
Abstract
O-Glycosyl donors, despite being one of the last successful donors to appear, have developed themselves into a burgeoning class of glycosyl donors. They can be classified in two main types: O‑alkyl and O‑aryl (or hetaryl) glycosyl donors. They share, however, many characteristics, they can be (1) synthesized from aldoses, either by modified Fisher glycosidation (O-alkyl) or by nucleophilic aromatic substitution (O-aryl or O‑hetaryl), (2) stable to diverse chemical manipulations, (3) directly used for saccharide coupling, and (4) chemoselectively activated. Among these, n-pentenyl glycosides stand apart. They were the first O‑alkyl glycosyl donors to be described and have paved the way to many conceptual developments in oligosaccharide synthesis. The development of the chemoselectivity-based “armed-disarmed” approach for saccharide coupling, including its stereoelectronic or torsional variants, now extended to other kinds of glycosyl donors, was first recognized in n-pentenyl glycosides. The chemical manipulation of the anomeric substituent in the glycosyl donor to induce reactivity differences between related species (sidetracking) was also introduced in n-pentenyl glycosides. An evolution of this concept, the “latent‐active” strategy for glycosyl couplings, first described in thioglycosyl donors (vide infra), has been elegantly applied to O‑glycosyl donors. Thus, allyl and vinyl glycosides, 2-(benzyloxycarbonyl)benzyl (BCB) glycosides and 2′-carboxybenzyl (CB) glycosides are useful “latent‐active” glycosyl pairs. Finally, unprotected 3‑methoxy-2‑pyridyl (MOP) glycosides have been used in glycosylation processes with moderate success.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others
Keywords
- 2′-carboxybenzyl (CB) glycosides
- 3‑methoxy-2‑pyridyl glycosides
- Armed–disarmed
- DISAL glycosyl donors
- Halonium ion transfer
- Latent‐active glycosylation
- n-Pentenyl glycosides
- O-heteroaryl glycosyl donors
- Oligosaccharide synthesis
- Vinyl glycosides
Introduction
From the early days , chemists involved in chemical glycosylation have been trying to develop successful glycosyl donors. In general, the characteristics of a successful glycosyl donor might include: (a) preparation under mild reaction conditions, (b) selective activation by reagents that would not interfere with the protecting and functional groups present in the donor and the glycosyl acceptor, and (c) good reactivity [1,2,3,4]. More recently, the advent of convergent block synthesis to tackle complex oligosaccharide preparations have also demanded that glycosyl donor building blocks (d) are sufficiently stable to be purified and stored for considerable periods of time, and (e) are resistant towards a wide range of reaction conditions [5,6,7,8,9]. According to this, O‑glycosyl donors (the topic of this chapter), because of their remarkable ``shelf-life`` and stability (conditions d, e) will be attractive candidates for oligosaccharide block synthesis, provided that conditions a, b, and c are also met.
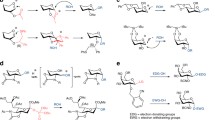
A chronology, displayed in Fig. 1, highlights the relatively recent arrival of O‑glycosyl donors to the assortment of relevant glycosyl donors. In fact, the first O‑alkyl glycosyl donor ( n-pentenyl glycoside ) [10], was introduced more than a century after the first glycosylation was described (synthesis of aryl glycosides from glycosyl chlorides) [11].
This late arrival is understandable on the basis of the outline in Scheme 1a, which makes it obvious that in situ transformation of one alkyl glycoside donor into a disaccharide (or into another alkyl glycoside) could be problematic. The acidic conditions normally used to cleave alkyl glycosides (1), generating oxocarbenium ion 2, could tamper with the newly formed intersaccharidic linkage in 3, notwithstanding the liberation of alkanol that might compete for glycosylation with the sought glycosyl acceptor, thus regenerating 1 (Scheme 1b). The successful implementation of the strategy represented in Scheme 1a would imply that: (a) the alkanols have to be released under a non‐nucleophilic form, and (b) the newly formed glycosidic linkage must be compatible with the promoter employed.

Chronology of selected glycosyl donors

O‑Alkyl-glycosyl donors in glycosylation
n-Pentenyl Glycosides
Introduction
The Origin of n-Pentenyl Glycosides (NPGs)
The discovery of n-pentenyl glycosides (NPGs) [12], was derived from an observation made by Mootoo and Fraser-Reid in a completely unrelated project [13]. Attempted formation of bromohydrin 5 by reaction of 4 with NBS in 1% aqueous acetonitrile led, instead to bromomethyl tetrahydrofuran 6 (Scheme 2) [14]. To rationalize this transformation (4→6), the authors invoked a 5-exo-cyclization [15] of the pyranosidic oxygen in 7 leading to cationic intermediate 8, and thence to oxocarbenium ion 9, that upon capture of H2O led to hemiacetal 6. The overall result of the process had been a, nonhydrolytic, electrophilic unravelling of the glycosidic-type bond in 4.

The origin of n-pentenyl glycosides
The overlap between structures 4 and 10 permitted the authors to design structure 11, as a candidate for testing electrophilic deprotection at the anomeric center of a pyranose (Scheme 3). It has now become clear, 20 years after this observation, that the correlation shown in Scheme 3 led to a breakthrough in glycoside synthesis.

The design of n-pentenyl glycosides
Chemoselective Liberation of the Anomeric Group in NPGs
To test the validity of their assumption, Mootoo and Fraser-Reid prepared NPGs 12–18 and treated them with NBS in 1% aqueous acetonitrile [16]. Their results, summarized in Table 1, showed that differently substituted NPGs could be chemoselectively liberated at the anomeric center to yield hemiacetals 19–24. Furthermore, benzylidene, silyl, p-methoxybenzyl (PMB), ethoxyethyl, and allyl protecting groups proved to be compatible with the conditions employed in the deprotection of the anomeric pent-4-enyl group. Diol 18, however, furnished a complex reaction mixture probably related to competing glycosylation processes, vide infra.
NPGs as Glycosyl Donors
To test the potential of NPGs as glycosyl donors, Fraser-Reid et al. first examined the reaction of compound 12 with NBS in MeCN-MeOH [10]. The reaction took place in 3 h, at room temperature yielding methyl glucoside 25 in 85% yield as a 1:3 (α:β) anomeric mixture (Table 2, entry i). The utilization of iodonium di-sym-collidine perchlorate (IDCP) [17] as a promoter resulted in a faster reaction (0.5 h), which maintained the previous anomeric mixture (Table 2, entry ii). The use of a 4:1 mixture of Et2O-CH2Cl2 as solvent, to favor α‑glucoside formation while solubilizing IDCP, resulted in a 3:1 (α:β) anomeric mixture of 25 (Table 2, entry iii). When CH2Cl2 was used as a solvent a 1.2:1 (α:β) anomeric mixture was obtained.
NPGs were next tested in the elaboration of disaccharides by glycosylation of monosaccharide acceptors. Gluco- (12), manno- (26), and 2-deoxy- (27) NPGs reacted with sterically demanding methyl glucoside 28, to give disaccharides 29–31 (Table 3). Gluco‐derivatives gave the best α versus β solvent dependence, MeCN favoring β, and Et2O favoring α (the same trend as noted for MeOH, Table 2). For the manno- and 2-deoxy donors 26 and 27, no consistent pattern of solvent dependence was noticed. 2-Deoxy-donor 27, gave appreciable α‑selectivities with secondary acceptors (Table 3, entries vii–ix). MeCN gave the lowest overall yield of disaccharide products (Table 3, entries i, iv, vii). Reactions of 2-deoxy NPG 27 (Table 3, entries vii–ix) were generally much faster than those of either 12 or 26, an observation that parallels the observed trends in acid lability of the three donors (Table 3, entries i–vi). With Et2O as solvent, reactions with primary alcohol acceptors displayed more stereoselectivity than reactions with secondary hydroxyl acceptors.
Acyl-Substituted NPG Donors
The results in Table 3 made it clear that the use of different solvents to induce (α versus β) stereoselectivity in glycosyl couplings of NPGs was only moderately successful, generally leading to anomeric mixtures [10]. As it has been established, good stereocontrol in the formation of 1,2-trans glycosidic linkages can be conveniently obtained with the assistance of a neighboring participating group , generally an acyl moiety [18]. In this context, Fraser-Reid and co-workers examined the glycosylation of NPG 32 for the preparation of 1,2-trans glycoside 33 (Scheme 4). Unfortunately, the reaction did not lead to glycoside 33, but to compounds resulting from addition across the terminal double bond of the pent-4-enyl moiety. Along this line, the authors had previously noticed that hydrolysis of acyl derivative 34 was considerably slower than that of 13 [16]. These results paralleled previous observations by Paulsen [1] on the deactivating effect of esters versus ether protecting groups upon differently substituted glycosyl halides. These observations, however, according to the state-of-the-art in glycosylation in 1988, only meant that acyl-NPGs would not be useful as glycosyl donors.

Reaction of acylated NPGs with halonium ions
Armed–Disarmed Strategy for Glycosyl Coupling
Fraser-Reid and co-workers, however, anticipated that the difference in reactivity found in differently substituted NPGs could be applied in a chemoselective protocol for glycosyl coupling [19]. The activated and the deactivated NPGs were termed “armed sugar” and “disarmed sugar”, respectively. Thus, as illustrated in Scheme 5a, coupling of 12 and 36, mediated by IDCP afforded a 62% yield of disaccharide 37 [19]. Therefore, the acyl groups of 36 indeed “disarmed” the NPG, thereby ensuring that 12 served as the only glycosyl donor. No evidence for a hexaacetyl disaccharide 38, arising from self‐condensation of 12 was found, nor of further reaction of (disarmed) disaccharide 37 with the acceptor 36.

Armed-disarmed strategy for chemoselective glycosyl couplings
The chemoselective coupling , however, is not the only quality of the armed-disarmed strategy for glycosyl assembly. An additional aspect of this strategy is the ability to “rearm” disarmed glycosyl donors for further glycosyl coupling. Thus, “disarmed” 37 was converted to “armed” disaccharide 39, (by replacing the acetyl groups with benzyl substituents) which could then glycosylate galacto- derivative 40, to yield trisaccharide 41, in 60% yield (Scheme 5b). An alternative way of “rearming” NPGs by increasing the potency of the promoter used for glycosylation was also introduced by the same authors [20]. According to that, an iodonium ion generated in situ from NIS and TfOH was able to promote the coupling of “disarmed” pent-4-enyl glycosides (e. g. α‑32, Scheme 5c) with acceptors to give 1,2-trans disaccharides, e. g. 43, via neighboring group participation [18].
The armed-disarmed concept takes advantage of reactivity differences induced by the ring substituents on the anomeric leaving group and, although originally described for NPG donors, it has been extended to various types of glycosyl donors. These include thioglycosides [21,22], glycals [23], glycosyl fluorides [24], selenoglycosides [25], glycosyl phosphoroamidates [26,27], glycosyl thioformimidates [28,29,30], and S‑benzoxazolyl glycosides [31,32,33].
Mechanistic Aspects of the Oxidative Hydrolysis of NPGs
The currently accepted mechanism for the reaction of NPGs (e. g. 11) with halonium ions is outlined in Scheme 6a. The oxygen in the acetal function participates in a favored 5-exo-tet ring opening of an intermediate cyclic halonium ion, 44 [15]. The ensuing furanilium ion 45, evolves by splitting off a non‐nucleophilic halotetrahydrofuran 47 [34], thus leading to oxo‐carbenium ion 46, that can trap the nucleophile (ROH). The overall result is the cleavage of the acetal moiety with the formation of a new glycosyl derivative, 48. Madsen and Fraser-Reid have demonstrated that even when the system NIS/TESOTf [20] is used to promote the cleavage of NPGs, the reaction is not acid catalyzed but still halonium ion catalyzed [35].
In this connection, the question of why the reaction of an NPG in the presence of water leads to an aldose 48 (R=H), rather than to a halohydrin 49 (R=H) was raised (Scheme 6b). In fact, the successful cleavage observed for NPGs rests on two issues: (a) the concentration of nucleophile (water in the case of hydrolysis), and (b) the rate of the 5-exo-tet cyclization, 44→45. Pertinent to question a the intramolecular reaction 44→45, is preferred to the bimolecular reaction with water leading to a halohydrin, 44→49, under the conditions used by the authors. An increase in the concentration of water would enhance the rate of the bimolecular reaction, without affecting the intramolecular process. Indeed added water led to the formation of bromohydrin 49 (R=H) [36]. Related to the second issue, Rodebaugh and Fraser-Reid examined the same reaction with allyl, butenyl, and hexenyl glycosides 50 (\( { n = 1, 2, 4 } \)), differing on the rate of cyclization compared to NPGs [37,38]. They found that, unlike NPGs, they all gave rise to isomeric halohydrins 51 and 52, upon treatment with NBS in aqueous MeCN (Scheme 6c).

Oxidative cleavage of NPGs
Evidence for Intermolecular Halonium-Ion Transfer
In a related experiment, hexenyl glycoside 50 (\( { n = 4 } \)), that has been found to react 2.3-times slower than NPG 12 [37,38], was made to compete with 12 for 1 equiv. of NBS. The hexenyl glycoside 50 (\( { n = 4 } \)) was recovered unchanged together with hemiacetal 19, arising from the hydrolysis of 12 (Scheme 7a). Rodebaugh and Fraser-Reid proved that this phenomenon was due to a diffusion‐controlled intermolecular halonium-ion transfer (a similar process has been previously noted by Brown and co-workers for “sterically encumbered olefins” [39]). Accordingly, bromonium species 53, obtained by irreversible reaction of 50 with NBS [37,38], would undergo a fast bromonium ion transfer to NPG 12 leading to halonium 54 (i. e. 44, Scheme 6a) which will undergo a fast transformation to 19. The general process, a classic example of Le Chatelier's principle, is represented in Scheme 7b. When two alkenes are made to compete for one equivalent of halonium ion X+, a steady-state regime can be envisaged whereby the faster (F) (e. g. NPG 12) reacts completely, and the slower (S) (e. g. 50, \( { n = 4 } \)) is recovered completely.

Intermolecular halonium-ion transfer
Intermolecular Halonium-Ion Transfer: A Key Factor in the Implementation of the Armed-Disarmed Protocol
This intermolecular halonium ion transfer had indeed been postulated earlier as the key factor to account for the absence of self-coupling product 38, when armed and disarmed NPGs, 12 and 36 respectively, were made to compete for one equivalent of NBS (Scheme 5a). The observed 6-fold difference in the hydrolysis rates of 36 and 12 should have resulted in the presence of 38, in at least 10% [40]. Irreversible reaction of NPGs 55 and 59 with NBS leads to halonium ions 56 and 60, respectively (Scheme 8). The transfer of halonium (e. g. 60→55) is reversible and rapid compared with the subsequent steps leading to glycoside formation (56→57→58). By corollary, the inherent reactivity of the glycosyl donors is thus revealed in the final product distribution. If the acceptor functionality is located in the less reactive component, selective glycosylations take place leading to a specific disaccharide.

Halonium ion transfer: a key factor in armed-disarmed couplings
Torsional Disarming of NPGs
Fraser-Reid and co-workers found that widely used cyclic acetals also affected anomeric reactivity [41]. They showed that these reactivity differences could be applied to an armed-disarmed protocol based on torsional, rather that electronic, effects. The measured experimental relative rates of oxidative hydrolysis for some pairs of galacto- (62, 63), manno- (64, 65), and gluco- (12, 13), acetalated and nonacetalated NPGs are displayed in Fig. 2. From these data, the authors were able to design the successful armed-disarmed couplings shown in Scheme 9. These reactivity differences were ascribed to the fact that trans-fused protection restricts the molecule from ring flexibility, thereby making it increasingly difficult to reach a half-chair transition state from a chair ground state.

Relative rates of oxidative hydrolysis for acetal‐protected NPGs

Armed-disarmed couplings based on torsional effects
Sidetracking of NPGs : A Reversal for the Armed-Disarmed Strategy
In the armed-disarmed protocol, the reactivity differences induced by the ring substituents upon the anomeric center were exploited for chemoselective couplings. In these protocols, the more reactive “armed” NPG always glycosylates the “disarmed” NPG. On the basis of chemical manipulation of the pent-4-enyl moiety, rather than the ring substituents, Fraser-Reid and co-workers showed that “disarmed” NPGs could be used to glycosylate “armed” NPGs [40]. Treatment of NPG 72 with bromine and tetra-n-butylammonium bromide (TBAF), in a bimolecular reaction (e. g. 44→48, Scheme 6b) yielded dibromo‐derivative (Scheme 10) 73. Glycosylation of the latter with “disarmed” 74, under the agency of NIS/TESOTf, yielded disaccharide 75a, which could be transformed to the pentenyl disaccharide 75b. Several methods proved to be successful for the restoration of the double bond from the dibromoderivative including, (a) Zn/TBAI in sonicating EtOH, (b) NaI in methyl ethyl ketone, and (c) SmI2 in THF [42]. The choice of the reagent will vary with the reactivity of the substrate, as well as the protecting groups thereon. More recently, a milder brominating system, the combination of CuBr2 and LiBr in MeCN:THF (3:1), has been used to brominate n-pentenyl glycosides containing O‑benzyl, O-p-methoxybenzyl, N‑phthaloyl, and N‑tetrachlorophthaloyl protecting groups [43].

