
Abstract
Plant proteolysis is an important process through which proteome quality and quantity surveillance is accomplished. Proteases regulate protein turnover and expand their functional diversity. Proteolysis is mostly known as a means to remove proteins but may also result in the production of new shorter proteins. Hence, proteolysis is directly linked with many aspects of the plant’s lifecycle. Here, we provide an overview of selected examples of the major proteolytic pathways known, digestive (autophagy, proteasome, ubiquitin-like pathways and N-end rule) and limited proteolysis in development.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
In plants, cellular and extracellular proteomes show remarkable functional flexibility, a result of different posttranslational fates, including a broad range of covalent modifications (Liu et al. 2020). Almost all proteins, regardless of their lifetime, biochemical properties and cellular function, are sooner or later subjected to proteolysis (Liu and Moschou 2018). The term “proteolysis” is erroneously associated usually with protein end-point degradation and thus loss-of-function. However, proteolysis is like a Swiss-knife and has many trades. Indeed, each protein thus has multiple proteolytic routes on which to embark, with proteolysis outcomes varying between two extremes: either complete degradation (digestive proteolysis) or specific cleavage of the polypeptide chain at one or a few sites (limited proteolysis). While the outcome of digestive proteolysis is usually protein destruction and functional loss, limited proteolysis can additionally lead to the maturation of proteins or peptides and gain- or switch-of-function (Liu and Moschou 2018). The substrate and products of many proteolytic modules are known; however, the corresponding protease remains unidentified. By contrast, many proteases have unknown substrates (orphan proteases).
In this chapter, we discuss proteolytic aspects with direct links to development. We also provide an overview of biological pathways that utilize specific proteolytic cleavage as a mechanism for tuning their outcomes. Due to the immense amount of data on the chapter’s topic, our discussion is succinct and by no means exhaustive. We thus occasionally refer the interested reader to excellent reviews that provide deeper insights.
1.1 The Basics of Limited and Digestive Proteolysis
While all proteins give in to proteolytic degradation, the proteome fraction subjected to limited proteolysis remains elusive. Often regarded as a mere degradative mechanism in the destruction of proteins or turnover in maintaining physiological homeostasis, recent research in the field of degradomics (i.e. the high-throughput study, usually by proteomics, that can detect proteolysis products and even cleavage sites) has led to the recognition of two main yet unexpected concepts. First, that targeted proteolytic cleavage events by a wide repertoire of proteases are pivotal regulators of most, if not all, developmental processes. Second, an unexpected in vivo abundance of stable cleaved proteins revealed pervasive, functionally relevant protein processing.
Below, we succinctly describe the two main proteolytic branches. As of note, as the C-end rule pathway has only recently emerged as a potential proteolytic branch and is as yet unknown in plants, we refer interest readers to, e.g., Lin et al. (2018).
1.2 Digestive Proteolysis
1.2.1 Autophagy
Autophagy carries out digestive proteolysis and usually requires endoproteases for full functionality. Autophagic protein degradation takes place in the vacuole and can be either bulk or selective, i.e. when certain cellular components are preferentially targeted for destruction (Fig. 1a; (Minina et al. 2017a)). Selective autophagy is determined by the interaction of ATG8 with specific cargoes regardless of their size. Readers may refer to Marshall and Vierstra (2018), which is an overview regarding selective autophagy. As of note, the repertoire of autophagy extends beyond proteolytic degradation; it also digests nucleic acids, lipids and carbohydrates but here we will focus on proteolysis.

Schematic representation of the major pathways in digestive and limited proteolysis. (a) Autophagy: Molecules and organelles are either enclosed in autophagosomes (macro-autophagy) or enter directly the lytic vacuole (micro-autophagy). (b) Ubiquitin-Proteasome System: Proteins are selectively ubiquitinated and recognized by proteasome leading to full degradation. (c) Limited proteolysis: Proteases recognize and cleave their protein-targets, creating new proteoforms with different properties such as new interactions or subcellular localization. Figure was created using BioRender (https://biorender.com)
Autophagic degradation is carried out via recognition of substrates by specific adaptors and their sequestration into a double-membrane vesicle, the autophagosome, which is then delivered to and degraded in the vacuole (Minina et al. 2017a). Autophagy-related (ATG) proteins are posttranslationally activated and modulate autophagosome biogenesis ((Marshall et al. 2019) and the references therein). Interestingly, autophagy is regulated by limited proteolysis that takes place in the early stages of the autophagy pathway. In particular, the small ubiquitin (Ub)-like protein ATG8 is cleaved C-terminally by the cysteine protease ATG4 at a highly conserved Gly and the exposed C-terminal glycine is conjugated with the growing autophagosomal membrane (Yoshimoto et al. 2004; Pérez-Pérez et al. 2021). This residue coincides with the penultimate residue in yeast, while in most ATG8s this conserved Gly does not occupy this position. ATG8 is then released from the mature autophagosome membrane by the same ATG4 protease (Pérez-Pérez et al. 2021).
1.2.2 Proteasome
Unlike autophagy, the Ub-proteasome system (UPS) is almost always selective degrading only Ub-tagged proteins. In the UPS pathways, Ub covalently conjugates to substrate lysines (K) through activating (E1), conjugating (E2) and ligating (E3) enzymes (Sadanandom et al. 2012; Santner and Estelle 2010; Spoel et al. 2009; Eckardt 2001; Emenecker et al. 2020; Lin et al. 2020). Ub is first activated by E1 and then transferred onto an E2 conjugating enzyme. Subsequently, E3 Ub-ligases interact simultaneously with a Ub-loaded E2 and the substrate and mediate the formation of isopeptide bonds between the Ub C-terminus and the acceptor K. Depending on the Ub modification nature (e.g. K48 or K63), Ub-carrying proteins have different fates, two prominent examples of which are degradation by the UPS (K48-linked Ub chains) and endocytosis from the plasma membrane to the vacuole (K63-linked chains) (Schwechheimer 2018). The single-subunit E3 ligases include proteins containing a conserved REALLY INTERESTING NEW GENE (RING) domain, a U-box domain, or a Homologous to the E6-AP Carboxyl Terminus (HECT) domain that mediates interaction with the E2-Ub.
1.2.3 Ubiquitin-Like Pathways
Other posttranslational modifications (PTMs) may associate with digestive proteolytic pathways. For example, an analogous enzymatic cascade to Ub is protein tagging via isopeptide bonds with SUMO (small Ub-like modifier) (Srivastava et al. 2016; Yates et al. 2016a, b). Other Ub-like modifiers (UBLs) include NEDD8 (NEURAL PRECURSOR CELL EXPRESSED, DEVELOPMENTALLY DOWN-REGULATED 8) or RUB1 (related to Ub 1) (Schwechheimer 2018), URM1 (Ub-related modifier-1) (Wang et al. 2019), ATG8/12 (autophagy 8/12), MUB (membrane-anchored Ub) (Dowil et al. 2011), UFM1 (Ub-fold modifier-1) (Sasakawa et al. 2006) and HUB1 (homology to Ub-1) (Downes and Vierstra 2005). Like in the case of Ub, UBL conjugation generally depends on an E1-E2-E3 cascade and targets the α-amino acid group of K residues. Although UBLs share a similar fold, their functions and properties differ from Ub. Typically, only a small fraction of a protein competent for SUMOylation is usually SUMOylated, while proteases may execute de-conjugation reactions, including those of the posttranslationally formed isopeptide bonds found in mono-, multi- and polyUb chains or the very similar small SUMOs (Yates et al. 2016b) and conjugates (Schmaler and Dubiel 2010).
In plants as well as in other eukaryotes, the number of E3 ligases for NEDD8 and of NEDD8-modified proteins is much smaller when compared to Ub-modified ones (Hakenjos et al. 2013). Little is known about the function or conjugation of UFM1, MUB and HUB1 in plants. While knowledge of UFM1 is completely missing in plants, all E1, E2 and E3 enzymes for UFM1 conjugation have already been characterized in mammals (Daniel and Liebau 2014). MUB, a UBL present in animals, fungi and plants, is anchored to the membrane owing to the presence of a prenylation signal at its C-terminus instead of the usual di-glycine motif (Nagels Durand et al. 2016). In yeast and mammals, HUB1 functions non-covalently and no E1-E2-E3 cascade has been identified.
1.2.4 N-End Rule Pathway
The N-end rule relates the in vivo half-life of a protein to the nature of its N-terminal (Nt)-residue, which, alongside other requisite features (an unstructured, exposed N-terminus and accessible downstream K), forms a degradation signal called the “N-degron”. Proteins are produced with a methionine (Met; or formylmethionine, fMet) at their Nt (Xu et al. 2015; Frottin et al. 2006; Linster et al. 2015). In most proteins, Nt-Met is cotranslationally cleaved by METHIONINE AMINO-PEPTIDASES (MetAPs), exposing new Nt-residues. The N-end rule pathway has been co-opted to the UPS, targeting proteins for destruction by the UPS through conjugation of a polyUb chain (Zhang et al. 2017a; Gibbs et al. 2011, 2014; 2016; Gibbs 2015). N-degrons are typically conditional, exposed and recognized by Ub-E3 ligases (N-recognins) only under certain conditions.
We know two divisions of the N-end rule pathway: (1) the arginylation (Arg/) N-end rule, which recognizes substrates with unmodified basic or hydrophobic residues and (2) the acetylation (Ac/) N-end rule, which targets proteins bearing certain Nt-acetylated residues (Zhang et al. 2017a; Holman et al. 2009; Vicente et al. 2017). The confirmed “N-recognins” of the Arg/N-end rule: PROTEOLYSIS1 (PRT1) and PRT6 bind to substrates bearing aromatic or basic Nt-residues, respectively (Graciet et al. 2009; Garzon et al. 2007). During protein synthesis, the α-amino group of Nt-residues can be cotranslationally acetylated by ribosome-associated Nt-acetyltransferases (NATs) (Varland et al. 2015). This acetylation occurs either directly on Nt-Met or the 2nd residue after Met-removal by MetAP. Three NATs (NATA, B and C) catalyse the majority of these modifications, with each having distinct substrate specificities. Posttranslational Nt-acetylation also probably occurs.
NAT loss-of-function mutations cause growth defects and reduced photosynthetic efficiency (Gibbs 2015). The NatB loss-of-function mutant tcu2 shows that Nt-acetylation regulates flowering time and leaf, inflorescence, flower, fruit and embryonic development. Furthermore, recent evidence shows that the N-end rule pathway is linked to meristematic activity in the shoot apical meristem in Arabidopsis. The molecular mechanism relates to the degradation of the MicroProtein LITTLE ZIPPER2 (ZPR2) which is degraded by the oxygen-dependent N-degron pathway and thus is stabilized at low O2 levels (Weits et al. 2019). Key to the hypoxia-mediated regulation are the ERF-VII transcription factors (White et al. 2017). Similar to the aforementioned ZPR2 in the shoot meristem, the O2-dependent N-end rule pathway degrades the ERF-VII factors which are thus stabilized at low O2 (Gibbs et al. 2011). As explained below, limited proteolysis depends on a cohort of proteases (Liu et al. 2020; Tornkvist et al. 2019; Liu and Moschou 2018), which may also generate variation in protein turnover by exposing new Nt that can undergo various modifications or amend activities and functions by removing regulatory domains. An example is the BIG BROTHER protein described in Sect. 4.
1.3 Limited Proteolysis
Limited proteolysis is executed by endoproteases which cleave proteins in internal sites, while exoproteases (amino- or carboxypeptidases) trim protein ends (Willems et al. 2017). Proteases enzymatically hydrolyse peptide bonds, resulting in a widespread, irreversible posttranslational modification of the protein's structure and biological function (Fig. 1c). Arabidopsis and rice (Oryza sativa sp.) have close to 800 putative proteases, classified into clans and families. Proteases cleave peptide bonds by polarizing a normally unreactive carbonyl group of the substrate. The carbonyl oxygen is stabilized in an oxyanion hole, making the carbon atom more vulnerable to attack by an activated nucleophile. Proteases are classified by catalytic type and, at present, these are grouped into those in which the activated nucleophile is a side-chain of an amino acid on the protease (“protein nucleophiles”) and those in which the nucleophile is an activated water molecule (“water nucleophiles”).
Protein nucleophiles can be serine-, threonine- or cysteine-type, while water nucleophiles can either be amino acids sidechains (aspartates or glutamates) or by metal ions bound by sidechains. The corresponding catalytic types of water nucleophiles are known as aspartyl-, glutamyl- or metallopeptidases. Furthermore, some proteases are orphan as their catalytic type remains either unknown or does not fall within any known category. Proteins not annotated as proteases may execute proteolysis. For example, in humans, the enzyme O-linked β-N-acetylglucosamine transferase (OGT) that has a canonical function in serine and threonine glycosylation also executes proteolytic maturation of the cell-cycle regulator host cell factor-1 (HCF-1) (Lazarus et al. 2013). Besides, not all proteases cleave proteins. For example, the Arabidopsis γ-glutamyl-transferase 1 and 2 (GGT1 and GGT2) hydrolyse the tripeptide glutathione, some members of the M20 protease family hydrolyse auxin-amino acid conjugates (Bartel and Fink 1995; Davies et al. 1999; Martin et al. 2007), while some carboxypeptidases show acyltransferase activity (sinapoyl-Glc accumulator 1 and 2) (Fraser et al. 2007). Recent evidence suggests that in animals the vast majority of deubiquitinases (i.e. proteases removing Ub from proteins) display isopeptidase and esterase activity (De Cesare et al. 2021). This finding is important, as many esterases could potentially also cleave proteins. We expect that this list of proteases that cleave other than peptide bonds will significantly expand soon.
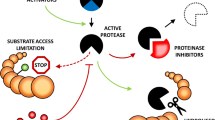
Among other processes, diverse inhibitors regulate proteases, keep them inactive and, in many cases, a single protease may have more than one inhibitor, while the inhibitors may target more than one protease (Kumar et al. 2015). For example, the Arabidopsis inhibitors Serpin1 and disulfide isomerase-5 both interact with and inhibit the cysteine protease RESPONSIVE TO DESICCATION-21 (RD21) (Lampl et al. 2010). The interaction with disulphide isomerase-5 keeps RD21 inactive and it also functions as a chaperone, escorting RD21 en route from the endoplasmatic reticulum via the Golgi to vacuoles (Ondzighi et al. 2008). The acidic vacuolar environment activates RD21, which then contributes to protein degradation in senescing leaves (Otegui et al. 2005; van der Hoorn and Kaiser 2012). Such complex relationships between proteases and inhibitors create complicated proteolytic networks. This is further perplexed by the different inhibitory types: suicidal or reversible.
1.4 The Interplay Between Proteolytic Pathways: The Case of Autophagy-Limited Proteolysis
The best example of the interplay between limited proteolysis and autophagy is ATG8 cleavage by the cysteine protease ATG4. ATG8 is a small Ub-like protein, essential for the elongation and closure of the autophagosomal membrane (Yamasaki et al. 2020). Upon induction of autophagy, ATG8 undergoes limited proteolysis followed by reversible lipidation. The C-terminus of ATG8 is cleaved by the protease ATG4 and the nascent C-terminal glycine is conjugated with a lipid phosphatidylethanolamine (PE) moiety via an amide bond, anchoring ATG8 in an autophagosomal membrane. Intriguingly, ATG8 is removed from the membrane of a mature autophagosome by the same ATG4 protease, which can hydrolyse the amide bond between ATG8 and PE. This ATG4 dual function is crucial for the formation and elongation of the autophagic membrane, proper localization and recycling of ATG8, and finally removal of ATG8 from the mature autophagosomes, which seems necessary for their subsequent fusion with the vacuole (Pérez-Pérez et al. 2021).
ATG4 is the only protease identified among known ATG proteins and is called also “autophagin” (Ding et al. 2018; Rawlings 2013; Vizovišek et al. 2018; Minina et al. 2017a). The balance between proteolytic and delipidating ATG4 activities defines ATG8-PE amount present in the cell. Unlike yeast, most of animal and plant genomes contain several orthologs of ATG4 and ATG8 genes, indicating potential sub- and/or neofunctionalization. To our knowledge, until very recently, only mammalian ATG8 orthologs had been shown to have different roles in certain types of selective autophagy (Lystad et al. 2014). While plant ATG8 orthologs have not yet been sufficiently characterized, assessment of the specificity of two autophagin orthologs towards nine ATG8s in Arabidopsis has revealed only slight selectivity but differences in their proteolytic activity and sensitivity (Seo et al. 2016). Moreover, recent advances have shown the involvement of different isoforms of ATG8 specifically in the regulation of selective autophagy pathways in plants (Zess et al. 2019).
Calpain-dependent cleavage of ATG5 (Yousefi et al. 2006) or caspase-dependent cleavage of ATG6 (Wirawan et al. 2010) in mammals yields protein fragments that cannot sustain autophagosomes formation, but instead activate apoptotic cell death in animals. N-terminal domain of the catalytically inactive protease ATG4D leads simultaneously to activation of the ATG4D and to release of the cytotoxic C-terminal domain of the protein (Betin and Lane 2009). Thus, limited proteolysis of these proteins switches cellular stress response from survival to death. Likewise, in Arabidopsis cleavage of the plant Bcl2 associated athanogene 6 (BAG6) by the aspartyl protease APCB1 induces autophagosome formation and restricts necrotic lesions spreading induced by the necrotrophic fungus Botrytis cinerea (Li et al. 2016b). Another example of an interplay between limited proteolysis and autophagy in plants is provided in Sect. 2.2.
