
Abstract
We now have a robust knowledge base for a role for oxidative stress and reactive oxygen and nitrogen species in the pathogenesis of kidney failure and disease. This has developed from recent advances in research, particularly from the use of new markers of oxidative stress in urine, serum, and tissue samples, in conjunction with kidney functional and structural deficit. While experimental models have, in general, been more successful in defining a role for oxidative stress in diseases of the kidney than human investigations, a variety of clinical trials has now also supported the data gained experimentally through the greatly improved diagnostic techniques. Kidney failure is the final limiting outcome of most kidney diseases and can occur short term, over days to weeks (acute kidney injury), or more insidiously over a long time (chronic kidney disease and end-stage kidney failure). A unifying hypothesis for development of these diseases is mitochondrial dysfunction, excess production of damaging reactive species, and a deficit in natural antioxidant activity. Although results from experimental models indicate benefit of boosting our natural cellular antioxidant defenses with antioxidant therapies, to date, these therapies per se have generally been disappointing in delivering renoprotection in humans. This chapter serves to address the general classifications of acute kidney injury and chronic kidney disease; indicate the function of the reactive species, particularly reactive oxygen species, in the development and progression of the diseases; present some examples of experimental and clinical studies on oxidative stress in kidney disease; and highlight some of the clinical benefits, but also the uncertainties, of antioxidant therapies.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
Oxidative stress, or the imbalance between excess free radicals produced by damaged cells in disease and the level of natural antioxidants that our cells have to combat the free radicals, is a process common to many diseases, including kidney diseases. Increased bioavailability of reactive oxygen species (ROS) occurs, primarily from the excess ROS generation or reduced antioxidant capacity in the damaged kidney. The kidney has the potential to develop many diseases, but the danger zone is reached for patients when kidneys fail. Kidney failure occurs in two general modes: acute or short term and chronic or long term. Both modes involve structural damage and deterioration of kidney function. Acute failure of the kidney occurs rapidly over a period of days to weeks and involves, in addition to a functional deficit, a complex set of structural changes that are now referred to as acute kidney injury (AKI) (Bellomo et al. 2012). The use of “AKI” reflects two basic changes in terminology: for ease of understanding the adjective “renal” is now often replaced with “kidney,” and the term “injury” reflects the complexity of the structural change previously and incorrectly referred to as “acute tubular necrosis.” AKI refers strictly to the injury associated with acute renal failure and not to the failure itself, but this term is now preferred for the acute disease syndrome. Chronic renal failure is now more often termed “chronic kidney disease” (CKD), a term that encompasses the continuum of change between normal kidney function and terminal kidney failure. CKD involves kidney damage and functional deterioration occurring over a longer period of time than AKI, from months to years. It has many causes, one being progression from incomplete healing after AKI, but the two most common in today’s society are probably diabetes mellitus (DM) and hypertension.
Unfortunately, even after years of research into processes, or mechanisms, involved in AKI and CKD, their complexity has meant that progress has been slow, or sometimes lacking, in finding realistic therapies that successfully treat each disease. The development of acute and chronic kidney dysfunction and the incidence of complications associated with the diseases are promoted by many factors, including cell death, inflammation, and their mediators. All have links with oxidative stress (Descamps-Latscha et al. 2001; Karamouzis et al. 2008). While it is well recognized that oxidative stress is unlikely to be the sole driver of AKI or CKD, as part of a complex cascade with multiple feedback loops and impacts, oxidative stress may be a promising target for therapeutic intervention (Koyner et al. 2008; Small et al. 2012). The integrated computational and experimental approaches of systems biology also offer an exciting means of identifying new, critical, regulators of the diseases and potential targets for therapy. This chapter briefly defines the syndromes of AKI and CKD, explains oxidative stress and indicates its role in the pathogenesis of AKI and CKD, discusses some examples of experimental and clinical studies on oxidative stress in kidney disease, and highlights some of the clinical benefits, but also the uncertainties, of antioxidant therapies. Note that referencing of original articles is minimal, but readers may find relevant original articles in the referenced overview articles presented within the text of this chapter.
Acute Kidney Injury
AKI is a condition, or syndrome, where kidney function rapidly becomes impaired or ceases, with a correlative and usually marked decrease in urinary output (termed oliguria or “production of a small amount of urine”). Glomerular filtration rate (GFR) decreases. Because kidney filtration is impaired and urine production decreases, waste products build up in the blood. Serum (or plasma) creatinine is one such waste product and is most commonly used to define AKI. For example, the AKI Network (AKIN) definition of AKI (Mehta et al. 2007) depends on one of the following:
-
An increase in serum creatinine by ≥0.3 mg/dL (≥26.5 μmol/L) within 48h
-
An increase in serum creatinine to ≥1.5 times baseline within the previous 7 days
-
Urine volume ≤0.5 mL/kg/h for 6h
In addition, the Kidney Disease: Improved Global Outcomes (KDIGO) Clinical Practice Guidelines suggest AKI should be staged according to severity (stages 1–3 depending on serum creatinine and urine output; see http://kdigo.org/clinical_practice_guidelines/AKI.php) (Khwaja 2012). Other clinically measurable characteristics of AKI are increased blood urea nitrogen (BUN), proteinuria, decreased urine osmolality, increased urinary sodium (the failed kidney cannot reabsorb sodium), increased serum phosphate (kidneys usually excrete phosphate), and decreased serum calcium (because serum phosphate is increased). However, the definitions of AKI and the KDIGO and AKIN classifications still rely heavily on elevated serum creatinine in patients, and this takes time to increase in the blood.
Urine or blood biomarkers (naturally occurring molecules or genes that characterize diseases) of AKI, that can be measured before serum creatinine is increased, are now being researched. Several urinary biomarker candidates (kidney injury molecule-1/KIM-1, albumin, total protein, β2-microglobulin, cystatin C, clusterin, and trefoil factor-3) were approved by the US Food and Drug Administration and the European Medicines Agency as markers of acute rodent kidney toxicity (Dieterle et al. 2010). In humans, urinary KIM-1 (Bonventre 2008) and neutrophil gelatinase-associated lipocalin (NGAL) (Haase et al. 2009) are rapidly finding favor to indicate the early presence of kidney failure and AKI. General usage of the biomarkers is increasing and other reliable ones are in investigation (Fassett et al. 2011).
AKI may be subclassified by the point of failure, determined by the flow of fluid to and through the kidney: “prerenal” occurs from hypotension, hypovolemia, shock, or reduced cardiac output; “intrarenal” is within the kidney (e.g., from drugs, toxins, immune responses, infection and sepsis, radiation, local lack of blood flow, glomerulopathy, vasculopathy); and “postrenal” occurs from blockage of urine flow at the ureter, bladder, or urethra. AKI is a very significant clinical problem and is a common, often fatal, complication of patients entering the intensive care units of hospitals where it has a mortality rate of approximately 30–80 % (Coca et al. 2009). The structural damage of AKI is typified by repairable cell injury but also cell death (apoptosis, necrosis, autophagic cell death). Interstitial cell activation and inflammation, and endothelial injury are also common (Bonventre and Yang 2011). The tubular epithelium usually regenerates over time, and treatment is designed to maintain the patient until the acute lesion is healed and normal kidney function resumes. Fluid intake and diet are regulated, and dialysis may be needed. The oliguric phase can last approximately 10 days to 3 weeks and is often followed by a diuretic phase where there is excessive water loss and subsequent change in electrolyte balance, changes that may also need to be treated.