Sidetracking of NPGs in saccharide synthesis
Conversion of NPGs to Other Glycosyl Donors
NPGs have been converted into different glycosyl donors.
Conversion to Glycosyl Bromides
Konradsson and Fraser-Reid [44] reported the conversion of NPGs into glycosyl bromides , e. g. 76, by treatment of the corresponding pentenyl glycoside with a dilute dichloromethane solution of bromine, conditions that favor unimolecular reaction. The reaction was shown to be compatible with acetals, benzyl, silyl, and allyl protecting groups in the NPG (Scheme 11a).
Conversion to Glycosyl Phosphates
Pale and Whitesides [45] described the synthesis of glycosyl phosphates 77 [46,47], by reaction of dibenzyl phosphate with NPG 12, with the use of either IDCP or NBS as promoters. The authors noted the influence of the solvent (MeCN, Et2O, CH2Cl2) and the promoter in the α/β selectivity of the glycosyl phosphates formed (Scheme 11b).
Conversion to Glycosyl Fluorides
Clausen and Madsen [48] reported the transformation of NPG 78 into glycosyl fluoride 79 by treatment with NBS and (diethylamino)-sulfur trifluoride (DAST) (Scheme 11c).
López et al. [49] described the preparation of glycosyl fluorides 80, by reaction of NPGs with bis(pyridinium) iodonium (I) tetrafluoroborate (IPy2BF4) in the presence of tetrafluoroboric acid (Scheme 11d). The process was compatible with the presence of silyl and benzyl groups in the NPG.
Chemoselective Liberation Followed by Anomeric Activation

Conversion of NPGs into different glycosyl donors
The ability to chemoselectively deprotect pent-4-enyl glycosides opens an avenue for a two-step transformation of NPGs into different glycosyl donors. In this context, NPGs can be transformed [50] into thioglycosides 81 [51,52,53], glycosyl trichloroacetimidates 82 [54], and glycosyl chlorides 83 [55] (Scheme 11e).
NPGs in the Stereocontrolled Assembly of α- and β- Glycoproteins
Pyranosylacetonitrilium Ions from NPGs
Ratcliffe and Fraser-Reid found that acetonitrile was able to trap glycosyl oxocarbenium ions (e. g. 46), arising from NPGs, to give acetonitrilium ions , e. g. 84 (Scheme 12a) [56]. The latter reacted with water to produce intermediate 85 that evolves to α‑amide 86, in a Ritter-type reaction [57] (Scheme 12a).

Reactions of pyranosylacetonitrilium ions arising from NPGs
This transformation was significant from a mechanistic standpoint. The formation of the α‑acetonitrilium ion was not expected on the basis of the reverse anomeric effect (originally defined as the tendency of positively charged substituents at C‑1 of a pyranose ring to adopt the equatorial orientation [58]). The authors, however, unambiguously established the α‑orientation of the amide and explained this result assuming the formation of the kinetically favored α‑d‑glucopyranosylacetonitrilium ion, 84 [59].
Synthesis of N-α-Linked Glycoproteins from Pyranosylacetonitrilium Ions
The synthetic value of the above‐mentioned transformation was considerably enhanced when a carboxylic acid, rather than water, was used to trap the pyranosylacetonitrilium ion (Scheme 12b) [59]. Reaction of 87 with aspartic acid derivative 88, in dry acetonitrile containing NBS, led to α‑imide 89 in 61% yield [60,61]. The acetonitrilium ion 90 was trapped by carboxylic acid 88, to give an imidic anhydride 91, which rearranged in situ to give the N,N‑diacyl derivative 89. The route to 2‑acetamido-1-N-(l-aspart-4-oyl)-2-deoxy-β‑d-glycopyranosyl-amine 92, was completed by selective N‑deacetylation of 89 with piperidine (Scheme 12c) [59].
Synthesis of N-β-Linked Glycoproteins from Pyranosylacetonitrilium Ions
More interestingly, the presence of a neighboring participating group at C2 induces the formation of β‑nitrilium ion intermediates, e. g. 94 (Scheme 12d), thus paving the way to β‑linked glycoproteins [62]. Accordingly, phthalimido NPG 93, reacted with aspartic acid derivative 88, in acetonitrile using NBS as promoter, via the β‑nitrilium intermediate 94, to give the β‑asparagine‐linked product 95 in 48% yield.
More recently, this method has been elaborated in a three‐component‐reaction (NPG, acetonitrile, carboxylic acid) route to N‑glycosylamines [63].
n-Pentenyl 2-Amino-2-Deoxy Glycoside Derivatives as Glycosyl Donors
Several pent-4-enyl 2-amino-2-deoxy glycoside derivatives were evaluated as glycosyl donors for the synthesis of 2-amino-2-deoxy oligosaccharides [64]. 2-Deoxy-2‑phthalimido 96, and 2‑anisylimino-2-deoxy-d-glucopyranosides 98, underwent IDCP-induced coupling with a variety of sugar alcohols to give β and α disaccharides 97 and 99, respectively, in moderate to good yields (Scheme 13a,b) [65,66]. 2-Deoxy-2-N‑tetrachlorophthaloyl NPGs , e. g. 100, 103, [67,68,69] are useful donors for the stereocontrolled access to 1,2-trans glycosides as exemplified in Scheme 13c,d. Good yields of disaccharide 102, and aminoacid 104 were obtained by the use of NIS/TESOTf as promoter. β‑N‑Linked glycopeptide 106 was prepared by treatment of 105 with acid 88 in dry MeCN containing NBS (Scheme 13e).

Glycoside formation from pent-4-enyl 2-amino-2-deoxy glycosides
Controversial results have been reported when 2-deoxy-2-azido NPGs were used as glycosyl donors (Scheme 14). Fraser-Reid and co-workers reported that 107 failed to give pseudo‐disaccharide 109 upon reaction with acceptor 108 under the agency of NIS/TESOTf (Scheme 14a) [70]. By contrast, good results were obtained with the benzylidenated derivative 110 (Scheme 14b). The authors ascribed this result to the conformational constraint imposed by the benzylidene ring, in keeping with their precedents [41].

Glycoside formation from pent-4-enyl 2-deoxy-2-azido NPGs

Pent-4-enyl 2‑allyloxycarbonyl-2-deoxy-d- and l‑glucopyranosides
Svarovsky and Barchi [71] observed a striking reactivity difference between pent-4-enyl β‑ and α‑2-azido-2-deoxy galactosides 113 and 116, respectively (Scheme 14c-X). Thus, whereas β‑NPGs 113a and 113b reacted with serine derivative 114 to give the sought α‑glycosyl aminoacids 115a,b, with complete stereocontrol, the corresponding α‑anomers 116a,b gave very poor yields of 115a,b (< 10%) and much slower reaction rates.
Fraser‐Reid's group advanced the synperiplanar lone-pair hypothesis (SLPH) , to account for the fact that β‑d-glycopyranosides hydrolyze ≈2–3 times faster [72] than the corresponding α‑anomers [73]. This theory advocates that as the reaction progresses synperiplanar lone-pair interactions in the energetically accessible half-chair conformation of the β‑anomer are equivalent to the antiperiplanar interactions in the half-chair of the α‑anomer (antiperiplanar lone pair hypothesis, ALPH ) [74]. On the other hand, the torsional effects associated with the conformational restraint imposed by the presence of the benzylidene ring might enhance this β/α reactivity difference to the point that the α‑anomer hardly reacts [75].
2‑Allyloxycarbonylamino-2-deoxy d- and l‑glucopyranosides 118 and 121, respectively, have been reported by Lafont and Boullanger [76], to successfully glycosylate 10‑tetradecyloxymethyl-3,6,9,12‑tetraoxahexacosanol (119) and 1,3-bis(undecyloxy)propan-2-ol (122), in the course on their studies on neoglycolipids for monolayers (Scheme 15). In this case, the chemoselectivity in the reaction of the anomeric pent-4-enyl moiety in the presence of the allyloxycarbonyl group is noteworthy. Related n-pentenoyl derivatives have been reported as efficient protecting groups for amines by its mild deprotection with iodine in THF-water [77].
Rojas and co-workers [78] described a novel synthetic route to α‑linked 2-deoxy-2‑mannosamine derivatives, which involved a stereocontrolled glycosidation step of NPG oxazolidinones (e. g. 124, Table 4) and N‑CBz NPGs (e. g. 127, Table 5). The authors found a striking difference in reactivity between α‑ and β‑anomers of oxazolidinones 124. α‑NPG oxazolidinones served as highly stereoselective donors (Table 4, entries iii, v–vii), whereas the β‑anomer was nearly inert (Table 4, entries ii, iv). However, regioselective N‑CBz oxazolidinone ring opening to 127, prior to glycosylation permitted elaboration of either NPG anomer to the desired α‑Man-NCBz products 128 (Table 5).
Semi-Orthogonal Couplings of NPGs
The term “semi‐orthogonality” between glycosyl donors (e. g. A and B, Scheme 16) was introduced by Demchenko [79]. It indicates that whereas selective activation of armed and disarmed glycosyl donor A can be effected in the presence of either armed or disarmed donor B (Scheme 16a), the opposite is not feasible. Thus, in semi‐orthogonal donors , the selective activation of disarmed glycosyl donor B in the presence of glycosyl donor A can not be accomplished (Scheme 16c).

Semi‐orthogonality of glycosyl donors
Semi-Orthogonality of O-Pentenyl and S-Ethyl Glycosides
Demchenko and De Meo found conditions for the selective activation of NPGs and ethyl 1‑thioglycosides (Scheme 17) [79]. They demonstrated that armed NPGs (e. g. β‑12) could be activated in the presence of thioglycosides (e. g. 131) with IDCP as the promoter (Scheme 17a). On the other hand, the use of methyl triflate (MeOTf) permitted the activation of disarmed thioglycosides (e. g. 133b) in the presence of armed or disarmed NPGs, 134 (Scheme 17b).

Semi‐orthogonality of O‑pentenyl and S-ethyl‐glycosides
Semi-Orthogonality of NPGs and Glycosyl Fluorides
López et al. reported the selective activation of armed NPGs (e. g. 136) in the presence of armed glycosyl fluorides (e. g. 137) on treatment with IDCP (Scheme 18a) [49]. On the other hand, armed and disarmed glycosyl fluorides 139, could be activated in the presence of armed NPGs (e. g. 140) on treatment with ytterbium triflate (Yb(OTf)3) (Scheme 18b).

Semi‐orthogonality of NPGs and glycosyl fluorides
n-Pentenyl Furanoside Donors
Chemoselective Deprotection of the Anomeric Center
Unlike n-pentenyl pyranosides, the corresponding furanosides have attracted comparatively little attention. Sharma and Rao reported the preparation of n-pentenyl d‑allo-, and d‑gulo- furanosides 143 and 146, respectively (Scheme 19) [55]. They made use of an efficient acid-induced rearrangement of diacetonides 142 and 145, in the presence of n-pentenyl alcohol. The ensuing pent-4-enyl diacetonides 143 and 146, were chemoselectively cleaved to hemiacetals 144 and 147.

Pent-4-enyl furanosides from d‑allose and d‑gulose diacetonides
Application to the Synthesis of Nucleosides
Chapeau and Marnett developed a synthetic route to purine nucleosides from n-pentenyl ribosides (Scheme 20) [80]. The authors used a Fischer glycosylation of d‑ribose with 4‑pentenol to produce pent-4-enyl β‑d-erythro‐pentofuranoside 148a, in 86% yield. Reaction of the latter with benzoyl chloride produced their glycosyl donor 148b, in 69% yield. Addition of TfOH to acetonitrile solutions containing 148b, the selected purine, and NIS, resulted in a rapid coupling to form the desired nucleosides 149, in a stereocontrolled manner with yields ranging from 50 to 70% (Scheme 20a). The absence of an acyl group at O2 in 2-deoxy NPG 150, enhanced its reactivity to iodonium sources so IDCP could be used as the promoter. Thus, reaction of 150 with 6‑chloropurine in acetonitrile was neither regio- nor stereo‐selective, yielding four coupling products 151α,β and 152α,β, in similar amounts (Scheme 20b).

Pentenyl ribofuranosides in the synthesis of purine nucleosides
n-Pentenyl Furanosides as Glycosyl Donors
Arasappan and Fraser-Reid described the preparation of n-pentenyl galactofuranosides and evaluated their prospects as glycosyl donors (Scheme 21) [81]. Fischer glycosidation of d‑galactose under kinetic conditions using n-pentenyl alcohol and DMSO as co-solvent [82] afforded an anomeric α/β (1:3) mixture of n-pentenyl galactofuranosides 153, (≈80–85% yield), contaminated with small amounts of the corresponding n-pentenyl galactopyranosides, 154 (Scheme 21a). Glycosylation with α‑ or β‑ pentenyl glycosides 155 was irrelevant to the product β/α ratio, both favoring the β‑furanoside β‑156 (Scheme 21b,c). Reactions with donors α‑ or β‑157 resulted in the β‑linkage product 158, exclusively (saccharide acceptors with free hydroxyl groups at C‑2, C‑4, and C‑6 were assayed), presumably due to the neighboring group participation of the C‑2 ester functionality (Scheme 21d).

Pentenyl galactofuranosides
Plusquellec and co-workers reported an improved method for the preparation of n-pentenyl furanosides [83] based on their previously described use of FeCl3 as a catalyst in Fischer-type glycosylations [84]. Accordingly, d‑glucose, d‑galactose, and d‑mannose upon treatment with FeCl3 and n-pentenyl alcohol followed by in situ acetylation, yielded pent-4-enyl d‑gluco-, d‑galacto-, and d‑mannofuranoside derivatives 157, 161, and 163, respectively in yields ranging from 50 to 75%. Glycosylation of glycerol diether 159 with these donors, promoted by NIS/TESOTf yielded glycolipids 160, 162, and 164 in high yields and with excellent 1,2-trans stereoselectivity (Scheme 22).

Synthesis of archaeol glycolipid analogues from pent-4-enyl furanosides
n-Pentenyl Arabinofuranosides in the Assembly of Oligoarabinans of Mycobacterium tuberculosis
Recent interest in oligoarabinans, have been triggered by their presence in the lipoarabinomannan polysaccharide component of the cell wall complex of mycobacteria [85]. Several research groups have employed n-pentenyl arabinofuranosides in their approaches to oligoarabinans.
n-Pentenyl β‑d-arabinofuranoside 166, readily prepared from 165, was employed as acceptor/donor in Gurjar's approach to arabinosyl pentasaccharide 171 (Scheme 23) [86]. Accordingly, 166 was glycosylated with S-(2‑pyridyl)-1‑thiofuranose 167 to yield, in a stereoselective manner, β‑disaccharide 168. The latter, itself a pentenyl donor, was then used as a glycosyl donor in two glycosylation events. First, IDCP-promoted glycosylation of silyl derivative 169 yielded trisaccharide 170a in 62% yield, and in a stereocontrolled manner. Desilylation of 170a furnished 170b, which then functioned as the acceptor in the second IDCP-induced glycosylation with 168 to produce pentasaccharide 171.

Synthesis of a pentaarabinofuranosyl structure motif of Mycobacterium tuberculosis

NPOEs in the synthesis of arabinofuranosyl donors
More recent studies by Fraser‐Reid's group, have focused on the use of NPOEs both as arabinofuranosyl donors, and as convenient starting materials for the preparation of n-pentenyl arabinofuranosyl acceptors [87]. TPSOTf‐induced rearrangement of NPOE 172, followed by desilylation afforded pentenyl glycoside 173b (Scheme 24). Glycosylation of pentenyl glycoside 173b with NPOE 172 was carried out using NIS/Yb(OTf)3 , a chemospecific promoter for NPOEs [88,89]. Iteration of the sequence permitted the preparation of the α‑1,5‑linked arabinan segment of the complex lipoarabinomanan cell wall array of Mycobacterium tuberculosis, 175.
The potency of this strategy relies ultimately in the sturdiness, and yet the possibility for chemoselective cleavage, of pentenyl arabinofuranosides (e. g. 175) [90]. Its value has been demonstrated recently with the synthesis of the pentenyl glycoside of mannose‐capped dodecafuranoarabinan of Mycobacterium species, 176a (Fig. 3). The final NPG→trichloroacetimidate transformation (176a→176b) made possible the coupling of this arabinan segment to an oligomannan acceptor, thus resulting in the synthesis of the largest heterooligosaccharide to date, a 28-mer arabinomannan [91].