2 Proteolysis in Reproductive Development and Embryogenesis
2.1 Reproductive Development
Flowers are the reproductive plant organs. Reproductive development is characterized by major metabolic changes. Through meiosis and mitosis, the male and female gametophytes also called the pollen grain and the embryo sac, respectively, are generated from anther tissue and ovules. Furthermore, a very important process for successful reproductive development is pollen germination followed by tube growth. This process is relatively fast and dynamic, characterized by vesicular trafficking and cytoskeletal changes, as well as highly active metabolism (Cameron and Geitmann 2018). These changes necessitate the use of proteolytic systems to remodel the proteome and thus it is hardly surprising that limited and digestive proteolysis regulate reproductive development.
The cysteine protease separase (known also as “Extra Spindle Poles”, ESP) executes the release of sister chromatid cohesion during meiosis and mitosis (Yang et al. 2009). ESP-dependent proteolytic cleavage of the α-kleisin subunit of the cohesin complex that holds sister chromatids together at the metaphase-to-anaphase transition is essential for the proper segregation of chromosomes (Minina et al. 2017b; Cromer et al. 2019). In Arabidopsis, meiotic-specific ESP RNA interference blocked cohesin removal from chromosomes and resulted in the presence of a mixture of fragmented chromosomes and intact bivalents leading to alterations in nonhomologous centromere association as well as disruption of the radial microtubule system after telophase II. Moreover, ESP RNA interference affects the proper establishment of nuclear-cytoplasmic domains, resulting in the formation of multinucleate microspores. Likewise, two Arabidopsis putative glycosylphosphatidylinositol (GPI)-anchored aspartic protease genes, A36 and A39, which are highly expressed in pollen and pollen tubes, play a role in reproductive development (Gao et al. 2017a, b). a36 a39 mutants show precocious cell death of pollen and are female gametophytic defected. GFP-A36 and A39 localize at the plasma membrane and cytoplasmic puncta, colocalizing with the GPI-anchored protein COBRA-LIKE10 which plays a role in the cell wall structure and pollen tube guidance (Li et al. 2013). Hence, in a36 a39, the abundance of highly methyl-esterified homogalacturonans and xyloglucans is significantly increased in the apical pollen tube wall.
Aspartic proteases also play vital roles in tapetum degeneration timing, which is crucial in sexual reproduction (Olsson et al. 2019; Yang et al. 2012; Cecchetti et al. 2008). In Arabidopsis and rice, the aspartic proteases AtUNDEAD and OsAP25/OsAP37, respectively, modulate tapetal PCD timing; their absence leads to pollen abortion (Phan et al. 2011). In rice, S5 participates in indica-japonica hybrid fertility and could stimulate ER stress, giving rise to PCD in the embryo sac (Chen et al. 2020; Yang et al. 2012). OsAP65 is essential for pollen germination and tube growth (Huang et al. 2013). In Arabidopsis, loss of function of the ER-localized PROMOTION OF CELL SURVIVAL1 (PCS1) aspartic protease causes gametophytic degeneration (Ge et al. 2005). Therefore, plant aspartic proteases may be implicated in the restriction of PCD in plant reproduction, although the underlying mechanism is not clear.
The MMS21 (HPY2) is a SUMO E3 ligase conserved in eukaryotes and required for DNA repair and chromosome integrity maintenance. In Arabidopsis, MMS21 loss-of-function causes defective meristems, dwarf phenotypes and gametophytic defects (Liu et al. 2014). SUMO E3 ligase MMS21/HPY2 represses the transition from the mitotic cycle into the endocycle in Arabidopsis (Ishida et al. 2012). The hpy2-1 mutant survives for only a few weeks under normal growth, but a few seedlings eventually form shoots that show fasciation and defects in phyllotaxis. Their root meristems contain abnormally enlarged cells and a higher proportion of cells in the endocycle. These endocyclic cells also contain higher DNA content (reaching 64C and 128C) and much larger nuclei compared to wild-type. Mutants of MMS21/HPY2 that survive through the reproductive stage display severe fertility defects exemplified by a much-reduced seed set and increased rate of seed abortion (Ishida et al. 2012; Liu et al. 2014). Reciprocal pollination experiments suggested that most of the reduced fertility of hpy2 mutants is likely due to pollen defects, although some defects in female gametophyte may also contribute to sterility but to a lesser extent. These and other experiments also suggested that hpy2 pollen tube growth was defective even in wild-type pistils. These results are consistent with MMS21/HPY2 expression in anther and pollen. Further characterization revealed that mms21/hpy2 anthers have morphological defects, are generally variable in size and shape and produce fewer and nonviable pollen grains compared with wild-type (Liu et al. 2014). Collectively, these results suggest that MMS21/HPY2 is required for male gametophyte development.
Regarding flowering, the SUMO ligase (siz1) and SUMO protease (esd4) mutations reduce the floral repressor FLOWERING LOCUS C (FLC) mRNA abundance and consequently enhance SOC1 expression which promotes flowering. Both mutants show compromised salicylate (SA) signalling; high SA levels associate with lower mRNA levels and accelerated flowering (Jin et al. 2008). Besides, SIZ1 may also activate FLC expression through an SA-independent pathway that requires the flowering time gene FLD (Jin et al. 2008). On the other hand, the FLC protein interacts with SIZ1, reducing the SUMOylation of FLC in vitro (Son et al. 2013). Furthermore, SIZ1 functions did not depend on SA levels because expressing nahG in siz1 did not restore ovule viability, suggesting that SIZ1 plays a direct role in ovule development. Reciprocal pollination and pollen viability analyses revealed that siz1 pollen is normal (Ling et al. 2012). On the other hand, siz1 female gametophyte could not support full fertilization of wild-type pollen and scanning electron microscopy revealed that pollen grains germinate and pollen tubes migrate through the style of siz1 plants but fail in the final stage of pollen tube guidance to reach the micropylar opening and hence fail to enter the embryo sac. Collectively, these findings suggest that SIZ1 is required for normal female gametophyte development. In the aforementioned cases, however, the role of these proteases in mechanistic terms is unclear.
Pollen tube growth is considered a costly and vigorous process because the cell needs to provide large amounts of the cell wall and membrane components, newly synthesized proteins and energy to fulfil growth demands. As described above, atg6 causes male sterility due to the lack of pollen germination, but knockdown of this gene leads to morphologically normal pollen with decreased germination rate in comparison with wild-type plants (Fujiki et al. 2007; Harrison-Lowe and Olsen 2008). In rice Osatg7 and Osatg9 mutants showed complete sporophytic male sterility and decreased anther dehiscence under normal growth conditions, indicating that autophagy is crucial in reproductive development (Kurusu et al. 2014). Furthermore, pollen of these mutants appeared premature due to defects in anther during maturation, while heterozygous plants have normal pollen. Besides, the pollination of the heterozygous plants with wild-type resulted in normal fertility. These findings suggest that the cause of immature pollen phenotype displayed by autophagy-defective mutants depends on defects in various organs or parental tissue. In accordance, analyses of Arabidopsis and maize (Zea mays) plants harbouring mutations in the autophagy genes (atg) indicated that autophagy contributes to nitrogen remobilization from vegetative to reproductive tissues, including seeds (Have et al. 2017).
2.2 Embryogenesis
In this section, we focus on seed-carrying plants. In a plant seed, the embryo lies dormant surrounded by nutritive endosperm while awaiting suitable conditions to germinate. A hydrophobic cuticle around the embryo protects it from water loss during the early days of growth. Seeds carry large amounts of seed storage proteins, which serve as the primary source of nitrogen for the growing seedling during germination. In developing dicot seeds, the most abundantly expressed storage proteins are members of the 2S albumin and the 7S and 11S globulin protein families. Precursor polypeptides of these storage protein classes are synthesized at the ER, and the mature (processed) polypeptides of all of these three protein classes accumulate inside specialized vacuoles, called protein storage vacuoles (PSVs) (Delgadillo et al. 2020).
In the developing seed, a bidirectional molecular dialogue between embryo and endosperm safeguards cuticle integrity before germination and involves a limited proteolytic pathway (Doll et al. 2020). In this pathway, the ABNORMAL LEAF SHAPE1 subtilase produces the TWISTED SEED1 (TWS1), a peptide that acts as a GASSHO ligand and is recognized by the GASSHO receptor-like kinases. Cuticle surveillance depends on the action of the subtilase, which, unlike the TWS1 precursor and the GASSHO receptors, is not produced in the embryo but the neighbouring endosperm. Active TWS1 precursor mediates the GASSHO-dependent cuticle reinforcement in the embryo.
Autophagic cell death is considered one of the major programmed cell death (PCD) types in eukaryotes (Bozhkov and Jansson 2007). The most characteristic example of autophagic PCD is the embryonic suspensor rupture. This organ is an early embryonic structure that links the embryo to the endosperm and its role is to mediate the transportation of nutrients and signalling molecules as well as to push the embryo through the endosperm (Peng and Sun 2018). In Norway spruce (Picea abies) the cysteine protease metacaspase type II (mcII-Pa) mediates lytic vacuole formation, thereby promoting suspensor death. mcII-Pa knockdown results in fewer autophagosomes in suspensor cells, however, the opposite is invalid, indicating that mcII-Pa activates autophagy and not vice versa (Minina et al. 2014; Reza et al. 2018). Moreover, ATG5 and ATG6 gene downregulation disrupts suspensor differentiation leading to alternative necrotic death and arrest of embryonic development. Yet, the molecular mechanism by which mcII-Pa modulates vacuole formation is unknown.
3 Proteolysis in Hormonal Regulation
Unlike hormonal regulation in animals, plant hormonal pathways depend on breaking points, i.e. proteins, with the ability to restrict a whole pathway. In this section, we provide examples of limited and digestive proteolysis and their effect on the regulation of development. Noteworthy, we present only a subset of direct functions that have been recently described as key regulators of hormonal signalling. The UPS of key regulatory proteins in hormonal pathways has been demonstrated or is at least likely, for all of the phytohormone response pathways.
3.1 Examples of Digestive Proteolysis in Hormonal Regulation
Here we provide selected examples of digestive proteolysis in hormonal regulation. More detailed discussion on the roles of UPS in hormonal pathways can be found in Kelley and Estelle (2012); Lopez-Obando et al. (2015) and autophagy in hormonal regulation in Gou et al. (2019); Liao and Bassham (2020).
3.1.1 Gibberellins
Plant embryos survive for a long time in a developmentally arrested state as dry seeds (i.e. dormancy), a state rapidly reversed during germination. The plant hormone gibberellin A (GA) promotes germination and dormancy loss when environmental conditions are favourable for germination and growth. GA stimulates plant growth and development by targeting the DELLA proteins for proteolytic degradation by the UPS. DELLA proteins are the key repressors of almost all GA responses. There are five DELLA proteins in Arabidopsis, the GA INSENSITIVE (GAI), REPRESSOR OF ga1-3 (RGA), RGA-LIKE 1 (RGL1) and RGL2 (Van De Velde et al. 2017; Salanenka et al. 2018).
A good example of GA functions is the growth modulation in different light conditions. Light promotes plant photomorphogenesis, giving rise to open and expanded cotyledons, and short hypocotyls in Arabidopsis seedlings. In the dark, seedlings undergo de-etiolation (Lyu et al. 2019), characterized by closed cotyledons and elongated hypocotyls (Lyu et al. 2019). A subset of basic helix-loop-helix (bHLH) transcription factors, known as phytochrome-interacting factors (PIFs), has a key role in etiolation and light-regulated plant development. Upon illumination, the photoactivated phytochromes trigger PIFs’ rapid phosphorylation and subsequently UPS-mediated degradation, leading to transcriptional changes that promote photomorphogenesis. DELLAs promote PIF degradation through UPS. When GA is present, GA receptor GID1 binds to DELLAs to form GID1-GA-DELLA complex, which then triggers DELLA proteins degradation by the UPS. DELLAs have a conserved DELLA domain (motif Asp-Glu-Leu-Leu-Ala) at the Nt, essential for GA-triggered protein degradation (Li et al. 2016a).
SUMO regulates GA signalling, for example through SLEEPY1 (SLY1) SUMOylation by SIZ1 (Mazur et al. 2019). SLY1 is an F-box protein component of an SCF complex (SCFSLY1) that mediates the interaction of GID1 with DELLA proteins in response to GA, thereby facilitating DELLA degradation (Wang et al. 2009). SIZ1 SUMOylates SLY1, this increases SLY1 stability and interaction with DELLA proteins, promoting growth due to enhanced DELLA degradation (Kim et al. 2015). Interestingly, GA induces SIZ1 expression and stimulates SLY1 SUMOylation. These data propose that GA stimulates growth by inducing SIZ1 expression, which in turn leads to SLY1 sumoylation, stabilization and interaction with DELLA proteins. SLY1 interaction with DELLA leads to subsequent DELLA degradation. This model implicates SUMO as a positive regulator of GA-mediated plant development.
3.1.2 Jasmonates
Jasmonic acid (JA) together with its precursors and derivatives, referred to as jasmonates (JAs), regulate – among other processes – the response to wounding, by inducing specialized metabolism (Nagels Durand et al. 2016). JAs further contribute to plant plasticity by regulating responses to abiotic stresses, thereby adjusting growth and productivity under adverse conditions. Finally, JAs are also important in the regulation of responses to light and development (Zhou et al. 2019).
The core JA-signalling module comprises (1) MYC2, a key transcription factor regulating expression of JA-responsive genes, (2) Jasmonate ZIM-domain (JAZ) proteins, repressors that inhibit MYC2 activity in the absence of the hormone and (3) CORONATINE INSENSITIVE 1 (COI1) that acts as the JA receptor in vivo and, in response to JA-Ile, targets the JAZ repressors for proteolytic degradation. The co-repressors Novel Interactor of JAZ (NINJA) and TOPLESS (TPL) mediate the repressing effect of JAZ proteins (Santner and Estelle 2007). In the absence of JAs, members of the JAZ protein family repress expression of JA-responsive genes by inhibiting MYC2 activity. Upon treatment with the hormone, JAZ proteins interact with COI and are degraded by UPS, thereby allowing JA-responsive genes activation. In the absence of JAs, members of the JAZ protein family repress expression of JA-responsive genes by inhibiting MYC2 activity. Upon treatment with the hormone, JAZ proteins are degraded by the UPS, thereby allowing transcriptional activation of JA-responsive genes. This degradation depends on the direct interaction between JAZ and COI1 (Nagels Durand et al. 2016). JAZ proteins contain three conserved domains: the zinc-fingers expressed in the inflorescence meristem (ZIM) domain, a region of weak homology at the N-terminus and the C-terminal Jas domain which is most strongly conserved. The Jas domain is essential for JAZ stability because it constitutes the interaction platform between JAZ and COI1 upon hormone treatment. The Jas domain of JAZ repressors is required for both the formation of the COI1-JA-Ile-JAZ co-receptor complex and the interaction with the JID domain of MYC TFs.
3.2 Examples of Limited Proteolysis in Hormonal Regulation
3.2.1 Auxin
The plant hormone indole-3-acetic acid (IAA or auxin) regulates many developmental processes and stress responses, acting as permissive or restrictive signal depending on concentrations of active auxin or tissue/cell context. Auxin promotes the recognition of the transcriptional repressors Aux/IAAs “degron” domain II (DII) by the E3 Ub-ligase proteolytic complex Skp1–Cullin1–F-box (SCF)TIR1/AFB, leading to their degradation in the nucleus. In the apical hook, a parallel cytoplasmic pathway commences at the cell membrane and involves TMK1 (Cao et al. 2019). TMK proteins contain an intracellular kinase domain, a single transmembrane pass and an extracellular domain with two leucine-rich repeats separated by a non-LRR region. tmk1 loss-of-function mutants have disrupted apical hook development. TMK1 shows a transient cytosolic and nuclear distribution at the concave side of the apical hook. The redistribution depends on the cleavage of TMK1 by an unknown pathway that coincides with local auxin maxima, releasing a fragment containing the intracellular kinase domain, dubbed as TMK1-C (Cao et al. 2019).
In strict contrast to the Skp1–Cullin1–F-box (SCF)TIR1/AFB digestive pathway, TMK1-C stabilizes IAA repressors (Cao et al. 2019). The stabilization step involves the translocation of TMK1-C from the plasma membrane to the cytosol and nucleus where it interacts with and phosphorylates IAA32/34. The two proteins lack the DII and thus are not degraded by SCFTIR1/AFB. In the tmk1, IAA32/34 proteins decreased, and auxin could not induce their accumulation. However, it is unclear what determines the stability of IAA32/34 and these proteins could contain cryptic degrons or are degraded by non-UPS pathways such as exoproteases.
Another example of a limited proteolytic pathway regulating auxin is the aforementioned Arabidopsis ESP (see Sect. 3.1). ESP regulates auxin gradient in the root meristem by adjusting PINFORMED (PINs) proteins which give rise to directional polar auxin transport and contribute to auxin levels regulation (Liu et al. 2017, 2020; Moschou et al. 2016). This regulation is accomplished through a collaboration between ESP and microtubule-binding kinesins (Kin7.3-clade), affecting microtubule stability and delivery of PINs to polar plasma membrane domains. There, PINs transport auxin contributing to a certain distribution. Although microtubules are important in the regulation of PIN polarity, the molecular mechanism by which ESP executes the polarization of PINs is unknown.