Chronic Kidney Disease
CKD is one of the most common chronic diseases in OECD (Organization for Economic Cooperation and Development) countries and is often the precursor to end-stage kidney disease (ESKD), the stage at which patients rely on kidney replacement therapies to survive. Since the introduction of angiotensin-converting enzyme inhibitors in the early 1980s and angiotensin-II receptor blockers in the mid-1990s as antihypertensive therapies, pharmacologic blockade of the renin-angiotensin-aldosterone system has become one of the most widespread therapeutic approaches in the management of CKD and associated cardiovascular disease (Hoogwerf 2010). However, the incidence of CKD continues to increase, indicating contributing mechanisms beyond the renin-angiotensin-aldosterone system. CKD has many causes: systemic causes, for example, hypertension or DM; obesity in Western societies and its links with type 2 DM; the increasingly aging population in many countries; an increase in smoking especially in females; and low nephron number at birth, one of the emerging causes. Many people develop CKD after incomplete recovery from single or multiple episodes of AKI caused by, for example, ischemia-reperfusion, nephrotoxic drugs, environmental toxins, radiation or other cancer therapy, or kidney stones (Venkatachalam et al. 2010; Bedford et al. 2012). Structurally, there is residual fibrosis from the attempt by the kidney to repair, and this residual fibrosis may progress to CKD (Boor et al. 2010). However, most people with diagnosed CKD having no knowledge of a primary insult.
The term CKD represents a continuum of chronic change in the kidney (Fig. 113.1) (Levy et al. 2009; Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group 2013). Histological features that might have originally indicated the nature of the primary insult are overwhelmed by nonspecific tissue changes of chronic progression, including chronic inflammation, tubulointerstitial fibrosis, glomerulosclerosis, and vascular rarefaction. The definition of CKD has been modified over several years and is now defined, generally, as abnormalities of kidney structure and function, present for greater than 3 months, with implications for health (Eknoyan et al. 2001; KDIGO 2013). The advocated formula for estimated GFR (eGFR) was developed by the Modification of Diet in Renal Disease (MDRD) group (Twomey and Reynolds 2006; Florkowski and Chew-Harris 2011). KDIGO 2013 recommends that dimensions of kidney function loss and kidney damage are used to categorize CKD, with cause, eGFR category, and albuminuria category mapping to different levels of CKD severity. The kidney damage involves destruction of kidney mass with loss of nephrons, apparently irreversible fibrosis, and progressive decline in GFR. From the stages of GFR and CKD in Table 113.1, one can see that CKD may not be detectable with decreased GFR until it is moderate (Stages 3a and b) or advanced (Stages 4 and 5). CKD is also closely aligned with greatly increased rates of cardiovascular disease, with CKD patients more likely to die from myocardial infarct than from kidney failure. There is a real need now to understand molecular mechanisms for early development and sustained progression of CKD and realistic modulatory factors that will decrease its incidence and slow its progression.

The complex continuum of development, progression, and complications of chronic kidney disease. This is a conceptual representation of the development of chronic kidney disease (CKD) over time. Complications of CKD develop from the many causes and include the important consideration of cardiovascular disease (CVD). Increased risk and damage to the kidney with time (large blue arrow) cause structural and functional deficit that is represented by a decreasing glomerular filtration rate (GFR). CKD progresses to kidney failure and eventually death if renal replacement therapies are not available (Adapted from Levy et al. 2009)
Oxidative Stress
If one considers the causes of AKI and CKD, each is likely to have a component of oxidative stress within its pathogenic mechanisms. As well as showing the short-term influence of oxidative stress in development of AKI, Fig. 113.2 also demonstrates the possible continual temporal contribution of oxidative stress in the pathogenesis of CKD. This next section explains the term “oxidative stress” in the context of production of ROS that are over and beyond the ability of our natural antioxidants to cope with ensuing disease development.

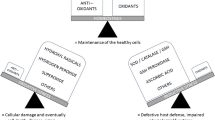
Acute kidney injury and chronic kidney disease induced by oxidative stress. Oxidative stress (lightning) participates in development and progression of acute kidney injury (AKI) and chronic kidney disease (CKD). AKI is subclassified by the point of injury to the kidney: 1. prerenal, caused by a sudden and severe drop in blood pressure (shock) or interruption in blood flow (hemorrhage, stenosis) to the kidneys; 2. intrarenal, caused by direct damage from toxins, local ischemia, inflammation, infection, and sepsis, for example; 3. postrenal, caused by sudden obstruction of urine flow from the kidney caused by kidney stones, bladder tumor, and enlarged prostate. In CKD, normal kidney parenchyma (top kidney) is gradually replaced with fibrosis in a continuum of change. The kidneys become smaller and have fewer functioning nephrons
The production of free radicals – the reactive species – is absolutely essential for normal cell energy supply, detoxification, chemical signaling, and immune function. It is the overproduction of the reactive species, via exposure to external oxidant substances, initiation of excess production of reactive species internally, or a failure in the antioxidant defense mechanisms, that damage essential biomolecules like DNA, RNA, proteins, and lipids. This is referred to as “oxidative stress.” Many of the pathological processes that occur in AKI and CKD, for example, cell death, inflammation, and/or fibrosis (Lieberthal et al. 1998; Edinger and Thompson 2004; Granata et al. 2009; Quiros et al. 2011) are induced by oxidative stress. The highly reactive ROS are capable of damaging various structures and functional pathways in the cells, whether the injury is acute or chronic. The main ROS are superoxide (O2 •−), the hydroxyl radical (OH•−), and hydrogen peroxide (H2O2). Mitochondria are considered the major source of ROS, with estimated levels of ROS within mitochondria being 5–10 times higher than cystolic and nuclear cellular compartments (Cadenas and Davies 2000). This is mostly due to the presence of the electron transport chain (ETC) within the mitochondrial inner membrane (Turrens 2003). One to three percent of inspired molecular oxygen is converted to the most common of the ROS, O2 •−, a powerful precursor of H2O2. H2O2 is stable but has the potential to interact with a variety of substrates to cause damage, especially in the presence of ferrous iron (Kurz et al. 2007), which leads to formation of the most reactive and damaging of the ROS, OH•−. In healthy cells, the production of H2O2 is countered by the catalyzing actions of mitochondrial or cystolic catalase (CAT) or thiol peroxidases into harmless water and molecular oxygen. The ETC consists of five multienzyme complexes responsible for maintaining mitochondrial membrane potential and cell energy (ATP generation). Each of these complexes presents a site of ROS generation, with complexes I and III being identified as primary sites of O2 •− generation (Raha et al. 2000).
Mitochondria possess their own pool of antioxidants to counteract their generation of ROS. The main antioxidant enzymes are superoxide dismutase (SOD), CAT, and glutathione peroxidase (GPX) (Raha et al. 2000). Mitochondrial manganese-SOD (Mn-SOD) converts O2 •− to H2O2 which is then decomposed to harmless H2O and O2 by CAT and GPX. Copper/zinc-SOD (Cu/Zn-SOD) may stabilize O2 •− within other cellular compartments, especially peroxisomes, and thus helps maintain the redox state of the whole cell. Among the various endogenous defenses against ROS, glutathione homeostasis is critical for a healthy cellular redox environment. Mitochondrial dysfunction, resulting in depleted ATP synthesis, has the potential to reduce the redox control of glutathione since the rate of glutathione synthesis is ATP dependent. Antioxidant networks in which there is cross talk and synergism to efficiently scavenge ROS may also exist, but these networks still need further definition because if they are found to be cell or tissue specific, these antioxidant networks could be harnessed to develop polytherapeutic antioxidant supplements to combat oxidant-related pathologies, like AKI and CKD.
