PG (1984) The current picture of the structure and assembly of tobacco mosaic virus. J Gen Virol 65:257–279
Acknowledgements
I wish to gratefully acknowledge Ms. Linda S. Nixon for her relentless dedication and assistance in developing graphics, and in assembling and editing of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Tomalia, D.A. (2013). Twenty-First Century Polymer Science After Staudinger: The Emergence of Dendrimers/Dendritic Polymers as a Fourth Major Architecture and Window to a New Nano-periodic System. In: Percec, V. (eds) Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize I. Advances in Polymer Science, vol 261. Springer, Cham. https://doi.org/10.1007/12_2013_252
Download citation
DOI: https://doi.org/10.1007/12_2013_252
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-01136-3
Online ISBN: 978-3-319-01137-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)