Mannose‐capped multibranched dodecafuranoarabinan of Mycobacterium species

Multibranched hexafuranoarabinan of Mycobacterium species
n-Pentenyl arabinofuranosides have also been used by Seeberger and co-workers in the final stages of their synthesis of a 12-mer component of Mycobacterium tuberculosis [92]. n-Pentenyl arabinan hexasaccharide 177a, was transformed to the corresponding trichloroacetimidate 177b and coupled with a mannan hexasaccharide acceptor to yield the sought arabinomannan dodecasaccharide (Fig. 4).
Intramolecular Aglycon Delivery from n-Pentenyl Glycofuranosides
An approach to α‑d-fucofuranosyl glycosides that makes use of the intramolecular aglycon delivery (IMAD) [93,94,95,96] starting from an n-pentenyl fucofuranoside has been described (Scheme 25) [97]. n-Pentenyl fucofuranoside 178 bearing a free 2-OH group was attached to a 4-O‑PMB‐protected galactopyranoside 179, upon treatment of the mixture with DDQ. The unstable tethered compound 180, could be activated with NIS, in the absence of even catalytic amounts of acid, to undergo an efficient p-methoxybenzyl‐assisted aglycon delivery [95] leading to the desired glycoside 181. The unusual structure 181, resulting from quenching of the benzylic cation with N‑succinimide, was then processed to α‑d-fucofuranoside 182.

NPGs as glycosyl donors in intramolecular aglycon delivery
Intramolecular C-Glycosylation of NPGs
The intramolecular C-glycosylation of NPGs has been studied by Martin's group in the course of their approaches to bergenin [98] and related natural products [99,100]. The treatment of pentenyl β‑d-glucopyranose 183 with IDCP promoted an internal, Friedel–Crafts type, C‑arylation reaction in excellent yield (Scheme 26a). The resulting product was exclusively the kinetically favored, cis-fused tricyclic system 184. Treatment of the latter with an oxophilic Lewis acid (BF3·OEt2) led to the trans-fused (β‑linked) 185. On the contrary, analogous reaction of α‑d-mannopyranoside 186 led to a mixture of trans- and cis-fused compounds 187, and 188 (5:1), where the major trans-fused (α‑linked) product 187, was this time the kinetic product (Scheme 26b). Treatment of the latter with BF3·OEt2 promoted the epimerization to the, more stable, 1,2-cis epimer 188, in 81% yield.

Intramolecular C-glycosylation of NPGs
NPGs of N-Acetylneuraminic Acids (Neu5Ac)
One report describing O‑sialylation of 4‑pentenyl glycosides of Neu5OAc, e. g. 189, has appeared (Table 6) [101]. Good α/β selectivity (11:1) was attained in the glycosylation of primary acceptor 40 in MeCN using NIS/TfOH as the promoter (Table 6, entry i), however, with the secondary acceptor 191 the α/β selectivity dropped to 4:1 (entry iii). The use of Et2O as solvent produced a 1:1 mixture of anomers (entry ii).
NPGs of l-Iduronic Acid as Glycosyl Donors
In their studies on heparin/heparin sulfate, and dermatan sulfate, Petitou, Sinaÿ and co-workers found that n-pentenyl glycosides of l‑Iduronic acid, e. g. 192, were efficient glycosyl donors [102]. In contrast, the corresponding thioglycosides, and glycosyl fluorides did not give the expected disaccharides. Reaction of n-pentenyl glycosyl donors 192 (α or β) with acceptors 194–197 (Table 7) was carried out in CH2Cl2 with NIS/TfOH to furnish the corresponding α‑disaccharides 193, in good yields.
More recently, Reichardt and Martín-Lomas have evaluated n-pentenyl glycosides of glucosamine α1→4l-iduronic acid disaccharide, as substrates for autocondensation in their approach to heparin oligosaccharide fragments. However, the NIS used as promoter, being itself a nucleophile, competes with the acceptor disaccharide in the polycondensation process, which results in fast chain-reaction termination and a low yield and degree of polymerization [103].
NPGs in Regioselective Couplings
The synthesis of branched saccharides by multiple glycosylations onto a central monosaccharide normally requires the use of orthogonal protecting groups in the acceptor. In this context, regioselective glycosylation of diols or polyols would ease the number of protection‐deprotection steps in these synthetic protocols.
The Role of the O-2 Substituent in Regioselective Couplings
In their studies on myo-inositol glycosylation, Fraser-Reid and co-workers made the observations summarized in Scheme 27 [104]. In the hope of achieving selective glycosidation of the equatorial-OH, they treated diol 198 with the armed n-pentenyl donor 64 (Scheme 27a). However, the major product was the mixture of α/β glycosides 199 from glycosidation at the axial-OH (Scheme 27a). In order to improve α anomeric stereoselectivity they selected the corresponding disarmed NPG 200, as the donor (Scheme 27b). Surprisingly, the only product obtained was the disaccharide 201 from glycosidation at the equatorial-OH.

Influence of the O‑2 protecting group in regioselective glycosylations
In a series of subsequent papers Fraser-Reid and co-workers confirmed these discrepancies, and showed that the O‑2 substituent in glycosyl donors, besides its recognized role for stereocontrol, exerts a profound influence in eliciting regioselective glycosyl couplings [105,106,107]. In most cases, 2-O‑acyl NPGs and NPOEs shared the same regiopreferences, which were usually different from the ones displayed by 2-O‑alkyl NPGs. The regiopreferences of the former were generally more pronounced or even exclusive.
Reciprocal Donor Acceptor Selectivity (RDAS)
The influence of the O‑2 substituent in regioselective couplings is not limited to pentenyl glycoside donors. Thioglycoside and trichloroacetimidate donors have shown the same tendency [108]. The glycosylation of allose diol 203 with donors 64 and 202a–f (Table 8) illustrates this point. NPOE 202a (that shows the same regiopreferences as disarmed NPGS), disarmed thiomannoside 202c, and disarmed trichloroacetimidate 202e, exhibited the same preference for the O3 of allose acceptor 203 (Table 8, entries i, iii, v). On the contrary, armed donors 64, 202c, and 202e furnished a 2:1 mixture of disaccharides 204b and 205. The above‐mentioned examples indicate that each donor expresses preference for one of the diol–OHs in the acceptor and vice versa. The authors coined the term Reciprocal Donor Acceptor Selectivity (RDAS) [109] to account for these findings.
In Situ Double Differential Glycosylations of Two Donors with One Acceptor
The practical utility of this concept was further demonstrated when diol acceptor 203, NPOE 202a, and armed-NPG 64 were treated with NIS/BF3·Et2O to give one single trisaccharide 206, in 57% yield (Scheme 28) [110]. It seemed that in the formation of the trisaccharide the regiopreferences of the NPOE 202a and the armed NPG 64, displayed in Table 8, have been followed.

In situ three‐component double differential glycosylation of two donors and one diol acceptor
Analogously, the regiopreferences (RDAS) of disarmed NPG 200, and armed NPG 64 vis a vis mannose diol 207, were evaluated (Scheme 29a,b). With the disarmed donor 200 mannosylation occurred at the (C6)-OH only to give 208 in 53% yield, and also the symmetrical trisaccharide 209 in 13% yield, but with no evidence for the dimannan resulting from glycosylation of the (C3)-OH (Scheme 29a). By contrast, the armed donor 64 gave a 38% yield of the O‑6 product, 210, but also 11% of the O3 regioisomer 211 (Scheme 29b). Analysis of these results according to conventional wisdom, dictates that the preference of both donors, 200 and 64, for the primary −OH was to be expected [1] but raised the question of the possible outcome of a three‐components double glycosylation when 200 and 64 compete for diol 207. Previous calculations had shown that the relative reactivity of these donors (k 64/k 200) is 3.2 [111]. Hence, it was expected that O6 mannosylation by the armed donor, 64, would predominate in any trimannan produced. Surprisingly, a single trimannan 212, in which the less reactive donor 200 ended up at O6 was obtained, even in the presence of 2 equiv. of the “more reactive” 64.

In situ three‐component double differential glycosylation of two donors and one diol acceptor
The Origin of Regioselectivity in Three-Component Couplings
In searching for the origin of the regioselectivity observed in the formation of trisaccharides 206 and 212 (Scheme 28 and Scheme 29) several factors were considered. The reactions in Scheme 28 and Scheme 29c were carried out with excess NIS promoter, conditions under which the intermolecular halonium ion transfer (responsible for the armed-disarmed effect) is not operative. A study of the three types of n-pentenyl donors indicated that their relative reactivities were in the order NPOE > armed > disarmed (e. g. 202a > 64 > 200) [111]. Therefore, the most and the least reactive donors have “chosen” their preferred −OH in the final trisaccharide. On the other hand, the most and the least reactive donors give rise to the highly delocalized, more stable intermediate 213, while the armed donor gives the less stable oxocarbenium ion 214 (Scheme 30) [112]. The conclusion was that in competitive glycosylations the more stable donor/intermediate (not the most reactive donor) controls regioselectivity, resulting in the formation of the single trisaccharides 206 and 212 and the single disaccharides 204a and 208.

Reactive intermediates from different glycosyl donors
In order to confirm this assumption the authors performed the experiments in Table 9 [113]. Equimolar amounts of armed and disarmed donors 64, and 200 or 202b were allowed to compete for one equivalent of acceptor 215 under the agency of NIS. When one equivalent of NIS was used the major product obtained was that of glycosylation of armed NPG 216, thus in agreement with a process of intermolecular halonium transfer and preferred reaction of the more reactive donor (Table 9, entries i, iii). When the amount of NIS was increased to three equivalents, the observed ratio of compounds 216 and 217 indicated enhanced coupling of the disarmed donor (Table 9, entries ii, iv), thus in agreement with the proposed rationalization for the regiopreferences observed in the three‐component reactions.
NPGs in Oligosaccharide Synthesis
Since their discovery, the unique properties of NPGs have allowed the preparation of several oligosaccharides. The pentenyl moiety may be installed early in the synthetic sequence and can survive many types of protecting group manipulations. Some selected syntheses of oligosaccharides are briefly discussed below.
The Pentasaccharide Core of the Protein Membrane Anchor Found in Trypanosoma brucei
Fraser-Reid and co-workers described a block (i. e., convergent), and a linear approach to the title compound, 226 (Scheme 31) [114]. The convergent approach, outlined in Scheme 31, makes use of the stereocontrolled glycosylation of inositol derivative 222 with 2-deoxy-2-imino NPG 221 (Scheme 31b). Protecting group manipulations led to acceptor 223 that was glycosylated with NPG 224, to furnish, after desilylation, the acceptor CDE block, 225. The donor counterpart 220, had been readily prepared by Koenigs–Knorr [115] coupling of NPG 219 with glycosyl bromide 218 (Scheme 31a). Finally, coupling of fragments AB (220) and CDE (225) promoted by NIS/TfOH led to pentasaccharide 226, in 73% as an α/β (2:3) mixture (Scheme 31c).

The pentasaccharide core of the protein membrane anchor of Trypanosoma brucei
The Nonamannan Component of High Mannose Glycoproteins
The concise approach to nonamannan 227 (Scheme 32), was greatly simplified with the sidetracking of NPGs [116,117] that allows the same NPG synthon to function as glycosyl donor or as glycosyl acceptor.

The nonamannan component of high mannose glycoproteins
In the retrosynthesis of 227, the authors identified three types of elements depending on the number of sugar units attached to them. Two components carried sugars at O3 and O6, four held substituents at O2, and the last three had no monosaccharides attached. According to that, the nonasaccharide target could be correlated with only two mannopyranose precursors 228 and 229, since synthon 228 could be used to access the last two kinds of sugars. The approach featured the final link of a pentasaccharide donor with a tetrasaccharide acceptor, as outlined in Scheme 32.
The synthesis of pentasaccharide donor 233 started with the mannosylation of sidetracked NPG 230 with disarmed NPG donor 228 (Scheme 33). The ensuing, stereoselectively formed disaccharide 231a, after unveiling of its C6-OH, underwent a second mannosylation with 228. Removal of the acetates in 232a with NH3/MeOH led to diol trisaccharide 232b, which was bis‐mannosylated with 228 to give pentasaccharide 233a in 59% yield. Regeneration of the pentenyl moiety in sidetracked 233a, with Zn/nBu4NI, granted access to pentasaccharide NPG donor, 233b.

Convergent synthesis of nonamannan 227. Synthesis of the pentasaccharide donor 233
The lowest antenna of 227 was built from 234 (Scheme 34). By taking advantage of the sidetracking concept, compound 228 could be used as a glycosyl donor or, after dibromination and deacetylation as the glycosyl acceptor 234, thereby facilitating the rapid assembly of trisaccharide fragment 235. Thus, coupling of 234 and 228 afforded the expected disaccharide in 73% yield, deacetylation and additional coupling with 228 led to trisaccharide 235a in 62% yield. The latter was transformed in glycosyl donor 235b by reductive elimination, and coupled with 230 to give sidetracked tetrasaccharide 236a. Dechloroacetylation of the latter led to 236b that was glycosylated by pentasaccharide donor 233b to give nonasaccharide 227 in 57% yield.

Convergent synthesis of nonamannan 227. Synthesis of the tetrasaccharide acceptor 236b and final assembly
Synthesis of NodRf-III (C18:1) (MeFuc)
Nodulation factors comprise a family of unique oligosaccharides composed substantially of glucosamine (2-amino-2-deoxy-d-glucose) units that are N‑acylated with acetic acid and fatty acids residues, the latter residing at the nonreducing terminus [118]. The block synthesis of NodRf-III (C18:1) (MeFuc) 237, a nod factor produced by Rhizobium fredii, is an illustrative example of the chemistry developed around 2-amino-2-deoxy-NPGs (Scheme 35) [119]. The key elements in this stereocontrolled synthesis are: (a) the use of the TCP protecting group, which provides a facile method for N‑differentiation in the glucosamine oligomer, (b) the assistance of the sidetracking methodology, (c) a solvent‐assisted stereoselective α‑fucosylation, (d) a β‑selective, neighboring group assisted, glycosidation, and (e) the use of FeCl3 for late-stage debenzylation of the oligosaccharide moiety [120,121]. In the retrosynthesis (Scheme 35), the authors selected a TCP as protecting group for the nitrogen atom that would bear the unique fatty acid, while the repeating unit would be a 2-deoxy-2-N‑phthaloyl NPG capable of acting as a glycosyl donor (e. g. 238). The reducing end retron was identified with benzyl glycoside 239.

Retrosynthesis of nodulation factor NodRf-III (C18:1) (MeFuc)
The disaccharide acceptor 239 was prepared in 85% yield by coupling (NIS/TESOTf ) of acceptor 241 with n-pentenyl fucoside 240 in Et2O:CH2Cl2, (5:1) (Scheme 36). The disaccharide donor 238 was assembled in 71% by coupling of NPG 242 with sidetracked acceptor 243 (NIS/TESOTf), followed by reinstating of the pent-4-enyl moiety from the dibromo pentenyl residue in 244. Final coupling (NIS/TESOTf) of donor 238 with acceptor 239 yielded tetrasaccharide 245 in 65% yield. The final stages in the preparation of 237 involved: (i) FeCl3 debenzylation, (ii) silylation of the resulting free –OHs, (iii) deprotection of the TCP and condensation with an activated fatty acid, (iv) removal of the phthalimido protecting groups, and (v) acylation, saponification, and desilylation.

Synthesis of nodulation factor NodRf-III (C18:1) (MeFuc)
Synthesis of Phosphorylated Rat Brain Thy-1 Glycosylphosphatidylinositol Anchor
Glycosylphosphatidylinositol (GPI) membrane anchors constitute a class of glycolipids that covalently link certain proteins to cell and virion surfaces [122,123]. A boost in their chemistry occurred in 1988 when Ferguson et al. reported the first covalent structure of a member of this family [124,125]. The first synthesis of a fully phosphorylated GPI, compound 246 (Fig. 5), was accomplished by Fraser‐Reid's group based entirely on NPG chemistry [126,127,128,129,130].