Several key organelles may contribute to oxidative stress. The mitochondria are presumed. Damage to mitochondria causes loss of cellular energy, loss of mitochondrial membrane potential, translocation of pro-apoptotic proteins like Bax at the membrane permeability transition pore, and leakage of toxic cytochrome c from the mitochondria (Small and Gobe 2012). Increased autophagy (literally, self-eating) of dysfunctional mitochondria may occur to promote cell survival, but this can also cause cell death through autophagic cell death if too many mitochondria are removed (Edinger and Thompson 2004). Almost counterintuitively, as a result of normal cellular metabolism and oxidative phosphorylation by the respiratory chain, mitochondrial production of ROS also means these organelles are targets for ROS-mediated damage, by virtue of the proximity of the ROS they produce. Other organelles that contribute to oxidative stress are the peroxisomes, endoplasmic reticulum, Golgi apparatus, and proteasomes (Kukan 2004; Malhotra and Kaufman 2007; Schrader and Fahimi 2006; Jiang et al. 2011). It is generally accepted that the mitochondria are the most important because of their key role in cellular energy metabolism by the generation of ATP, regulation of calcium homeostasis, tissue oxygen gradients, cell death in forms of apoptosis and necrosis, and cell signaling.
Free radicals have extremely short half-lives and so are difficult to measure. Much of the basic research has been carried out in cell culture, where a cause (say, H2O2) and effect (say, cell death, cell senescence, and autophagy) are detectable. Reactive species can be measured directly by electron paramagnetic resonance or various spin-trapping methods that present some practical limitations, especially in humans, because these methods are costly, and their safety and efficiency have not been proven. Oxidative stress biomarkers are available (Halliwell and Whiteman 2004). Assays for oxidative stress or antioxidant status and some biomarkers are shown in Table 113.2. For kidney disease, the gold standard is considered to be 8-isoprostane (Small et al. 2012).
Acute Kidney Injury and Oxidative Stress
The kidney is highly energetic and therefore relies heavily on aerobic metabolism for the production of ATP by oxidative phosphorylation (Beeson et al. 2010). It is very susceptible to ischemic/hypoxic and toxic damage, some of the contributing factors being (1) the elaborate ion transport mechanisms in the kidney which lead to generation of toxic metabolites, (2) the large surface area of the tubular epithelium which then allows toxin interaction and uptake, and (3) the high kidney blood flow and the highly metabolic state of tubular cells, especially the proximal tubular epithelium that is extremely sensitive to ischemic and drug-induced damage. Ischemia-reperfusion injury, one of the most common causes of AKI, occurs from the combined effects of hypoxia (ischemia) and ROS-mediated oxidative damage (reperfusion), affecting multiple cellular components, including DNA, membrane lipids, and cellular proteins. The links between ischemic/hypoxic and toxic causes of AKI are very close, and the structural deficit is also similar (Gobe and Endre 2003). Kidney oxygenation markedly declines after administration of many nephrotoxins, for example, contrast media where normally low oxygen tension in medullary structures drops to as low as 10 mmHg after delivery of the contrast media (Heyman et al. 1999).
Various pathological processes are known to involve oxidative stress in AKI (Noiri et al. 2001), but it is likely that cell death and inflammation are the most important. The initial insult, often involving hypoxia or toxin-induced mitochondrial dysfunction and a drastic loss of ATP, initiates cell death (apoptosis, necrosis). Chemical messages are sent from the damaged cells to initiate an acute inflammatory response, initially a necessary part of healing but also able to exacerbate the injury. Highly reactive ROS are produced by dysfunctional mitochondria in the damaged cells and also by the acute inflammatory cells, and these further damage various structures and functional pathways in cells. In consequence, a recurring cycle of oxidant damage cause and effect develops that is difficult to break.
In a healthy cell or tissue, ROS are countered by endogenous natural antioxidant defenses. When damage is at a level seen in AKI, an imbalance between ROS and antioxidants tips the scales to a continuing state of oxidative stress. However, the oxidant “imbalance” theory as an explanation for the contribution of ROS to the pathogenesis of AKI is very basic. The term oxidative stress should incorporate the effects on various crucial pathways and cell metabolites that are also controlled by the interplay between oxidants and antioxidants. Upstream transcriptional gene regulation of oxidant injury is becoming increasingly recognized, and this not only provides insight into the physiological role of oxidative stress but also presents regulatory systems that are possibly prone to deregulation (Hybertson et al. 2011). The rationale for antioxidant therapies lies in restoring imbalances in the redox environment of cells and/or restoring normality to the cellular pathways. Figure 113.3 gives a diagrammatic representation of the peak in oxidative stress, and the return to normality, in AKI and the progressive buildup of oxidative damage with CKD.

Patterns of oxidative stress and antioxidant defense in AKI and CKD. In healthy cells, a balance exists between the production of reactive species and natural antioxidants (grey box). Over our life, but especially as we age, the natural antioxidant defenses decrease. In acute kidney injury (AKI) with full repair, there is a peak in reactive species and oxidative dame that is reversible. In chronic kidney disease (CKD), there is likely to be a progressive increase in oxidative stress and damage. Delivery of antioxidant therapy may modulate cell damage from the oxidant stress in AKI and CKD
The decision by kidney cells to undergo apoptosis, necrosis, or even autophagic cell death may depend on the extent or severity of the cause of AKI and also on the particular sensitivities to injury of different cell types in the heterogeneous kidney cell milieu. For example, with reference to the main zone of injury after ischemia-reperfusion injury in the kidney, the outer stripe of the outer medulla, the relative contributions of necrosis or apoptosis are different. This may depend on the severity of ischemia, with prolonged ischemia tending to cause necrosis because of the resultant negation of the cells’ energy versus partial ischemia tending to cause apoptosis, and the supersensitivity of the straight segment of the proximal tubule to ischemia-reperfusion (necrosis > apoptosis) compared with the higher cellular tolerance to ischemic conditions of the thick ascending limb (apoptosis > necrosis) (Gobe and Johnson 2007).
Selected Examples of Oxidative Stress and Its Treatment in Acute Kidney Injury
Research into the molecular basis of oxidative stress in AKI must of necessity combine basic approaches with translational science in humans, in a broad-based effort to develop new and realistic treatment strategies. Many experimental studies start at the cell culture level. The whole organ applicability of these experiments is always questioned, but they do form the basis for future whole animal experiments and human trials. Selected examples, including those indicating the benefits of antioxidant therapies, are given in the following paragraphs.
A cell culture model of ischemia-reperfusion injury in the kidney was used to define reasons for the particular sensitivities of the proximal and distal tubular epithelium to oxidant stress (Cuttle et al. 2001). These cell types were examined for cell death and related molecular pathways after treatment with H2O2. In particular, a family of pro- and antiapoptotic proteins, called the Bcl-2 family, was studied for their mitochondrial expression and localization. Damaged and dying proximal tubular cells had increased pro-apoptotic Bax, and surviving cells in the treated distal tubular epithelium showed translocation of antiapoptotic Bcl-X(L) from cytosol to the mitochondria. These results demonstrated that oxidative stress is a direct cause of cell death in the sensitive proximal tubular epithelium but also that it could initiate molecular pathways for cell survival in cells more tolerant of the stress, like the distal tubular epithelium. The potential benefit to the kidney in AKI is that the distal tubular epithelium is a source of reparative cytokines, and protection by the antiapoptotic Bcl-2 proteins may allow time for these reparative proteins to be synthesized and secreted, thereby creating a secondary protective or regenerative environment.