3.2.2 Ethylene
Ethylene is a volatile plant hormone involved in many plant processes such as ripening, ageing and senescence, and is produced in all plant organs. Ethylene is perceived at the ER membrane (Chao et al. 1997). When ethylene is absent, specific ethylene receptors form complexes with the protein kinase CONSTITUTIVE TRIPLE RESPONSE-1 (CTR1) and the integral membrane protein ethylene insensitive 2 (EIN2), a process which leads to EIN2 phosphorylation by CTR1 (Hua and Meyerowitz 1998).
EIN2 is a general regulator of the ethylene signalling pathway. When ethylene is present, it acts as an inverse agonist by inhibiting its receptors. EIN2 is, then, released, dephosphorylated and cleaved releasing a fragment with two functions. On the one hand, the EIN2 C-terminal part is translocated to the nucleus, where it stabilizes the EIN3 transcription factor via the EIN2 nuclear-associated protein 1 (ENAP1), resulting in the activation of ethylene response genes (Wen et al. 2012). On the other hand, the EIN2-C is bound, occasionally, to EBF1 and EBF2 mRNAs, where this complex is associated with processing bodies in the cytoplasm, leading to degradation. Degradation of EBF1 and EBF2 releases, among others, EIN3 and results thus in more ethylene signalling. Interestingly, the protease executing EIN2 cleavage is unknown.
3.3 Peptidic Hormone-Like Molecules
Small posttranslationally modified peptides are signalling molecules involved in many aspects of plant growth and development (Stührwohldt et al. 2020). One of the posttranslational modifications that peptides undergo is proteolytic cleavage. As exemplified in Sect. 3.2, subtilisins (SBTs) are amongst the most probable candidates for diffusible peptide production. SBTs are usually produced as inactive proteins, activated upon removal of their inhibitory leading peptides through autoprocessing. Once auto-processed, they cleave substrates in the apoplast (Vartapetian et al. 2016).
The discovery of SBT substrates and their functional studies have been obscured by extensive redundancies within the large SBT family (56 members in Arabidopsis; (Schardon et al. 2016). A notable exception of SBTs’ “redundancy rule” is SBT5.2 that modulates the density of stomata in high levels of CO2, by producing the mature peptide EPIDERMAL PATTERNING FACTOR 2 (EPF2). In the apoplast, EPF2 inhibits epidermal cells from adopting a meristemoid mother cell fate (MMC; (Engineer et al. 2014). By combining expression profiling with proteomic studies, Engineer et al. identified SBT5.2 as the potential protease cleaving EPF2. EPF2 cleavage by SBT5.2 was confirmed in vitro and the double loss-of-function mutant epf2sbt5.2 had no additive phenotypes compared with the single mutants, suggesting that EPF2 and SBT5.2 act together. EPF2 overexpression in the sbt5.2 background reduced stomata to a lesser extent than in the wild-type, indicating SBT5.2 involvement in EPF2-peptide maturation.
Peptides can mature in a stepwise manner in different cell compartments. The peptides CLEL6 and CLEL9 (also known as GOLVEN 1 and 2) control gravitropic responses of the shoot and the root by modulating auxin distribution (Stührwohldt et al. 2020). Several proteolytic processing events located in consecutive compartments of the secretory pathway are required for maturing CLEL peptides. Using an inhibitor-based approach for loss-of-function analysis, targeting protease function at the level of enzyme activity it was shown that the stepwise maturation of CLEL peptides is mediated by SBTs. Following the cleavage of the signal peptide upon entry into the ER, the CLEL6 and 9 precursors are processed at two sites by SBT6.1. Cleavage by SBT6.1 in the cis-Golgi allows for the continued passage of the partially processed (pre-activated) precursors through the secretory pathway and is thus a prerequisite for subsequent posttranslational modifications including tyrosine sulfation and proline hydroxylation within the Golgi, and proteolytic maturation after exiting the Golgi (Stührwohldt et al. 2020). The activation of CLEL6 and CLEL9 by SBTs in the trans-Golgi network or other post-Golgi compartments depends on the Nt aspartate of the mature peptides.
As mentioned earlier, extensive redundancies within SBTs (or other protease families) make them refractory to functional studies. To overcome this limitation, Schardon et al. expressed heterologous SBT inhibitors from Phytophthora infestans, called extracellular proteinase inhibitors (EPIs; EPI1 and EPI10), under specific promoters active during floral organ abscission. EPI expression blocked the production of the diffusible, (IDA) mature form (mIDA), which controls floral abscission in Arabidopsis (petals, sepals and stamens), and a similar pathway functions in root cap sloughing (Shi et al. 2018). The SBT4.12, SBT4.13 and SBT5.2, expressed during abscission, could cleave IDA in vitro. Although these data are insufficient to conclude how many SBTs are involved in the abscission, they provide evidence for SBTs involvement in this process.
4 Proteolysis in Leaf Development and Endoreduplication
During leaf development, the balance between cell proliferation and differentiation is highly coordinated even before the new leaf emerges and grows. Endoreplication permits multiple rounds of DNA replication without subsequent cell division (cytokinesis), leading to the successive doubling of the nuclear DNA content of the cell (Dissmeyer et al. 2009). The transition from a mitotic cell cycle into an endocycle is often associated with the switch of meristematic cells from division to expansion and differentiation. Proteolysis ensures the unidirectional progression of the cell cycle by causing irreversible changes in key factors that direct this process. The cyclin-dependent kinase inhibitor KRP2 is considered a crucial component to whether the cell will undergo endoreplication through the regulation of mitotic Cyclin-Dependent Kinase (CDK)A;1 complex (De Schutter et al. 2007). More specifically, KRP2 expression levels are regulated posttranslationally via proteolysis in a phosphorylation-dependent way and the abundance of this protein determines the inhibition of CDKA;1, thus, resulting in mitotic progression or endocycle entrance.
Previous studies showed that regulated proteolysis, especially UPS, is considered an important factor for leaf growth. Two E3 ligases, BIGBROTHER (BB) and HISTONE MONOUBIQUITINATION (HUB1, and its homolog HUB2) define cell proliferation phase length and rate in leaves by tagging with Ub the proteins that need to undergo degradation. More specifically, HUB1 can mono-ubiquitinate histone H2B in vitro and the corresponding knockdown mutants showing increased cell cycle duration in young leaves. That led to reduced rosette biomass, pale leaves and changes in leaf shape (Fleury et al. 2007). Regarding the BB protein, changes in expression levels lead to different organ sizes and BB abundance correlates with cell proliferation (Cattaneo and Hardtke 2017). The Arabidopsis Ub-activated protease DA1 limits the duration of cell proliferation during organ growth. DA1 is activated by BB and another RING E3 ligase and the DA2. The BB and DA2 are cleaved by DA1 and this cleavage leads to their destabilization (Dong et al. 2017). The cleavage of BB leads to destabilization via the N-end rule pathway-related protein PRT1. DA1 protease also cleaves the de-ubiquitinase UBP15 and the transcription factors TEOSINTE BRANCHED 1/CYCLOIDEA/PCF 15 (TCP15) and TCP22 which repress endoreduplication and promote cell proliferation. Furthermore, another example of cell cycle regulation through proteolysis during leaf development is the Anaphase-Promoting Complex/Cyclosome (APC/C) which is a Ub E3 ligase that targets mitotic cyclins for degradation by the UPS (Kondorosi et al. 2005). Eloy et al. overexpressed the subunit 10 of this complex and observed enhanced leaf size due to increased APC/C activity that led to higher cell division rates, while knockout mutant showed strong defects in female gametogenesis (Eloy et al. 2011).
Finally, another CDK activity repression pathway includes the SIAMESE(SIM) gene that encodes the founding member of the SIAMESE-RELATED family. While normally, the trichomes of Arabidopsis undergo three to four endoreplication cycles, in sim mutants this does not occur due to endocycle repression, leading them to divide mitotically (De Veylder et al. 2011). Furthermore, the SIM homolog SMR1/LOSS OF GIANT CELLS FROM ORGANS (LGO) is upregulated in developing leaves and sepals, indicating a more general role in modulation of endoreplication transition during organ emergence (Roeder et al. 2012).
5 Proteolysis in Senescence
Senescence is the last developmental phase that eventually results to death of single cells, tissues or even the whole plant. This stage is highly regulated and accompanied by an increased rate of protein degradation. It can also function as a recycling process by which the nutrients accumulated in senescing tissues are remobilized to be used for the production of new vegetative, reproductive or storage organs. While the mRNA levels of most genes decline, those of “senescence-associated genes” (SAGs) increase during senescence. As protein degradation is a vital process for successful nutrient recycling, it is not surprising that many of the upregulated genes in senescing tissues encode proteases (Bhalerao et al. 2003).
Among the many classes of plant proteases, mostly serine and cysteine-dependent proteases associate with senescence across different species (Diaz and Martinez 2013). Serine proteases are the largest protease class in plants and evidence shows that they are the dominant proteases during senescence (van der Hoorn 2008). For example, two SBTs (P1 and P2) are activated during dark and nitrogen-starvation induced senescence in wheat and execute ribulose 1,5-bisphosphate carboxylase/oxygenase (Rubisco) degradation in developing grains (Roberts et al. 2011).
At least three cysteine proteases are induced in the vacuole during dark-induced senescence in wheat (Martínez et al. 2007). Another cysteine protease is the SAG12, a papain-like protease. However, the physiological role of SAG12 is not known; the loss-of-function sag12 mutant neither fails to develop senescence-associated vacuoles nor shows any morphological phenotype. The Vacuolar Processing Enzyme (VPE) cysteine protease is responsible for vacuolar protein maturation and promotes cell death during stress and development (Nakaune et al. 2005). In Arabidopsis, the isoforms αVPE and γVPE are upregulated in vegetative organs during senescence (Sanmartin et al. 2005). Finally, plant metacaspases are upregulated in Arabidopsis during senescence in leaves (MC6 and MC9) and flowers (MC3 and MC9), although their exact mechanistic function remains elusive (Breeze et al. 2011).
The class of aspartic proteases is also linked to senescence, although their role is less documented than cysteine protease. Proteomic studies in different species such as oilseed rape (Brassica napus) and rice showed high levels of aspartic proteases, throughout senescence (Desclos et al. 2009). The most characteristic example of aspartic protease role in plant senescence is CND41 in tobacco (Kato et al. 2004, 2005). This protease localizes in chloroplast and degrades Rubisco in vitro at physiological pH. Besides, silencing of CND41 resulted in Rubisco accumulation in older leaves and delayed senescence suggesting deficient remobilization machinery, while overexpression led to accelerated senescence and higher Rubisco degradation in senescent leaves.
Metalloproteases have a role in senescence but to a lesser extent. The most studied metalloprotease family in plants is the FtsH, which is a family of membrane-bound ATP-dependent proteases that contain a zinc-binding domain. Twelve genes are encoding FtsHs in the nuclear genome of Arabidopsis, and nine of them localize in chloroplasts while the others are found in the thylakoid membrane (Kato and Sakamoto 2010). Many FtsHs are upregulated during senescence in Arabidopsis (Guo et al. 2004). Furthermore, studies in Brassica napus senescence under nitrogen starvation led to the induction of the chloroplast-localized FtsH protease at the early stages of senescence while decreasing at the end, indicating a role in early protein degradation in chloroplasts (Desclos et al. 2009).
With regard to digestive proteolysis, in both plants and mammals, autophagy has direct effects in triggering and executing cellular senescence. As mentioned above, recycling of inefficient photosynthetic organs provides plants with nutrient supplies to progress their life cycle at the whole-plant level and sustains seed quality via efficient seed filling. More specifically, leaf senescence is a dynamic process that responds to source/sink demands of the plant regulated by stress responses, nutrients sensing and phytohormones (Buchanan-Wollaston 1997; Lim et al. 2003; Guiboileau et al. 2010). Autophagy plays an important role during senescence as defects in autophagy generally have almost normal development but are more sensitive in nitrogen and carbon starvation or progress to early senescence in low light conditions (Yoshimoto et al. 2014; Thompson et al. 2005; Chung et al. 2010). One exception is the loss-of-function atg6 knockout mutant which shows defective pollen germination (Fujiki et al. 2007) and therefore there can be no homozygous mutants. On the other hand, in mammals, knockout of ATG genes may lead to embryo lethality while from a molecular point of view, elevated levels of autophagy activation may induce cell death, whereas decreased autophagy triggers cellular senescence (Kuma et al. 2005; Rajendran et al. 2019). Furthermore, loss-of-function atg9 mutants in Arabidopsis showed early leaf chlorosis (degradation of chlorophyll) under nitrogen or carbon-starvation conditions (Zhuang et al. 2017).
6 Organellar Proteolysis and Development
6.1 Mitochondria
In mitochondria, protein turnover is regulated externally via anterograde/retrograde communications with the nucleus and internally via proteolysis and chaperones that act posttranslationally on the proteome. Mitochondrial protein turnover is controlled by ATP-dependent proteases involved in the degradation of misfolded proteins or limited proteolytic cleavage of pre-sequences, resulting in protein maturation. ATP-dependent proteases have two conserved domains, an AAA+ domain which enables the protease to bind ATP and performs as a chaperone and a proteolytic domain with a Serine-K catalytic dyad (Lee and Suzuki 2008; Rigas et al. 2009). Among others, long-filament phenotype-1 (LON1), caseinolytic protease (CLPP) and filamentous temperature-sensitive H (FTSH) are the most dominant and conserved components of the proteolytic networks in eukaryotes (van Wijk 2015).
CLPP is an energy-dependent serine-type protease that plays a role in protein quality control. The CLPP protease has an active Serine-Histidine-Aspartate catalytic triad and is present widely amongst bacterial species as well as fungal, mammalian and plant mitochondria (Bhandari et al. 2018). In plants, knockouts of the single gene for CLP protease subunit, clpp2, did not result in growth or development defects, nuclear transcripts were unaffected, whereas mitochondrial genes encoding oxidative phosphorylation protein complexes were abundant (Petereit et al. 2020).
LON1 is a highly conserved mitochondrial protein family in eukaryotes acting as a chaperone facilitating the proper folding of newly synthesized or imported proteins in the mitochondria and as a protease removing protein aggregates (Li et al. 2017). Mitochondrial LΟΝ1 loss impairs oxidative phosphorylation complexes and tricarboxylic acid cycle (TCA) enzymes. Phenotypically, LON depletion in Arabidopsis restricts root growth and leads to deleterious, developmental phenotypes (Rigas et al. 2009). When LON1 is disrupted, oxidative phosphorylation proteins are less abundant, resulting in heat shock proteins and prohibitin accumulation in the mitochondria (Solheim et al. 2012).
FtSH proteases are membrane-bound metalloproteases family abundant in eubacteria, animals and plants. So far, we know four FtSH genes in Arabidopsis mitochondria; FtSH3 and FtSH10 are mitochondrial AAA proteases, while FtSH4 and FtSH11 function as mitochondrial inner membrane AAA proteases (Kato and Sakamoto 2010). More specifically, FtSH4 is involved through reactive oxygen species accumulation, in phytohormone signalling and transcriptional regulation (Zhang et al. 2017b). FtSH4 substrates include the inner membrane translocase Pam18-2 and mitochondrial pyruvate transporter 4 (MPC4) (Opalińska et al. 2017). In Arabidopsis FtSH4 loss of function results in abnormalities of rosette leaves when grown under short-day conditions, structural alterations in mitochondria, accompanied with increased reactive oxygen species levels, carbonylated mitochondrial proteins, reduced cardiolipin contents (Gibala et al. 2009), oxidative phosphorylation subunits alterations (Heidorn-Czarna et al. 2018) and Hsp70 and prohibitins accumulation (Gibala et al. 2009). Additionally, evidence shows that FtSH4 is involved in auxin homeostasis, regulating plant growth and development (Zhang et al. 2014), as well as the regulation of autophagic-PCD and senescence through SA homeostasis (Huang et al. 2018). Moreover, FtSH4 is essential for oxidative phosphorylation subunits biogenesis and, thus, biogenesis of mitochondria during germination (Heidorn-Czarna et al. 2018). Overall, though, recent studies show that mitochondrial AAA proteases, despite their importance in translation and mitochondrial proteome surveillance, do not seem to have serious morphological modifications in plants grown under normal conditions (Kolodziejczak et al. 2018).
6.2 Chloroplasts
Nucleus-encoded proteins constitute a large part of the organellar proteome. These proteins are imported by multiprotein translocases (TOC) in chloroplast envelope membranes. Prokaryotic-type proteases control internal chloroplast protein turnover. So far, we know three procaryotic-like proteases modulating chloroplast protein turnover: FtsH, Deg and Clp. Overall, we so far know 20 chloroplast proteolytic types. Specifically, processing peptidases and energy-driven processive proteases are the major players in chloroplast proteome biogenesis, remodelling and maintenance.