Rat brain Thy-1 GPI anchor 1, 246
The retrosynthetic analysis dictated a heptasaccharide 251, with all free hydroxyls in the final product benzylated, and the three sites of phosphorylation all differentially protected, so that all three can be manipulated separately for maximal flexibility. The free amine of glucosamine is protected as an azide, which can be taken through multiple transformations and will only be unmasked at the end of the synthesis (Scheme 37). The heptasaccharide is in turn put together in three portions, a galatosaminylmannose 249 being coupled to azidoglucosylinositol 250 and then a trimannose, 247, being coupled to that moiety. Coupling of glycosyl donor 249 with the disaccharide acceptor 250 was carried out with NIS/TESOTf to give α‑linked tetrasaccharide 248 in 66% yield. Notably, the allyl protecting group survived the treatment with NIS. The O6 of the mannose residue was deprotected by removal of the chloroacetate moiety with thiourea and glycosylated with pentenyl trimannoside 247 to give the fully protected heptasaccharide 251 in 39% yield. The three positions (marked with arrows) were then deprotected and phosphorylated according to the following sequence: dechloroacetylation with thiourea, saponification of the acetate with methoxide, and deallylation with PdCl2. Complete debenzylation, then culminated the synthesis of 246.

Synthesis of rat brain Thy-1 GPI anchor 1
Synthesis of the Glycopeptidolipid of Micobacterium avium Serovar 4

Synthesis of the glycopeptidolipid of Micobacterium avium Serovar 4, 252
Heidelberg and Martin described the first synthesis of the “polar mycoside C” 252 (Scheme 38) [131]. The synthesis was based on the disconnection of the final structure into three saccharidic building blocks, an l‑rhamnosyl pseudodipeptide 254, a 6-deoxy-l-talosyl dipeptide 255, and a pentenyl trisaccharide donor 257. The key steps were the creation of the glycosidic linkage between the trisaccharide donor 257, and the 6-deoxy-l‑talose unit 255 (IDCP, 60% yield), and the final coupling of the two glycopeptide fragments. Other pentenyl mediated couplings were the glycosylation of orthoester 253 leading to 254 (NIS/TMSOTf, 81%) and the stereoselective α‑coupling of disarmed NPG 256 to give glycodipeptide 255 (NIS/TMSOTf, 70%).
Synthesis of Oligogalacturonates Based on NPGs

Retrosynthesis of oligogalacturonates for conjugation to bovine serum albumin
Madsen and co-workers described a concise approach to oligogalacturonates (e. g. 258, Scheme 39) conjugated to bovine serum albumin (BSA) based on NPGs. They synthesized several oligogalacturonates, which were linked to the BSA by reductive amination via an aldehyde spacer at the reducing end (Scheme 39) [48,132,133]. Their strategy called for two orthogonal protecting groups (P1 and P2), and three different monomeric building blocks: a spacer galactoside C to serve as glycosyl acceptor for the reducing end, and two glycosyl donors A and B, the former for the nonreducing end and the latter for the galacturonic repeating unit. p-Methoxyphenyl (PMP) and acetyl groups were used as protecting groups. The methodology was then based on the repeated coupling of galactose donors onto galactose acceptors followed by deprotection at O6, as in 259, which permitted the oxidation of these primary positions to either the carboxylic acids or methyl esters.
The attaching of the spacer to galactose (i. e. 261, building block C) was carried out by glycosylation, under Lemieux conditions [134], of a glycosyl bromide readily obtained from NPG, 260 (Scheme 40). Coupling of NPG 260 with galactosyl bromide 262 (AgOTf, 71%) led to pentenyl disaccharide 263 that glycosylated acceptor 261 (NIS/TESOTf, 71%) to give trisaccharide 264a. Deprotection of the latter to 264b, and glycosylation with glycosyl donor 263 (NIS/TESOTf, 90%) led to pentasaccharide 265a. Glycosyl assembly to hexasaccharide 267, included the glycosylation of pentenyl donor 266 with acceptor 265b (NIS/TESOTf, 69%), obtained by NaOMe treatment of 265a. The final stages of the synthesis included CAN-mediated deprotection of the p-methoxyphenyl groups, Dess–Martin oxidation and esterification.

Synthesis of oligogalacturonates
Miscellaneous
Arasappan and Fraser-Reid reported an NPG-based methodology for the stereoselective construction of the tetrasaccharyl cap portion of Leishmania Lipophosphoglycan [135].
Kuzuhara and co-workers have reported the use of a NPG disaccharidic synthon as the chain elongating unit in the synthesis of amphiphilic chitopentaose and chitoheptaose derivatives [136].
Toshida and co-workers have described the synthesis of a set of di- and tri-sulfated galabioses by using an n-pentenyl galactoside donor and IDCP as the catalyst [137].
NPGs in Solid-Phase Oligosaccharide Synthesis
Solid-phase oligosaccharide synthesis has received considerable attention in the last years [138]. Some of the approaches that involve NPGs are discussed below.
Glycosylation of Supported Alcohol Acceptors with NPG Donors
Fraser-Reid and co-workers designed a photolabile o-nitrobenzylic linker 268, which was used in the synthesis of a branched trimannan oligosaccharide 271 (Scheme 41) [139]. Differentially protected NPG 269 was coupled to the resin via linker 268. Selective removal of the C6 chloroacetyl and C3 acetyl groups, followed each by mannosylation (NIS/TESOTf) with NPG 64, afforded trimannan 271, in 42% overall yield after photolytic cleavage.

NPGs in solid-phase oligosaccharide synthesis
In a related approach, Fraser-Reid and co-workers used Chiron's polystyrene‐grafted “crowns” with Rich's photocleavable o-nitrobenzyl linker [140] and NPG donors in the synthesis of trisaccharide 277 (Scheme 42) [141]. After attachment of the first aminoglucosyl moiety to the linker, via its corresponding NPG, the C6 dinitrobenzoyl (DNB) group was removed to give 272b. Coupling with mannose donor 273, deprotection of the O2 chloroacetyl group, and galactosylation with NPG 275 furnished trisaccharide 276. Global deprotection followed by peracetylation and photolytic cleavage from the solid support provided trisaccharide 277.

NPGs in solid-phase oligosaccharide synthesis
Pentenyl Glycoside-Based Linkers
Seeberger and co-workers developed a new linker concept in solid-phase oligosaccharide synthesis. They designed a new NPG-based linker that upon deprotection rendered an oligosaccharide NPG suitable for further glycosylations in fragment couplings (Scheme 43) [142]. The first carbohydrate moiety (e. g. 278) was connected via a glycosidic bond to octenediol‐functionalized Merrifield's resin, 279. Resins with loadings of up to 0.65 mmol/g were obtained and employed in oligosaccharide synthesis. Glycosylation events can now take place in deprotected saccharide 280 to yield oligosaccharide 282. The octenediol linker was then cleaved by olefin cross metathesis using Grubbs' catalyst under an atmosphere of ethylene to afford fully protected oligosaccharides in the form of NPGs, e. g. 283. A further refinement of this strategy is that it can be made compatible with glycosyl donors that require electrophiles as activators by sidetracking the linker to the corresponding dibromooctane derivative (e. g. 284) [143]. Seeberger and co-workers have illustrated the potency of this strategy with several oligosaccharide syntheses [144,145,146,147].

Seeberger's NPG-based linkers for oligosaccharide synthesis
Mogemark et al. described a fluorinated selenide linker 285, for solid-phase synthesis of NPGs (Scheme 44) [148]. The resin-bound linker could be glycosylated both with trichloroacetimidates and glycosyl fluorides to give anchored saccharides, e. g. 286, that can be submitted to glycosylation once deprotected. After oxidation to a selenoxide with t-BuOOH the linker undergoes β‑elimination upon heating, and releases the NPG 287, in excellent yield.

A fluorinated selenide linker for solid-phase synthesis of NPGs
Miscellaneous Uses of NPGs
The versatility of NPGs has been further enhanced by chemical modifications of the pent-4-enyl moiety itself. In this context, the pentenyl moiety has been transformed in many spacer functionalities [149,150], used as a handle to incorporate amino-acid moieties [151,152,153,154], used as a monomer in copolymerization strategies [155,156,157], used in the formation of dendrimers [158], and converted to dimeric and trimeric structures for multivalent presentations [159,160]. These applications fall beyond the scope of this chapter.
Preparation of NPGs
NPGs, being normal O‑glycosides, can be readily obtained by application of the standard procedures for preparing such derivatives [12]. They can be obtained by Fischer glycosidation (Scheme 45a) [82,161,162]. An obvious advantage of this procedure is that the n‑pentenyl group can be installed right at the outset of the synthesis; however, the formation of α/β anomers might sometimes be a drawback. Use of the Koenigs–Knorr coupling [115] permits the stereocontrolled preparation of NPGs (Scheme 45b). SnCl4 facilitates the formation of NPGs from acetyl mannosides (Scheme 45c) [117]. The most useful method for the preparation of NPGs is, arguably, the acid‐catalyzed rearrangement of NPOEs, prepared under Lemieux–Morgan conditions (Scheme 45d,e) [163]. This method permits the stereoselective synthesis of NPGs with different protecting groups [50,164]. Rousseau and Martin described the rearrangement of acetyl NPOEs with TMSOTf (Scheme 45d) [99], and Fraser-Reid and co-workers have used TBSOTf [165] or ytterbium triflate [166] to rearrange benzoyl‐substituted NPOEs (Scheme 45e).

Methods for the preparation of NPGs
Enol Ether-Type Glycosides
Early Contributions

Synthesis of 1′-substituted vinyl glucosides
De Raadt and Ferrier were the first to report the preparation and attempted glycosylation of 1′-substituted-vinyl glycosides [167]. Reaction of tetra-O‑acetyl-α‑d-glucopyranosyl bromide (288) with bis(acetonyl)mercury derivatives 289a–c in refluxing chloroform afforded vinyl-, isopropenyl-, and styryl-β‑d-glucosides 290a–c in excellent yields (Scheme 46). However, when 290a–c were each treated with either NBS or bromine/AgClO4 in the presence of methanol no glycosides were formed, the products in each case being mixed stereoisomers of the glycosyl acetals 291a–c.

Synthesis of 1′-substituted vinyl glycosides
Schmidt and co-workers described the preparation of vinyl glucosides 292 from the reaction of tetra-O‑benzyl glucose with ethyl phenyl propiolate under the agency of sodium hydride (Scheme 47) [168]. The reaction of 292, as an anomeric mixture, with various acceptors was examined in acetonitrile at −40 ℃ in the presence of TMSOTf as catalyst. Reaction of 2 with 6-OH and 4-OH methyl glucosides as acceptors gave the corresponding disaccharides in 61 and 67% yield and as 85:15 and 75:25 β/α mixtures, respectively. Similar results were obtained for tetra-O‑benzyl galactose.
Isopropenyl Glycosides
Sinaÿ and co-workers described the synthesis of isopropenyl glycosides [169] by reaction of the corresponding anomeric acetates with the Tebbe reagent [170]. Reaction of 1-O‑acetyl-2,3,4,6-tetra-O‑benzyl-d-glucopyranose (294) with a solution of Tebbe reagent in toluene gave the isopropenyl glycosides 295, in 87–90% yields (Scheme 48a). Likewise, isopropenyl galactoside 297 (α:β ≈ 1:1) was prepared from the corresponding acetate 296 by Tebbe methylenation in 88% yield (Scheme 48b). Treatment of 295 (α:β = 4:1) in MeCN at −25 ℃ with the primary hydroxyl acceptor 298, in the presence of TMSOTf gave the disaccharides 299 (68%) with an excellent β‑selectivity (20:1) (Scheme 48c). The condensation of 295 with the secondary alcohol 300 in MeCN at −25 ℃ in the presence of BF3·Et2O afforded the disaccharide 301 in good yield, albeit with reduced stereoselectivity (β:α = 5:1) (Scheme 48d). When the same glycosylation was carried out in CH2Cl2 instead of MeCN the disaccharide 301 was obtained in limited yield (Scheme 48e). The successful glycosylation of phenyl 1-thio‐glycoside 302 with 295 in the presence of TMSOTf illustrates the usefulness of isopropenyl glycosides in the synthesis of thiophenyl disaccharides (e. g. 303, Scheme 48f). The authors found no significant variations on yield or stereoselectivity by the use of either mainly α or mainly β isopropenyl derivatives. The best results for galactosylation were achieved in CH2Cl2 with TMSOTf as promoter (Scheme 48g).

Synthesis and glycosylation reactions of isopropenyl glycosides
Chenault and co-workers reported the use of O‑isopropenyl glycosides bearing ester protecting groups [171,172]. These compounds are stable at room temperature and can be readily purified by column chromatography on silicagel, moreover their glycosylation would proceed to give β‑glycosides via neighboring group participation. The reaction of bis(acetonyl)mercury [173] with glycopyranosyl halides proved to be a good method for the preparation of isopropenyl β‑glycopyranosides (e. g. 305, Scheme 49a). The authors described routes to O‑isopropenyl 2,3,4,6-tetra-O‑pivaloyl-α, and β‑d‑glucopyranosides α‑307 and β‑307, respectively. Reaction of 2,3,4,6-tetra-O‑pivaloyl-α‑d-glucopyranosyl bromide (306) with diacetonyl mercury led to β‑307 (Scheme 49b), whereas regioselective methylidenation [174] of 309 (prepared stereoselectively by acid‐catalyzed exchange of the anomeric pivaloyloxy group of penta-O‑pivaloyl-β‑d-glucopyranose, 308) generated α‑307 as the only product (Scheme 49c). The β‑isomer, however, exhibited greater shelf life than the latter.

Synthesis of pivaloyl isopropenyl glycosides
On the basis of the reaction of NPGs with electrophiles, Chenault et al. considered the possible activation of isopropenyl glycosides with electrophiles. The mechanism of activation was expected to involve initial capture of the electrophile (E+) by the vinyl ether double bond of 310 leading to the formation of cation 311 or 312 (Scheme 50). Collapse of 311 or 312 to form glycosyl oxocarbenium ion 313 and acetone derivative 314 would be followed by nucleophilic attack on 313 to generate glycoside 315. An alternative reaction would involve direct nucleophilic attack on 311 or 312 to generate the addition product 316.

Activation of isopropenyl glycopyranosides
The authors found that “armed ” and “disarmed” isopropenyl glycosides displayed different behavior towards electrophiles (Scheme 51). Armed isopropenyl glycoside β‑294, glycosylated acceptor 40 to give disaccharide 317 under the agency of IDCP, a relatively weak electrophile, in a nonpolar solvent (CH2Cl2) (Scheme 51a). On the other hand, disarmed glycoside β‑307, led under the same conditions to the electrophilic addition product 318. Use of a more potent electrophile (NIS/TfOH) in CH2Cl2 also resulted in the formation of the addition product 318 (Scheme 51b). However, NIS/TfOH in more polar MeCN successfully promoted the glycosidic coupling (Scheme 51c). Apparently, the relatively electron‐releasing ethereal protecting groups lower the energy barrier to oxocarbenium ion formation from armed β‑294 relative to that from disarmed glycoside β‑307. In general, factors which favor the formation of the glycosyl oxocarbenium ion (strong electrophile, polar solvent, electron‐releasing protecting groups on the glycosyl donor) lead to transglycosylation. Factors which retard the formation of the glycosyl cation (weak electrophile, nonpolar solvent, electron‐withdrawing protecting groups on the glycosyl donor) lead to addition across the isopropenyl ether double bond.

Reaction of armed and disarmed isopropenyl glycopyranosides
The ability of various electrophiles to promote transglycosylation of disarmed isopropenyl glycosides is outlined in Table 10. NIS/TfOH, TMSOTf, and Tf2O in MeCN, all led to the formation of disaccharide 319 in good yield (Table 10, entries i–iii). Reactions were carried out at 0 ℃ and were complete within 2–5 min. With silver triflate (AgOTf) the reaction was slower and gave a lower yield of disaccharide 319 (Table 10, entry iv). When TfOH, NIS, or NBS were used alone β‑307 failed to react and the glycosyl donor was recovered unchanged (Table 10, entries vi, viii). Thus, neither NIS/TfOH, TMSOTf, Tf2O, nor AgOTf seem to activate isopropenyl glycosides by acting as a source of TfOH. Dimethyl(methylthio)-sulfonium triflate (DMTST) was the only promoter that led exclusively to the formation of disaccharide 319 from β‑307 when CH2Cl2 was used as the solvent.
In terms of glycosyl donors, either α‑307 or β‑307 gave the same results in terms of yields. Likewise, isopropenyl galactopyranosides reacted in a similar manner to glucopyranosides. Acylated isopropenyl donors gave lower yields than pivaloyl analogs, presumably because of complications due to orthoester formation [175].
Isopropenyl glycosides could be activated selectively in the presence of armed NPGs, and that allowed a one-pot synthesis of trisaccharide 322 involving the successive glycosyl coupling of a vinyl glycoside β‑307, and an NPG, 321 (Scheme 52).