Two examples are given of preclinical animal models. Cisplatin is a drug often given to treat cancers, but its use is limited by its selective nephrotoxicity. Knowing if oxidative stress is a mechanism of injury may mean that antioxidant therapy, given with the cisplatin, could protect the kidneys but allow the cancers to be killed. In an animal model of cisplatin injury and AKI, diminished kidney function (increased serum creatinine and BUN) and increased tubular damage were compared with urinary and tissue 8-hydroxy-deoxyglutathione (8-OHdG) and malondialdehyde (MDA), markers of oxidative stress (Zhou et al. 2006). Increased urinary MDA correlated significantly with serum creatinine and the tubular damage score over 5 days of AKI, by which time readings were normalizing. The conclusion was that oxidative stress is a key mechanism of cisplatin nephrotoxicity, and the damaged cells could be targeted for antioxidant therapy. Mercuric chloride, a toxic environmental heavy metal with known nephrotoxicity, was investigated in a rat model. The drug melatonin, delivered prior to mercuric chloride toxicity, reduced the extent of tubular injury (Nava et al. 2000). Kidney content of MDA was decreased, and the activities of the natural antioxidants GPX and CAT were increased. Thus, the beneficial effects of pharmacological doses of melatonin were demonstrated in an environment of toxic injury in the kidney, and the benefit of the drug was likely to be due to its antioxidant properties.
Reports on human/clinical trials are less frequent than the experimental models. One important example is given here. Himmelfarb et al. (2004) investigated patients with AKI and failure, arguing that dysregulated inflammation and altered metabolism may increase oxidative stress in these patients. The patients in kidney failure had to meet the selection criteria of the Program to Improve Care in Acute Renal Disease (PICARD) study. They underwent plasma protein oxidation and plasma cytokine measurements and were compared with critically ill patients without kidney failure, patients with ESKD, and healthy subjects. Plasma protein thiol oxidation and carbonyl content were markedly different in kidney failure patients compared with healthy subjects, end-stage, and critically ill patients (P < 0.001 in all cases). Plasma protein thiol oxidation in kidney failure patients improved with dialysis. They concluded that increased oxidative stress occurred in these kidney failure patients and that this may be an important target for nutritional and pharmacologic therapy in such patients.
Chronic Kidney Disease and Oxidative Stress
Chronic diseases of the kidney possess various commonalities which can be linked through pathways controlled by oxidative stress. Vascular, cellular, and biochemical factors all contribute, and chronic inflammation is an obvious key factor (Vlassara et al. 2009). Chronic inflammation may follow acute inflammation, but in many cases of cardiovascular disease, it is likely that it begins as a low-grade response with no initial manifestation of an acute reaction. There are links between visceral obesity, increased secretion of inflammatory mediators seen in visceral fat, oxidative stress, and CKD (Tanner et al. 2012). Proinflammatory cytokines are produced by adipocytes and also cells in the adipose stroma. The links with oxidative stress as an endogenous driver of CKD become immediately obvious when one recognizes the close association between oxidative stress and inflammation. However, an improved understanding of the precise molecular mechanisms by which chronic inflammation modifies CKD is required before the full implications of its presence, including links with persistent oxidative stress, as a cause of CKD, can be realized.
Another important mechanism for CKD is the increase in serum uric acid levels (hyperuricemia) seen when kidneys fail (reviewed in Badve et al. 2011). Hyperuricemia has harmful systemic effects, not the least being development of CKD. It is also closely linked with the development of cardiovascular disease. Retention of uremic toxins promotes inflammation and oxidative stress, by priming acute inflammatory cells, like neutrophils, and activating proinflammatory and pro-fibrotic cytokines. Additionally, uric acid synthesis can promote oxidative stress directly through the activity of xanthine oxidoreductase, an enzyme that is converted to xanthine oxidase and, in doing so, loses its capacity to bind NADH and, instead, generates O2 •− (Moorhouse et al. 1987). The inhibition of xanthine oxidase forms, in part, the basis for treatment of CKD, and other oxidative stress-induced kidney diseases like AKI, with allopurinol.
The kidney is a vital source of l-arginine, a precursor for nitric oxide (NO). A reduction in kidney mass in CKD can, therefore, reduce l-arginine and subsequently NO activity, which is vital for regulation of the vascular endothelium. Decreased endothelial function has the potential to exacerbate CKD development because of vascular loss. This complex picture of cause and effect is common in CKD pathogenesis. CKD is associated with increased levels of asymmetric N(G),N(G)-dimethylarginine (ADMA), which induces endothelial dysfunction through competitive inhibition of l-arginine. ADMA may also reduce NO production via decreased phosphorylation of endothelial NO synthase. There is now also a novel link in the pathogenesis of oxidative stress-induced CKD through a functional mitochondrial angiotensin system (Abadir et al. 2011). Angiotensin type II receptors were co-localized with angiotensin on the inner mitochondrial membrane of human mononuclear cells and mouse kidney tubular cells. This system was found to modulate mitochondrial NO production and respiration, thereby contribution to stress from increased reactive species.
Cell loss by apoptosis, necrosis, and autophagic cell death and decreased ability to regenerate after damage occur in CKD. All may be controlled by mitochondrial health. Damage to the mitochondria likely plays a key pathogenic role in CKD, but the strategic points of mitochondrial regulation still need definition if they are to be targeted to modulate the disease. These points of regulation include mitochondrial fusion, fission, and motility (mitochondrial dynamics); mitochondrial turnover and degradation (biogenesis); as well as mitochondrial respiration and biochemistry. A novel consideration is that mitochondrial regulation may be determined by an individual’s genetic makeup. Hung et al. (2010) recently investigated C-reactive protein (CRP) polymorphisms and progression of CKD in African Americans. CRP is a biomarker of inflammation and is increased even in early stages of CKD. This study found that CRP polymorphisms were more common in African Americans with hypertensive CKD. The African Americans, like the Australian Aborigines and Pima Indians, have higher incidence of CKD than most Caucasian populations (Lillioja et al. 1993; Hoy et al. 2010). Genetics, and predisposition to oxidative stress, are factors that need further investigation.
Evidence for links between oxidative stress and CKD is plentiful in the literature. The ANZDATA registry (from Kidney Health Australia, www.kidney.org.au) has reported that aging (>50 years) per se has one of the closest correlations with CKD, perhaps indicating that an insidious but unnoticed accumulation of damaging oxidants with age contributes significantly to CKD development. Other evidence comes from investigations of systemic diseases such as hypertension, DM, and hypercholesterolemia; radiocontrast agents; kidney and systemic infections; use of antibiotics, chemotherapeutics, and other drugs; and environmental toxins, occupational chemicals, smoking, and alcohol consumption. All of these causes of CKD can induce oxidative stress in the kidney. In the next subsections, ROS and aging, and oxidative stress as a mechanism for development of DM, are now reviewed.
ROS and Aging: Evidence for End-Organ Damage
Increasing age has dire implications for kidney health. O’Hare et al. (2007) reported on a large national cohort of US veterans (209,622) who fulfilled criteria for stage 3 or higher CKD. They investigated the prognostic implications of eGFR for death and ESKD and found this varied greatly depending on the age of the patient. Patients aged 75 years or older at baseline comprised 47 % of the overall cohort and accounted for 28 % of the 9227 cases of ESKD seen at follow-up. Among patients of all ages, rates of death and ESKD were related inversely to eGFR at baseline, but in those with comparable levels of eGFR, older patients had higher rates of death and lower rates of ESKD than younger patients. Among those 85 years or older, the risk of death always exceeded the risk of ESKD in this cohort.
Links between oxidative stress and CKD have been demonstrated in humans with aging (Karamouzis et al. 2008). Here, a total of 116 patients with CKD (85 pre-dialysis patients divided into groups according to CKD stage and 31 patients with ESKD on hemodialysis) were compared with 29 healthy subjects. Plasma levels of 15-F(2t)-isoprostane, as well as total antioxidant capacity and serum levels of vitamin E, were measured in all participants. Plasma isoprostane levels were higher in pre-dialysis and ESKD patients compared to healthy subjects, with progressive increases with advancing CKD stages. Total antioxidant capacity was similar between healthy subjects and pre-dialysis patients, and ESKD patients had a small reduction. Vitamin E levels were higher in healthy subjects compared to any other group. Thus, the balance, as CKD progresses, swings towards increasing oxidative stress.