6.2.1 The CHLORAD Pathway
The most well-studied example of proteolysis in chloroplasts is photosystem turnover regulation during excess light which can damage the photosynthetic mechanism. More specifically, photosystem II reversible phosphorylation and dephosphorylation lead to the E3-dependent ubiquitinilation and degradation of its D1 domain. This D1 domain has to be synthesized again de novo so that the photosystem II complex can perceive light and participate again in the photosynthetic pathway (Nath et al. 2013). On the other hand, targeted protein turnover at the chloroplast outer membrane is perhaps mediated by cytoplasmic proteolytic pathways. For example, the outer membrane CDC48 is an Omp85-type channel and SP2 is a cytosolic AAA+ chaperone that fulfils conductance and motor functions, respectively, in the retrotranslocation of target proteins from chloroplasts. This D1 domain has to be synthesized again de novo so that the photosystem II complex can perceive light and participate again in the photosynthetic pathway (Nath et al. 2013). CDC48 extricates proteins from the chloroplast membrane (they are integral membrane proteins). This process exposes outer membrane proteins to the UPS, of the substrates tagged by the chloroplast-localized E3 ligase SP1. In accordance, SP1, SP2 and CDC48 physically interact and form a complex at the chloroplast membrane. This proteolytic system is known as “chloroplast-associated protein degradation”, or CHLORAD (Ling et al. 2019).
Yet, we anticipate that many more proteins are CHLORAD components and possible links to chlorophagy (see below), a specialized type of autophagy targeting chloroplasts (van Wijk 2015; Izumi and Nakamura 2018; Ding et al. 2018; Tornkvist et al. 2019; Have et al. 2017). We should note, for example, that in mammalian cells, Ub-tagging largely acts as a trigger of autophagic removal of dysfunctional organelles (Marmor-Kollet et al. 2020). For example, during mitophagy (i.e. mitochondrial autophagy), dysfunctional mitochondria are tagged by the E3 ligase Parkin, allowing for the autophagic removal of tagged mitochondria (MacVicar et al. 2019; Jin and Youle 2012). We should note, for instance, that in mammalian cells, Ub-tagging largely acts as a trigger of autophagic removal of dysfunctional organelles (Marmor-Kollet et al. 2020).
6.2.2 The Autophagy-Rubisco Degradation Pathway
Chloroplasts have specialized proteolytic machineries for protein degradation during senescence. Whereas stromal proteins decrease during the earlier stages of leaf senescence in wheat or barley (Hordeum vulgare), chloroplast numbers/cell decrease later (Kubínová et al. 2013). Therefore, stromal proteins can degrade without the breakdown of the chloroplast. Immuno-electron microscopy (EM) analysis of Rubisco degradation in senescing wheat (Triticum aestivum) leaves revealed the presence of cytosol-localized small vesicles (~1 μm) that contained Rubisco, but not thylakoid proteins (Izumi and Nakamura 2018). While stromal proteins decrease during the earlier stages of leaf senescence in wheat or barley (Hordeum vulgare), chloroplast numbers/cell decrease later (Kubínová et al. 2013). Therefore, stromal proteins appear to degrade either inside or outside the chloroplast without the breakdown of the entire chloroplast. These vesicles are frequently surrounded by autophagosome-like double membranes and were originally referred to as Rubisco-containing bodies (RCBs). In Arabidopsis and rice leaves expressing stroma-targeted green fluorescent protein (GFP) or GFP-labeled Rubisco showed that RCBs are not produced in the atg5 or atg7 mutants and that RCBs co-localized with the autophagosomal marker, GFP-ATG8. These observations reveal that RCBs are a type of autophagic body that delivers a portion of the stromal proteins into the vacuole (Otegui 2017).
ESCRT is part of an evolutionarily conserved system responsible for endosomal membrane remodelling. In Arabidopsis, the endosomal sorting complex required for transport (ESCRT)-III paralogs charged multivesicular body protein 1A (CHMP1A) and CHMP1B and delivered RCBs to the vacuole (Spitzer et al. 2015). In loss-of-function chmp1a chmp1b mutants, RCBs accumulated in the cytoplasm; therefore, CHMP1 proteins sort RCBs. How a portion of the stroma is separated as RCBs, and how RCBs are then recruited for autophagic transport remains unclear.
The RCB pathway is particularly active in sugar-starved, excised Arabidopsis leaves in darkness or the presence of photosynthesis inhibitors. The starchless mutants, phosphoglucomutase (pgm) and ADP-glucose pyrophosphorylase1 (adg1), lacking starch, showed enhanced RCBs production (Izumi et al. 2010). Moreover, starchless and atg double mutants have reduced growth and enhanced senescence compared to the respective single mutants. These findings indicate that the RCB pathway mediates nitrogen remobilization from older leaves that cannot acquire sufficient light due to the shading of developing leaves by upper tissues. Analyses of Arabidopsis and maize (Zea mays) plants with atg mutations showed that autophagy contributes to nitrogen remobilization from vegetative tissues to reproductive tissues and seeds (Li et al. 2015). However, such a role for autophagy in rice plants was not evaluated, because autophagy-deficient rice plants exhibit male sterility due to impaired pollen maturation (Hanamata et al. 2019; Sera et al. 2019).
Some isolated vacuoles from the darkened leaves of wild-type plants contained chloroplasts that exhibited chlorophyll autofluorescence signals. These findings suggest that shrunken chloroplasts, which are produced through the active separation of their components in the RCB pathway, become the targets of autophagic transport as entire organelles, a process known as chlorophagy (Yamauchi et al. 2019). ATG8-interacting protein 1 (ATI1) and ATI2 were identified in a yeast two-hybrid screen as candidates that interact with the Arabidopsis ATG8 isoform, ATG8f (Wu et al. 2021; Michaeli et al. 2014). These proteins associate with plastids (in addition to the ER) as small vesicles of approximately 1 μm in diameter, which are referred to as ATI bodies. These delivery cargos differ from those of the RCBs that specifically contain a portion of stroma; however, the vacuolar transport of plastid-associated ATI bodies is an autophagy-dependent process, as ATI was not produced in atg5 mutants (Michaeli et al. 2014). Therefore, ATI bodies represent a distinct form of autophagy vesicles that transport some stroma, thylakoid and envelope components into the vacuole. Plastid-associated ATI bodies are also observed inside the chloroplast, and ATI1 interacts with some thylakoid proteins in vivo. It is thus conceivable that plastid-associated ATI bodies form in chloroplasts and are then delivered into the vacuole via autophagosome-mediated transport, although how such bodies are evacuated from chloroplasts remains unclear.
6.2.3 The SAV Pathway
Senescing leaves of Arabidopsis, soybean (Glycine max) and tobacco (Nicotiana tabacum), and perhaps other plants, show small, lytic senescence-associated vacuoles (SAVs). SAVs are stained by the R-6502 dye, emitting strong fluorescence upon the hydrolytic activity of cysteine proteases (Martínez et al. 2008). SAVs form in the peripheral cytoplasmic region of mesophyll cells and are much smaller than the central vacuole, but with much greater lytic activity. SAVs contain SAG12, stromal proteins such as Rubisco and glutamine synthetase, but not thylakoid proteins such as LHCII and the photosystem II reaction centre D1 (Martínez et al. 2008). As atg7 mutants had SAVs, they may be an autophagy-independent, extra-chloroplastic route for stromal protein degradation in senescing leaves. The trafficking pathway of stromal proteins into the SAVs, as well as their composition in proteases, remains uncertain.
6.2.4 The CV Pathway
Another type of organelle related to protein degradation is chloroplast vesiculation (CV), which depends on a protein produced by the CV gene. In Arabidopsis, CV-GFP expression under the dexamethasone-inducible promoter caused the formation of a type of chloroplast-derived vesicle showing a strong signal referred to as CV-containing vesicles (CCV) (Kamranfar et al. 2018). CCVs are around 1 μm and contain stroma, envelope and thylakoid proteins, as shown by immunoblot analysis of some chloroplast proteins, co-immunoprecipitation assays and detection of fluorescently tagged microscopy of stroma markers. CCVs do not associate with GFP-ATG8a and SAVs, and the atg5 mutation does not affect CCVs. Thus, CCVs are part of a vacuolar degradation process for chloroplasts that is independent of autophagy and SAVs.
6.2.5 “N-end rule” Pathway in Chloroplasts
Similar pathways to N-end rule may also function in organelles. Nuclear-encoded proteins comprise > 95% of the chloroplast proteome and are targeted to the plastid by an Nt-chloroplast transit peptide (cTP). Upon delivery to the chloroplast, the cTP is removed by the stromal processing peptidase (SPP) to expose new Nt-amino acids, which can then be further modulated by one of at least seven aminopeptidases (van Wijk 2015). SPP cleaves at a range of different sites, and single or multiple positions; this enzymatic promiscuity coupled with subsequent amino-peptidase activity may ensure the removal of unfavourable (potentially destabilizing) Nt-residues (Rowland et al. 2015). Furthermore, plastid-encoded proteins initiate with Nt-fMet and undergo cotranslational de-formylation followed by Nt-Met excision, both essential for normal plastid development (Giglione et al. 2014). Interestingly Met-retention on chloroplast proteins associates with protein instability (Giglione et al. 2003), whilst fMet can act as a destabilizing residue in bacteria, and possibly also chloroplasts (Piatkov et al. 2015). Cotranslational and posttranslational Nt-acetylation also occurs on chloroplastic proteins, which appears to enhance protein stability (Bienvenut et al. 2011); a nuclear-encoded chloroplast-targeted NAT probably catalyses this modification (Disch et al. 2006).
6.3 Peroxisomes
Peroxisomes are organelles, abundant in all eukaryotes and essential for development and stress responses. Some of their important cellular functions are fatty acid β-oxidation, photorespiration, ureide and polyamine metabolism, biosynthesis of plant hormones and reactive oxygen/nitrogen species production (Moschou et al. 2008; van Wijk 2015). Peroxisomes metabolize the auxin indole-3-butyric acid (IBA) into the active auxin indole-3-acetic acid (IAA) (reviewed in Strader and Bartel (2011)). Mutants with dysfunctional peroxisomes often display impaired IBA-to-IAA conversion (Strader and Bartel 2008) and dampened IBA responsiveness (Zolman et al. 2008).
Arabidopsis peroxisomes contain several proteases, e.g. LON2 (Lon protease 2), DEG15 (Degradation of periplasmic protein 15), SCPL20 (Serine carboxypeptidase-like protein 20), RDL1 (Response to drought21A-like 1) and PXM16 (Peroxisomal M16 metalloprotease) (Pan and Hu 2018). LON2 facilitates sustained matrix protein import in mature peroxisomes and the degradation of matrix proteins during peroxisome remodelling (Adebesin et al. 2018). LON2 facilitates sustained matrix protein import in mature peroxisomes and the degradation of matrix proteins during peroxisome remodelling (Adebesin et al. 2018). In watermelon, DEG15 can be a processing peptidase as a dimer to cleave PTS2 from PTS2-containing proteins or a general protease in its monomeric form (Helm et al. 2007). SCPL20 is involved in β-oxidation and plant pathogen response (Quan et al. 2010, 2013), and RDL1 plays a role in β-oxidation, seed viability and stress response (Quan et al. 2013). The exact role of RDL1, SCLP20 and PXM16 remains to be elucidated.
Plant peroxisomal proteome varies to some extent in different developmental stages and tissue types. During germination, enzymes in peroxisomes catalyse β-oxidation and the glyoxylate cycle, thereby allowing lipids stored in seeds to be used as energy before photosynthetic machinery commences. As phototrophy is established several days after germination, peroxisomes are remodelled to carry out the glycolate pathway required for photorespiration. This remodelling most likely involves the simultaneous action of several processes, such as LON2-mediated degradation of the glyoxylate cycle enzymes and a specialized autophagy type called “pexophagy”. LON2 dysfunction results in glyoxylate cycle enzymes stabilization (Farmer et al. 2013; Goto-Yamada et al. 2014). Besides its protease activity, LON2 also has chaperone activity that suppresses pexophagy and peroxisome remodelling (Goto et al. 2014). Interestingly, the protease activity of LON2 seems to interfere with its chaperone-dependent inhibition of pexophagy and thus helps to control the protection of peroxisomes from autophagy. Remodelling of the peroxisomal proteome is also attributed to light- and sugar-dependent transcriptional changes, and possibly other proteases, the UPS and proteins involved in peroxisome biogenesis (Goto et al. 2015).
All examined Arabidopsis lon2 mutant alleles are resistant to IBA-induced lateral root formation but respond normally to auxins not requiring β-oxidation (Lingard and Bartel 2009; Burkhart et al. 2012), suggesting reduced IBA-to-IAA conversion. Additionally, lon2 mutants display age-dependent defects in PTS2 processing that are accompanied by defects in peroxisomal matrix protein import, indicating that LON2 is necessary for the sustained import of matrix proteins. Despite these defects in peroxisome physiology, disrupting Arabidopsis LON2 does not appear to result in matrix protein stabilization.
A second peroxisomal protease with a role in Arabidopsis matrix protein degradation is DEG15 (Schuhmann et al. 2012). The family of Deg proteases (for degradation of periplasmic proteins), also known as HtrA proteases (for high-temperature requirement A), are one important group of these proteolytic enzymes (Schuhmann et al. 2012). Degs are ATP-independent serine endopeptidases found in all domains of life, including Bacteria, Archaea and Eukarya. DEG15 processes PTS2 proteins into their mature forms by removing the Nt PTS2-containing (Helm et al. 2007; Schuhmann et al. 2008), but DEG15 is not required for isocitrate lyase or malate synthase degradation (Lingard and Bartel 2009). Similarly, the peroxisomal M16 metalloprotease PXM16 is not required for degradation of glyoxylate cycle enzymes; pxm16 and lon2 pxm16 mutants efficiently degrade isocitrate lyase (Lingard and Bartel 2009).
Pexophagy is involved in peroxisomal quality control and removes damaged catalase aggregates and clustered peroxisomes (Kim et al. 2013; Shibata et al. 2013; Yoshimoto et al. 2014; Hackenberg et al. 2013). The plant pexophagy receptor for ATG8 remains elusive. A bioinformatics approach named hfAIM (high fidelity ATG8 interacting motif) identified 9 peroxisomal PEX proteins in Arabidopsis that contain putative hfAIM, among which PEX6 and PEX10 were further verified by bimolecular fluorescence complementation (Xie et al. 2016). Yeast two-hybrid screen identified PEX10 as an ATG8-interacting protein, suggesting that PEX10 is a promising candidate for a receptor in pexophagy (Marshall et al. 2019). A recent study showed that autophagy mediates glucose-promoted peroxisomal degradation in roots and that ATG8 physically interacts with a peptide of PXA1 (Peroxisomal ABC transporter 1)/CTS (Comatose)/PED3 (Peroxisome defective 3) (Huang et al. 2019), making PXA1 another possible receptor for pexophagy. Whether full-length PXA1 interacts with ATG8 in planta has not been shown. More rigorous studies will determine whether any of these ATG8-interacting proteins function as true pexophagy receptors.
6.4 Endoplasmic Reticulum
The ER environment is very sensitive and can result in cytotoxic conditions known as ER stress, which can compromise the balance between protein folding and synthesis. ER stress is under continuous surveillance and proteolytic pathways execute the removal of misfolded proteins. Processes including the unfolded protein response (UPR), ER-associated degradation (ERAD) and autophagy play important roles in restoring ER homeostasis. For excellent reviews on ERAD and UPR, we refer readers to Chen et al. (2020); Bao and Howell (2017).
In plants, we know two ER stress sensor branches: the inositol requiring enzyme 1 (IRE1) and the activating TF 6 (ATF6). IRE1 pathway orchestrates UPR and is the most evolutionary conserved branch of the UPR. The ATF6 (basic leucine zipper protein (bZIP)) are ER transmembrane proteins with a cytosolic Nt containing bZIP transcription factor domain and an ER C-terminal domain containing ER-retention signals (Vitale et al. 2019; Iwata et al. 2008). When cells encounter ER stress, the ATF6 complex translocates to the Golgi where it is activated via regulated intramembrane proteolysis (RIP) (Brown et al. 2000). RIP releases ATF6. This biological process is mediated via proteases and results in the release of the now active bZIPTF domain of the ATF6 complex to the cytosol which is then translocated to the nucleus, regulating ER stress responses (Pastor-Cantizano et al. 2020).
IRE1 modulates UPR in plants and is a single-pass transmembrane protein that has a lumenal domain and cytoplasmic domain, conferring different functions at both ER membrane sides. IRE1 contains both RNase and protein kinase domains, and the RNase activity, but not the protein kinase activity, of IRE1 is critical for ER stress-induced autophagy. In addition to clearing unfolded proteins under stress, ERAD also functions in the turnover of normal proteins to regulate cell development, such as HMG COENZYME A REDUCTASE (HMGCR) in mammals (Menzies et al. 2018). In development, ERAD functions seem conserved from mammals to plants. The mouse homolog of Ubc6p, UBE2J1, is required for spermiogenesis, evidenced by UBE2J1−/− mice sterility (Koenig et al. 2014). Interestingly, in tomato (Solanum lycopersicum) SlUBC32 loss of function homolog of UBE2J1 results in the late-ripening of tomato fruit (Wang et al. 2014). Besides, in rice OsDER1 endosperm-specific knockdown induced UPR and resulted in abnormal phenotypes of seed size and starch granules. OsDER1 associates with OsHRD1, OsHRD3 and OsCDC48, suggesting that the HRD1-mediated ERAD pathway controls rice seed development (Qian et al. 2018).