One-pot synthesis of trisaccharide 322
3-Butene-2-yl Glycosides as Precursors for Vinyl Glycosides
Boons and co-workers introduced stable allyl glycosides (e. g. 323, Scheme 53), which are converted to the enol ether-type glycosides 324, prior to glycosylation [176].
Latent-Active Glycosylation Strategy
The allyl glycoside 323, can be considered a “latent” [177] form of a glycosyl donor which can be efficiently isomerized to the “active” vinyl glycoside, 324. The isomerization reaction was performed by a rhodium catalyst obtained by treating the Wilkinson's catalyst, (Ph3)P3RhCl, with BuLi [178]. Base labile functionalities in the molecule are compatible with these isomerization conditions [179]. The “active” vinyl glycoside 324, undergoes Lewis acid‐catalyzed glycosylation reactions with “latent” allyl glycoside 325, to give “latent” disaccharide 326 (Scheme 53). Unlike isopropenyl glycosides, which require stoichiometric amounts of Lewis acids for activation [169], the reaction of Boons' vinyl glycosides only demands catalytic amounts of TMSOTf. The higher reactivity of the substituted vinyl glycoside was ascribed to the additional methyl substituent of the vinyl moiety that makes the double bond more electron rich. Although racemic 3-buten-2-ol could be used for the preparation of 323 without affecting its reactivity, the use of diastereomeric allyl glycosides can be avoided with the use of optically pure 3-buten-2-ol, easily obtainable in multigram amounts.

Vinyl glycoside-based latent‐active strategy for glycosyl coupling
The use of neighboring participating groups permits the formation of 1,2-trans glycosides (e. g. 326, Scheme 53). The choice of solvent and, to some extent, the choice of activator, was used to control the α/β ratio in glycosyl donors without participating groups at O2. TMSOTf‐promoted condensation of 327 with 328 in MeCN gave disaccharide 329 as the β‑anomer mainly (α/β = 1:8) (Table 11, entry i). An improved α‑selectivity was obtained (73%, α/β = 3:1) when the coupling was performed in ether/dichloroethane (Table 11, entry iv).
Preparation of Trisaccharide Libraries
Linear Trisaccharide Libraries
Boons et al. described an approach to combinatorial synthesis of trisaccharide libraries based on their latent‐active glycosylation strategy [180]. One major building block, 330 (i. e. B 1, Scheme 54) can be converted into a glycosyl donor 331 (i. e. D 1) and a glycosyl acceptor 332 (i. e. A 1). Coupling of compounds 331 and 332 gives disaccharide 333a in excellent yield (the anomeric ratio can be greatly influenced by changes in the temperature: α/β = 1:20 at low temperature; α/β = 1:1 at ambient temperature). The latter can be converted into a glycosyl acceptor 333b (i. e. D 1 A 1) by removing the acetyl protecting group and into a glycosyl donor by isomerizing the allyl moiety. These compounds can be used in oligosaccharide synthesis, as outlined in Scheme 54, for example by coupling 333b with 332 to give trisaccharide 334 (i. e. D 1 D 1 A 1). Application of this strategy to four allyl building blocks (B 1→4) would lead to four vinyl glycosyl donors (D 1→4) and four allyl glycosyl acceptors (A 1→4). Individual glycosylations of each donor with each acceptor will furnish 16 disaccharides (D 1→4 A 1→4) (if glycosylations are stereoselective, or 32 disaccharides if conditions are met for 1:1 anomeric selectivity). Next, the disaccharides can be mixed, and removal of the acetyl groups will give an assortment of acceptors. The pool of compounds can be split, and in combinatorial steps each pool of glycosyl acceptors can be coupled with a particular glycosyl donor (D 1→4) resulting in four libraries of 32 (or 64, as above) trisaccharides each.

Preparation of linear trisaccharide libraries
Branched Trisaccharide Libraries
Biologically important oligosaccharides often contain more complex features such as branching points and further functional groups. In this context, Boons and co-workers, using the latent‐active strategy, designed a synthetic method to create orthogonally protected saccharides (acetyl and p-methoxybenzyl groups were used as orthogonal protecting groups) that could be easily further derivatized [181]. Thus, a common allyl glycoside building block (e. g. 335, Scheme 55) can be converted to two vinyl glycoside donors bearing orthogonal protecting groups (e. g. 336 and 338), and to an allyl glycosyl acceptor 337, bearing one free hydroxyl and one selectively removable PMB ether. The latter will be coupled with donor 336 bearing an acetyl protecting group to give an orthogonally protected disaccharide, 339. Compound 339 can be elaborated into linear or branched trisaccharides 342 and 343. Thus, deprotection of the acetyl group in 339, and glycosylation with vinyl donor 336 will yield linear trisaccharide 342, whereas removal of the PMB group and coupling with 337 will produce orthogonally protected branched trisaccharide 343.

Preparation of branched trisaccharide libraries
3-Buten-2-yl 2-amino-2-deoxy Glycosides as Glycosyl Donors
Boons and co-workers studied the use of 3-buten-2-yl 2-azido-2-deoxy, and 2-deoxy-2‑phthalimido glycosides, as building blocks for the preparation of sugar containing oligosaccharides [182]. Vinyl glycoside donors 344, 346, and 348, were uneventfully prepared by isomerization of the corresponding 3-buten-2-yl glycosides with (Ph3)P3RhCl/BuLi in yields exceeding 90%. Several glycosyl acceptors were used in the study, although representative data in Scheme 56 refer solely to acceptor 300. The glycosylation with azido donor 344, in MeCN using TMSOTf as the promoter at −30 ℃, proceeded with high β‑selectivity (Scheme 56a), whereas NIS/TMSOTf in a dioxane/toluene mixture gave good α‑selectivities (Scheme 56c). 2-Buten-2-yl 2-deoxy-2‑phthalimido glycosides 346 and 348 reacted in CH2Cl2 in the presence of a catalytic amount of TMSOTf to give only the β‑linked disaccharides 347 and 349, respectively.

3-Buten-2-yl derivatives for the synthesis of amino sugar containing disaccharides
An Approach for Heparin Synthesis Based on 3-Buten-2-yl Glycosides
Haller and Boons described an approach fully based on 3-buten-2-yl glycosides for the synthesis of trisaccharide 350 and sulfated disaccharide 351 (Scheme 57) [183]. In their strategy the glucuronic acid moieties were introduced at a late stage of the synthetic sequence by selective oxidation of primary hydroxyl groups with TEMPO and NaOCl. “Latent” allyl glycoside 353 functioned as an acceptor for the reducing end in compounds 350 and 351, and was also transformed to “active” vinyl glycoside 352, for the nonreducing unit of 350. The 2‑acetamido-2-deoxy unit in 350, was retrosynthetically correlated with 2-azido-2-deoxy glycosyl donor 354.

3-Buten-2-yl derivatives for the synthesis of amino sugar containing oligosaccharides
Conversion of 2-Buten-2-yl Glycosides to Other Glycosyl Donors
Treatment of “active” vinyl glycosides with NIS/TMSOTf in CH2Cl2 in the presence of dibenzyl phosphate gives good yield of glycosyl phosphates [184].
2-Buten-2-yl glycosides can also be transformed to glycosyl fluorides and trichloroacetimidates by hydrolysis to the corresponding hemiacetal (HgO, HgBr2, aq. acetone) followed by standard treatment (CCl3CN, DBU, CH2Cl2 or DAST, THF, respectively) [183].
Synthesis of 3-Buten-2-yl Glycosides
Being normal alkyl glycopyranosides, 3-buten-2-yl glycosides can be prepared by standard glycosylation methods, as previously mentioned for NPGs.
Oxathiines: Vinyl Glycosyl Donors for the Synthesis of 2-Deoxy Glycosides
Cycloadduct 357, readily available by cycloaddition of tri-O‑benyzl glucal (355) with the electron-poor 3‑thioxopentane-2,4-dione (356) [185] has been used by two research groups as precursor glycosyl for vinyl glycosyl donors 358, 361, and 363 (Scheme 58). Franck and co-workers showed that glycoside 358, prepared by methylenation of 357, underwent β‑selective glycosylation with a variety of glycosyl acceptors in the presence of TfOH to give glycosides 359, in good yields [186]. Moreover, Raney nickel desulfurization of 359 granted access to 2-deoxy‐glycosides 360 [187]. Capozzi and co-workers reported that acetyl [188], and silyl [189] derivatives 361 and 363, also functioned as glycosyl donors in reactions catalyzed by MeOTf in nitromethane and TMSOTf in CH2Cl2, respectively. The timing in the quenching of the reactions is crucial for obtaining completely selective β‑glycosylations, and prolonged reaction times led to α/β anomeric mixtures. The total β‑stereoselectivity of the coupling was ascribed by Capozzi and co-workers to an S N 2 type reaction (361→362, Scheme 59) that induces β‑stereospecific glycosylation [188]. The observed subsequent α/β–equilibration presumably proceeds through an oxonium intermediate 364 (Scheme 59).

Oxathiines: vinyl glycoside donors for the synthesis of 2-deoxy glycosides

Stereocontrol in the formation of 2-deoxy glycosides from oxathiines
DISAL Glycosyl Donors
Synthesis and Glycosylation Reactions
Petersen and Jensen reasoned that glycosides of phenols (e. g. 366) carrying sufficiently electron‐withdrawing substituents could possibly serve as O‑glycosyl donors under neutral or mildly basic conditions (Scheme 60) [190,191]. Carbohydrate hemiacetals have been used as nucleophiles in aromatic substitutions using activated fluoroarenes [192,193]. Accordingly, glycosides of methyl 2‑hydroxy-3,5‑dinitrobenzoate (DISAL, a DInitroSALicylic acid derivative), e. g. 367 (Scheme 61), and methyl 4‑hydroxy-3,5‑dinitrobenzoate (para-isomer) were prepared by reaction of carbohydrate hemiacetals with the corresponding activated fluoroarenes in the presence of a base. The use of 4-(N,N-dimethylamino)pyridine (DMAP) gave an α/β ratio similar to the starting 1-OH, i. e. predominantly α. In contrast, the formation of β‑DISAL donors was favored using 1,4‑dimethylpiperazine as base. The fluoroarenes were prepared by nitration of 2‑fluoro- or 4‑fluoro‐benzoic acid.

Glycosides of phenols with electron‐withdrawing substituents as glycosyl donors
The preparation of disaccharides, from benzyl‐protected DISAL donors (e. g. 367, Scheme 61), was best carried out in 1‑methylpyrrolidin-2-one (NMP), a high polar, aprotic solvent, at 40 ℃, in the absence of Lewis acids (Scheme 61a,b). The fact that glycosylations also occurred in the presence of base (e. g. Et3N, 2,6‑lutidine) indicated that the glycosylations were not auto‐catalytically promoted by the released phenol. Under these conditions, galactose derivative 40 was glycosylated with DISAL donor 367 (1.5 equiv.) to give disaccharide 368 in 90% yield (α/β = 2.4:1). Glycosylation of a secondary hydroxyl group with DISAL donor 367 required increasing the temperature to 60 ℃, and resulted in the formation of disaccharide 370 as the α‑glycoside in 74% yield (Scheme 61b). The para-glycosyl donor, (vide supra) also proved effective in analogous glycosylations.

Glycosylation with DISAL glycosyl donors
Unlike benzyl‐protected DISAL donors, benzoyl‐protected donors, e. g. 371, did not give the expected glycosides under these neutral conditions, in part due to trapping of intermediates as the orthoesters (Scheme 61c). Lewis acids, such as BF3·Et2O or TMSOTf, activated the acylated DISAL donor 367, albeit diisopropylidene acceptors 40 and 369 were not stable in the reaction media [194]. More robust benzyl‐protected acceptors were glycosylated with alkylated and acylated DISAL donors in the presence of BF3·Et2O to give disaccharides 372 and 373 in 82 and 46% yield, respectively (Scheme 61d,e). Interestingly, LiClO4 was found to be an efficient additive for activation of DISAL donors in nonpolar solvents, giving significantly higher yields of disaccharides than BF3·Et2O (Scheme 61f). Acylated DISAL donor 371 did not give good yield of disaccharides when reacting with secondary hydroxyl acceptors (Scheme 61g). More recently, Jensen and co-workers have shown that high‐temperature glycosylation of DISAL donors using precise microwave heating results in improved yield of disaccharides (Scheme 61h) [195].
DISAL Donors in Solid-Phase Synthesis
This approach was extended to solid-phase glycosylation of d‑glucosamine derivatives anchored by the 2-amino group through a Backbone Amide Linker to a solid support [196].
Intramolecular Glycosylation Approach to the Synthesis of 1,4-Linked Disaccharides
The DISAL donor concept was developed further to allow intramolecular glycosylations [197]. The glycosyl donor and acceptor were linked through the DISAL leaving group positioned to facilitate intramolecular glycosyl transfer to 4-OH by a 1,9‑glycosyl shift (Scheme 62a). The tethered glycoside 381 underwent intramolecular transglycosylation to form the 1,4‑linked mannoside 382 as an anomeric mixture (α/β = 3.7:1) in moderate yield (Scheme 62b).

DISAL-Based intramolecular glycosylation approach to 1,4-linked disaccharides
Application of DISAL Donors to Oligosaccharide Synthesis
Jensen and co-workers reported the synthesis of hexasaccharide 383, a starch‐related hexasaccharide (Scheme 63) [198]. Their approach was based on the use of DISAL disaccharides 384 and 385, readily obtained from the corresponding disaccharide hemiacetals, for sequential glycosylations. Glycosylation of phenyl 1-thio disaccharide 386 with DISAL donor 385 took place with good yield and excellent α‑selectivity in CH3NO2 in the presence LiClO4 and Li2CO3. The trityl group that have survived the coupling, was next removed and the ensuing tetrasaccharide glycosylated with DISAL donor 384 (LiClO4, Li2CO3, (CH2Cl2)2, 35 ℃, 38% yield, α/β = 3:2).
DISAL donors have also been used in the preparation of phenazine natural products and analogs [199].

Synthesis of hexasaccharide 383 based on DISAL donors
2-Deoxy-2-amino Derivatives as DISAL Donors
Jensen and co-workers evaluated the behavior of different glucosamine‐derived DISAL donors in glycosylation reactions [200]. N‑tetrachlorophthaloyl (TCP), N‑trifluoroacetyl (TFAc), and N‑trichloroethoxycarbonyl (Troc) DISAL donors 387, 388, and 389 and 390, respectively, were prepared from the corresponding hemiacetals (Fig. 6). Glycosylation of cyclohexanol, in NMP at 60 ℃, with these donors took place with yields ranging from 35 to 76%. The N‑TCP protected donor 387, was the least reactive. N‑Troc protected donors 389 and 390, gave the highest glycosylation yields with monosaccharides (63–71% yield), although they displayed lower selectivities with primary hydroxyl acceptors (α/β ratio, from 1:1 to 1:7). A secondary hydroxyl acceptor was glycosylated with N‑Troc DISAL donor 389 under microwave heating (130 ℃, CH3NO2, LiClO4) to give the corresponding disaccharide in 38% yield (β‑anomer only). N‑TFAc DISAL donor 388 gave even lower yields on coupling reactions with primary hydroxyl acceptors (35–45%) although β‑disaccharides were obtained exclusively.