ROS generation from mitochondrial complexes, and/or oxidative stress, increases with age (Choksi et al. 2007; Granata et al. 2009; Percy et al. 2009). Braidy et al. (2011) have also demonstrated, in aging rats, that the cofactor NAD+, a key regulator of metabolism, stress resistance, and longevity in the kidney, plays a key role in CKD development. Apart from its role as an important redox carrier, NAD+ serves as the sole substrate for NAD-dependent enzymes, including poly(ADP-ribose) polymerase, an important DNA nick sensor, and the NAD-dependent histone deacetylases, sirtuins, which act in a wide variety of processes, including senescence, apoptosis, differentiation, and aging. Thus, there is developing evidence for increasing effects of oxidative stress in the aging kidney, probably contributing to development of CKD
Diabetes Mellitus as an Example of Oxidative Stress and Chronic Kidney Disease
The last few decades have seen an explosion in the occurrence of type 2 DM, which is characterized by dramatic pathologies in the small vessels causing nephropathy (CKD), retinopathy, and neuropathy (see cardinal studies in the Pima Indians in Lillioja et al. 1993). DM is also closely linked with cardiovascular disease. There is substantial proven data supporting a role for oxidative stress as a pathogenic mechanism for DM, mainly through induction of O2 •− by hyperglycemia (Evans et al. 2002). The pathways include increased flux through the polyol pathway, in which glucose is reduced to sorbitol, reducing levels of both NADPH and reduced glutathione; increased formation of advanced glycation end products; activation of protein kinase C with proinflammatory and pro-fibrotic outcomes; and increased shunting of excess glucose through the hexosamine pathway, mediating increased transcription of genes for inflammatory cytokines. Hyperglycemia drives excess production of electron donors (mainly NADH/H+) from the tricarboxylic acid cycle, and the excess results in transfer of single electrons to oxygen, producing O2 •− and other ROS.
The subsequent oxidative stress undoubtedly contributes to deregulated apoptosis which has been described in humans and animal models of diabetic nephropathy, which is manifested particularly in glomeruli and vascular endothelium. A unifying hypothesis for several molecular pathways involved in DM nephropathy has been the mitochondrial production of ROS in response to chronic hyperglycemia. Lifestyle modification such as exercise may attenuate diabetic nephropathy via attenuation of the mitochondrial apoptotic pathway and oxidative damage. Antioxidant therapy does hold much potential for reducing diabetic nephropathy (Golbidi et al. 2011). However, it remains a mystery as to why many antioxidant therapies produce minimal renoprotection in humans despite positive preclinical research findings.
Selected Examples of Treatment of Chronic Kidney Disease with Antioxidants
Vitamin E (α-tocopherol) has been used as an antioxidant therapy in CKD patients (Ramos et al. 2011). Vitamin E, or α-tocopherol, is a lipid-soluble antioxidant that incorporates into the plasma membrane of cells, thereby scavenging free radicals, mainly the peroxyl radical, and halting lipid peroxidation chain reactions (Kagan et al. 1990). A benefit of α-tocopherol is its ability to restore its antioxidant capacity from its oxidized form following free radical scavenging and incorporate back into the plasma membrane. Vitamin C (ascorbic acid) is able to directly reduce α-tocopherol, and intracellular glutathione and lipoic acid can restore α-tocopherol indirectly by restoring vitamin C (quantitative approach presented in Fujisawa et al. 2006). This is a prime example of a cellular antioxidant network prone to dysregulation. One of the concerns is that high-dose α-tocopherol may affect coagulation and cause increased bleeding. This has occurred in normal healthy humans due to adverse interactions between α-tocopherol and vitamin K-dependent carboxylase. There is also a lack of consensus as to whether vitamin E therapy induces an overall benefit. It is known that patients with CKD stage 4 display the largest decrease in serum α-tocopherol levels following a progressive decline from stage 1 indicating an increased need for α-tocopherol in the CKD population (Karamouzis et al. 2008). A positive correlation of serum α-tocopherol levels and GFR was found. However, vitamin E supplementation had no significant benefit. The use of α-tocopherol in CKD patients is not without controversy. Miller and colleagues (2005) concluded that high-dose (≥400 IU/day) vitamin E supplementation may increase all cause mortality which may be due to α-tocopherol displacing gamma-(γ)-tocopherol and delta-(δ)-tocopherol in the body. However, this study was highly criticized owing to a bias in data analysis and numerous methodological flaws (see July issue, Ann Intern Med 2005, for multiple responses). The results from the Selenium and Vitamin E Cancer Prevention Trial (SELECT) are of real concern. They suggest that vitamin E supplementation (here, used for cancer therapy) significantly increases the risk of prostate cancer for young healthy men (Klein et al. 2011). The apparent disconnect between benefits, or not, of vitamin E supplementation and associated kidney outcomes stems largely from differences in trial design and failure in reports to specify the form of tocopherol used.
N-acetylcysteine (NAC) is a potent cell antioxidant that can interact directly with ROS and nitrogen species because it is a scavenger of oxygen free radicals (Zafarullah et al. 2003; Renke et al. 2008). NAC also acts as an essential precursor to many endogenous antioxidants involved in the decomposition of peroxides (Zhang et al. 2011). It has shown some benefit in kidney cells under oxidative stress and has been used with mixed success in CKD patients (some original investigation reports in Renke et al. 2008; Hsu et al. 2010; Moist et al. 2010). It seems to exert the greatest antioxidant and anti-inflammatory properties when used against the greatest injury, such as in end-stage kidney disease patients receiving either hemodialysis or peritoneal dialysis. In those cases, NAC reduced gold standard biomarkers of oxidative stress, like serum 8-isoprostane.
Inflammation and fibrosis are causes, as well as consequences, of oxidative stress. Fish oils, including long-chain omega-3 fatty acids, possess a variety of anti-inflammatory properties that should lead to antioxidant secondary effects (Kim and Chung 2007). They are also known to enhance endogenous antioxidant defense systems such as γ-glutamyl-cysteinyl ligase and glutathione reductase. In vivo studies have now confirmed an improvement in kidney function and structure using EPA/DHA supplementation, with reduced oxidative stress, inflammation, and tubulointerstitial fibrosis through the reversal of inflammatory and oxidant pathways (original investigations, An et al. 2009; Peake et al. 2011). Omega-3 fatty acid treatment of peripheral blood mononuclear cells from pre-dialysis CKD patients reduced inflammatory biomarkers, like the interleukins, tumor necrosis factor-α, and C-reactive protein, to levels observed in healthy subjects (Mayer et al. 2003). Recently, a highly beneficial outcome of fish oil supplementation was found with heart failure patients with comorbid diabetes (Kazemian et al. 2012). An overview of the benefits, or otherwise, of use of omega-3 polyunsaturated fatty acids in kidney disease is given in Fassett et al. 2010.
Allopurinol, an inhibitor of xanthine oxidase, is used typically to reduce hyperuricemia in gout, but it also acts, dependent on the dose, as an antioxidant. Allopurinol and its metabolite, oxypurinol, act as competitive substrates for xanthine oxidase. They enhance urinary urate excretion and block uric acid reabsorption by urate transporters in the proximal tubule, thereby facilitating enhanced uric acid excretion (El-Sheikh et al. 2008; Riegersperger et al. 2011). Allopurinol may also have antioxidant activities in addition to its enzyme inhibitory activities, by scavenging OH•− as well as chlorine dioxide and HOCl (Moorhouse et al. 1987). Inhibition of xanthine oxidase-dependent production of ROS provides allopurinol an indirect mechanism for decreasing oxidative stress in hyperuricemic CKD patients. Interventional studies of use of allopurinol in kidney disease have shown improved uric acid levels, GFR, cardiovascular outcomes, and delayed CKD progression, although in some cases urate and C-reactive protein levels were decreased, without improvement in proteinuria (Kanbay et al. 2007; Goicoechea et al. 2010). Allopurinol can be used in patients with poor kidney function but must be used with care, and large, long-term, interventional studies investigating kidney function in the CKD populations are needed to fully determine if allopurinol is protective to the kidney via antioxidant mechanisms.