ERAD may regulate auxin signalling. ER stress triggers the accumulation of AUX/IAA transcriptional repressors by downregulating auxin receptors and transporters in Arabidopsis (Chen et al. 2020). More research will determine ERAD involvement in other phytohormone-dependent growth regulation (Zhao et al. 2014). The same ERAD component, CER9/DOA10A/SUD1 (ECERIFERUM 9/DOA10A/SUPPRESSOR OF DRY2 DEFECTS1) that functions in both drought response and seedling development mediated by ABA signalling suggests that ERAD is essential in mediating the balance between plant growth and stress adaptation.
Furthermore, although selective degradation of ER by autophagy termed “ER-phagy” may function in plants, it is yet unclear whether it has direct roles in development. For more information on this topic, we refer readers to Bao and Bassham (2020). C53 protein is an ER-phagy receptor shared by plants and mammals. C53 harbours a unique shuffled ATG8 interacting motif (AIM) with ATG8 and is recruited to autophagosomes upon ER stress. C53 senses ER-associated stalled ribosomes and forms a tripartite receptor complex with the UFMylation ligase components UFL and DDRGK1, resulting in the degradation of nascent ER proteins. The corresponding c53 mutants are therefore more ER stress-sensitive (Stephani et al. 2020). C53 thus provides an alternative quality control pathway for the maintenance of ER homeostasis.
7 Closing Remarks
The previous proteolytic pathways highlight proteases’ importance in the regulation of aspects of plant development (Fig. 2). The scarcity of information on relevant protease substrates is the major bottleneck in fully elucidating proteolytic pathways affecting development. Although proteolytic pathways have been traditionally linked to intrinsically destructive processes, we believe that this might be an oversimplification of a more profound and general role of such pathways throughout the life span of a cell. The endeavour of “de-orphanizing” proteases, finding their substrates may include high-resolution expression analyses and proteomics (e.g. as was done for the stomatal regulator SBT5.2 (Engineer et al. 2014)). Many proteases, however, are posttranslationally regulated, and therefore quantitative proteomics may not reveal their activity maxima. This problem can be solved by activity profiling methods with specific probes that can reveal the spatiotemporal protease activity (Pružinská et al. 2017). Moreover, expressing heterologous protease inhibitors from pathogens through careful selection of promoters may prove an elegant strategy to overcome genetic redundancies (e.g. SBTs in floral organ abscission; (Schardon et al. 2016)). Methods, such as positional proteomics, can be used to discover in vivo protease substrates and associated products of limited or digestive proteolysis. For example, the high-throughput proteomic approach dubbed COMBINED FRACTIONAL DIAGONAL CHROMATOGRAPHY (COFRADIC) revealed in vivo substrates of METACASPASE 9 (Wrzaczek et al. 2015; Tsiatsiani et al. 2011, 2012, 2013; Tsiatsiani and Heck 2015). Furthermore, information on protease localization is of paramount importance in inferring possible functions of proteases and relevance of the identified interactome, allowing researchers also to focus positional proteomics in a subset of the proteome, increasing the discovery rate of functionally relevant substrates. Such frameworks will streamline future efforts to decipher proteolytic pathways in an unthinkable depth, creating opportunities for practical applications.

Schematic representation of the most important developmental procedures, in which limited and digestive plant proteolysis are involved and described in brief in this chapter. Figure was created using BioRender (https://biorender.com)
Author Contributions
I.H.H., A.M. and P.N.M. writing-original draft preparation; I.H.H., A.M. and P.N.M. review and editing; P.N.M. funding acquisition; all authors have read and agreed to the published version of the chapter.
Funding
This research was funded by a grant to P.N.M. from the Hellenic Foundation for Research & Innovation (HFRI), “Always strive for excellence-Theodoros Papazoglou” NESTOR project grant number 1426.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
Adebesin F, Widhalm JR, Lynch JH, McCoy RM, Dudareva N (2018) A peroxisomal thioesterase plays auxiliary roles in plant β-oxidative benzoic acid metabolism. Plant J 93(5):905–916. https://doi.org/10.1111/tpj.13818
Bao Y, Bassham DC (2020) ER-phagy and its role in ER homeostasis in plants. Plants 9(12):1771
Bao Y, Howell SH (2017) The unfolded protein response supports plant development and defense as well as responses to abiotic stress. Front Plant Sci 8(344). https://doi.org/10.3389/fpls.2017.00344
Bartel B, Fink GR (1995) ILR1, an amidohydrolase that releases active indole-3-acetic acid from conjugates. Science 268(5218):1745–1748
Betin VM, Lane JD (2009) Atg4D at the interface between autophagy and apoptosis. Autophagy 5(7):1057–1059. https://doi.org/10.4161/auto.5.7.9684
Bhalerao R, Keskitalo J, Sterky F, Erlandsson R, Björkbacka H, Birve SJ, Karlsson J, Gardeström P, Gustafsson P, Lundeberg J, Jansson S (2003) Gene expression in autumn leaves. Plant Physiol 131(2):430–442. https://doi.org/10.1104/pp.012732
Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, Houry WA (2018) The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem Biol 13(6):1413–1425. https://doi.org/10.1021/acschembio.8b00124
Bienvenut WV, Espagne C, Martinez A, Majeran W, Valot B, Zivy M, Vallon O, Adam Z, Meinnel T, Giglione C (2011) Dynamics of post-translational modifications and protein stability in the stroma of Chlamydomonas reinhardtii chloroplasts. Proteomics 11(9):1734–1750. https://doi.org/10.1002/pmic.201000634
Bozhkov P, Jansson C (2007) Autophagy and cell-death proteases in plants: two wheels of a funeral cart. Autophagy 3(2):136–138
Breeze E, Harrison E, McHattie S, Hughes L, Hickman R, Hill C, Kiddle S, Y-s K, Penfold CA, Jenkins D, Zhang C, Morris K, Jenner C, Jackson S, Thomas B, Tabrett A, Legaie R, Moore JD, Wild DL, Ott S, Rand D, Beynon J, Denby K, Mead A, Buchanan-Wollaston V (2011) High-resolution temporal profiling of transcripts during Arabidopsis leaf senescence reveals a distinct chronology of processes and regulation. Plant Cell 23(3):873. https://doi.org/10.1105/tpc.111.083345
Brown MS, Ye J, Rawson RB, Goldstein JL (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100(4):391–398. https://doi.org/10.1016/S0092-8674(00)80675-3
Buchanan-Wollaston V (1997) The molecular biology of leaf senescence. J Exp Bot 48(2):181–199. https://doi.org/10.1093/jxb/48.2.181
Burkhart S, Lingard M, Bartel B (2012) Genetic dissection of peroxisome-associated matrix protein degradation in Arabidopsis thaliana. Genetics:193. https://doi.org/10.1534/genetics.112.146100
Cameron C, Geitmann A (2018) Cell mechanics of pollen tube growth. Curr Opin Genet Dev 51:11–17. https://doi.org/10.1016/j.gde.2018.03.008
Cao M, Chen R, Li P, Yu Y, Zheng R, Ge D, Zheng W, Wang X, Gu Y, Gelova Z, Friml J, Zhang H, Liu R, He J, Xu T (2019) TMK1-mediated auxin signalling regulates differential growth of the apical hook. Nature 568(7751):240–243. https://doi.org/10.1038/s41586-019-1069-7
Cattaneo P, Hardtke CS (2017) BIG BROTHER uncouples cell proliferation from elongation in the arabidopsis primary root. Plant Cell Physiol 58(9):1519–1527. https://doi.org/10.1093/pcp/pcx091
Cecchetti V, Altamura MM, Falasca G, Costantino P, Cardarelli M (2008) Auxin regulates Arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell 20(7):1760–1774. https://doi.org/10.1105/tpc.107.057570
Chao Q, Rothenberg M, Solano R, Roman G, Terzaghi W, Ecker JR (1997) Activation of the ethylene gas response pathway in Arabidopsis by the nuclear protein ETHYLENE-INSENSITIVE3 and related proteins. Cell 89(7):1133–1144
Chen Q, Yu F, Xie Q (2020) Insights into endoplasmic reticulum-associated degradation in plants. New Phytol 226(2):345–350. https://doi.org/10.1111/nph.16369
Chung T, Phillips AR, Vierstra RD (2010) ATG8 lipidation and ATG8-mediated autophagy in Arabidopsis require ATG12 expressed from the differentially controlled ATG12A AND ATG12B loci. Plant J 62(3):483–493. https://doi.org/10.1111/j.1365-313X.2010.04166.x
Cromer L, Jolivet S, Singh DK, Berthier F, De Winne N, De Jaeger G, Komaki S, Prusicki MA, Schnittger A, Guérois R, Mercier R (2019) Patronus is the elusive plant securin, preventing chromosome separation by antagonizing separase. Proc Natl Acad Sci:201906237. https://doi.org/10.1073/pnas.1906237116
Daniel J, Liebau E (2014) The ufm1 cascade. Cell 3(2):627–638. https://doi.org/10.3390/cells3020627
Davies RT, Goetz DH, Lasswell J, Anderson MN, Bartel B (1999) IAR3 encodes an auxin conjugate hydrolase from Arabidopsis. Plant Cell 11(3):365–376
De Cesare V, Carbajo Lopez D, Mabbitt PD, Fletcher AJ, Soetens M, Antico O, Wood NT, Virdee S (2021) Deubiquitinating enzyme amino acid profiling reveals a class of ubiquitin esterases. Proc Natl Acad Sci 118(4):e2006947118. https://doi.org/10.1073/pnas.2006947118
De Schutter K, Joubes J, Cools T, Verkest A, Corellou F, Babiychuk E, Van Der Schueren E, Beeckman T, Kushnir S, Inze D, De Veylder L (2007) Arabidopsis WEE1 kinase controls cell cycle arrest in response to activation of the DNA integrity checkpoint. Plant Cell 19(1):211–225. https://doi.org/10.1105/tpc.106.045047
De Veylder L, Larkin JC, Schnittger A (2011) Molecular control and function of endoreplication in development and physiology. Trends Plant Sci 16(11):624–634. https://doi.org/10.1016/j.tplants.2011.07.001
Delgadillo MO, Ruano G, Zouhar J, Sauer M, Shen J, Lazarova A, Sanmartin M, Lai LTF, Deng C, Wang P, Hussey PJ, Sanchez-Serrano JJ, Jiang L, Rojo E (2020) MTV proteins unveil ER- and microtubule-associated compartments in the plant vacuolar trafficking pathway. Proc Natl Acad Sci U S A 117(18):9884–9895. https://doi.org/10.1073/pnas.1919820117
Desclos M, Etienne P, Coquet L, Jouenne T, Bonnefoy J, Segura R, Reze S, Ourry A, Avice J-C (2009) A combined 15 N tracing/proteomics study in Brassica napus reveals the chronology of proteomics events associated with N remobilisation during leaf senescence induced by nitrate limitation or starvation. Proteomics 9:3580–3608. https://doi.org/10.1002/pmic.200800984
Diaz I, Martinez M (2013) Plant C1A cysteine peptidases in germination and senescence. In: Handbook of proteolytic enzymes, vol 2, pp 1852–1858. https://doi.org/10.1016/B978-0-12-382219-2.00417-8
Ding X, Zhang X, Otegui MS (2018) Plant autophagy: new flavors on the menu. Curr Opin Plant Biol 46:113–121. https://doi.org/10.1016/j.pbi.2018.09.004
Disch S, Anastasiou E, Sharma VK, Laux T, Fletcher JC, Lenhard M (2006) The E3 ubiquitin ligase BIG BROTHER controls arabidopsis organ size in a dosage-dependent manner. Curr Biol 16(3):272–279. https://doi.org/10.1016/j.cub.2005.12.026
Dissmeyer N, Weimer AK, Pusch S, De Schutter K, Alvim Kamei CL, Nowack MK, Novak B, Duan GL, Zhu YG, De Veylder L, Schnittger A (2009) Control of cell proliferation, organ growth, and DNA damage response operate independently of dephosphorylation of the Arabidopsis Cdk1 homolog CDKA;1. Plant Cell 21(11):3641–3654. https://doi.org/10.1105/tpc.109.070417
Doll NM, Royek S, Fujita S, Okuda S, Chamot S, Stintzi A, Widiez T, Hothorn M, Schaller A, Geldner N, Ingram G (2020) A two-way molecular dialogue between embryo and endosperm is required for seed development. Science 367(6476):431–435. https://doi.org/10.1126/science.aaz4131
Dong H, Dumenil J, Lu FH, Na L, Vanhaeren H, Naumann C, Klecker M, Prior R, Smith C, McKenzie N, Saalbach G, Chen L, Xia T, Gonzalez N, Seguela M, Inze D, Dissmeyer N, Li Y, Bevan MW (2017) Ubiquitylation activates a peptidase that promotes cleavage and destabilization of its activating E3 ligases and diverse growth regulatory proteins to limit cell proliferation in Arabidopsis. Genes Dev 31(2):197–208. https://doi.org/10.1101/gad.292235.116
Dowil RT, Lu X, Saracco SA, Vierstra RD, Downes BP (2011) Arabidopsis membrane-anchored ubiquitin-fold (MUB) proteins localize a specific subset of ubiquitin-conjugating (E2) enzymes to the plasma membrane. J Biol Chem 286(17):14913–14921. https://doi.org/10.1074/jbc.M110.158808
Downes B, Vierstra RD (2005) Post-translational regulation in plants employing a diverse set of polypeptide tags. Biochem Soc Trans 33(Pt 2):393–399. https://doi.org/10.1042/bst0330393
Eckardt NA (2001) Auxin and the power of the proteasome in plants. Plant Cell 13(10):2161–2163
Eloy NB, de Freitas LM, Van Damme D, Vanhaeren H, Gonzalez N, De Milde L, Hemerly AS, Beemster GTS, Inzé D, Ferreira PCG (2011) The APC/C subunit 10 plays an essential role in cell proliferation during leaf development. Plant J 68(2):351–363. https://doi.org/10.1111/j.1365-313X.2011.04691.x
Emenecker RJ, Holehouse AS, Strader LC (2020) Emerging roles for phase separation in plants. Dev Cell 55(1):69–83. https://doi.org/10.1016/j.devcel.2020.09.010
Engineer CB, Ghassemian M, Anderson JC, Peck SC, Hu H, Schroeder JI (2014) Carbonic anhydrases, EPF2 and a novel protease mediate CO2 control of stomatal development. Nature 513(7517):246–250. https://doi.org/10.1038/nature13452
Farmer LM, Rinaldi MA, Young PG, Danan CH, Burkhart SE, Bartel B (2013) Disrupting autophagy restores peroxisome function to an Arabidopsis lon2 mutant and reveals a role for the LON2 protease in peroxisomal matrix protein degradation. Plant Cell 25(10):4085–4100. https://doi.org/10.1105/tpc.113.113407
Fleury D, Himanen K, Cnops G, Nelissen H, Boccardi TM, Maere S, Beemster GTS, Neyt P, Anami S, Robles P, Micol JL, Inzé D, Van Lijsebettens M (2007) The Arabidopsis thaliana homolog of yeast BRE1 has a function in cell cycle regulation during early leaf and root growth. Plant Cell 19(2):417–432. https://doi.org/10.1105/tpc.106.041319
Fraser CM, Thompson MG, Shirley AM, Ralph J, Schoenherr JA, Sinlapadech T, Hall MC, Chapple C (2007) Related Arabidopsis serine carboxypeptidase-like sinapoylglucose acyltransferases display distinct but overlapping substrate specificities. Plant Physiol 144(4):1986–1999. https://doi.org/10.1104/pp.107.098970
Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T (2006) The proteomics of N-terminal methionine cleavage. Mol Cell Proteomics 5(12):2336–2349. https://doi.org/10.1074/mcp.M600225-MCP200
Fujiki Y, Yoshimoto K, Ohsumi Y (2007) An Arabidopsis homolog of yeast ATG6/VPS30 is essential for pollen germination. Plant Physiol 143(3):1132. https://doi.org/10.1104/pp.106.093864