Glucosamine‐derived DISAL donors

2′-Carboxybenzyl (CB) glycosides
2′-Carboxybenzyl (CB) Glycosides

2′-Carboxybenzyl glycoside-based “latent‐active” strategy for glycosyl coupling
Kim and co-workers introduced a novel type of O‑glycosyl donor, the 2′-carboxybenzyl (CB) glycoside 391b, readily available by selective hydrogenolysis of the benzyl ester functionality of 2-(benzyloxycarbonyl)benzyl (BCB) glycosides , 391a [201,202,203]. Lactonization of the glycosyl triflate 392, which was derived from the CB glycoside 391b, is the driving force for the facile generation of the oxocarbenium ion 394 (Scheme 64). Reaction of 394 with the glycosyl acceptor (Sugar–OH) would give the desired saccharide 395. In the course of the transformation, a non‐nucleophilic phthalide 393 is extruded. Treatment of CB glycosides with Tf2O in the presence of di-tert‐butylmethylpyridine (DTBMP) at −78 ℃ and subsequent addition of the glycosyl acceptor afforded the expected disaccharides in excellent yields.
β-d-Mannosylation Employing 2′-Carboxybenzyl Glycosyl Donors
The stereospecific formation of β‑mannopyranosyl linkages is a challenging task in oligosaccharide synthesis [204]. Crich and co-workers found that 4,6-O‑benzylidene‐protected glycosyl sulfoxides or thioglycosides are useful donors in the construction of β‑mannopyranosyl linkages [205,206,207,208,209]. Kim and co-workers have shown that CB glycosides with a 4,6‑benzylidene group can also be applied for stereoselective β‑mannopyranosylation . Glycosylations of primary alcohol acceptors, 398 and 399, in CH2Cl2 were completed in 1 h at −78 ℃ to afford only β‑mannosides in high yields (Table 12 entries i, iii). Toluene was also found to be a good solvent (Table 12, entry ii). This high β‑selective mannosylation was also achieved with secondary alcohols, e. g. 369, 400, and with hindered tertiary alcohol 129 (Table 12, entries iv–vi). Glucosyl CB donors possessing the 4,6‑benzylidene group gave high yields of α‑glucosides.
Latent-Active Glycosylation Strategy
A remarkable feature of 2′-carboxybenzyl glycosides (e. g. 391b, Scheme 64) is that they can be used as a latent‐active pair, together with their synthetic precursors 2-(benzyloxycarbonyl)benzyl (BCB) glycosides (e. g. 391a, Scheme 64). The successful mannosylation of “latent” BCB‐glycoside 401 with “active” CB glycoside 396, to give disaccharide 402a indicated that a sequential glycosylation strategy for oligosaccharide synthesis would be possible (Scheme 65). Thus, BCB disaccharide 402a was readily converted into the active CB disaccharide 402b by selective hydrogenolysis (92%, in the presence of benzyl and benzylidene groups), which upon treatment with Tf2O/DTBMP glycosylated the latent BCB glycoside 401 to yield trisaccharide 403 in 72% yield.
Stereoselective Construction of 2-Deoxyglycosyl Linkages
Kim and co-workers have developed a highly α‑ and β‑ stereoselective (dual stereoselective) [210] method for the synthesis of 2‑deoxyglycosides by employing CB 2‑deoxyglycosides as glycosyl donors. Glycosylation of the 4,6-O‑benzylidene‐protected glycosyl donor 404 with secondary alcohols afforded predominantly β‑glycosides (Table 13, entries iii–v). Complete reversal of the stereoselectivity, from β to α, was observed in the glycosylation of secondary alcohols with benzyl‐protected glycosyl donor 406 (Table 14, entries iii–v). On the other hand, glycosylation of primary hydroxyl acceptors with both donors did not show appreciable stereoselectivity (Table 13 and Table 14, entries i, ii). The authors suggested that the secondary hydroxyl acceptors formed β‑disaccharides by S N 2-like displacement of an α‑triflate favored in 4,6-O‑benzylidene derivatives, as previously mentioned in the formation of β‑mannosides of 4,6-O‑benzylidene derivatives. No or poor β‑selectivity in the reaction of 404 with primary alcohols was interpreted assuming that the more reactive primary alcohols reacted both with the α‑triflate and an oxocarbenium ion.
2′-Carboxybenzyl Furanosyl Donors . Acceptor-Dependent Stereoselective β-d‑Arabinofuranosylation
Kim and co-workers reported recently that CB tribenzyl-d-arabino furanoside 409 (easily available from methyl tribenzyl-d-arabinofuranoside) could be efficiently applied in stereoselective β‑arabinofuranosylation processes [211]. They found that the presence of acyl‐protective groups on the glycosyl acceptors was essential for attaining β‑stereoselective glycosyl couplings . Thus, reaction of donor 409 with acceptor 398 having benzoyl‐protective groups afforded a β‑disaccharide almost exclusively (β/α = 99:1) in 97% yield (Table 15, entry i), while the same reaction with acceptor 42 having benzyl‐protective groups gave a mixture of α‑ and β‑disaccharides (β/α = 7:1) (Table 15, entry ii). Further examples in Table 15 clearly showed that the protective groups in the acceptors, regardless of pyranoses or furanoses and of primary alcohols or secondary alcohols, were the crucial factor for the outcome of the stereochemistry in glycosylations with 409. This observed stereoselectivity was also donor dependent, since glycosylation with 2‑benzyl-3,5‑dibenzoyl CB arabinofuranoside was not as stereoselective [211].
Synthesis of an Octaarabinofuranoside Based on Stereoselective β-d-Arabinofuranosylation
The authors applied this acceptor‐dependent β‑arabinofuranosylation method to the synthesis of octaarabinofuranoside 417. Their retrosynthesis of compound 417 led to three components, a linear methyl trisaccharide 416, a branched BCB trisaccharide 415, and to CB furanosyl donor 409. Levulinyl protective groups were chosen in fragments 415 and 416 for selective deprotection prior to furanosyl coupling (Scheme 66). Three arabinose building blocks were used in the assembly.

Retrosynthesis of octaarabinose 417. A component of lipoarabinomannan in mycobacterial cell wall

Synthesis of octaarabinose 417
Arabinofuranosyl donor 418 glycosylated acceptor 419, to yield after levulinyl‐deprotection and repetitive glycosylation with 419, the linear trisaccharide 420 (Scheme 67a). Coupling of latent BCB donor 422, with active CB donor 421, led after deprotection of the levulinyl groups to diol 423 (Scheme 67b). The crucial double β‑arabinofuranosylation of diol 423 with 3.7 equiv. of the arabinofuranosyl donor 424, paved the way to pentaarabinofuranoside 425a (82% yield) with complete β‑selectivity. The latent BCB arabinofuranoside 425a was converted into the active CB arabinoside 425b. Finally, coupling of the latter with triarabinofuranosyl acceptor 420, afforded octaarabinofuranoside 417, in 83% yield.
2′-(Allyloxycarbonyl)benzyl (ACB) Glycosides: New “Latent” Donor for the Preparation of “Active” 2-Azido-2-deoxy BC Glycosyl Donors
Kim and co-workers introduced 2′-(allyloxycarbonyl)benzyl (ACB) glycosides , e. g. 426a, as new “latent” glycosyl donors for 2-azido-2-deoxy‐glucosides [212]. Introduction of the new ACB group in the place of the previously used BCB group was necessary because the azide functionality at C‑2 was also reduced during the conversion of the BCB group into the CB group under the normally used hydrogenolysis conditions (Pd/C, H2, NH4OAc, MeOH). 2‑Azido-2-deoxy ACB glycosides could be converted into active CB glycosyl donors (e. g. 426b, Scheme 68) without affecting the azide functionality on treatment with a catalytic amount of Pd(Ph3P)4 in the presence of morpholine [213].

2′(Allyloxycarbonyl)benzyl (ACB) glycosides
Synthesis of Oligosaccharides Based on BC Glycosyl Donors
The CB glycoside methodology by means of the “latent” BCB (or ACB) glycoside and the “active” CB glycoside has proved itself as a reliable method for the synthesis of complex oligosaccharides.
Synthesis of Trisaccharide 431, the Repeat Unit of the O-Antigen Polysaccharide from Danish Helicobacter pylori Strains
Kim and co-workers synthesized the repeat unit of the O‑antigen polysaccharide from Danish Helicobacter pylori strains, 431 (Scheme 69) [214]. Coupling of donor CB l‑rhamnoside 427 and acceptor BCB d‑rhamnoside 428 gave α‑disaccharide 429a in 88% yield. Selective hydrogenolysis of “latent” BCB disaccharide afforded “active” CB disaccharide 429b in 92% yield. Finally, glycosylation of 3-C-methyl mannoside, 430, with 429b yielded the target α‑trisaccharide 431, along with its β‑anomer in 7:1 ratio in 80% yield. A result that indicated that neighboring group participation is operative in CB glycosides.

Synthesis of trisaccharide 431
Synthesis of Tetrasaccharide 438
The CB methodology was also applied to the synthesis of protected tetrasaccharide 438, an analogue of the tetrasaccharide repeat unit of the O‑antigen polysaccharide from the E. coli lipopolysaccharide (Scheme 70) [215]. Coupling of “latent” BCB acceptor 433 with “active” CB glycosyl donor 432 gave a mixture of α‑disaccharide 434a along with its β‑isomer (4:1) in 74% yield (Scheme 70a). Glycosylation of acceptor 436 with donor 435 gave β‑mannoside 437a, that after removal of the PMB protecting group led to 437b (Scheme 70b). Finally, coupling of the latter with active donor 434b, prepared from latent 434a, yielded tetrasaccharide 438, in 75% yield (Scheme 70c).

Synthesis of tetrasaccharide 438
Synthesis of Tetrasaccharide Repeat Unit from E. coli O77
A route to a tetrasaccharide 439 was reported, which made use of the previously mentioned “latent” 2′-(allyloxycarbonyl)benzyl (ACB) glycosides in combination with “latent” and “active” BCB and CB glycosides, respectively [212]. The retrosynthesis is outlined in Scheme 71. All glycosyl couplings were based on the “latent‐active” methodology, and all were stereoselective. A slightly modified synthesis of 439 has been reported including one CB mediated coupling [216].

Retrosynthesis of tetrasaccharide 439
Total Synthesis of Agelagalastatin
The total synthesis of agelagalastatin , an antineoplastic glycosphingolipid , has been described by Kim and co-workers [217]. The retrosynthesis, outlined in Scheme 72, involved a β‑d-galactofuranosylation, an α‑d-galactofuranosylation, and a final α‑d-galactopyranosylation. The β‑d-galactofuranosylation was achieved in 79% yield via neighboring group participation of the pivaloyl group at O2 in compound 445. The α‑d-galactofuranosylation to 441, took place with 91% yield with a nonparticipant benzyl group at O2 in donor 443. The final α‑d-galactopyranosylation (Scheme 73) was carried out with CB trisaccharide donor 441 furnishing compound 447 in 77% yield as a 1.4:1 (α/β) mixture of saccharides. The efficiency of this coupling was improved by conversion of the CB trisaccharide donor to glycosyl fluoride 448. Treatment of 441 with TF2O/DTBMP followed by HF-pyridine, as a source of fluoride, yielded glycosyl fluoride 448. The glycosylation of acceptor 442 with glycosyl fluoride 448, then gave the target protected agelagalastatin 447, in 72% yield as the pure α‑isomer.

Retrosynthesis of Agelagalastatin 440

Transformation of CB glycoside 441 into glycosyl fluoride 448 and final coupling of Agelagalastatin
Conversion of 2′-Carboxybenzyl Glycosides into Other Glycosyl Donors
CB glycosides have been converted to phenyl 1-thio glycosides and glycosyl fluorides in one-pot operations [218]. Thus, treatment of CB glycosyl donors with TF2O/DTBMP in CH2Cl2 at −78 ℃ for 10 min followed by addition of PhSH furnished thioglycosides, e. g. 451, 453 (Scheme 74), whereas treatment with DAST or HF-pyridine (see Scheme 73) yielded the corresponding glycosyl fluorides, e. g. 449, 452 (Scheme 74). The high β‑selectivity observed in the formation of glycosyl fluoride 452 and thioglycoside 453 from 4,6-O‑benzylidenemannopyranoside 396 was ascribed to the presence of a highly reactive 4,6-O‑benzylidenemannopyranosyl α‑triflate , in keeping with previously mentioned findings.

Conversion of CB glycosides to glycosyl fluorides and thiophenyl glycosides
2′-Carboxybenzyl Glycosides as Glycosyl Donors for C-Glycosylation
Glycosylation of various glycosyl acceptors (NuH or NuTMS, Scheme 75) with manno- and gluco- CB glycosyl donors 450 and 454, respectively afforded α‑C-glycosides 455, exclusively or predominantly in good yields [218]. Experimentally these reactions were carried out by addition of the donor to a solution of the acceptor, DTBMP, and Tf2O in CH2Cl2 at −78 ℃. These modified conditions led to increased yields of C-glycosides and minimized the amount of self‐condensed esters 456.

CB glycosides in the synthesis of α‑C-glycosides
O-Heteroaryl Glycosyl Donors
Glycosides of some heterocycles have also been investigated as glycosyl donors.
2-Pyridyl 2,3,4,6-tetra-O-benzyl-d-glucosides
The first example, reported by Nikolaev and Kochetkov [219], dealt with the use of 2‑pyridyl 2,3,4,6-tetra-O‑benzyl-β‑d-glucoside in glycosylation. This heteroaryl glycoside was prepared by glycosylation of 2(1H)-pyridinone by the corresponding sugar chloride, and was activated by electrophiles, such as MeOTf and Et3O·BF4, to give mixtures of cis- and trans-glycosides.
O-Hetaryl Glycosides by Schmidt's Group
Schmidt and co-workers [168,220,221] reported the preparation, and use in glycosylation reactions of several O‑hetaryl glycosides, e. g. 458, conveniently prepared by anomeric O‑hetarylation of hexoses , e. g. 457, with the corresponding electron‐deficient heteroaromatic/heterocyclic systems (Scheme 76). The best results in terms of glycosylation were obtained with tetrafluoropyridyl glycosides 460 and 462, obtained by reaction of hexoses 459 and 461 with 2,3,4,5,6‑pentafluoro pyridine (Scheme 77a,b). Under TMSOTf catalysis, in CH2Cl2 at room temperature, they furnished the corresponding α‑ and β‑disaccharides 463 and 464 in 98 and 74% yield, respectively (Scheme 77c,d).

O‑Hetaryl glycosides synthesized by Schmidt's group

2,3,5,6‑Tetrafluoro-4-(2,3,4,6-tetra-O‑acetyl or O‑benzyl-β‑d-glucopyranosyloxy) pyridine as glycosyl donors
3-Methoxy-2-pyridyl (MOP) Glycosides
On the basis of the concept of remote activation [222], first applied to pyridine thioglycosides [223], Hanessian and co-workers introduced 3‑methoxy-2‑pyridyl (MOP) glycosides [224,225]. They first reported the usefulness of ribofuranosyl MOP donor 465 in the coupling with silylated pyrimidine bases, by activation with TMSOTf, to give 1,2-cis nucleosides, 466, with high selectivity (Scheme 78) [226]. These glycosides react in MeOTf-, Cu(OTf)2-, TfOH-, or Yb(OTf)3-promoted reactions to give disaccharides [227].

MOP Ribofuranosyl donors in the synthesis of 1,2-cis furanosyl nucleosides
Coupling of Unprotected MOP Glycosyl Donors
Interestingly, unprotected MOP glycosides could also be used as donors. In fact, when using an excess of glycosyl acceptor (≈10 equiv.), disaccharides are obtained in reasonable yields, as illustrated in Scheme 79.

Disaccharide synthesis with unprotected MOP donors
It was also found that introduction of any protecting group on the unprotected MOP glycosyl donors resulted in a significant decrease of the reactivity. This deactivation was considerable when p-fluorobenzoates (FBz) were used as protecting groups, and it was applied to the synthesis of disaccharides, and to iterative oligosaccharide synthesis (Scheme 80).

Selective activation of unprotected- versus protected-MOP glycosides
Esterification and Phosphorylation of Unprotected MOP Glycosides
MOP glycosyl donors have been used in stereocontrolled esterification and phosphorylation, leading to glycosyl 1,2-cis-1‑carboxylates or glycosyl 1,2-cis-glycosyl-1‑phosphates in one step. Treatment of MOP donor 475 in acetonitrile or DMP with an excess (20–200 equiv.) of a carboxylic acid under anhydrous conditions led to the corresponding d‑glycosyl carboxylate 477, in excellent yields [228]. Moreover, treatment of 6-O‑tert‐butyldiphenylsilyl MOP donor 478 with only 1.5 equiv. of the corresponding carboxylic acid in CH2Cl2 resulted in the formation of 1,2-cis-glycosyl carboxylate 479 (Scheme 81a). Treatment of β‑d-galactopyranosyl, and 2-azido-2-deoxy-α‑d-galactopyranosyl MOP donors 475 and 471, respectively with 7 equiv. of phosphoric acid or dibenzyl phosphate in DMF led to the corresponding α‑glycosyl phosphates 480 and 481, respectively. The same donors are also capable of transferring glucopyranosyl (e. g. 467, Scheme 81d) and galactopyranosyl units to UDP-free acid 482, to afford the corresponding uridine 5′ diphosphosugars (e. g. 483) in one step [229].

Stereocontrolled synthesis of glycosyl 1‑carboxylates and 1‑phosphates
MOP Glycosides in Oligosaccharide Synthesis
A solid-phase oligosaccharide synthesis based on the MOP donor/acceptor methodology was developed by Hanessian and co-workers [224]. Thus, an O‑unprotected polymer-phase bound MOP donor is coupled with an excess of a partially esterified MOP acceptor. Selective removal of the ester (or related protecting groups) from the new saccharides generates a new O‑unprotected MOP donor to engage in a subsequent iteration.
Hanessian and co-workers also illustrated the usefulness of the MOP methodology with some syntheses of oligosaccharides [230,231]. They reported a concise synthesis of a Galα1→3Galβ1→4GlcNAcOR trisaccharide 489, outlined in Scheme 82 [232]. Treatment of MOP donor 484 with 3‑benzyloxycarbonylamino 1‑propanol in the presence of HBF4·Et2O in CH2Cl2 led to the expected β‑glycoside 485. Protecting group manipulation and glycosylation with MOP galactopyranosyl 486 in the presence of Cu(TfO)2 as activator gave the intended β‑disaccharide 487. Final glycosylation of 487 with β‑galacto MOP donor 488 (Cu(OTf)2) or Yb(OTf)3, as promoters) led to protected trisaccharide 489.

Synthesis of a trisaccharide using MOP glycosyl donors
6-Nitro-2-benzothiazolyl Glycosides
Mukaiyama et al. described glycosyl 6-nitro-2‑benzothiazoates (e. g. 490) as useful glycosyl donors [233]. They are prepared by reaction of glycose derived hemiacetals (e. g. 459) with 2‑chloro-6-nitro-2‑benzothiazoate (Scheme 83a). The purified α‑isomer α‑490, reacted with primary hydroxyl acceptors in the presence of catalytic TfOH at −78 ℃ to give mainly β‑glucosides, e. g. 492 (Scheme 83b). Although, a highly stereoselective α‑glucosylation (α/β = 88:12) was carried out in high yield using 20 mol% of HClO4 in tert-BuOMe (Scheme 83c). 6-Nitro-2‑benzothiazolyl α‑mannosides (e. g. 494) effected stereoselective β‑mannosylation with several glycosyl acceptors [234,235]. The highest β‑stereoselectivity was achieved when tetrakis(pentafluorophenyl)boric acid [HB(C6F5)4] [236] was employed as catalyst (Scheme 83d). BF3·Et2O, a weaker Lewis acid, showed a reversed stereoselectivity [237] (Scheme 83e). The β‑selective coupling was employed by Mukaiyama and co-workers in the formation of the β‑Man(1→4)GlcN linkage, e. g. 498, that exists in N‑linked glycans (Scheme 83f) [238,239].

Glycosyl 6-nitro-2‑benzothiazoate as a glycosyl donor
Miscellaneous O-Glycosyl Donors
Noyori and Kurimoto [240] described that hydroxyl‐protected and -unprotected glycosyl aryloxides reacted with alcohols under mild electrolytic conditions to give the corresponding glycosides. They hypothesized that the glycosylation reaction proceeded via oxocarbenium ion intermediates generated from the radical cation of the easily oxidizable aryloxy substrate (Scheme 84).

Electrochemical glycosylation of glycosyl aryloxides
A combination of trimethylsilyl bromide and zinc triflate promoted the glycosylation of benzyl-, isopropyl-, and methyl glycosides with several glycosyl acceptors in moderate to good yields [241,242].
2‑Deoxyglycosides , e. g. 501, were obtained by DDQ oxidation of 3,4‑dimethoxybenzyl glycosides 500, in MeCN in the presence of primary, secondary, and tertiary alcohols (Scheme 85) [243].

3,4-Dimethoxybenzyl 2-deoxy glycosides as glycosyl donors
Davis and co-workers [244] examined the self‐activating properties of unprotected and acetylated bromobutyl glycosides 505 and 508, respectively (Scheme 86). These readily available compounds reacted with galactose acceptor 40 (1 equiv.) in the presence of a halophilic Lewis acid promoter (AgOTg) to give disaccharides 506 and 509 in moderate yields. The suggested reaction pathway involved a spontaneous, or acid, triggered, 5-exo-tet cyclization of the bromobutyl glycoside, e. g. 508→510, to form an anomeric furanosyl cation 510, which would evolve to give non‐nucleophilic volatile tetrahydrofuran, and oxocarbenium ion 511. The latter will then react with acceptor 40 to furnish the disaccharide.

Bromobutyl glycosides as glycosyl donors
Hung and co-workers have reported on the use of 2‑allyloxyphenyl mannoside 512 as a useful glycosyl donor [245]. Mannoside 512 reacted in the presence of NIS/TfOH in CH2Cl2 at room temperature, with a series of primary and secondary hydroxyl acceptors to give α‑mannosides, e. g. 513, in good yields (Scheme 87). The proposed mechanism for the formation of the oxocarbenium ion 517, outlined in Scheme 87, implies a 6-exo-tet cyclization on halonium ion 514, and the ejection of the non‐nucleophilic species 516.

2‑Allyloxyphenyl mannosides as glycosyl donors
Hotha and Kashyap have identified propargyl glycosides 518, as new glycosyl donors (Scheme 88) [246,247]. Various aglycones, including primary and secondary alcohols, reacted with propargyl glycosides in the presence of 3 mol% of AuCl3 in MeCN at 60 ℃, to give α/β‑mixtures of glycosides and disaccharides in good yields. The α,β‑ratio of the transglycosylation products was found to be independent of the anomeric ratio of the donor. per-O‑Acylated propargyl glycosides did not give transglycosylation products. A possible reaction pathway to the generation of an intermediate oxocarbenium ion, based on the alkynophilicity of gold catalysts, was advanced by the authors (Scheme 88). Coordination of AuCl3 to the glycosyl donor 518 would be followed by formation of the cyclopropyl gold carbene intermediate (520) that could evolve to intermediate 521, which would lead to oxocarbenium ion 522, and alkenyl gold complex 523. The latter upon protodemetalation will generate AuCl3 and cyclopropanone 525 via intermediate 524.

Propargyl glycosides as glycosyl donors
Abbreviations
- BCB:
-
2-(benzyloxycarbonyl)benzyl
- CAN:
-
cerium ammonium nitrate
- CB:
-
2′-carboxybenzyl
- DAST:
-
(diethylamino)-sulfur trifluoride
- DDQ:
-
2,3‑dichloro-5,6‑dicyano-p-benzoquinone
- DISAL:
-
a dinitrosalicylic acid glycoside derivative
- DMAP:
-
4-(N,N-dimethylamino)pyridine
- DTBMP:
-
di-tert‐butylmethylpyridine
- IDCP:
-
iodonium di-sym-collidine perchlorate
- MOP:
-
3‑methoxy-2‑pyridyl
- NBS:
-
N‑bromosuccinimide
- NIS:
-
N‑iodosuccinimide
- NMP:
-
1‑methylpyrrolidin-2-one
- NPG:
-
n-pentenyl glycosides
- NPhth:
-
N‑phthaloyl
- NPOE:
-
n-pentenyl orthoester
- PMB:
-
p-methoxybenzyl
- PMP:
-
p-methoxyphenyl
- TBAF:
-
tetra-n-butylammonium bromide
- TBAI:
-
tetra-n-butylammonium iodide
- TBSOTf:
-
tert‐butyldimethylsilyl trifluoromethanesulfonate
- TESOTf:
-
triethylsilyl trifluoromethanesulfonate
- Tf2O:
-
trifluoromethanesulfonic anhydride
- TfOH:
-
trifluoromethanesulfonic acid
- THF:
-
tetrahydrofuran
- TMSOTf:
-
trimethylsilyl trifluoromethanesulfonate
- TPSOTf:
-
tert‐butyldiphenylsilyl trifluoromethane sulfonate
- Troc:
-
N-trichloroethoxycarbonyl
- TTCP:
-
N-tetrachlorophthaloyl
References
Paulsen H (1982) Angew Chem Int Ed Engl 21:155
Schmidt RR (1986) Angew Chem Int Ed Engl 25:212
Paulsen H (1990) Angew Chem Int Ed Engl 29:823
Toshima K, Tatsuta K (1993) Chem Rev 93:1503
Schmidt RR (1986) Angew Chem Int Ed Engl 25:212
Boons GJ (1996) Tetrahedron 52:1095
Boons GJ (1996) Contemp Org Synth 3:173
Davis BG (2000) J Chem Soc Perkin Trans 1 2137
Demchenko AV (2005) Lett Org Chem 2:580
Fraser-Reid B, Konradsson P, Mootoo DR, Udodong U (1988) J Chem Soc Chem Commun 823
Michael A (1879) Am Chem J 1:305
Fraser-Reid B, Udodong UE, Wu Z, Ottoson H, Merrit JR, Rao CS, Roberts C, Madsen R (1992) Synlett 927
Mootoo DR, Date V, Fraser-Reid B (1988) J Chem Soc Chem Commun 1462
Mootoo DR, Fraser-Reid B (1986) J Chem Soc Chem Commun 1570
Baldwin JE (1976) J Chem Soc Chem Commun 734
Mootoo DR, Date V, Fraser-Reid B (1988) J Am Chem Soc 110:2662
Lemieux RU, Morgan AR (1965) Can J Chem 43:2190
Goodman L (1967) Adv Carbohydr Chem Biochem 22:109
Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B (1988) J Am Chem Soc 110:5583
Konradsson P, Mootoo DR, McDevitt RE, Fraser-Reid B (1990) J Chem Soc Chem Commun 270
Veeneman GH, van Boom JH (1990) Tetrahedron Lett 31:275
Veeneman GH, van Leeuwen SH, van Boom JH (1990) Tetrahedron Lett 31:1331
Friesen RW, Danishefsky SJ (1989) J Am Chem Soc 111:6656
Barrena MI, Echarri R, Castillón S (1996) Synlett 675
Cheung MK, Douglas NL, Hinzen B, Ley SV, Pannecoucke X (1997) Synlett 257
Hashimoto S, Sakamoto H, Honda T, Ikegami S (1997) Tetrahedron Lett 38:5181
Hashimoto S, Sakamoto H, Honda T, Abe H, Nakamura S, Ikegami S (1997) Tetrahedron Lett 38:8969
Mukaiyama T, Chiba H, Funasaka S (2002) Chem Lett 392
Chiba H, Funasaka S, Kiyota K, Mukaiyama T (2002) Chem Lett 746
Chiba H, Funasaka S, Mukaiyama T (2003) Bull Chem Soc Jpn 76:1629
Demchenko AV, Kamat MN, De Meo C (2003) Synlett 1287
Kamat MN, Demchenko AV (2005) Org Lett 7:3215
Bongat AFG, Kamat MN, Demchenko AV (2007) J Org Chem 72:1480
Llera JM, López JC, Fraser-Reid B (1990) J Org Chem 55:2997
Madsen R, Fraser-Reid B (1995) J Org Chem 60:772
Fraser-Reid B, Merrit RJ, Handlon AJ, Andrews CW (1993) Pure Appl Chem 65:779
Rodebaugh R, Fraser-Reid B (1994) J Am Chem Soc 116:3155
Rodebaugh R, Fraser-Reid B (1996) Tetrahedron 52:7663
Brown RS (1997) Acc Chem Res 30:131
Fraser-Reid B, Wu Z, Udodong UE, Ottosson H (1990) J Org Chem 55:6068
Fraser-Reid B, Wu Z, Andrews CW, Skowronski E, Bowen JP (1991) J Am Chem Soc 113:1434
Merrit JR, Debenham JS, Fraser-Reid B (1996) J Carbohydr Chem 15:65
Rodebaugh R, Debenham JS, Fraser-Reid B, Snyder JP (1999) J Org Chem 64:1758
Konradsson P, Fraser-Reid B (1988) J Chem Soc Chem Commun 1124
Pale P, Whitesides GM (1991) J Org Chem 56:4547
Plante OJ, Andrade RB, Seeberger PH (1999) Org Lett 1:211
Hashimoto S, Honda T, Ikegami S (1999) J Chem Soc Chem Commun 685
Clausen MH, Madsen R (2003) Chem Eur J 9:3281
López JC, Uriel C, Guillamón‐Martín A, Valverde S, Gómez AM (2007) Org Lett 9:2759
Fraser-Reid B, Lu J, Jayaprakash KN, López JC (2006) Tetrahedron Asymmetry 17:2449
Ferrier RJ, Hay RW, Vethaviyasar N (1973) Carbohydr Res 27:55
Fügedi P, Garegg PJ, Lönn H, Norberg T (1987) Glycoconjugate J 4:97
Codée JDC, Litjens REJN, van den Bos LJ, Overkleeft HS, van der Marel GA (2005) Chem Soc Rev 34:769
Schmidt RR, Michel J (1980) Angew Chem Int Ed Engl 19:731
Sharma GVM, Rao SM (1992) Tetrahedron Lett 33:2365
Ratcliffe AJ, Fraser-Reid B (1989) J Chem Soc Perkin Trans 1 1805
Ritter JJ, Minieri PP (1948) J Am Chem Soc 70:4045
Lemieux RU, Morgan AR (1965) Can J Chem 43:2205
Ratcliffe AJ, Fraser-Reid B (1990) J Chem Soc Perkin Trans 1 747
Ratcliffe AJ, Konradsson P, Fraser-Reid B (1990) J Am Chem Soc 112:5665
Ratcliffe AJ, Konradsson P, Fraser-Reid B (1991) Carbohydr Res 216:323
Handlon AL, Fraser-Reid B (1993) J Am Chem Soc 115:3796
Nair LG, Fraser-Reid B, Szardenings AK (2001) Org Lett 3:317
Banoub J, Boullanger P, Lafont D (1992) Chem Rev 92:1167
Mootoo DR, Fraser-Reid B (1989) Tetrahedron Lett 30:2363
Klaffke W, Warren CD, Jeanloz RW (1992) Carbohydr Res 244:171
Debenham JS, Fraser-Reid B (1994) XVIIth International Carbohydrate Symposium, Ottawa, Canada
Debenham JS, Madsen R, Roberts C, Fraser-Reid B (1995) J Am Chem Soc 117:3302
Debenham JS, Fraser-Reid B (1996) J Org Chem 61:432
Fraser-Reid B, Anilkumar G, Nair LG, Olsson L, García Martín M, Daniels JK (2000) Isr J Chem 40:255
Svarovsky SA, Barchi JJ (2003) Carbohydr Res 338:1925
Feather MS, Harris JF (1965) J Org Chem 30:153
Andrews CW, Fraser-Reid B, Bowen JP (1993) Involvement of nσ* interactions in glycoside cleavage. In: Thatcher GRJ (ed) The anomeric effect and associated stereoelectronic effects. ACS Symposium Series 539. American Chemical Society, Washington, DC, p 114
Deslongchamps P (1983) Stereoelectronic effects in organic chemistry. Pergamon Press, New York, pp 30–35
Ratcliffe AJ, Mootoo DR, Andrews CW, Fraser-Reid B (1989) J Am Chem Soc 111:7661
Lafont D, Boullanger P (2006) Tetrahedron Assymmetry 17:3368
Madsen R, Roberts C, Fraser-Reid B (1995) J Org Chem 60:7920
Bodner R, Marcellino BK, Severino A, Smenton AL, Rojas CM (2005) J Org Chem 70:3988
Demchenko AV, De Meo C (2002) Tetrahedron Lett 43:8819
Chapeau M‑C, Marnett LJ (1993) J Org Chem 58:7258
Arasappan A, Fraser-Reid B (1995) Tetrahedron Lett 36:7967
Konradsson P, Roberts C, Fraser-Reid B (1991) Recl Trav Chim Pays-Bas 110:23
Velty R, Benvegnu T, Plusquellec D (1996) Synlett 817
Ferrières V, Bertho J‑N, Plusquellec D (1995) Tetrahedron Lett 36:2749
Brennan PJ, Nikaido H (1995) Annu Rev Biochem 64:29
Mereyala HB, Hotha S, Gurjar MK (1998) 685
Lu J, Fraser-Reid B (2004) Org Lett 6:3051
Jayaprakash KN, Radhakrishnan KV, Fraser-Reid B (2002) Tetrahedron Lett 43:6955
Jayaprakash KN, Fraser-Reid B (2004) Synlett 301
Lu J, Fraser-Reid B (2005) Chem Commun 862
Fraser-Reid B, Lu J, Jayaprakash KN, López JC (2006) Tetrahedron Asymmetry 17:2449
Hölemann A, Stocket BL, Seeberger PH (2006) J Org Chem 71:8071
Barresi F, Hindsgaul O (1991) J Am Chem Soc 113:9337
Stork G, Kim G (1992) J Am Chem Soc 114:1087
Ito Y, Ogawa T (1994) Angew Chem Int Ed Engl 33:1765
Krog-Jensen C, Oscarson S (1996) J Org Chem 61:4512
Gelin M, Ferrières V, Lefeuvre M, Plusquellec D (2003) Eur J Org Chem 1285
Hay JE, Haynes LJ (1958) J Chem Soc 2231
Rousseau C, Martin OR (2000) Tetrahedron Asymmetry 11:409
Girard N, Rousseau C, Martin OR (2003) Tetrahedron Lett 44:8971
Ikeda K, Fukuyo J, Sato K, Sato M (2005) Chem Pharm Bull 53:1490
Tabeur C, Machetto F, Mallet J‑M, Duchaussoy P, Petitou M, Sinaÿ P (1996) Carbohydr Res 281:253
Reichardt N‑C, Martín-Lomas M (2005) Arkivoc ix:133
Anilkumar G, Nair LG, Fraser-Reid B (2000) Org Lett 2587–2589
Fraser-Reid B, López JC, Radhakrishnan KV, Mach M, Schlueter U, Gómez AM, Uriel C (2002) J Am Chem Soc 124:3198
Fraser-Reid B, Anilkumar GN, Nair LG, Radhakrishnan KV, López JC, Gómez AM, Uriel C (2002) Aust J Chem 55:123
Fraser-Reid B, López JC, Radhakrishnan KV, Mach M, Schlueter U, Gómez AM, Uriel C (2002) Can J Chem 80:1075
López JC, Gómez AM, Uriel C, Fraser-Reid B (2003) Tetrahedron Lett 44:1417
Fraser-Reid B, López JC, Gómez AM, Uriel C (2004) Eur J Org Chem 1387
Fraser-Reid B, López JC, Radhakrishnan KV, Nandakumar MV, Gómez AM, Uriel C (2002) Chem Común 2104
Wilson BG, Fraser-Reid B (1995) J Org Chem 60:317
Fraser-Reid B, Grimme S, Piacenza M, Mach M, Schlueter U (2003) Chem Eur J 9:4687
Uriel C, Gómez AM, López JC, Fraser-Reid B (2003) Synlett 2203
Mootoo DR, Konradsson P, Fraser-Reid B (1989) J Am Chem Soc 111:8540
Koenigs W, Knorr E (1901) Ber 34:957
Merritt JR, Fraser-Reid B (1992) J Am Chem Soc 114:8334
Merritt JR, Naisang E, Fraser-Reid B (1994) J Org Chem 59:4443
Lerouge P (1994) Glycobiology 4:127
Debenham JS, Rodebaugh R, Fraser-Reid B (1996) J Org Chem 61:6478
Rodebaugh R, Debenham JS, Fraser-Reid B (1996) Tetrahedron Lett 37:5477
Park MH, Takeda R, Nakanishi K (1987) Tetrahedron Lett 28:3823
Ferguson MAJ (1991) Curr Opin Struct Biol 1:52
Ferguson MAJ, Low MG, Cross GAM (1985) J Biol Chem 260:14547
Ferguson MAJ, Homans SW, Dwek RA, Rademacher TW (1988) Science 239:753
Homans SW, Ferguson MAJ, Dwek RA, Rademacher TW, Anand R, Williams AF (1988) Nature 333:269
Campbell AS, Fraser-Reid B (1995) J Am Chem Soc 117:10387
Roberts C, Madsen R, Fraser-Reid B (1995) J Am Chem Soc 117:1546
Madsen R, Udodong UE, Roberts C, Mootoo DR, Konradsson P, Fraser-Reid B (1995) J Am Chem Soc 117:1554
Udodong UE, Madsen R, Roberts C, Fraser-Reid B (1993) J Am Chem Soc 115:7886
Bridges AJ (1996) Chemtracts Org Chem 9:215
Heidelberg T, Martin OR (2004) J Org Chem 69:2290
Clausen MH, Madsen R (2004) Carbohydr Res 339:2159
Clausen MH, Jørgensen MR, Thorsen J, Madsen R (2001) 543
Lemieux RU, Hendricks KB, Stick RV, James K (1975) J Am Chem Soc 97:4056
Arasappan A, Fraser-Reid B (1996) J Org Chem 61:2401
Hinou H, Umino A, Matsuoka K, Terunuma D, Takahashi S, Esumi Y, Kuzuhara H (2000) Bull Chem Soc Jpn 73:163
Yoshida T, Chiba T, Yokochi T, Onozaki K, Sugiyama T, Nakashima I (2001) Carbohydr Res 335:167
Seeberger PH, Haase WH (2000) Chem Rev 100:4349
Rodebaugh R, Fraser-Reid B, Geysen HM (1997) Tetrahedron Lett 44:7653
Rich DH, Gurwara SK (1975) J Am Chem Soc 97:1575
Rodebaugh R, Joshi S, Fraser-Reid B, Geysen HM (1997) J Org Chem 62:5660
Andrade RB, Plante OJ, Melean LG, Seeberger PH (1999) Org Lett 1:1811
Melean LG, Haase WC, Seeberger PH (2000) Tetrahedron Lett 41:4329
Hewitt MC, Seeberger PH (2001) J Org Chem 66:4233
Ratner DM, Plante OJ, Seeberger PH (2002) Eur J Org Chem 826
Hewitt MC, Zinder DA, Seeberger PH (2002) J Am Chem Soc 124:13434
Palmacci ER, Plante OJ, Hewitt MC, Seeberger PH (2003) Helv Chim Acta 86:3975
Mogemark M, Gustafsson L, Bengtsson C, Elofsson M, Kihlberg J (2004) Org Lett 6:4885
Buskas T, Söderberg E, Konradsson P, Fraser-Reid B (2000) J Org Chem 65:958
Bindschädler P, Noti C, Castagnetti E, Seeberger PH (2006) Helv Chim Acta 89:2591
Allen JR, Allen JG, Zhang X‑F, Williams LJ, Zatorski A, Ragupathi G, Livingston PO, Danishefsky SJ (2000) Chem Eur J 6:1366
Allen JR, Danishefsky SJ (2000) J Prakt Chem 342:736
Allen JR, Ragupathi G, Livingston PO, Danishefsky SJ (1999) J Am Chem Soc 121:10875
Allen JR, Harris CR, Danishefsky SJ (2001) J Am Chem Soc 123:1890
Nishimura SI, Matsuoka K, Furuike T, Ishii S, Kurita K, Nishimura KM (1991) Macromolecules 24:4236
Nishimura SI, Furuike T, Matsuoka K, Maruyama K, Nagata K, Kurita K, Nishi N, Tokura S (1994) Macromolecules 27:4876
Miyagawa A, Kurosawa H, Watanabe T, Koyama T, Terunuma D, Matsuoka K (2004) Carbohydr Polymers 57:441
Yamada A, Hatano K, Koyama T, Matsuoka K, Esumi Y, Terunuma D (2006) Carbohydr Res 341:467
Rele SM, Iyer SS, Baskaran S, Chaikof EL (2004) J Org Chem 69:9159
Lu J, Fraser-Reid B, Chowda G (2005) Org Lett 7:3841
Fischer E (1893) Ber 26:2400
Fischer E (1895) Ber 28:1145
Lemieux RU, Morgan AR (1965) Can J Chem 43:2199
Lu J, Jayaprakash KN, Schlueter U, Fraser-Reid B (2004) J Am Chem Soc 126:7540
Mach M, Schlueter U, Mathew F, Fraser-Reid B, Hazen KC (2002) Tetrahedron 58:7345
Jayaprakash KN, Fraser-Reid B (2004) Org Lett 6:4211
de Raadt A, Ferrier RJ (1991) Carbohydr Res 216:93
Vankar YD, Vankar PS, Behrendt M, Schmidt RR (1991) Tetrahedron 47:9985
Marra A, Esnault J, Veyrières A, Sinaÿ P (1992) J Am Chem Soc 114:6354
Tebbe FN, Parshall GW, Reddy GS (1978) J Am Chem Soc 100:3611
Chenault HK, Castro A (1994) Tetrahedron Lett 35:9145
Chenault HK, Castro A, Chafin LF, Yang Y (1996) J Org Chem 61:5024
Lutsenko IF, Khomutov RV (1955) Doklady Akad Nauk SSSR 102:97
Petasis NA, Bzowej EI (1990) J Am Chem Soc 112:6392
Kunz H, Harreus A (1982) Liebigs Ann Chem 41
Boons GJ, Isles S (1994) Tetrahedron Lett 35:3593
Roy R, Andersson FO, Letellier M (2002) Tetrahedron Lett 33:6053
Boons GJ, Burton A, Isles S (1996) J Chem Soc Chem Commun 141
Boons GJ, Isles S (1996) J Org Chem 61:4262
Boons GJ, Heskamp B, Hout F (1996) Angew Chem Int Ed Engl 35:2845
Johnson M, Aries C, Boons GJ (1998) Tetrahedron Lett 39:9801
Bai Y, Boons GJ, Burton A, Johnson M, Haller M (2000) J Carbohydr Chem 19:939
Haller M, Boons GJ (2001) J Chem Soc Perkin Trans 1 814
Boons GJ, Burton A, Wyatt P (1996) Synlett 310
Capozzi G, Dios A, Franck RW, Geer A, Marzabadi C, Menichetti S, Nativi C, Tamarez M (1996) Angew Chem Int Ed Engl 35:777
Marzabadi CH, Franck RW (1996) Chem Commun 2651
Marzabadi CH, Franck RW (2000) Tetrahedron 56:8385
Capozzi G, Mannocci F, Menichetti S, Nativi C, Paoletti S (1997) Chem Commun 2291
Bartolozzi A, Capozzi G, Menichetti S, Nativi C (2001) Eur J Org Chem 2083
Petersen L, Jensen KJ (2001) J Org Chem 66:6268
Jensen KJ (2002) J Chem Soc Perkin Trans 1 2219
Koeners HJ, de Kok AJ, Romers C, van Boom JH (1980) Recl Trav Chim Pays-Bas 99:355
Sharma SK, Corrales G, Penadés S (1995) Tetrahedron Lett 36:5627
Petersen L, Jensen KJ (2001) J Chem Soc Perkin Trans 1 2175
Larsen K, Worm-Leonhard K, Olsen P, Hoel A, Jensen KJ (2005) Org Biomol Chem 3:3966
Tolborg JF, Jensen KJ (2000) Chem Commun 147
Laursen JB, Petersen L, Jensen KJ (2001) Org Lett 3:687
Petersen L, Laursen JB, Larsen K, Motawia MS, Jensen KJ (2003) Org Lett 5:1309
Laursen JB, Petersen L, Jensen KJ, Nielsen J (2003) Org Biomol Chem 1:3147
Grathe S, Thygessen MB, Larsen K, Petersen L, Jensen KJ (2005) Tetrahedron Asymmetry 16:1439
Kim KS, Lee YJ, Kim HY, Kang SS, Kwon SY (2001) J Am Chem Soc 123:8477
Kim KS, Lee ME, Cho JW (2004) Bull Korean Chem Soc 25:139
Kim KS, Jeon HB (2007) In: Demchenko AV (ed) Frontiers in modern carbohydrate chemistry. ACS Symposium Series, vol 960, Ch 9. ACS, Washington, DC
Demchenko AV (2003) Curr Org Chem 7:35
Crich D, Sun S (1997) J Am Chem Soc 119:11217
Crich D, Sun S (1996) J Org Chem 61:4506
Crich D, Sun S (1997) J Org Chem 62:1198
Crich D, Sun S (1998) Tetrahedron 54:8321
Crich D, Smith M (2000) Org Lett 2:4067
Kim KS, Park J, Lee YJ, Seo YS (2003) Angew Chem Int Ed 42:459
Lee YJ, Lee K, Jung EH, Jeon HB, Kim KS (2005) Org Lett 7:3263
Lee R, Jeon JM, Jung JH, Jeon HB, Kim KS (2006) Can J Chem 84:506
Kunz H, Waldmann H (1984) Angew Chem Int Ed Engl 23:71
Kwon YT, Lee YJ, Lee K, Kim KS (2004) Org Lett 6:3901
Kim KS, Kang SS, Seo YS, Kim HJ, Lee YJ, Jeong KS (2003) Synlett 1311
Lee BY, Baek JY, Jeon HB, Kim KS (2007) Bull Korean Chem Soc 28:257
Lee YJ, Lee BY, Jeon HB, Kim KS (2006) Org Lett 8:3971
Lee YJ, Baek JY, Lee BY, Kang SS, Park HS, Jeon HB, Kim KS (2006) Carbohydr Res 341:1708
Nikolaev AV, Kochetkov NK (1986) Izv Akad Nauk SSSR Ser Khim 2556
Huchel U, Schmidt C, Schmidt RR (1995) Tetrahedron Lett 36:9457
Huchel U, Schmidt C, Schmidt RR (1998) Eur J Org Chem 1353
Hanessian S (1997) In: Hanessian S (ed) Preparative carbohydrate chemistry. Marcel Dekker Inc., New York, p 381
Hanessian S, Bacquet C, Lehong N (1980) Carbohydr Res 80:C17
Hanessian S, Lou B (2000) Chem Rev 100:4443
Lou B, Reddy GV, Wang H, Hanessian S (1997) In: Hanessian S (ed) Preparative carbohydrate chemistry. Marcel Dekker Inc., New York, p 389
Hanessian S, Condé JJ, Lou B (1995) Tetrahedron Lett 36:5865
Lou B, Huynh HK, Hanessian S (1997) In: Hanessian S (ed) Preparative carbohydrate chemistry. Marcel Dekker Inc., New York, p 413
Hanessian S, Mascitti V, Lu PP, Ishida H (2002) Synthesis 1959
Hanessian S, Lu PP, Ishida H (1998) J Am Chem Soc 120:13296
Lou B, Eckhardt E, Hanessian S (1997) In: Hanessian S (ed) Preparative carbohydrate chemistry. Marcel Dekker Inc., New York, p 449
Hanessian S, Huynh HK, Reddy GV, Duthaler RO, Katopodis A, Streiff MB, Kinzy W, Oehrlein R (2001) Tetrahedron 57:3281
Hanessian S, Saavedra OM, Mascitti V, Marterer W, Oehrlein R, Mak CP (2001) Tetrahedron 57:3267
Mukaiyama T, Hashihayata T, Mandai H (2003) Chem Lett 32:340
Hashihayata T, Mandai H, Mukaiyama T (2003) Chem Lett 32:442
Hashihayata T, Mukaiyama T (2003) Heterocycles 61:51
Jona H, Mandai H, Chavasri W, Takeuchi K, Mukaiyama T (2002) Bull Chem Soc Jpn 75:291
Hashihayata T, Mandai H, Mukaiyama T (2004) Bull Chem Soc Jpn 77:169
Mandai H, Mukaiyama T (2005) Chem Lett 34:702
Mandai H, Mukaiyama T (2006) Bull Chem Soc Jpn 79:479
Noyori R, Kurimoto I (1986) J Org Chem 51:4320
Higashi K, Nakayama K, Shioya E, Kusama T (1992) Chem Pharm Bull 40:1042
Higashi K, Susaki H (1992) Chem Pharm Bull 40:2019
Inanaga J, Yokoyama Y, Hanamoto T (1993) Chem Lett 85
Davis BG, Word SD, Maughan MAT (2002) Can J Chem 80:555
Lee JC, Pan GR, Kulkarni SS, Luo SY, Liao CC, Hung SC (2006) Tetrahedron Lett 47:1621
Hotha S, Kashyap S (2006) J Am Chem Soc 128:9620
Hotha S, Kashyap S (2006) J Am Chem Soc 128:17153
Acknowledgements
The author thanks the Dirección General de Enseñanza Superior (Grant CTQ2006-15279-C03-02) for financial support.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Springer-Verlag Berlin Heidelberg New York
About this entry
Cite this entry
López, J. (2008). O-Glycosyl Donors. In: Fraser-Reid, B.O., Tatsuta, K., Thiem, J. (eds) Glycoscience. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-540-30429-6_13
Download citation
DOI: https://doi.org/10.1007/978-3-540-30429-6_13
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-540-36154-1
Online ISBN: 978-3-540-30429-6
eBook Packages: Chemistry and Materials ScienceReference Module Physical and Materials ScienceReference Module Chemistry, Materials and Physics