Other drugs that have been used or are currently under trial for CKD are bardoxolone methyl and coenzyme Q10 (CoQ10). With bardoxolone methyl [2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO-Me)], a different approach has been investigated, modulating pathways that respond to oxidative stress rather than targeting ROS by directly increasing endogenous antioxidants. Bardoxolone methyl is a potent activator of the Nrf2/Keap1 pathway that acts in inducing the expression of antioxidant genes and currently shows promise for halting the progressive decline of GFR in type 2 diabetic CKD patients (Pergola et al. 2011). The kidneys contain the some of the highest endogenous levels of CoQ10, likely due to the respective reliance on aerobic metabolism and high density of mitochondria in the intrinsic functioning cells from this organ (Lass and Sohal 2000; Ishikawa et al. 2011). CoQ10 is a fundamental lipid-soluble component of all cell membranes including those enclosing subcellular compartments. The physiological roles of CoQ10 act mostly within the mitochondria where it has three well-characterized functions: (1) the transfer of electrons from complexes I and II to complex III along the ETC of the inner mitochondrial membrane and subsequent membrane polarization and ATP generation, (2) the prooxidant generation of O2 •− and H2O2, and (3) the antioxidant quenching of free radicals. It is imperative that endogenous CoQ10 levels are maintained to ensure mitochondrial health, and this forms the rationale for CoQ10 therapy (James et al. 2004).
Polypharmacotherapy with Antioxidants
Different antioxidants may work together to improve whole cell and organ function through a targeted polypharmaceutical approach to decrease oxidative stress. This has been investigated in preclinical and clinical trials, with disparate results. Korish (2010) demonstrated in a 5/6 nephrectomy CKD rat model that l-arginine improved the effects of l-carnitine, catechin, and vitamins E and C on blood pressure, dyslipidemia, inflammation, and kidney function. In a murine model of diabetic nephropathy, the benefits of NAC, l-ascorbic acid (vitamin C), and α-tocopherol caused decreased lipid peroxidation, BUN, serum creatinine, and blood glucose, mainly due to a reduction in the inflammatory response induced by hyperglycemia (Park et al. 2011). Mosca et al. (2002) demonstrated that daily intake of NAC, α-tocopherol, CoQ10, and 3 other antioxidants (l-carnitine, selenomethionine, and α-lipoic acid) successfully increased plasma CAT, GPX, and total antioxidant capacity while decreasing lipid peroxides and ROS generation by lymphocyte mitochondria, but in healthy human participants. Thus, the results should not be extrapolated to the CKD population. However, a prospective trial investigating oral supplementation of mixed tocopherols and α-lipoic acid in stage 3 and 4 CKD patients revealed disappointing outcome: over 2 months, supplementation did not reduce (biomarkers of) oxidative stress (F2-isoprostanes and protein thiol concentration) or inflammation (C-reactive protein and interleukin-6) (Ramos et al. 2011). The 2 months of intervention may have been too brief, however, and longer trials need to be carried out.
Conclusion
Despite years of research, mortality and morbidity associated with AKI have not decreased substantially. CKD is a progressive disease with increasing incidence. Pharmacologic blockade of the renin-angiotensin-aldosterone system and use of statins have only been somewhat successful in treating and slowing progression of this disease. Cell death, inflammation, and fibrosis all contribute to both disease syndromes, and all have some aspect of oxidative stress in their causes or mechanisms. Although antioxidant therapies for AKI and CKD have proved successful in many preclinical trials, often these results do not translate to human disease, and it is vital for the progression of antioxidant therapy research in AKI and CKD that measures of oxidative stress are taken. These could then be compared with pathophysiological outcome, especially in connection with any antioxidant therapies that may be delivered with or without more conventional therapies. Early detection of the diseases is also necessary if application of extrinsic antioxidants that may modulate the cause of the injury and/or stimulate reparative molecules is to be successful. The signaling pathways associated with oxidative stress also need definition, for specific targeting by antioxidants. Given the complex nature of oxidative stress and its molecular pathways, antioxidants may need to be given as a polypharmacotherapy to target each aberrant pathway. In some other chronic diseases where inflammation and fibrosis participate in pathogenesis, for example, rheumatoid arthritis, combination treatment with disease-modifying drugs has been used very successfully, after monotherapy was tested and found wanting. An antioxidant polypharmacotherapy may be necessary to expedite the successful disease modification of AKI and CKD.
References
Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B, Walston JD (2011) Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci USA 108:14849–14854
An WS, Kim HJ, Cho KH, Vaziri ND (2009) Omega-3 fatty acid supplementation attenuates oxidative stress, inflammation, and tubulointerstitial fibrosis in the remnant kidney. Am J Physiol Renal Physiol 297:F895–F903
Badve SV, Brown F, Hawley CM, Johnson DW, Kanellis J, Rangan GK, Perkovic V (2011) Challenges of conducting a trial of uric-acid-lowering therapy in CKD. Nat Rev Nephrol 7:295–300
Bedford M, Farmer C, Levin A, Ali T, Stevens P (2012) Acute kidney injury and CKD: chicken or egg? Am J Kidney Dis 59:485–491
Beeson CC, Beeson GC, Schnellmann RG (2010) A high-throughput respirometric assay for mitochondrial biogenesis and toxicity. Anal Biochem 404:75–81
Bellomo R, Kellum JA, Ronco C (2012) Acute kidney injury. Lancet 380:756–766.