Gao H, Li R, Guo Y (2017a) Arabidopsis aspartic proteases A36 and A39 play roles in plant reproduction. Plant Signal Behav 12(4):e1304343. https://doi.org/10.1080/15592324.2017.1304343
Gao H, Zhang Y, Wang W, Zhao K, Liu C, Bai L, Li R, Guo Y (2017b) Two membrane-anchored aspartic proteases contribute to pollen and ovule development. Plant Physiol 173(1):219–239. https://doi.org/10.1104/pp.16.01719
Garzon M, Eifler K, Faust A, Scheel H, Hofmann K, Koncz C, Yephremov A, Bachmair A (2007) PRT6/At5g02310 encodes an Arabidopsis ubiquitin ligase of the N-end rule pathway with arginine specificity and is not the CER3 locus. FEBS Lett 581(17):3189–3196. https://doi.org/10.1016/j.febslet.2007.06.005
Ge X, Dietrich C, Matsuno M, Li G, Berg H, Xia Y (2005) An Arabidopsis aspartic protease functions as an anti-cell-death component in reproduction and embryogenesis. EMBO Rep 6(3):282–288. https://doi.org/10.1038/sj.embor.7400357
Gibala M, Kicia M, Sakamoto W, Gola EM, Kubrakiewicz J, Smakowska E, Janska H (2009) The lack of mitochondrial AtFtsH4 protease alters Arabidopsis leaf morphology at the late stage of rosette development under short-day photoperiod. Plant J 59(5):685–699. https://doi.org/10.1111/j.1365-313X.2009.03907.x
Gibbs DJ (2015) Emerging functions for N-terminal protein acetylation in plants. Trends Plant Sci 20(10):599–601. https://doi.org/10.1016/j.tplants.2015.08.008
Gibbs DJ, Lee SC, Isa NM, Gramuglia S, Fukao T, Bassel GW, Correia CS, Corbineau F, Theodoulou FL, Bailey-Serres J, Holdsworth MJ (2011) Homeostatic response to hypoxia is regulated by the N-end rule pathway in plants. Nature 479(7373):415–U172. https://doi.org/10.1038/nature10534
Gibbs DJ, Md Isa N, Movahedi M, Lozano-Juste J, Mendiondo GM, Berckhan S, Marin-de la Rosa N, Vicente Conde J, Sousa Correia C, Pearce SP, Bassel GW, Hamali B, Talloji P, Tome DF, Coego A, Beynon J, Alabadi D, Bachmair A, Leon J, Gray JE, Theodoulou FL, Holdsworth MJ (2014) Nitric oxide sensing in plants is mediated by proteolytic control of group VII ERF transcription factors. Mol Cell 53(3):369–379. https://doi.org/10.1016/j.molcel.2013.12.020
Gibbs DJ, Bailey M, Tedds HM, Holdsworth MJ (2016) From start to finish: amino-terminal protein modifications as degradation signals in plants. New Phytol 211(4):1188–1194. https://doi.org/10.1111/nph.14105
Giglione C, Vallon O, Meinnel T (2003) Control of protein life-span by N-terminal methionine excision. EMBO J 22:13–23. https://doi.org/10.1093/emboj/cdg007
Giglione C, Fieulaine S, Meinnel T (2014) N-terminal protein modifications: Bringing back into play the ribosome. Biochimie 114. https://doi.org/10.1016/j.biochi.2014.11.008
Goto S, Mano S, Nishimura M (2014) The role of peroxisomes in plant reproductive processes. In: Sexual reproduction in animals and plants, pp 419–429. https://doi.org/10.1007/978-4-431-54589-7_35
Goto S, Mano S, Yamada K, Kazusato O, Hosokawa Y, Hara-Nishimura I, Nishimura M (2015) Dynamics of the light-dependent transition of plant peroxisomes. Plant Cell Physiol 56(7):1264–1271. https://doi.org/10.1093/pcp/pcv081
Goto-Yamada S, Mano S, Nakamori C, Kondo M, Yamawaki R, Kato A, Nishimura M (2014) Chaperone and protease functions of LON protease 2 modulate the peroxisomal transition and degradation with autophagy. Plant Cell Physiol 55(3):482–496. https://doi.org/10.1093/pcp/pcu017
Gou W, Li X, Guo S, Liu Y, Li F, Xie Q (2019) Autophagy in plant: a new orchestrator in the regulation of the phytohormones homeostasis. Int J Mol Sci 20(12). https://doi.org/10.3390/ijms20122900
Graciet E, Walter F, O'Maoileidigh DS, Pollmann S, Meyerowitz EM, Varshavsky A, Wellmer F (2009) The N-end rule pathway controls multiple functions during Arabidopsis shoot and leaf development. Proc Natl Acad Sci U S A 106(32):13618–13623. https://doi.org/10.1073/pnas.0906404106
Guiboileau A, Sormani R, Meyer C, Masclaux-Daubresse C (2010) Senescence and death of plant organs: nutrient recycling and developmental regulation. C R Biol 333:382–391. https://doi.org/10.1016/j.crvi.2010.01.016
Guo Y, Cai Z, Gan S (2004) Transcriptome of Arabidopsis leaf senescence. Plant Cell Environ 27:521–549. https://doi.org/10.1111/j.1365-3040.2003.01158.x
Hackenberg T, Juul T, Auzina A, Gwizdz S, Malolepszy A, Van Der Kelen K, Dam S, Bressendorff S, Lorentzen A, Roepstorff P, Lehmann Nielsen K, Jorgensen JE, Hofius D, Van Breusegem F, Petersen M, Andersen SU (2013) Catalase and NO CATALASE ACTIVITY1 promote autophagy-dependent cell death in Arabidopsis. Plant Cell 25(11):4616–4626. https://doi.org/10.1105/tpc.113.117192
Hakenjos JP, Bejai S, Ranftl Q, Behringer C, Vlot AC, Absmanner B, Hammes U, Heinzlmeir S, Kuster B, Schwechheimer C (2013) ML3 is a NEDD8- and ubiquitin-modified protein. Plant Physiol 163(1):135–149. https://doi.org/10.1104/pp.113.221341
Hanamata S, Sawada J, Toh B, Ono S, Ogawa K, Fukunaga T, Nonomura K-I, Kurusu T, Kuchitsu K (2019) Monitoring autophagy in rice tapetal cells during pollen maturation. Plant Biotechnol (Tokyo) 36(2):99–105. https://doi.org/10.5511/plantbiotechnology.19.0417a
Harrison-Lowe N, Olsen L (2008) Autophagy protein 6 (ATG6) is required for pollen germination in Arabidopsis thaliana. Autophagy 4(3). https://doi.org/10.4161/auto.5629
Have M, Marmagne A, Chardon F, Masclaux-Daubresse C (2017) Nitrogen remobilization during leaf senescence: lessons from Arabidopsis to crops. J Exp Bot 68(10):2513–2529. https://doi.org/10.1093/jxb/erw365
Heidorn-Czarna M, Domanski D, Kwasniak-Owczarek M, Janska H (2018) Targeted proteomics approach toward understanding the role of the mitochondrial protease FTSH4 in the biogenesis of OXPHOS during arabidopsis seed germination. Front Plant Sci 9:821
Helm M, Lück C, Prestele J, Hierl G, Huesgen P, Fröhlich T, Arnold GJ, Adamska I, Görg A, Lottspeich F, Gietl C (2007) Dual specificities of the glyoxysomal/peroxisomal processing protease Deg15 in higher plants. Proc Natl Acad Sci U S A 104:11501–11506. https://doi.org/10.1073/pnas.0704733104
Holman TJ, Jones PD, Russell L, Medhurst A, Úbeda Tomás S, Talloji P, Marquez J, Schmuths H, Tung S-A, Taylor I, Footitt S, Bachmair A, Theodoulou FL, Holdsworth MJ (2009) The N-end rule pathway promotes seed germination and establishment through removal of ABA sensitivity in Arabidopsis. Proc Natl Acad Sci 106(11):4549–4554. https://doi.org/10.1073/pnas.0810280106
Hua J, Meyerowitz EM (1998) Ethylene responses are negatively regulated by a receptor gene family in Arabidopsis thaliana. Cell 94(2):261–271
Huang J, Zhao X, Cheng K, Jiang Y, Ouyang Y, Xu C, Li X, Xiao J, Zhang Q (2013) OsAP65, a rice aspartic protease, is essential for male fertility and plays a role in pollen germination and pollen tube growth. J Exp Bot 64(11):3351–3360. https://doi.org/10.1093/jxb/ert173
Huang X-X, Zhu G-Q, Liu Q, Chen L, Li Y-J, Hou B-K (2018) Modulation of plant salicylic acid-associated immune responses via glycosylation of dihydroxybenzoic acids. Plant Physiol 176(4):3103–3119. https://doi.org/10.1104/pp.17.01530
Huang L, Yu L-J, Zhang X, Fan B, Wang F-Z, Dai Y-S, Qi H, Zhou Y, Xie L-J, Xiao S (2019) Autophagy regulates glucose-mediated root meristem activity by modulating ROS production in Arabidopsis. Autophagy 15(3):407–422. https://doi.org/10.1080/15548627.2018.1520547
Ishida T, Yoshimura M, Miura K, Sugimoto K (2012) MMS21/HPY2 and SIZ1, two Arabidopsis SUMO E3 ligases, have distinct functions in development. PLoS One 7(10):e46897. https://doi.org/10.1371/journal.pone.0046897
Iwata Y, Fedoroff NV, Koizumi N (2008) Arabidopsis bZIP60 is a proteolysis-activated transcription factor involved in the endoplasmic reticulum stress response. Plant Cell 20(11):3107–3121. https://doi.org/10.1105/tpc.108.061002
Izumi M, Nakamura S (2018) Chloroplast protein turnover: the influence of extraplastidic processes, including autophagy. Int J Mol Sci 19(3). https://doi.org/10.3390/ijms19030828
Izumi M, Wada S, Makino A, Ishida H (2010) The autophagic degradation of chloroplasts via rubisco-containing bodies is specifically linked to leaf carbon status but not nitrogen status in Arabidopsis. Plant Physiol 154(3):1196–1209. https://doi.org/10.1104/pp.110.158519
Jin SM, Youle RJ (2012) PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 125(4):795–799. https://doi.org/10.1242/jcs.093849
Jin JB, Jin YH, Lee J, Miura K, Yoo CY, Kim WY, Van Oosten M, Hyun Y, Somers DE, Lee I, Yun DJ, Bressan RA, Hasegawa PM (2008) The SUMO E3 ligase, AtSIZ1, regulates flowering by controlling a salicylic acid-mediated floral promotion pathway and through affects on FLC chromatin structure. Plant J 53(3):530–540. https://doi.org/10.1111/j.1365-313X.2007.03359.x
Kamranfar I, Xue GP, Tohge T, Sedaghatmehr M, Fernie AR, Balazadeh S, Mueller-Roeber B (2018) Transcription factor RD26 is a key regulator of metabolic reprogramming during dark-induced senescence. New Phytol 218(4):1543–1557. https://doi.org/10.1111/nph.15127
Kato Y, Sakamoto W (2010) New insights into the types and function of proteases in plastids. Int Rev Cell Mol Biol 280:185–218. https://doi.org/10.1016/S1937-6448(10)80004-8
Kato Y, Murakami S, Yamamoto Y, Chatani H, Kondo Y, Nakano T, Yokota A, Sato F (2004) The DNA-binding protease, CND41, and the degradation of ribulose-1,5-biphosphate carboxylase/oxygenase in senescent leaves of tobacco. Planta 220:97–104. https://doi.org/10.1007/s00425-004-1328-0
Kato Y, Yamamoto Y, Murakami S, Sato F (2005) Post-translational regulation of CND41 protease activity in senescent tobacco leaves. Planta 222:643–651. https://doi.org/10.1007/s00425-005-0011-4
Kelley DR, Estelle M (2012) Ubiquitin-mediated control of plant hormone signaling. Plant Physiol 160(1):47–55. https://doi.org/10.1104/pp.112.200527
Kim J, Lee H, Lee HN, Kim S-H, Shin KD, Chung T (2013) Autophagy-related proteins are required for degradation of peroxisomes in arabidopsis hypocotyls during seedling growth. Plant Cell 25(12):4956. https://doi.org/10.1105/tpc.113.117960
Kim S-I, Park Bong S, Kim Do Y, Yeu Song Y, Song Sang I, Song Jong T, Seo Hak S (2015) E3 SUMO ligase AtSIZ1 positively regulates SLY1-mediated GA signalling and plant development. Biochem J 469(2):299–314. https://doi.org/10.1042/BJ20141302
Koenig PA, Nicholls PK, Schmidt FI, Hagiwara M, Maruyama T, Frydman GH, Watson N, Page DC, Ploegh HL (2014) The E2 ubiquitin-conjugating enzyme UBE2J1 is required for spermiogenesis in mice. J Biol Chem 289(50):34490–34502. https://doi.org/10.1074/jbc.M114.604132
Kolodziejczak M, Skibior-Blaszczyk R, Janska H (2018) m-AAA complexes are not crucial for the survival of arabidopsis under optimal growth conditions despite their importance for mitochondrial translation. Plant Cell Physiol 59(5):1006–1016. https://doi.org/10.1093/pcp/pcy041
Kondorosi E, Redondo-Nieto M, Kondorosi A (2005) Ubiquitin-mediated proteolysis. To be in the right place at the right moment during nodule development. Plant Physiol 137(4):1197–1204. https://doi.org/10.1104/pp.105.060004
Kubínová Z, Janáček J, Lhotáková Z, Kubínová L, Albrechtová J (2013) Unbiased estimation of chloroplast number in mesophyll cells: advantage of a genuine three-dimensional approach. J Exp Bot 65(2):609–620. https://doi.org/10.1093/jxb/ert407
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N (2005) The role of autophagy during the early neonatal starvation period. Nature 432:1032–1036. https://doi.org/10.1038/nature03029
Kumar MN, Hsieh YF, Verslues PE (2015) At14a-Like1 participates in membrane-associated mechanisms promoting growth during drought in Arabidopsis thaliana. Proc Natl Acad Sci U S A 112(33):10545–10550. https://doi.org/10.1073/pnas.1510140112
Kurusu T, Koyano T, Hanamata S, Kubo T, Noguchi Y, Yagi C, Nagata N, Yamamoto T, Ohnishi T, Okazaki Y, Kitahata N, Ando D, Ishikawa M, Wada S, Miyao A, Hirochika H, Shimada H, Makino A, Saito K, Kuchitsu K (2014) OsATG7 is required for autophagy-dependent lipid metabolism in rice postmeiotic anther development. Autophagy 10:878–888. https://doi.org/10.4161/auto.28279
Lampl N, Budai-Hadrian O, Davydov O, Joss TV, Harrop SJ, Curmi PMG, Roberts TH, Fluhr R (2010) Arabidopsis AtSerpin1, crystal structure and in vivo interaction with its target protease RESPONSIVE TO DESICCATION-21 (RD21). J Biol Chem 285(18):13550–13560. https://doi.org/10.1074/jbc.M109.095075
Lazarus MB, Jiang JY, Kapuria V, Bhuiyan T, Janetzko J, Zandberg WF, Vocadlo DJ, Herr W, Walker S (2013) HCF-1 is cleaved in the active site of O-GlcNAc transferase. Science 342(6163):1235–1239. https://doi.org/10.1126/science.1243990
Lee I, Suzuki CK (2008) Functional mechanics of the ATP-dependent Lon protease- lessons from endogenous protein and synthetic peptide substrates. Biochim Biophys Acta 1784(5):727–735. https://doi.org/10.1016/j.bbapap.2008.02.010
Li S, Ge FR, Xu M, Zhao XY, Huang GQ, Zhou LZ, Wang JG, Kombrink A, McCormick S, Zhang XS, Zhang Y (2013) Arabidopsis COBRA-LIKE 10, a GPI-anchored protein, mediates directional growth of pollen tubes. Plant J 74(3):486–497. https://doi.org/10.1111/tpj.12139
Li F, Chung T, Pennington JG, Federico ML, Kaeppler HF, Kaeppler SM, Otegui MS, Vierstra RD (2015) Autophagic recycling plays a central role in maize nitrogen remobilization. Plant Cell 27(5):1389–1408. https://doi.org/10.1105/tpc.15.00158
Li K, Yu R, Fan LM, Wei N, Chen H, Deng XW (2016a) DELLA-mediated PIF degradation contributes to coordination of light and gibberellin signalling in Arabidopsis. Nat Commun 7:11868. https://doi.org/10.1038/ncomms11868
Li Y, Kabbage M, Liu W, Dickman MB (2016b) Aspartyl protease-mediated cleavage of BAG6 is necessary for autophagy and fungal resistance in plants. Plant Cell 28(1):233–247. https://doi.org/10.1105/tpc.15.00626
Li L, Millar A, Huang S (2017) Mitochondrial Lon1 has a role in homeostasis of the mitochondrial ribosome and pentatricopeptide repeat proteins in plants. Plant Signal Behav 12:00–00. https://doi.org/10.1080/15592324.2016.1276686
Liao CY, Bassham DC (2020) Combating stress: the interplay between hormone signaling and autophagy in plants. J Exp Bot 71(5):1723–1733. https://doi.org/10.1093/jxb/erz515
Lim P, Woo H, Nam H (2003) Molecular genetics of leaf senescence in Arabidopsis. Trends Plant Sci 8:272–278. https://doi.org/10.1016/S1360-1385(03)00103-1
Lin HC, Yeh CW, Chen YF, Lee TT, Hsieh PY, Rusnac DV, Lin SY, Elledge SJ, Zheng N, Yen HS (2018) C-terminal end-directed protein elimination by CRL2 ubiquitin ligases. Mol Cell 70(4):602–613.e603. https://doi.org/10.1016/j.molcel.2018.04.006
Lin Y-L, Chung C-L, Chen M-H, Chen C-H, Fang S-C (2020) SUMO protease SMT7 modulates ribosomal protein L30 and regulates cell-size checkpoint function. Plant Cell:tpc.00301.02019. https://doi.org/10.1105/tpc.19.00301
Ling Q, Huang W, Baldwin A, Jarvis P (2012) Chloroplast biogenesis is regulated by direct action of the ubiquitin-proteasome system. Science 338(6107):655–659. https://doi.org/10.1126/science.1225053
Ling Q, Broad W, Trösch R, Töpel M, Demiral Sert T, Lymperopoulos P, Baldwin A, Jarvis RP (2019) Ubiquitin-dependent chloroplast-associated protein degradation in plants. Science 363(6429):eaav4467. https://doi.org/10.1126/science.aav4467
Lingard MJ, Bartel B (2009) Arabidopsis LON2 is necessary for peroxisomal function and sustained matrix protein import. Plant Physiol 151(3):1354. https://doi.org/10.1104/pp.109.142505
Linster E, Stephan I, Bienvenut WV, Maple-Grodem J, Myklebust LM, Huber M, Reichelt M, Sticht C, Moller SG, Meinnel T, Arnesen T, Giglione C, Hell R, Wirtz M (2015) Downregulation of N-terminal acetylation triggers ABA-mediated drought responses in Arabidopsis. Nat Commun 6:7640. https://doi.org/10.1038/ncomms8640
Liu C, Moschou PN (2018) Cutting in the middleman: hidden substrates at the interface between proteases and plant development. New Phytol 218(3):916–922. https://doi.org/10.1111/nph.14501
Liu M, Shi S, Zhang S, Xu P, Lai J, Liu Y, Yuan D, Wang Y, Du J, Yang C (2014) SUMO E3 ligase AtMMS21 is required for normal meiosis and gametophyte development in Arabidopsis. BMC Plant Biol 14(1):153. https://doi.org/10.1186/1471-2229-14-153
Liu C, Stael S, Gevaert K, Van Breusegem F, Bozhkov PV, Moschou PN (2017) The proteolytic landscape of an Arabidopsis separase-deficient mutant reveals novel substrates associated with plant development. bioRxiv:140962. https://doi.org/10.1101/140962
Liu C, Tornkvist A, Charova S, Stael S, Moschou PN (2020) Proteolytic proteoforms: elusive components of hormonal pathways? Trends Plant Sci 25(4):325–328. https://doi.org/10.1016/j.tplants.2020.01.002
Lopez-Obando M, Ligerot Y, Bonhomme S, Boyer F-D, Rameau C (2015) Strigolactone biosynthesis and signaling in plant development. Development 142(21):3615–3619. https://doi.org/10.1242/dev.120006
Lystad AH, Ichimura Y, Takagi K, Yang Y, Pankiv S, Kanegae Y, Kageyama S, Suzuki M, Saito I, Mizushima T, Komatsu M, Simonsen A (2014) Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep 15(5):557–565. https://doi.org/10.1002/embr.201338003
Lyu M, Shi H, Li Y, Kuang K, Yang Z, Li J, Chen D, Li Y, Kou X, Zhong S (2019) Oligomerization and photo-deoligomerization of HOOKLESS1 controls plant differential cell growth. Dev Cell 51(1):78–88.e73. https://doi.org/10.1016/j.devcel.2019.08.007
MacVicar T, Ohba Y, Nolte H, Mayer FC, Tatsuta T, Sprenger H-G, Lindner B, Zhao Y, Li J, Bruns C, Krüger M, Habich M, Riemer J, Schwarzer R, Pasparakis M, Henschke S, Brüning JC, Zamboni N, Langer T (2019) Lipid signalling drives proteolytic rewiring of mitochondria by YME1L. Nature. https://doi.org/10.1038/s41586-019-1738-6
Marmor-Kollet H, Siany A, Kedersha N, Knafo N, Rivkin N, Danino YM, Olender T, Cohen N, Moens T, Higginbottom A, Knock JC, Eitan C, Cohen BT, Van Den Bosch L, Anderson P, Ivanov P, Geiger T, Hornstein E (2020) Spatio-temporal proteomic analysis of stress granule disassembly using APEX reveals regulation by SUMOylation and links to ALS pathogenesis. bioRxiv. https://doi.org/10.1101/2020.01.29.830133
Marshall RS, Vierstra RD (2018) Autophagy: the master of bulk and selective recycling. Annu Rev Plant Biol 69(1):173–208. https://doi.org/10.1146/annurev-arplant-042817-040606
Marshall RS, Hua Z, Mali S, McLoughlin F, Vierstra RD (2019) ATG8-binding UIM proteins define a new class of autophagy adaptors and receptors. Cell 177(3):766–781.e724. https://doi.org/10.1016/j.cell.2019.02.009
Martin MN, Saladores PH, Lambert E, Hudson AO, Leustek T (2007) Localization of members of the gamma-glutamyl transpeptidase family identifies sites of glutathione and glutathione S-conjugate hydrolysis. Plant Physiol 144(4):1715–1732. https://doi.org/10.1104/pp.106.094409
Martínez DE, Bartoli CG, Grbic V, Guiamet JJ (2007) Vacuolar cysteine proteases of wheat (Triticum aestivum L.) are common to leaf senescence induced by different factors. J Exp Bot 58(5):1099–1107. https://doi.org/10.1093/jxb/erl270
Martínez DE, Costa ML, Gomez FM, Otegui MS, Guiamet JJ (2008) ‘Senescence-associated vacuoles’ are involved in the degradation of chloroplast proteins in tobacco leaves. Plant J 56(2):196–206. https://doi.org/10.1111/j.1365-313X.2008.03585.x
Mazur MJ, Kwaaitaal M, Mateos MA, Maio F, Kini RK, Prins M, van den Burg HA (2019) The SUMO conjugation complex self-assembles into nuclear bodies independent of SIZ1 and COP1. Plant Physiol 179(1):168–183. https://doi.org/10.1104/pp.18.00910
Menzies SA, Volkmar N, van den Boomen DJ, Timms RT, Dickson AS, Nathan JA, Lehner PJ (2018) The sterol-responsive RNF145 E3 ubiquitin ligase mediates the degradation of HMG-CoA reductase together with gp78 and Hrd1. Elife 7:e40009. https://doi.org/10.7554/eLife.40009
Michaeli S, Honig A, Levanony H, Peled-Zehavi H, Galili G (2014) Arabidopsis ATG8-INTERACTING PROTEIN1 is involved in autophagy-dependent vesicular trafficking of plastid proteins to the vacuole. Plant Cell 26(10):4084–4101. https://doi.org/10.1105/tpc.114.129999
Minina EA, Bozhkov PV, Hofius D (2014) Autophagy as initiator or executioner of cell death. Trends Plant Sci 19(11):692–697. https://doi.org/10.1016/j.tplants.2014.07.007
Minina EA, Moschou PN, Bozhkov PV (2017a) Limited and digestive proteolysis: crosstalk between evolutionary conserved pathways. New Phytol 215(3):958–964. https://doi.org/10.1111/nph.14627
Minina EA, Reza SH, Gutierrez-Beltran E, Elander PH, Bozhkov PV, Moschou PN (2017b) The Arabidopsis homolog of Scc4/MAU2 is essential for embryogenesis. J Cell Sci 130(6):1051–1063. https://doi.org/10.1242/jcs.196865
Moschou PN, Sanmartin M, Andriopoulou AH, Rojo E, Sanchez-Serrano JJ, Roubelakis-Angelakis KA (2008) Bridging the gap between plant and mammalian polyamine catabolism: a novel peroxisomal polyamine oxidase responsible for a full back-conversion pathway in Arabidopsis. Plant Physiol 147(4):1845–1857. https://doi.org/10.1104/pp.108.123802
Moschou PN, Gutierrez-Beltran E, Bozhkov PV, Smertenko A (2016) Separase promotes microtubule polymerization by activating CENP-E-related kinesin Kin7. Dev Cell 37(4):350–361. https://doi.org/10.1016/j.devcel.2016.04.015
Nagels Durand A, Pauwels L, Goossens A (2016) The ubiquitin system and Jasmonate signaling. Plants 5(1):6. https://doi.org/10.3390/plants5010006
Nakaune S, Yamada K, Kondo M, Kato T, Tabata S, Nishimura M, Hara-Nishimura I (2005) A vacuolar processing enzyme, δVPE, is involved in seed coat formation at the early stage of seed development. Plant Cell 17(3):876. https://doi.org/10.1105/tpc.104.026872
Nath K, Jajoo A, Poudyal RS, Timilsina R, Park YS, Aro E-M, Nam HG, Lee CH (2013) Towards a critical understanding of the photosystem II repair mechanism and its regulation during stress conditions. FEBS Lett 587(21):3372–3381. https://doi.org/10.1016/j.febslet.2013.09.015
Olsson V, Joos L, Zhu S, Gevaert K, Butenko MA, De Smet I (2019) Look closely, the beautiful may be small: precursor-derived peptides in plants. Annu Rev Plant Biol 70(1):153–186. https://doi.org/10.1146/annurev-arplant-042817-040413
Ondzighi CA, Christopher DA, Cho EJ, Chang SC, Staehelin LA (2008) Arabidopsis protein disulfide isomerase-5 inhibits cysteine proteases during trafficking to vacuoles before programmed cell death of the endothelium in developing seeds. Plant Cell 20(8):2205–2220. https://doi.org/10.1105/tpc.108.058339
Opalińska M, Parys K, Janska H (2017) Identification of physiological substrates and binding partners of the plant mitochondrial protease FTSH4 by the trapping approach. Int J Mol Sci 18:2455. https://doi.org/10.3390/ijms18112455
Otegui MS (2017) Vacuolar degradation of chloroplast components: autophagy and beyond. J Exp Bot 69(4):741–750. https://doi.org/10.1093/jxb/erx234
Otegui MS, Noh YS, Martinez DE, Vila Petroff MG, Staehelin LA, Amasino RM, Guiamet JJ (2005) Senescence-associated vacuoles with intense proteolytic activity develop in leaves of Arabidopsis and soybean. Plant J 41(6):831–844. https://doi.org/10.1111/j.1365-313X.2005.02346.x
Pan R, Hu J (2018) Proteome of plant peroxisomes. In: del Río LA, Schrader M (eds) Proteomics of peroxisomes: identifying novel functions and regulatory networks. Springer, Singapore, pp 3–45. https://doi.org/10.1007/978-981-13-2233-4_1
Pastor-Cantizano N, Ko DK, Angelos E, Pu Y, Brandizzi F (2020) Functional diversification of ER stress responses in Arabidopsis. Trends Biochem Sci 45(2):123–136. https://doi.org/10.1016/j.tibs.2019.10.008
Peng X, Sun M-X (2018) The suspensor as a model system to study the mechanism of cell fate specification during early embryogenesis. Plant Reprod 31(1):59–65. https://doi.org/10.1007/s00497-018-0326-5
Pérez-Pérez ME, Lemaire SD, Crespo JL (2021) The ATG4 protease integrates redox and stress signals to regulate autophagy. J Exp Bot. https://doi.org/10.1093/jxb/erab063
Petereit J, Duncan O, Murcha MW, Fenske R, Cincu E, Cahn J, Pružinská A, Ivanova A, Kollipara L, Wortelkamp S, Sickmann A, Lee J, Lister R, Millar AH, Huang S (2020) Mitochondrial CLPP2 assists coordination and homeostasis of respiratory complexes. Plant Physiol 184(1):148. https://doi.org/10.1104/pp.20.00136
Phan HA, Iacuone S, Li SF, Parish RW (2011) The MYB80 transcription factor is required for pollen development and the regulation of tapetal programmed cell death in Arabidopsis thaliana. Plant Cell 23(6):2209–2224. https://doi.org/10.1105/tpc.110.082651
Piatkov K, Vu T, Hwang C-S, Varshavsky A (2015) Formyl-methionine as a degradation signal at the N-termini of bacterial proteins. Microbial Cell 2:376–393. https://doi.org/10.15698/mic2015.10.231
Pružinská A, Shindo T, Niessen S, Kaschani F, Tóth R, Millar AH, van der Hoorn RAL (2017) Major Cys protease activities are not essential for senescence in individually darkened Arabidopsis leaves. BMC Plant Biol 17(1):4. https://doi.org/10.1186/s12870-016-0955-5
Qian D, Chen G, Tian L, Qu LQ (2018) OsDER1 is an ER-associated protein degradation factor that responds to ER stress. Plant Physiol 178(1):402–412. https://doi.org/10.1104/pp.18.00375
Quan S, Switzenberg R, Reumann S, Hu J (2010) In vivo subcellular targeting analysis validates a novel peroxisome targeting signal type 2 and the peroxisomal localization of two proteins with putative functions in defense in Arabidopsis. Plant Signal Behav 5(2):151–153. https://doi.org/10.4161/psb.5.2.10412
Quan S, Yang P, Cassin-Ross G, Kaur N, Switzenberg R, Aung K, Li J, Hu J (2013) Proteome analysis of peroxisomes from etiolated Arabidopsis seedlings identifies a peroxisomal protease involved in β-oxidation and development. Plant Physiol 163(4):1518–1538. https://doi.org/10.1104/pp.113.223453
Rajendran P, Alzahrani AM, Hanieh HN, Kumar SA, Ben Ammar R, Rengarajan T, Alhoot MA (2019) Autophagy and senescence: a new insight in selected human diseases. J Cell Physiol 234(12):21485–21492. https://doi.org/10.1002/jcp.28895
Rawlings ND (2013) Protease families, evolution and mechanism of action. In: Brix K, Stöcker W (eds) Proteases: structure and function. Springer, Vienna, pp 1–36. https://doi.org/10.1007/978-3-7091-0885-7_1
Reza S, Delhomme N, Street N, Ramachandran P, Dalman K, Nilsson O, Minina A, Bozhkov P (2018) Transcriptome analysis of embryonic domains in Norway spruce reveals potential regulators of suspensor cell death. Plos One 13:e0192945. https://doi.org/10.1371/journal.pone.0192945
Rigas S, Daras G, Laxa M, Marathias N, Fasseas C, Sweetlove L, Hatzopoulos P (2009) Role of Lon1 protease in post-germinative growth and maintenance of mitochondrial function in Arabidopsis. New Phytol 181:588–600. https://doi.org/10.1111/j.1469-8137.2008.02701.x
Roberts IN, Caputo C, Kade M, Criado MV, Barneix AJ (2011) Subtilisin-like serine proteases involved in N remobilization during grain filling in wheat. Acta Physiol Plant 33(5):1997–2001. https://doi.org/10.1007/s11738-011-0712-1
Roeder AHK, Cunha A, Ohno CK, Meyerowitz EM (2012) Cell cycle regulates cell type in the Arabidopsis sepal. Development 139(23):4416. https://doi.org/10.1242/dev.082925
Rowland E, Kim J, Bhuiyan NH, van Wijk KJ (2015) The Arabidopsis chloroplast stromal N-terminome: complexities of amino-terminal protein maturation and stability. Plant Physiol 169(3):1881–1896. https://doi.org/10.1104/pp.15.01214
Sadanandom A, Bailey M, Ewan R, Lee J, Nelis S (2012) The ubiquitin-proteasome system: central modifier of plant signalling. New Phytol 196(1):13–28. https://doi.org/10.1111/j.1469-8137.2012.04266.x
Salanenka Y, Verstraeten I, Lofke C, Tabata K, Naramoto S, Glanc M, Friml J (2018) Gibberellin DELLA signaling targets the retromer complex to redirect protein trafficking to the plasma membrane. Proc Natl Acad Sci U S A 115(14):3716–3721. https://doi.org/10.1073/pnas.1721760115
Sanmartin M, Jaroszewski L, Raikhel N, Rojo E (2005) Caspases. Regulating death since the origin of life. Plant Physiol 137:841–847. https://doi.org/10.1104/pp.104.058552
Santner A, Estelle M (2007) The JAZ proteins link jasmonate perception with transcriptional changes. Plant Cell 19(12):3839–3842. https://doi.org/10.1105/tpc.107.056960
Santner A, Estelle M (2010) The ubiquitin-proteasome system regulates plant hormone signaling. Plant J 61(6):1029–1040. https://doi.org/10.1111/j.1365-313X.2010.04112.x
Sasakawa H, Sakata E, Yamaguchi Y, Komatsu M, Tatsumi K, Kominami E, Tanaka K, Kato K (2006) Solution structure and dynamics of Ufm1, a ubiquitin-fold modifier 1. Biochem Biophys Res Commun 343(1):21–26. https://doi.org/10.1016/j.bbrc.2006.02.107
Schardon K, Hohl M, Graff L, Pfannstiel J, Schulze W, Stintzi A, Schaller A (2016) Precursor processing for plant peptide hormone maturation by subtilisin-like serine proteinases. Science 354(6319):1594–1597. https://doi.org/10.1126/science.aai8550
Schmaler T, Dubiel W (2010) Control of deneddylation by the COP9 signalosome. Subcell Biochem 54:57–68. https://doi.org/10.1007/978-1-4419-6676-6_5
Schuhmann H, Huesgen PF, Gietl C, Adamska I (2008) The DEG15 serine protease cleaves peroxisomal targeting signal 2-containing proteins in Arabidopsis. Plant Physiol 148(4):1847. https://doi.org/10.1104/pp.108.125377
Schuhmann H, Huesgen PF, Adamska I (2012) The family of Deg/HtrA proteases in plants. BMC Plant Biol 12:52–52. https://doi.org/10.1186/1471-2229-12-52
Schwechheimer C (2018) NEDD8-its role in the regulation of Cullin-RING ligases. Curr Opin Plant Biol 45(Pt A):112–119. https://doi.org/10.1016/j.pbi.2018.05.017
Seo E, Woo J, Park E, Bertolani SJ, Siegel JB, Choi D, Dinesh-Kumar SP (2016) Comparative analyses of ubiquitin-like ATG8 and cysteine protease ATG4 autophagy genes in the plant lineage and cross-kingdom processing of ATG8 by ATG4. Autophagy 12(11):2054–2068. https://doi.org/10.1080/15548627.2016.1217373
Sera Y, Hanamata S, Sakamoto S, Ono S, Kaneko K, Mitsui Y, Koyano T, Fujita N, Sasou A, Masumura T, Saji H, Nonomura K-I, Mitsuda N, Mitsui T, Kurusu T, Kuchitsu K (2019) Essential roles of autophagy in metabolic regulation in endosperm development during rice seed maturation. Sci Rep 9(1):18544. https://doi.org/10.1038/s41598-019-54361-1
Shi C-L, von Wangenheim D, Herrmann U, Wildhagen M, Kulik I, Kopf A, Ishida T, Olsson V, Anker MK, Albert M, Butenko MA, Felix G, Sawa S, Claassen M, Friml J, Aalen RB (2018) The dynamics of root cap sloughing in Arabidopsis is regulated by peptide signalling. Nat Plants 4:596–604. https://doi.org/10.1038/s41477-018-0212-z
Shibata M, Oikawa K, Yoshimoto K, Kondo M, Mano S, Yamada K, Hayashi M, Sakamoto W, Ohsumi Y, Nishimura M (2013) Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 25(12):4967. https://doi.org/10.1105/tpc.113.116947
Solheim C, Li L, Hatzopoulos P, Millar AH (2012) Loss of Lon1 in Arabidopsis changes the mitochondrial proteome leading to altered metabolite profiles and growth retardation without an accumulation of oxidative damage. Plant Physiol 160(3):1187. https://doi.org/10.1104/pp.112.203711
Son GH, Park BS, Song JT, Seo HS (2013) FLC-mediated flowering repression is positively regulated by sumoylation. J Exp Bot 65(1):339–351. https://doi.org/10.1093/jxb/ert383
Spitzer C, Li F, Buono R, Roschzttardtz H, Chung T, Zhang M, Osteryoung KW, Vierstra RD, Otegui MS (2015) The endosomal protein CHARGED MULTIVESICULAR BODY PROTEIN1 regulates the autophagic turnover of plastids in Arabidopsis. Plant Cell 27(2):391–402. https://doi.org/10.1105/tpc.114.135939
Spoel SH, Mou Z, Tada Y, Spivey NW, Genschik P, Dong X (2009) Proteasome-mediated turnover of the transcription coactivator NPR1 plays dual roles in regulating plant immunity. Cell 137(5):860–872. https://doi.org/10.1016/j.cell.2009.03.038
Srivastava AK, Zhang C, Yates G, Bailey M, Brown A, Sadanandom A (2016) SUMO is a critical regulator of salt stress responses in rice. Plant Physiol 170(4):2378–2391. https://doi.org/10.1104/pp.15.01530
Stephani M, Picchianti L, Gajic A, Beveridge R, Skarwan E, Sanchez de Medina Hernandez V, Mohseni A, Clavel M, Zeng Y, Naumann C, Matuszkiewicz M, Turco E, Loefke C, Li B, Dürnberger G, Schutzbier M, Chen HT, Abdrakhmanov A, Savova A, Chia K-S, Djamei A, Schaffner I, Abel S, Jiang L, Mechtler K, Ikeda F, Martens S, Clausen T, Dagdas Y (2020) A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. Elife 9:e58396. https://doi.org/10.7554/eLife.58396
Strader LC, Bartel B (2008) A new path to auxin. Nat Chem Biol 4(6):337–339. https://doi.org/10.1038/nchembio0608-337
Strader LC, Bartel B (2011) Transport and metabolism of the endogenous auxin precursor indole-3-butyric acid. Mol Plant 4(3):477–486. https://doi.org/10.1093/mp/ssr006
Stührwohldt N, Scholl S, Lang L, Katzenberger J, Schumacher K, Schaller A (2020) The biogenesis of CLEL peptides involves several processing events in consecutive compartments of the secretory pathway. Elife 9:e55580. https://doi.org/10.7554/eLife.55580
Thompson AR, Doelling JH, Suttangkakul A, Vierstra RD (2005) Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol 138(4):2097. https://doi.org/10.1104/pp.105.060673
Tornkvist A, Liu C, Moschou P (2019) Proteolysis and nitrogen: emerging insights. J Exp Bot. https://doi.org/10.1093/jxb/erz024
Tsiatsiani L, Heck AJ (2015) Proteomics beyond trypsin. FEBS J 282(14):2612–2626. https://doi.org/10.1111/febs.13287
Tsiatsiani L, Van Breusegem F, Gallois P, Zavialov A, Lam E, Bozhkov PV (2011) Metacaspases. Cell Death Differ 18(8):1279–1288. https://doi.org/10.1038/cdd.2011.66
Tsiatsiani L, Gevaert K, Van Breusegem F (2012) Natural substrates of plant proteases: how can protease degradomics extend our knowledge? Physiol Plant 145(1):28–40. https://doi.org/10.1111/j.1399-3054.2011.01534.x
Tsiatsiani L, Timmerman E, De Bock PJ, Vercammen D, Stael S, van de Cotte B, Staes A, Goethals M, Beunens T, Van Damme P, Gevaert K, Van Breusegem F (2013) The Arabidopsis metacaspase9 degradome. Plant Cell 25(8):2831–2847. https://doi.org/10.1105/tpc.113.115287
Van De Velde K, Ruelens P, Geuten K, Rohde A, Van Der Straeten D (2017) Exploiting DELLA signaling in cereals. Trends Plant Sci 22(10):880–893. https://doi.org/10.1016/j.tplants.2017.07.010
van der Hoorn RAL (2008) Plant proteases: from phenotypes to molecular mechanisms. Annu Rev Plant Biol 59(1):191–223. https://doi.org/10.1146/annurev.arplant.59.032607.092835
van der Hoorn RA, Kaiser M (2012) Probes for activity-based profiling of plant proteases. Physiol Plant 145(1):18–27. https://doi.org/10.1111/j.1399-3054.2011.01528.x
van Wijk KJ (2015) Protein maturation and proteolysis in plant plastids, mitochondria, and peroxisomes. Annu Rev Plant Biol 66(1):75–111. https://doi.org/10.1146/annurev-arplant-043014-115547
Varland S, Osberg C, Arnesen T (2015) N-terminal modifications of cellular proteins: the enzymes involved, their substrate specificities and biological effects. Proteomics 15(14):2385–2401. https://doi.org/10.1002/pmic.201400619
Vartapetian AC, Nina, Galiullina R, Mochalova L, Trusova S, Sobri Z, Gallois P (2016) Arabidopsis thaliana phytaspase: identification and peculiar properties. Funct Plant Biol
Vicente J, Mendiondo GM, Movahedi M, Peirats-Llobet M, Juan YT, Shen YY, Dambire C, Smart K, Rodriguez PL, Charng YY, Gray JE, Holdsworth MJ (2017) The Cys-Arg/N-end rule pathway is a general sensor of abiotic stress in flowering plants. Curr Biol 27(20):3183–3190.e3184. https://doi.org/10.1016/j.cub.2017.09.006
Vitale M, Bakunts A, Orsi A, Lari F, Tadè L, Danieli A, Rato C, Valetti C, Sitia R, Raimondi A, Christianson JC, van Anken E (2019) Inadequate BiP availability defines endoplasmic reticulum stress. Elife 8:e41168. https://doi.org/10.7554/eLife.41168
Vizovišek M, Vidmar R, Drag M, Fonović M, Salvesen GS, Turk B (2018) Protease specificity: towards in vivo imaging applications and biomarker discovery. Trends Biochem Sci 43(10):829–844. https://doi.org/10.1016/j.tibs.2018.07.003
Wang F, Zhu D, Huang X, Li S, Gong Y, Yao Q, Fu X, Fan L-M, Deng XW (2009) Biochemical insights on degradation of Arabidopsis DELLA proteins gained from a cell-free assay system. Plant Cell 21(8):2378–2390. https://doi.org/10.1105/tpc.108.065433
Wang Y, Wang W, Cai J, Zhang Y, Qin G, Tian S (2014) Tomato nuclear proteome reveals the involvement of specific E2 ubiquitin-conjugating enzymes in fruit ripening. Genome Biol 15(12):548. https://doi.org/10.1186/s13059-014-0548-2
Wang L, Cai X, Xing J, Liu C, Hendy A, Chen X-L (2019) URM1-mediated ubiquitin-like modification is required for oxidative stress adaptation during infection of the rice blast fungus. Front Microbiol 10:2039–2039. https://doi.org/10.3389/fmicb.2019.02039
Weits DA, Kunkowska AB, Kamps NCW, Portz KMS, Packbier NK, Nemec Venza Z, Gaillochet C, Lohmann JU, Pedersen O, van Dongen JT, Licausi F (2019) An apical hypoxic niche sets the pace of shoot meristem activity. Nature 569(7758):714–717. https://doi.org/10.1038/s41586-019-1203-6
Wen X, Zhang CL, Ji YS, Zhao Q, He WR, An FY, Jiang LW, Guo HW (2012) Activation of ethylene signaling is mediated by nuclear translocation of the cleaved EIN2 carboxyl terminus. Cell Res 22(11):1613–1616. https://doi.org/10.1038/cr.2012.145
White MD, Klecker M, Hopkinson RJ, Weits DA, Mueller C, Naumann C, O’Neill R, Wickens J, Yang J, Brooks-Bartlett JC, Garman EF, Grossmann TN, Dissmeyer N, Flashman E (2017) Plant cysteine oxidases are dioxygenases that directly enable arginyl transferase-catalysed arginylation of N-end rule targets. Nat Commun 8:14690. https://doi.org/10.1038/ncomms14690
Willems P, Ndah E, Jonckheere V, Stael S, Sticker A, Martens L, Van Breusegem F, Gevaert K, Van Damme P (2017) N-terminal proteomics assisted profiling of the unexplored translation initiation landscape in Arabidopsis thaliana. Mol Cell Proteomics 16(6):1064–1080. https://doi.org/10.1074/mcp.M116.066662
Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, Roelandt R, De Rycke R, Verspurten J, Declercq W, Agostinis P, Vanden Berghe T, Lippens S, Vandenabeele P (2010) Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis 1(1):e18. https://doi.org/10.1038/cddis.2009.16
Wrzaczek M, Vainonen JP, Stael S, Tsiatsiani L, Help-Rinta-Rahko H, Gauthier A, Kaufholdt D, Bollhöner B, Lamminmäki A, Staes A, Gevaert K, Tuominen H, Van Breusegem F, Helariutta Y, Kangasjärvi J (2015) GRIM REAPER peptide binds to receptor kinase PRK5 to trigger cell death in Arabidopsis. EMBO J 34(1):55–66. https://doi.org/10.15252/embj.201488582
Wu J, Michaeli S, Picchianti L, Dagdas Y, Galili G, Peled-Zehavi H (2021) ATI1 (ATG8-interacting protein 1) and ATI2 define a plant starvation-induced reticulophagy pathway and serve as MSBP1/MAPR5 cargo receptors. Autophagy:1–14. https://doi.org/10.1080/15548627.2021.1872886
Xie Q, Tzfadia O, Levy M, Weithorn E, Peled-Zehavi H, Van Parys T, Van de Peer Y, Galili G (2016) hfAIM: a reliable bioinformatics approach for in silico genome-wide identification of autophagy-associated Atg8-interacting motifs in various organisms. Autophagy 12(5):876–887. https://doi.org/10.1080/15548627.2016.1147668
Xu F, Huang Y, Li L, Gannon P, Linster E, Huber M, Kapos P, Bienvenut W, Polevoda B, Meinnel T, Hell R, Giglione C, Zhang Y, Wirtz M, Chen S, Li X (2015) Two N-terminal acetyltransferases antagonistically regulate the stability of a nod-like receptor in Arabidopsis. Plant Cell 27(5):1547–1562. https://doi.org/10.1105/tpc.15.00173
Yamasaki A, Alam JM, Noshiro D, Hirata E, Fujioka Y, Suzuki K, Ohsumi Y, Noda NN (2020) Liquidity is a critical determinant for selective autophagy of protein condensates. Mol Cell 77(6):1163–1175 e1169. https://doi.org/10.1016/j.molcel.2019.12.026
Yamauchi S, Mano S, Oikawa K, Hikino K, Teshima KM, Kimori Y, Nishimura M, Shimazaki K-I, Takemiya A (2019) Autophagy controls reactive oxygen species homeostasis in guard cells that is essential for stomatal opening. Proc Natl Acad Sci:201910886. https://doi.org/10.1073/pnas.1910886116
Yang X, Boateng KA, Strittmatter L, Burgess R, Makaroff CA (2009) Arabidopsis separase functions beyond the removal of sister chromatid cohesion during meiosis. Plant Physiol 151(1):323–333. https://doi.org/10.1104/pp.109.140699
Yang J, Zhao X, Cheng K, Du H, Ouyang Y, Chen J, Qiu S, Huang J, Jiang Y, Jiang L, Ding J, Wang J, Xu C, Li X, Zhang Q (2012) A killer-protector system regulates both hybrid sterility and segregation distortion in rice. Science 337(6100):1336–1340. https://doi.org/10.1126/science.1223702
Yates G, Srivastava A, Orosa B, Sadanandom A (2016a) Expression, purification, and enzymatic analysis of plant SUMO proteases. Methods Mol Biol 1450:125–133. https://doi.org/10.1007/978-1-4939-3759-2_10
Yates G, Srivastava AK, Sadanandom A (2016b) SUMO proteases: uncovering the roles of deSUMOylation in plants. J Exp Bot 67(9):2541–2548. https://doi.org/10.1093/jxb/erw092
Yoshimoto K, Hanaoka H, Sato S, Kato T, Tabata S, Noda T, Ohsumi Y (2004) Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell 16(11):2967–2983. https://doi.org/10.1105/tpc.104.025395
Yoshimoto K, Shibata M, Kondo M, Kazusato O, Sato M, Toyooka K, Shirasu K, Nishimura M, Ohsumi Y (2014) Quality control of plant peroxisomes in organ specific manner via autophagy. J Cell Sci 127. https://doi.org/10.1242/jcs.139709
Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU (2006) Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 8(10):1124–1132. https://doi.org/10.1038/ncb1482
Zess EK, Jensen C, Cruz-Mireles N, De la Concepcion JC, Sklenar J, Stephani M, Imre R, Roitinger E, Hughes R, Belhaj K, Mechtler K, Menke FLH, Bozkurt T, Banfield MJ, Kamoun S, Maqbool A, Dagdas YF (2019) N-terminal β-strand underpins biochemical specialization of an ATG8 isoform. PLoS Biol 17(7):e3000373. https://doi.org/10.1371/journal.pbio.3000373
Zhang S, Wu J, Yuan D, Zhang D, Huang Z, Xiao L, Yang C (2014) Perturbation of auxin homeostasis caused by mitochondrial FtSH4 gene-mediated peroxidase accumulation regulates arabidopsis architecture. Mol Plant 7(5):856–873. https://doi.org/10.1093/mp/ssu006
Zhang H, Gannon L, Hassall KL, Deery MJ, Gibbs DJ, Holdsworth MJ, van der Hoorn RAL, Lilley KS, Theodoulou FL (2017a) N-terminomics reveals control of Arabidopsis seed storage proteins and proteases by the Arg/N-end rule pathway. New Phytol. https://doi.org/10.1111/nph.14909
Zhang T, Tan P, Wang L, Jin N, Li Y, Zhang L, Yang H, Hu Z, Zhang L, Hu C, Li C, Qian K, Zhang C, Huang Y, Li K, Lin H, Wang D (2017b) RNALocate: a resource for RNA subcellular localizations. Nucleic Acids Res 45(D1):D135–D138. https://doi.org/10.1093/nar/gkw728
Zhao H, Zhang H, Cui P, Ding F, Wang G, Li R, Jenks MA, Lü S, Xiong L (2014) The putative E3 ubiquitin ligase ECERIFERUM9 regulates abscisic acid biosynthesis and response during seed germination and postgermination growth in Arabidopsis. Plant Physiol 165:1255–1268
Zhou W, Lozano-Torres JL, Blilou I, Zhang X, Zhai Q, Smant G, Li C, Scheres B (2019) A Jasmonate signaling network activates root stem cells and promotes regeneration. Cell 177(4):942–956.e914. https://doi.org/10.1016/j.cell.2019.03.006
Zhuang X, Chung KP, Cui Y, Lin W, Gao C, Kang B-H, Jiang L (2017) ATG9 regulates autophagosome progression from the endoplasmic reticulum in Arabidopsis. Proc Natl Acad Sci 114(3):E426. https://doi.org/10.1073/pnas.1616299114
Zolman BK, Martinez N, Millius A, Adham AR, Bartel B (2008) Identification and characterization of Arabidopsis indole-3-butyric acid response mutants defective in novel peroxisomal enzymes. Genetics 180(1):237–251. https://doi.org/10.1534/genetics.108.090399
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
Communicated by Francisco M. Cánovas
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hatzianestis, I.H., Mentzelopoulou, A., Moschou, P.N. (2021). Plant Proteolysis in Development: Insights and Functions. In: Lüttge, U., Cánovas, F.M., Risueño, MC., Leuschner, C., Pretzsch, H. (eds) Progress in Botany Vol. 83. Progress in Botany, vol 83. Springer, Cham. https://doi.org/10.1007/124_2021_54
Download citation
DOI: https://doi.org/10.1007/124_2021_54
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12781-6
Online ISBN: 978-3-031-12782-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)