Bonventre JV (2008) Kidney Injury Molecule-1 (KIM-1): a specific and sensitive biomarker of kidney injury. Scand J Clin Lab Invest 241:78–83
Bonventre JV, Yang L (2011) Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121:4210–4221
Boor P, Ostendorf T, Floege J (2010) Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol 6:643–656
Braidy N, Guillemin GJ, Mansour H, Chan-Ling T, Poljak A, Grant R (2011) Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in Wistar rats. PLoS One 6:e19194. doi:10.1371/journal.pone.0019194
Cadenas E, Davies KJ (2000) Mitochondrial free radical generation, oxidative stress, and aging. Free Radic Biol Med 29:222–230
Choksi KB, Nuss JE, Boylston WH, Rabek JP, Papaconstantinou J (2007) Age-related increases in oxidatively damaged proteins of mouse kidney mitochondrial electron transport chain complexes. Free Radic Biol Med 43:1423–1438
Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR (2009) Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53:961–973
Cuttle L, Zhang XJ, Endre ZH, Winterford C, Gobe GC (2001) Bcl-X(L) translocation in renal tubular epithelial cells in vitro protects distal cells from oxidative stress. Kidney Int 59:1779–1788
Descamps-Latscha B, Drüeke T, Witko-Sarsat V (2001) Dialysis-induced oxidative stress: biological aspects, clinical consequences, and therapy. Semin Dial 14:193–199
Dieterle F, Sistare F, Goodsaid F, Papaluca M, Ozer JS, Webb CP, Baer W, Senagore A, Schipper MJ, Vonderscher J, Sultana S, Gerhold DL, Phillips JA, Maurer G, Carl K, Laurie D, Harpur E, Sonee M, Ennulat D, Holder D, Andrews-Cleavenger D, Gu YZ, Thompson KL, Goering PL, Vidal JM, Abadie E, Maciulaitis R, Jacobson-Kram D, Defelice AF, Hausner EA, Blank M, Thompson A, Harlow P, Throckmorton D, Xiao S, Xu N, Taylor W, Vamvakas S, Flamion B, Lima BS, Kasper P, Pasanen M, Prasad K, Troth S, Bounous D, Robinson-Gravatt D, Betton G, Davis MA, Akunda J, McDuffie JE, Suter L, Obert L, Guffroy M, Pinches M, Jayadev S, Blomme EA, Beushausen SA, Barlow VG, Collins N, Waring J, Honor D, Snook S, Lee J, Rossi P, Walker E, Mattes W (2010) Renal biomarker qualification submission: a dialog between the FDA-EMEA and predictive safety testing consortium. Nat Biotechnol 28:455–462
Edinger AL, Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol 16:663–669
Eknoyan G, Levin NW, Eschbach JW, Golper TA, Owen WF Jr, Schwab S, Steinberg EP, Baylor College of Medicine, Renal Research Institute, Beth Israel Hospital Center, Minor & James Medical Center, Vanderbilt University, Duke University Medical Center, Covance Health Economics and Outcomes Services, Johns Hopkins University (2001) Continuous quality improvement: DOQI becomes K/DOQI and is updated. National Kidney Foundation’s Dialysis Outcomes Quality Initiative. Am J Kidney Dis 37:179–194
El-Sheikh AA, van den Heuvel JJ, Koenderink JB, Russel FG (2008) Effect of hypouricaemic and hyperuricaemic drugs on the renal urate efflux transporter, multidrug resistance protein 4. Br J Pharmacol 155:1066–1075
Evans JL, Goldfine ID, Maddux BA, Grodsky GM (2002) Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23:599–622
Fassett RG, Gobe GC, Peake JM, Coombes JS (2010) Omega-3 polyunsaturated fatty acids in the treatment of kidney disease. Am J Kidney Dis 56:728–742
Fassett RG, Venuthurupalli SK, Gobe GC, Coombes JS, Cooper MA, Hoy WE (2011) Biomarkers in chronic kidney disease: a review. Kidney Int 80:806–821
Florkowski CM, Chew-Harris JS (2011) Methods of estimating GFR – different equations including CKD-EPI. Clin Biochem Rev 32:75–79
Fujisawa S, Ishihara M, Atsumi T, Kadoma Y (2006) A quantitative approach to the free radical interaction between alpha-tocopherol or ascorbate and flavonoids. In Vivo 20:445–452
Gobe GC, Endre ZH (2003) Cell death in toxic nephropathies. Semin Nephrol 23:416–424
Gobe GC, Johnson DW (2007) Distal tubular epithelial cells of the kidney: potential support for proximal tubular cell survival after renal injury. Int J Biochem Cell Biol 39:1551–1561
Goicoechea M, de Vinuesa SG, Verdalles U, Ruiz-Caro C, Ampuero J, Rincón A, Arroyo D, Luño J (2010) Effect of allopurinol in chronic kidney disease progression and cardiovascular risk. Clin J Am Soc Nephrol 5:1388–1393
Golbidi S, Ebadi SA, Laher I (2011) Antioxidants in the treatment of diabetes. Curr Diabetes Rev 7:106–125
Granata S, Zaza G, Simone S, Villani G, Latorre D, Pontrelli P, Carella M, Schena FP, Grandaliano G, Pertosa G (2009) Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics 10:388
Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A, NGAL Meta-analysis Investigator Group (2009) Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 54:1012–1024
Halliwell B, Whiteman M (2004) Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol 142:231–255
Heyman SN, Reichman J, Brezis M (1999) Pathophysiology of radiocontrast nephropathy: a role for medullary hypoxia. Invest Radiol 34:685–691
Himmelfarb J, McMonagle E, Freedman S, Klenzak J, McMenamin E, Le P, Pupim LB, Ikizler TA, The PICARD Group (2004) Oxidative stress is increased in critically ill patients with acute renal failure. J Am Soc Nephrol 15:2449–2456
Hoogwerf BJ (2010) Renin-angiotensin system blockade and cardiovascular and renal protection. Am J Cardiol 105(Suppl 1):30A–35A
Hoy WE, Kincaid-Smith P, Hughson MD, Fogo AB, Sinniah R, Dowling J, Samuel T, Mott SA, Douglas-Denton RN, Bertram JF (2010) CKD in aboriginal Australians. Am J Kidney Dis 56:983–993
Hsu SP, Chiang CK, Yang SY, Chien CT (2010) N-acetylcysteine for the management of anemia and oxidative stress in hemodialysis patients. Nephron Clin Pract 116:c207–c216
Hung AM, Crawford DC, Griffin MR, Brown-Gentry K, Lipkowitz MS, Siew ED, Cavanaugh K, Lewis JB, Ikizler TA, AASK Study Group (2010) CRP polymorphisms and progression of chronic kidney disease in African Americans. Clin J Am Soc Nephrol 5:24–33
Hybertson BM, Gao B, Bose SK, McCord JM (2011) Oxidative stress in health and disease: the therapeutic potential of Nrf2 activation. Mol Aspects Med 32:234–246
Ishikawa A, Kawarazaki H, Ando K, Fujita M, Fujita T, Homma Y (2011) Renal preservation effect of ubiquinol, the reduced form of coenzyme Q10. Clin Exp Nephrol 15:30–33
James AM, Smith RA, Murphy MP (2004) Antioxidant and prooxidant properties of mitochondrial Coenzyme Q. Arch Biochem Biophys 423:47–56
Jiang Z, Hu Z, Zeng L, Lu W, Zhang H, Li T, Xiao H (2011) The role of the Golgi apparatus in oxidative stress: is this organelle less significant than mitochondria? Free Radic Biol Med 50:907–917
Kagan VE, Serbinova EA, Packer L (1990) Recycling and antioxidant activity of tocopherol homologs of differing hydrocarbon chain lengths in liver microsomes. Arch Biochem Biophys 282:221–225
Kanbay M, Ozkara A, Selcoki Y, Isik B, Turgut F, Bavbek N, Uz E, Akcay A, Yigitoglu R, Covic A (2007) Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearance, and proteinuria in patients with normal renal functions. Int Urol Nephrol 39:1227–1233
Karamouzis I, Sarafidis PA, Karamouzis M, Iliadis S, Haidich AB, Sioulis A, Triantos A, Vavatsi-Christaki N, Grekas DM (2008) Increase in oxidative stress but not in antioxidant capacity with advancing stages of chronic kidney disease. Am J Nephrol 28:397–404
Kazemian P, Kazemi-Bajestani SM, Alherbish A, Steed J, Oudit GY (2012) The use of omega-3 poly-unsaturated fatty acids in heart failure: a preferential role in patients with diabetes. Cardiovasc Drugs Ther 26:311–320
Khwaja A (2012) KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract 120:c179–c184
Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group (2013) KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int 3:1–150
Kim YJ, Chung HY (2007) Antioxidative and anti-inflammatory actions of docosahexaenoic acid and eicosapentaenoic acid in renal epithelial cells and macrophages. J Med Food 10:225–231
Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, Klotz L, Parsons JK, Chin JL, Darke AK, Lippman SM, Goodman GE, Meyskens FL Jr, Baker LH (2011) Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306:1549–1556
Korish AA (2010) Multiple antioxidants and L-arginine modulate inflammation and dyslipidemia in chronic renal failure rats. Ren Fail 32:203–213
Koyner JL, Sher Ali R, Murray PT (2008) Antioxidants. Do they have a place in the prevention or therapy of acute kidney injury? Nephron Exp Nephrol 109:e109–e117
Kukan M (2004) Emerging roles of proteasomes in ischemia-reperfusion injury of organs. J Physiol Pharmacol 55(1 Pt 1):3–15
Kurz T, Terman A, Brunk UT (2007) Autophagy, ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys 462:220–230
Lass A, Sohal RS (2000) Effect of coenzyme Q(10) and alpha-tocopherol content of mitochondria on the production of superoxide anion radicals. FASEB J 14:87–94
Levy AS, Stevens LA, Coresh J (2009) Conceptual model of CKD: applications and implications. Am J Kidney Dis 53(S3):S4–S16
Lieberthal W, Koh JS, Levine JS (1998) Necrosis and apoptosis in acute renal failure. Semin Nephrol 18:505–518
Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, Bennett PH, Bogardus C (1993) Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. Prospective studies of Pima Indians. N Engl J Med 329:1988–1992
Malhotra JD, Kaufman RJ (2007) Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxid Redox Signal 9:2277–2293
Mayer K, Meyer S, Reinholz-Muhly M, Maus U, Merfels M, Lohmeyer J, Grimminger F, Seeger W (2003) Short-time infusion of fish oil-based lipid emulsions, approved for parenteral nutrition, reduces monocyte proinflammatory cytokine generation and adhesive interaction with endothelium in humans. J Immunol 171:4837–4843
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A, Acute Kidney Injury Network (2007) Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 11:R31
Miller ER 3rd, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E (2005) Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med 142:37–46
Moist L, Sontrop JM, Gallo K, Mainra R, Cutler M, Freeman D, House AA (2010) Effect of N-acetylcysteine on serum creatinine and kidney function: results of a randomized controlled trial. Am J Kidney Dis 56:643–650
Moorhouse PC, Grootveld M, Halliwell B, Quinlan JG, Gutteridge JM (1987) Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett 213:23–28
Mosca L, Marcellini S, Perluigi M, Mastroiacovo P, Moretti S, Famularo G, Peluso I, Santini G, De Simone C (2002) Modulation of apoptosis and improved redox metabolism with the use of a new antioxidant formula. Biochem Pharmacol 63:1305–1314
Nava M, Romero F, Quiroz Y, Parra G, Bonet L, Rodríguez-Iturbe B (2000) Melatonin attenuates acute renal failure and oxidative stress induced by mercuric chloride in rats. Am J Physiol Renal Physiol 279:F910–F918
Noiri E, Nakao A, Uchida K, Tsukahara H, Ohno M, Fujita T, Brodsky S, Goligorsky MS (2001) Oxidative and nitrosative stress in acute renal ischemia. Am J Physiol Renal Physiol 281:F948–F957
O’Hare AM, Choi AI, Bertenthal D, Bacchetti P, Garg AX, Kaufman JS, Walter LC, Mehta KM, Steinman MA, Allon M, McClellan WM, Landefeld CS (2007) Age affects outcomes in chronic kidney disease. J Am Soc Nephrol 18:2758–2765
Park NY, Park SK, Lim Y (2011) Long-term dietary antioxidant cocktail supplementation effectively reduces renal inflammation in diabetic mice. Br J Nutr 106:1514–1521
Peake JM, Gobe GC, Fassett RG, Coombes JS (2011) The effects of dietary fish oil on inflammation, fibrosis and oxidative stress associated with obstructive renal injury in rats. Mol Nutr Food Res 55:400–410
Percy CJ, Brown L, Power DA, Johnson DW, Gobe GC (2009) Obesity and hypertension have differing oxidant handling molecular pathways in age-related chronic kidney disease. Mech Ageing Dev 130:129–138
Pergola PE, Krauth M, Huff JW, Ferguson DA, Ruiz S, Meyer CJ, Warnock DG (2011) Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am J Nephrol 33:469–476
Quiros Y, Vicente-Vicente L, Morales AI, López-Novoa JM, López-Hernández FJ (2011) An integrative overview on the mechanisms underlying the renal tubular cytotoxicity of gentamicin. Toxicol Sci 119:245–256
Raha S, McEachern GE, Myint AT, Robinson BH (2000) Superoxides from mitochondrial complex III: the role of manganese superoxide dismutase. Free Radic Biol Med 29:170–180
Ramos LF, Kane J, McMonagle E, Le P, Wu P, Shintani A, Ikizler TA, Himmelfarb J (2011) Effects of combination tocopherols and alpha lipoic acid therapy on oxidative stress and inflammatory biomarkers in chronic kidney disease. J Ren Nutr 21:211–218
Renke M, Tylicki L, Rutkowski P, Larczynski W, Aleksandrowicz E, Lysiak-Szydlowska W, Rutkowski B (2008) The effect of N-acetylcysteine on proteinuria and markers of tubular injury in non-diabetic patients with chronic kidney disease. A placebo-controlled, randomized, open, cross-over study. Kidney Blood Press Res 31:404–410
Riegersperger M, Covic A, Goldsmith D (2011) Allopurinol, uric acid, and oxidative stress in cardiorenal disease. Int Urol Nephrol 43:441–449
Schrader M, Fahimi HD (2006) Peroxisomes and oxidative stress. Biochim Biophys Acta 1763:1755–1766
Small DM, Gobe GC (2012) Cytochrome c: potential as a noninvasive biomarker of drug-induced acute kidney injury. Expert Opin Drug Metab Toxicol 8:655–664
Small DM, Coombes JS, Bennett N, Johnson DW, Gobe GC (2012) Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology (Carlton) 17:311–321
Tanner RM, Brown TM, Muntner P (2012) Epidemiology of obesity, the metabolic syndrome, and chronic kidney disease. Curr Hypertens Rep 14:152–159
Turrens JF (2003) Mitochondrial formation of reactive oxygen species. J Physiol 552(Pt 2):335–344
Twomey PJ, Reynolds TM (2006) The MDRD formula and validation. QJM 99:804–805
Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK (2010) Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298:F1078–F1094
Vlassara H, Torreggiani M, Post JB, Zheng F, Uribarri J, Striker GE (2009) Role of oxidants/inflammation in declining renal function in chronic kidney disease and normal aging. Kidney Int 114:S3–S11
Zafarullah M, Li WQ, Sylvester J, Ahmad M (2003) Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 60:6–20
Zhang F, Lau SS, Monks TJ (2011) The cytoprotective effect of N-acetyl-l-cysteine against ROS-induced cytotoxicity is independent of its ability to enhance glutathione synthesis. Toxicol Sci 120:87–97
Zhou H, Kato A, Miyaji T, Yasuda H, Fujigaki Y, Yamamoto T, Yonemura K, Takebayashi S, Mineta H, Hishida A (2006) Urinary marker for oxidative stress in kidneys in cisplatin-induced acute renal failure in rats. Nephrol Dial Transplant 21:616–623
Further Reading
Gobe G, Harmon B (2008) Apoptosis: morphological criteria and other assays. Encyclopedia of life sciences. Nature-Scientific American Publishing Group. eLS Web site. Published online DOI:10.1002/9780470015902.a0002569
Kehrer JP (1993) Free radicals as mediators of tissue injury and disease. Crit Rev Toxicol 23:21–48
Miyata T, Eckardt K-U, Nangaka M (eds) (2011) Studies on renal disorders, 1st edn, Oxidative stress in applied basic research and clinical practice. Humana Press, New York, pp 71–92, 161–178
Taal M, Chertow G, Marsden P, Skorecki K, Yu A, Brenner B (eds) (2011) Brenner and Rector’s The Kidney, 9th edn. Elsevier/Saunders, Philadelphia, pp 1044–1052, 1073–1075; 1411–1419; 1437–1438
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this entry
Cite this entry
Gobe, G.C. (2014). Oxygen, Free Radicals, and Renal Function. In: Laher, I. (eds) Systems Biology of Free Radicals and Antioxidants. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-642-30018-9_113
Download citation
DOI: https://doi.org/10.1007/978-3-642-30018-9_113
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-642-30017-2
Online ISBN: 978-3-642-30018-9
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences