
Abstract
Inflammation underlies chronic diseases frequently associated with aging, such as cancer, diabetes, obesity, Alzheimer’s disease, and atherosclerosis, among others. Macrophages are cells of the innate immune response, and they play a crucial role during the inflammatory process. In the early stages of inflammation, they are involved in the destruction of bacteria, parasites, viruses, and tumor cells that damage surrounding tissues. In the late stages, they participate in the resolution of inflammation by promoting tissue repair. In addition, macrophages present antigens to T lymphocytes, thus initiating the adaptive immune response. Aging impairs macrophage functions, thereby contributing to the immunosenescence of adaptive and innate immunity. Here we summarize data about the effects of aging on macrophages and discuss the molecular events underlying this process, such as the shortening of telomeres, the production of reactive oxygen species, and DNA repair.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

Keywords
Macrophages
Introduction
Macrophages are phagocytic cells involved in a number of complex functions in disease and health. They are critical for the establishment of the immune response against invading pathogens and for the maintenance of homeostasis – the latter achieved by promoting angiogenesis and tissue remodeling and repair. In addition, these cells are responsible for scavenging cellular debris and apoptotic cells (Locati et al. 2013).
Monocytes are the precursors of macrophages, and it has traditionally been claimed that they originate from undifferentiated stem cells in the bone marrow. Differentiation is induced in response to growth factors such as macrophage colony-stimulating factor (M-CSF), interleukin (IL)-3, and granulocyte macrophage colony-stimulating factor (GM-CSF). However, the only growth factor specific to macrophages is M-CSF because the M-CSF receptor (CSF1R, M-CSF-R, or CD115 is encoded by the c-fms proto-oncogene) is present only in the monocyte/macrophage lineage. Monocytes are carried to body tissues through the blood, and when they reach their destination, they become macrophages (Epelman et al. 2014). In response to various factors, including cytokines, cell-cell contacts, and extracellular matrix interactions, macrophages differentiate into various specialized cells depending on the tissue. For example, in the bone they become osteoclasts, in the liver Kupffer cells, in the skin Langerhans cells, in the bone bone marrow macrophages, and in the intestine crypt macrophages. The activities of differentiated macrophages vary in function of the tissue. The interaction of macrophages with factors such as GM-CSF, IL-4, and transforming growth factor (TGF)-β induces differentiation to dendritic cells, which are key elements in antigen presentation in the lymph node – a process that induces T cell activation and the acquired immune response. Under homeostasis, most macrophages are eliminated through apoptosis and renewed by the monocytes produced in the bone marrow or locally in tissues.
In recent years, it has been claimed that tissue macrophages do not originate in the bone marrow but rather from local stem cells in tissues. Experimental data are controversial, in some cases showing that resident macrophages can be maintained by local proliferation in healthy tissues, including the skin, lungs, central nervous system, and spleen (Guilliams et al. 2014). Although there are discrepancies about the origin of macrophages in several tissues, there is consensus that microglia of the brain are produced locally (Sheng et al. 2015).
The differentiation process is regulated by the combined action of several transcription factors (Valledor et al. 1998). Of these, PU.1, CCAAT-enhancer-binding proteins (C/EBP), and acute myeloid leukemia/core-binding factor (AML1/CFB)β play a crucial role in regulating the myeloid-specific expression of the M-CSF and GM-CSF receptors required for the differentiation, proliferation, and survival of macrophages. In response to growth factors, these cells proliferate; however, the presence of microbial agents, cytokines, or inflammatory molecules blocks this process and induces functional activities (Xaus et al. 1999). This activation leads to the release of toxic metabolites and to the elimination of microbes by phagocytosis.
Macrophages are key cells in innate and adaptive immune function. They can act directly by engulfing and digesting bacteria, parasites, viruses, and tumor cells or indirectly by releasing mediators such as IL-1 and tumor necrosis factor-α (TNF-α), which can regulate other cells. Macrophages are also responsible for processing antigens and presenting peptides to T lymphocytes, as well as for repairing tissue damage. In humans, mice, and rats, old age is accompanied by alterations in macrophage functions, which in turn contribute to the immunosenescence of adaptive and innate immunity. In this regard, dysfunctions in the following processes have been reported in aged macrophages: phagocytic activity, cytokine and chemokine secretion, antibacterial defenses such as the production of reactive oxygen and nitrogen intermediates, infiltration and wound repair in the late phase of inflammatory response, and antigen presentation (Herrero et al. 2001). Such changes lead to the impairment of the first line of immune defense and a decreased capacity to contribute to the development of specific immune responses by presenting antigens to T cells and by producing regulatory cytokines. Since macrophage activity is essential for the proper function of the immune system, studies addressing the effects of aging on the biology of these phagocytic cells and the molecular mechanisms involved in this process may provide insights into aging and immunosenescence. In addition, in order to fully understand the how macrophages age, it is of great interest to distinguish between the indirect (i.e., interactions with other cells) and direct (i.e., genome modifications) effects of aging on the biology of these cells.
Macrophages and Inflammation
The immune system responds to stress induced by chemical, physical, infectious agents by producing inflammation. In this process, macrophages play a key role, initially through their capacity to remove bacteria or parasites. To achieve this function, these cells use many components that can indirectly damage the surrounding tissues. After this pro-inflammatory activity (also called classical activation or M1), macrophages remove all the damaged tissue and initiate reconstruction. In this anti-inflammatory phase (so-called alternative activation or M2 ), macrophages prompt the synthesis of the extracellular matrix and also cell growth (Fig. 1).

Macrophages originate from HSCs in the bone marrow and migrate to body tissues where they become differentiated. Once in the tissues, they may proliferate or become activated during an inflammatory process. However, when not activated, most macrophages die by apoptosis
Under physiological conditions, mouse monocytes show a Ly6C− (an adhesion molecule) and CD43++ (a sialoglycoprotein) phenotype, while the equivalent cells in humans show a CD14− (lipopolysaccharide (LPS) receptor) and CD16++ (Fcγ receptor III) one (Ziegler-Heitbrock et al. 2010). Localized around blood vessels, these cells serve to patrol healthy tissues. When homeostasis is altered, they enter the affected tissues to initiate an early immune response. This activity requires the integrin LFA-1 and the chemokine receptor CX(3)CR1. During the first 24 h of this response, inflammatory loci are invaded by neutrophils, followed by monocytes with the Ly6C++ CD43− phenotype in mice and the CD14++ CD16− phenotype in humans. These monocytes trigger the pro- and anti-inflammatory activities mentioned earlier. Ly6C++ monocytes require the expression of selective chemokines (CCL7 and CCL2) and leukocyte-endothelial adhesion molecules (CD62E, β1-integrins, and VCAM-1). Once in the tissues, monocytes mature and become macrophages. The same macrophages that are polarized to pro-inflammatory action become anti-inflammatory a few days later (Arnold et al. 2007).
Macrophages play a key role in both innate and adaptive immunity. They recognize and destroy invading pathogens and apoptotic cells and modulate the immune response by producing cytokines and chemokines. Moreover, as antigen presenters, macrophages are involved in regulating the differentiation and activation of T cells. In addition to these functions, they play a crucial role in the resolution of inflammation and in tissue repair, by promoting the synthesis of the extracellular matrix, fibroblast proliferation, angiogenesis, and elimination of cellular debris (Wynn and Vannella 2016). Finally, these phagocytic cells eliminate modified proteins, oxidized low density lipoproteins, apoptotic cells, and other components from tissues. This action is achieved via the expression of scavenger receptors.
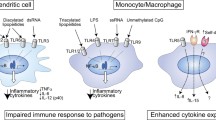
Macrophages reaching inflammatory loci in the early stages of the immune response kill any remaining microorganisms and remove cell debris and apoptotic bodies. They then go on to repair damaged tissues (Arnold et al. 2007) (Fig. 2). Cytokines or bacterial products trigger the activation of macrophages, a process in which these cells undergo a series of biochemical, morphological, and functional modifications. Recognized by specific receptors called Toll-like receptors (TLRs), Th1-type cytokines such as interferon-gamma (IFN-γ) – which interacts with its specific receptor – or bacterial products such as LPS, Gram-positive bacteria and yeast cell wall components, double-strand (ds) RNAs, bacterial flagellin, and CpG oligodeoxynucleotides induce classical activation (also known as M1) of these phagocytic cells. This activation leads to inflammation and elimination of the pathogen, and it is characterized by the expression of inducible nitric oxide synthase (NOS2) and by the biosynthesis and release of pro-inflammatory cytokines, including TNF-α, IL-1, and IL-6. Moreover, IFN-γ leads to the expression of several genes that regulate many aspects of macrophage biology. It induces the expression of the Fc high affinity receptors (FcγRI) on the cell surface, thus triggering increased antibody-dependent cytotoxicity; it enhances the phagocytic activity of macrophages; and it induces a respiratory burst (generation of nitric oxide (NO) and reactive oxygen species (ROS)) and the expression of lysosomal enzymes promoting the destruction of the pathogen (Gordon 2016). IFN-γ inhibits M-CSF-dependent proliferation and protects macrophages from apoptosis induced by glucocorticoids or M-CSF withdrawal. The protective effect of IFN-γ is mediated by p21waf1 expression and blockade of the cell cycle at the G1/S boundary (Xaus et al. 1999). In addition to modulating innate immunity, IFN-γ governs adaptive immunity by regulating the expression of major histocompatibility complex (MHC) class II genes at several levels – genes that are crucial for presenting antigens to T lymphocytes and for initiating an immune response.

Macrophages play a key role during the inflammatory process. In the initial phase, they are activated in a Th1 context, leading to the release of inflammatory mediators (pro-inflammatory cytokines and chemokines, NO, ROS, etc.). In the resolution phase, macrophages become alternatively activated by Th2-type cytokines and participate in tissue repair and remodeling through the production of polyamines and proline
In addition to this classical activation, several cytokines, such as IL-4 and IL-13, induce a distinct alternative activation program, also known as M2, in which the expression of arginase 1 is triggered, together with the upregulation of the mannose receptor (MR) and several other markers (Locati et al. 2013). Alternatively activated macrophages exert immune regulatory functions, drive Th2-type responses, and participate in tissue remodeling.
Curiously, arginine is the substrate for NOS2 and for arginase 1, and the system that transports this amino acid is induced by both types of cytokine, thus providing the cell with more arginine (Yeramian et al. 2006; Sans-Fons et al. 2013). NOS2 degrades arginine to produce the “killer” molecule NO, while arginase produces ornithine and polyamines, which are “repair” molecules.
Aged Macrophages
As early as 1908, Metchnikoff hypothesized that phagocytes/macrophages drive degenerative diseases associated with aging (Metchnikoff and Mitchell 1908). Macrophages from aged humans and mice display several functional defects with age. Many studies have focused on the effects of aging on macrophage biology but have yielded conflicting and sometimes opposing results. This may be due to factors such as the strain and sex of the experimental subjects, distinct macrophage origins (bone marrow, peritoneum, spleen, or alveolus), and differences in experimental conditions (culture, stimulant used, etc.). Furthermore, in the case of humans, it is difficult to define the term “healthy elderly subject” and would necessarily involve exhaustive health screening procedures. Moreover, most of the studies on humans in this field have been performed with monocytes, which generally provide a limited view of tissue macrophages. In addition, most studies addressing macrophage aging have reported modifications in the functional activities of these cells; however, few have attempted to explain the underlying causes of this dysfunction.
Differentiation and Maturation of Macrophages
The immune system is maintained by the generation of immune cells from hematopoietic stem cells (HSCs). These cells reside in the bone marrow and provide lifelong production of progenitors and peripheral blood cells. Simultaneously, HSCs must be able to maintain the stem cell pool through self-renewing divisions. Increasing experimental evidence supports the premise that HSCs become aged and have a limited functional lifespan (Denkinger et al. 2015). The first studies to suggest stem cell aging involved serial transplantation of whole bone marrow, which allowed only four to five rounds of transplantation. Given that the HSC compartment facilitates this regeneration, these findings point to the exhaustion of the stem cell pool. In fact, there is ample evidence that stem cell quality decreases with each self-renewing division. In both mice and humans, the proportion of differentiated blood cells arising from just a few HSC clones increases with age, suggesting that the number of active, functional HSCs declines with age (Jaiswal et al. 2014). The discrepancy between immunophenotypically and functionally defined HSCs has been interpreted as a compensatory increase in HSCs in response to declining function and a decreased capacity to differentiate. Results from studies comparing HSCs in various mouse strains indicate that the functional decline of this cell population can be correlated with lifespan. In addition, a negative correlation has also been shown between lifespan and proliferative capacity.
But how does the aging of HSCs affect the generation of macrophages? To date, it is not clear whether the generation of macrophages from their precursors is impaired with aging. In humans, aging brings about a reduction of CD68+ cells , which are markers of macrophage population (Ogawa et al. 2000). The percentage of CD68+ cells is high in children (first and second decades) and then decreases as the individual gets older. Moreover, it has been hypothesized that this reduction in the macrophage population negatively contributes to the reduction of HSC proliferation and influences the induction of apoptosis in the bone marrow of elderly people, probably via reduced production of growth factors and cytokines.
Mice have yielded conflicting data. It has been reported that the macrophage population is enhanced in the bone marrow, as shown by an increase in Mac1+ cells. This is reflected by an increase in the macrophage colony-forming unit (M-CFU) with age. Moreover, macrophages from the bone marrow of old mice generate less TNF-α than those from young mice. This observation would suggest that the increase in the number of macrophages reflects a compensation for their reduced function. However, it was found that the number of macrophage precursors, their size, their DNA content, and cell surface markers expressed during macrophage maturation, such as Mac1 or adhesion molecules, are similar in macrophages from aged and young mice (Herrero et al. 2001). It has been demonstrated that the accumulation of damaged DNA has a profound impact on the functional capacity of HSCs with age, leading to loss of reconstitution and proliferative potential, diminished self-renewal, increased apoptosis, and, ultimately, functional exhaustion (Rossi et al. 2007). In transplantation experiments, mice receiving HSCs from counterparts deficient in several genomic maintenance pathways show a marked decrease in the reconstitution of B cells, T cells, and myeloid cells. Moreover, these authors provided evidence that endogenous DNA damage increases with age in wild-type stem cells. This observation suggests that an impaired functional capacity of HSCs with accumulated DNA damage leads to the deficient generation of blood cells.
In the spleen, marginal zones (MZs) are architecturally organized for removing blood-borne antigens. MZ macrophages (MZMs) and MZ B cells are particularly important in host defense against T-independent pathogens and may be crucial for preventing diseases such as streptococcal pneumonia, which is devastating in older patients. Immunocytochemistry has revealed gross architectural changes in the MZs of aged mice, including a decrease in the number of MZMs. In the spleens, intravenously injected dextran showed less deposition of particles within the MZ of these animals (Birjandi et al. 2011).
Effects of Aging on Macrophage Functions
A great number of macrophage functions, including phagocytosis, antibacterial defenses, chemotaxis, wound repair, and activation, are altered in humans, rats, and mice during aging, thereby contributing to the immunosenescence of adaptive and innate immunity (Table 1).
Phagocytosis
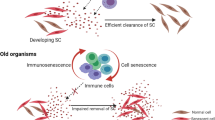
Phagocytosis is the first step of immune defense against invading pathogens; however, under physiological conditions, it also plays a critical role in scavenging apoptotic bodies, cells debris, etc. Tissue macrophages, alveolar macrophages, and polymorphonuclear leukocytes in the blood all exert phagocytic activity. In general, the data available on the effect of aging on the phagocytic function of macrophages and monocytes show a decrease in such activity. Several reports using murine models indicate a decline in the adherence, opsonization, tumor cell killing, and phagocytosis by peritoneal macrophages. In addition, the phagocytic activity of macrophages from aged individuals declines in parallel with a reduced production of macrophage-derived chemokines. It has recently been described that aging impairs phagocytosis by tissue-resident peritoneal macrophages but not by bone marrow-derived macrophages/monocytes (Linehan et al. 2014). This observation suggests that age-related defects in macrophage phagocytosis are caused by extrinsic factors in the tissue microenvironment.
The phagocytosis of apoptotic or necrotic cells by macrophages in aged mice is reduced compared with the same process in young mice (Takahashi et al. 2015). This process can release self-antigens, which may induce lymphocyte activation and autoantibody production, which in turn would result in the development of autoimmune diseases frequently associated with aging. Altered expression, function, and signal transduction of receptors involved in phagocytosis may explain the reduced phagocytic capacity observed in aging models. However, the effect of aging on these proteins has not been reported.
Chemotaxis
To eliminate invading pathogens, monocytes must migrate toward the site of inflammation in a process controlled by chemotactic stimuli. The main chemotactic factors are chemokines such as macrophage chemotactic and activating factor (MCAF), macrophage inflammatory protein (MIP)-1α, MIP-1β, RANTES, and IL-8 secreted by the endothelium, neutrophils, T cells, monocytes, and macrophages, as well as complement products such as C5a, C3a, and C4a. Aged mice show a decrease in macrophage production of MIP-1α and MIP-1β (Ashcroft et al. 1998). One study collected cutaneous punch biopsies of wounds from 138 healthy subjects aged 19–96, at fixed time points from day 1 up to 3 months post-wounding (Ashcroft et al. 1998). Using quantitative imaging, those authors demonstrated that the arrival of monocytes/macrophages and lymphocytes to the site was delayed in aged individuals. Thus, these data suggest that aged macrophages show an impaired chemotactic response, which may contribute to delayed pathogen clearance in healthy elderly individuals.
Activation of Macrophages
There are a number of observations suggesting that aging is associated with the anti-inflammatory phenotype (Sharma et al. 2014). Impaired macrophage polarization in the elderly may dysregulate the development of the host response, making them more susceptible to infectious diseases (Mahbub et al. 2012). Lymphoid organs of 27-month-old C57BL/6 J mice contain significantly more suppressive, IL-10-secreting, M2 macrophages than young adult mice (Jackaman et al. 2013). These M2 macrophages in aged mice are likely to support tumor growth by inhibiting T cell function (Ly et al. 2010). Age-related macular degeneration has been attributed to macrophages switching from a pro- to an anti-inflammatory type in response to the Rho-associated kinase (Zandi et al. 2015). Using Cx3cr1GFP/+ transgenic mice, it has been shown that the macrophage phenotype changes with advancing age, showing an increased number of fibrogenic genes, which have important implications for cardiac tissue injury responses and aging-associated cardiac fibrosis (Pinto et al. 2014). Therefore, the increase in the anti-inflammatory phenotype of macrophages with age may be related to various diseases.
The effects of many aspects of classical activation in aging macrophages have been widely studied. In this regard, there is contradictory data with respect to IFN-γ activation in aged macrophages. Human monocytes from the elderly show stronger generation of NO and ROS, which contribute to stress and damage reactions (Suchy et al. 2014). Also, mice show enhanced NO production under resting conditions, thus contributing to a reduction in the time needed to fully activate senescent macrophages following exposure to LPS (Smallwood et al. 2011). It was observed that macrophages from aged mice show increased susceptibility to oxidants and an accumulation of intracellular ROS (Sebastian et al. 2009).
Moreover, activated macrophages from aged humans and mice produce higher amounts of prostaglandin E2 (PGE2) than younger individuals. The age-related increase in PGE2 and cyclooxygenase (COX)-2 seems to be the result of the downregulation of the nuclear receptor retinoid X receptor (RXR)α, which suppresses nuclear factor-kappa B (NF-кB) activity (Chen et al. 2013).
In addition to microbicide activity, IFN-γ induces the expression of MHC class II molecules, which are involved in the initiation of the adaptive immune response. Antigen presentation by macrophages is decreased with age both in humans and mice, possibly due to diminished expression of MHC class II molecules (Herrero et al. 2001). It was found that the expression of MHC class II antigen IA molecules on the cell surface of bone marrow macrophages from aged mice is halved when stimulated with IFN-γ (Herrero et al. 2001). I-Aβ mRNA expression is also lower in aged macrophages as a result of a smaller number of transcription factors that bind to the W and X boxes of the MHC class II gene promoter.
Activation by LPS is also altered in aged macrophages. The LPS-induced production of inflammatory cytokines in peritoneal macrophages from rats and mice decreases with age. The stimulation of macrophages from aged rodents with LPS results in a significantly lower production of IL-1, TNF-α, and IL-6, as well as chemokines such as MIP-1α and MIP-1β. A study using human monocytes revealed a reduced production of IL-6 and TNF-α, which was related to decreased phosphorylation of external regulated kinase (ERK) 1/2 (Nyugen et al. 2010). Also, dynamic changes in circulating monocytes occur during aging in humans. In this regard, the proportions and numbers of CD14+CD16+ monocytes are significantly increased with age (Nyugen et al. 2010; Seidler et al. 2010).
The lower production of pro-inflammatory cytokines in aged mice has been attributed to IL-6 since aged KO mice for this cytokine recover the capacity to be activated by LPS (Gomez et al. 2010). The production of oxidative radicals in response to LPS also appears to decline with age, and the expression of NOS2 and the production of NO are reduced in macrophages from aged rodents.
Recently, it has been shown that tumor-exposed macrophages from elderly subjects but not those derived from young individuals produce high levels of IL-4, thus blocking T cell IFN-γ production. However, this situation can be rescued by exposing to tumor macrophages stimulated with IL-2 the T cells from elderly subjects that then induces the release of IFN-γ (Jackaman et al. 2014).
Several interpretations have been put forward to explain the decline in the production of inflammatory cytokines and oxidative radicals in response to LPS stimulation. The lack of consensus derives from the material used for experiments, as well as the experimental conditions. There is substantial evidence supporting the notion of the age-associated dysregulation of TLR, which would result in inappropriate activation and impairment of TLR signaling. The mechanisms underlying these changes remain unknown but are likely to be multifactorial, arising through expression at transcriptional and posttranscriptional levels, as well as changes in specific pathways that regulate TLR signal transduction (Shaw et al. 2011;Sharma et al. 2014).
A microarray analysis of RNA from resting and LPS-stimulated macrophages from aged and control mice demonstrated that immune response (pro-inflammatory chemokines, cytokines, and their receptors) and signal transduction genes (TLR and MAPK pathways) are specifically reduced in macrophages from aged mice. Moreover, many chemokines involved in innate immunity and inflammation, specifically in the chemotaxis of neutrophils, macrophages, and eosinophils, are decreased in these cells (Chelvarajan et al. 2006). All these observations correlate with the reduction in the overall inflammatory response of spleens of these animals. Furthermore, a variety of chemokines and receptors that affect CD4 and CD8 T cell migration and T helper cell type 1 development are reduced in aged mouse macrophages (Chelvarajan et al. 2006). Several components of the TLR pathway [TNF receptor-associated factor 6 (TRAF6), CD14, Rel, RelB, and some subunits of the NF-κB transcription factor] have reduced levels in LPS-stimulated aged macrophages. As this pathway is known to be critical for the production of chemokines and pro-inflammatory cytokines, it was proposed that the reduced levels of the components of this pathway explain the impaired production of several cytokines and chemokines in these macrophages (Chelvarajan et al. 2006). In addition to TLR pathway, those authors also detected an increase in the expression and phosphorylation of p38 mitogen-activated protein kinase (MAPK) in these cells. Low doses of a p38 MAPK inhibitor enhanced pro-inflammatory cytokine production by macrophages, thereby indicating that p38 MAPK activity participates in cytokine dysregulation in aged mouse macrophages.
More recently, a variety of explanations have been postulated on the decreased production of inflammatory cytokines and oxidative radicals in response to LPS stimulation. One study proposes that an increase in the phosphoinositide-3-kinase-Akt (pi3k-AKT) signaling pathway underlies cytokine dysregulation (Fallah et al. 2011). However, this was contradicted in another study that showed a decrease in pi3k-AKT (Verschoor et al. 2014). The decrease in IL-1β and IL-6 expression of aged macrophages after LPS treatment has been attributed to the dysregulated expression of miR-146a (Jiang et al. 2012), and the lower DNA methylation in the TNF promoter may contribute to the age-related increase in the production of this cytokine (Gowers et al. 2011).
The cellular origin of age-related inflammation has been related to the accumulation of cellular damage, which occurs as a result of exhaustion of the endogenous macrophagic mechanisms responsible for clearing damage-associated molecular patterns (DAMPs). The accumulation of DAMPs, such as necrotic cells and extracellular ATP, is sensed by pattern recognition receptors (PRRs) in macrophages, which trigger the chronic low-grade inflammation seen during aging (Youm et al. 2013). The primary sensor of diverse DAMPs is the NLRP3 inflammasome, which initiates a sterile inflammatory cascade. This inflammasome activates caspase-1, which in turn controls the secretion of IL-1β and IL-18. Interestingly, the ablation of the NLRP3 inflammasome prevents age-related inflammation and functional decline (Youm et al. 2013). Growth hormone receptor (GH-R)-dependent downregulation of this inflammasome in macrophages is linked to pro-longevity effects that maintain immune system homeostasis in aging. The downregulation of GH-R-mediated signals in macrophages represents an endogenous regulatory “break” mechanism that limits age-related inflammation by downregulating NLRP3 inflammasome activation (Spadaro et al. 2016).
The pro-inflammatory signature of macrophages in aged subjects can have deleterious effects. For example, in contrast to young mice, systemic cancer immunotherapy regimes or LPS administration to aged mice results in rapid and lethal toxicities correlating with heightened systemic pro-inflammatory cytokines. Prior in vivo depletion of macrophages in aged mice leads to lower cytokine levels and increased survival. Both aged TNF knockout mice and the treatment in vivo of TNF blockade in aged mice result in significant increases in survival and lessened pathology. Importantly, TNF blockade in tumor-bearing aged mice receiving cancer immunotherapy has significant antitumor effects (Bouchlaka et al. 2013). These data demonstrate the critical role of macrophages in the age-associated hyper-inflammatory cytokine responses to systemic immune stimulation.
Also, the reduced expression of cytokines may be important in immune defense. Pneumococcal infections continue to be a leading cause of death in people >65 years of age. A major defense mechanism is the production of IFN-β by macrophages. The initiation of type I IFN responses to Streptococcus pneumoniae is mediated by a cytosolic DNA-sensing pathway that involves the intracellular recognition of bacterial DNA by the adaptor molecule stimulator of IFN genes (STING) and phosphorylation of transcription factor IFN regulatory factor 3 (IRF3). Recently, it was described that during S. pneumoniae infection, age enhances endoplasmic reticulum (ER) stress and contributes to increased expression of autophagy-related gene 9, which inhibits STING-mediated IFN-β production (Mitzel et al. 2014).
There are few data regarding the way in which aging affects the alternative activation of macrophages. Arginase expression, which plays a crucial role in M1/M2 polarization, is decreased with age, as shown in mice infected with helminth (Sugawara et al. 2011). Also, IL-10 secretion in peritoneal macrophages is increased (Liscovsky et al. 2011). However, further studies are required to examine whether changes in macrophage polarization with aging are responsible for some aspects of immunosenescence.
Wound Repair
In addition to their crucial role in the initial phases of the inflammatory response, macrophages secrete angiogenic and fibrogenic growth factors and thus have key functions in the removal and regeneration of damaged tissue. These cells have been shown to be both anti- and pro-angiogenic, and their role in regulating angiogenesis at sites of tissue injury is critical and complex. Studies in humans and in rodents have revealed an age-related decline in coetaneous wound repair, which affects the inflammatory response and the growth phase of the repair process. These changes include enhanced platelet aggregation and delays in many processes, including reepithelialization, angiogenesis, collagen deposition, turnover and remodeling, healing strength, and infiltration and function of macrophages, as well as decreased wound strength. Using a murine model of excision wound repair, it was shown that repair and reepithelialization processes are delayed significantly in aged mice and that the rate of wound repair can be partially restored by the addition of peritoneal macrophages from young mice. In addition, the rates of collagen synthesis and angiogenesis are delayed in aged animals. TLRs 2, 4, 7, and 9 and adenosine A (2A) receptors mediate macrophage production of vascular endothelial growth factor (VEGF) and other angiogenic factors. Hence, the decrease in TLR function observed in aging may contribute to delay wound healing. Furthermore, the expression of cell adhesion molecules on the vascular endothelium is decreased in the elderly, and responsiveness (receptor expression) to VEGF and epithelial growth factor (EGF) is reduced (Danon et al. 1989).
Excess antiproliferative activity of macrophages may underlie several pathologies. Macrophage dysfunction plays a critical role during neovascular proliferation in diseases related to aging. In the eye, choroidal neovascularization (CNV) causes blindness in patients with age-related macular degeneration (AMD). Increased IL10 in senescent eyes activates STAT3 signaling, which induces the alternative activation of macrophages and vascular proliferation. Targeted inhibition of both IL10 receptor-mediated signaling and STAT3 activation in macrophages reverses the aging phenotype. In addition, adoptive transfer of STAT3-deficient macrophages into the eyes of aged mice significantly reduces the amount of CNV. Systemic and CD163+ eye macrophages obtained from AMD patients also activate STAT3 (Nakamura et al. 2015). This study demonstrated that impaired SOCS3 feedback leads to permissive IL10/STAT3 signaling that promotes alternative macrophage activation and pathological neovascularization. Subsequent to laser injury to the retina, IL-10 is upregulated, and Fas ligand, IL-12, and TNF-α are downregulated in ocular macrophages of old mice, thus revealing an anti-inflammatory phenotype.
In uveal melanoma in aged mice, tumor progression was found to be determined by the presence of macrophages, as local depletion of these cells prevented tumor outgrowth. This finding thus indicates that the macrophages of these mice exerted strong tumor-promoting activity. These macrophages carried M2-type characteristics, as shown by CD163 and peroxisome proliferator-activated receptor γ expression, and multiple angiogenic genes were heavily overrepresented in the tumors of these animals (Ly et al. 2010). Thus, communication between tissue cells and the innate immune system appears to be impaired, thus contributing to the functional deficiencies observed in tissue repair.
Muscle aging is associated with increases in anti-inflammatory macrophage population, which can enhance muscle fibrosis. Bone marrow cells from young mice transplanted into old mice showed fewer anti-inflammatory macrophages and less accumulation of collagen. Thus, the increase in anti-inflammatory macrophages in aging muscle and the associated muscle fibrosis are determined in part by the age of bone marrow cells (Wang et al. 2015).
Following bleomycin-induced lung injury, aged mice develop more lung fibrosis and exhibit increased morbidity and mortality when compared to young mice. Bleomycin-exposed aged NLRP3−/− mice show reduced fibrosis compared to their wild-type age-matched counterparts. Bone marrow-derived and alveolar macrophages from aged mice display higher levels of NLRP3 inflammasome activation and caspase-1-dependent IL-1β and IL-18 production (Stout-Delgado et al. 2016). These increases are associated with altered mitochondrial function and increased production of ROS. That study demonstrated that age-dependent increases in mitochondrial reactive oxygen species (mtROS) production and NLRP3 inflammasome activation by alveolar macrophages contribute to the development of experimental fibrosis.
The injured arteries in aging rats develop thicker neointimas than those in younger animals, and this greater thickness was found to be significantly correlated with a higher number of tissue macrophages and increased vascular IL-18. The depletion of macrophages in aged rats by means of treatment with clodronate liposomes ameliorates the vascular accumulation of IL-18 and significantly decreases neointimal formation. In addition, the injured arteries of these rats accumulate more fibrinogen-γ than those of young animals (Rodriguez-Menocal et al. 2014).
Metabolic Modifications
Advancing age is increasingly accompanied by obesity, the accumulation of visceral adipose tissue, and inflammation. The term obesity is not defined by excess weight only but also by the accumulation of a large amount of fat. While the source of the inflammation in aging has not been identified conclusively, the activity of macrophages in adipose tissue has been shown to be associated with higher inflammation in aged mice. Recently, there has been increasing interest in the field of age-induced obesity and inflammation; however, how age-induced obesity influences leukocyte-mediated inflammation is not clear. It should be noted that there are inconsistencies in the literature and in some cases contradictory results. These are attributable to the varying sources of cells (i.e., human or mouse), culture conditions, and experimental protocols used in the studies. In fact, it is difficult to analyze the cause and consequences of all the modifications described below.
Aging is accompanied by increases in visceral fat similar to those seen in young obese (ob/ob or diet-induced obese) mice. The pro-inflammatory cytokine levels and organ pathologies of both young ob/ob- and diet-induced obese mice are comparable to those in aged ad libitum mice after systemic immunotherapy, and they culminate in death (Mirsoian et al. 2014). Aging brings about qualitative changes in adipose tissue macrophages, which show a shift toward a pro-inflammatory environment. The mechanism behind this shift appears to be related to a decrease in peroxisome proliferator-activated receptor-γ (PPRγ) expression in adipose tissue macrophages. This observation reveals a unique inflammatory cell signature in the physiological context of aging adipose tissue that differs from that induced in a setting of diet-induced obesity (Lumeng et al. 2011).
Abnormal polarization in older macrophages is caused by programmatic changes that lead to reduced expression of the ATP-binding cassette transporter (ABCA)1 . Downregulation of ABCA1 by microRNA-33 impairs the ability of macrophages to effectively efflux intracellular cholesterol, which in turn leads to higher levels of free cholesterol within senescent macrophages. Elevated intracellular lipids polarize older macrophages to an abnormal, alternatively activated phenotype, thereby promoting pathologic vascular proliferation. Mice deficient for Abca1 are characterized by an accelerated aging phenotype, whereas restoration of the cholesterol efflux using liver X receptor agonists or miR-33 inhibitors reverses it. Monocytes from older humans with AMD show similar alterations (Sene et al. 2013).
Ghrelin is the only circulating orexigenic hormone known to increase obesity and insulin resistance. The expression of the ghrelin receptor, growth hormone secretagogue receptor (GHS-R) , increases in adipose tissues during aging, and aged Ghsr−/− mice exhibit a lean and insulin-sensitive phenotype. These animals show a reduction in macrophage infiltration, the M1/M2 ratio, and pro-inflammatory cytokine expression (Lin et al. 2016). These studies demonstrate that ghrelin signaling plays an important role in macrophage polarization and adipose tissue inflammation during aging.
Adipose tissue inflammation and insulin resistance in diet-associated obesity have been correlated with aberrant ER stress. An elevated ER stress response has been reported in the adipose tissue of old compared to young mice. Similar experiments with adipose tissue macrophages revealed an elevated ER stress response when induced with thapsigargin. Treatment with chemical chaperone 4-phenyle-butyric acid alleviates ER stress in adipose tissue macrophages and attenuates the production of TNF-α in both young and old mice. Finally, old mice fed 4-phenyle-butyric acid show reduced expression of ER stress and inflammatory cytokine genes (Ghosh et al. 2015). These data suggest that an exaggerated ER stress response in aging adipose tissue contributes to age-associated inflammation and that this stress response can be mitigated by treatment with chemical chaperones.
Monocytes play a crucial role in atherosclerosis by differentiating into foam cells (lipid-laden macrophages) and producing atherogenic pro-inflammatory cytokines. Ex vivo foam cell formation is enhanced in monocytes from older individuals by both extrinsic and intrinsic mechanisms (Angelovich et al. 2016).
Effect of Aging on Tissue-Resident Macrophages
In addition to studies on the effect of aging on macrophage biology, many reports have focused on the impact of aging on certain tissue-specific macrophages. Thus, alterations in the function of these macrophages may contribute to the pathologies observed in these tissues during aging. Macrophages are dispersed throughout the body. Some take up residence in particular tissues, where they become fixed macrophages that serve various functions and they are named to reflect their tissue location: alveolar macrophages in the lung, thymic macrophages in the thymus, histiocytes in connective tissues, Kupffer cells in the liver, mesangial cells in the kidney, osteoclasts in bones, Langerhans cells (LCs) in the skin, and microglia in the brain.
LCs were originally described as an epidermal macrophage population containing large granules and capable of phagocytosis. Later, they were classified as immature dendritic cells, since after activation they can migrate from the skin to regional lymph nodes, a hallmark characteristic of dendritic cells. Although macrophages and LCs belong to the myeloid lineage, the precise lineage relationship between them is not clear. The number of epidermal LCs and their function are diminished in humans and mice as a result of aging. However, it is not clear whether this defect is caused by a decrease in the production of bone marrow precursors.
Cutaneous delayed-type hypersensitivity (DTH) responses to recall antigens are significantly decreased in older individuals (Agius et al. 2009). These subjects show defective activation of dermal blood vessels as a result of decreased TNF-α secretion by macrophages. This decrease prevents memory T cell entry into the skin after antigen challenge. However, isolated cutaneous macrophages from these subjects can be induced to secrete TNF-α after stimulation with TLR 1/2 or TLR 4 ligands in vitro, thereby indicating that the defect is reversible. The decreased conditioning of tissue microenvironments by macrophage-derived cytokines can therefore lead to defective immune surveillance by memory T cells (Agius et al. 2009). These age-related changes may contribute to altered coetaneous immune function, such as poor or variable contact hypersensitivity to allergens in the elderly.
The lungs of aged mice have elevated levels of pro-inflammatory cytokines and a resident population of highly activated pulmonary macrophages that are refractory to further activation by IFN-γ. These macrophages secrete more pro-inflammatory cytokines in response to Mycobacterium tuberculosis infection than similar cells from young mice. The nonsteroidal anti-inflammatory drug ibuprofen reverses lung and macrophage inflammatory signatures (Canan et al. 2014).
Age-dependent dysfunction in macrophages is associated with poor activation of NF-κB and MAPK following TLR stimulation. In this regard, alveolar macrophages of aged mice show TNF-α-mediated induction of A20, a cytosolic and homeostatic suppressor of the NF-κB and MAPK signaling cascades that deubiquitinates (i.e., inactivates) the common upstream signaling molecule TRAF6 (Hinojosa et al. 2014).
Microglial cells are small, highly ramified, immune sentinels of the brain. They are distributed throughout the brain parenchyma and are continuously monitoring the microenvironment in order to detect injuries or pathogens. After activation, microglia initiate an innate immune response by producing pro-inflammatory cytokines.
There are many differences between aged and young microglia, including morphology, cell number, and dynamics (Harry 2013). Aged microglia display slower acute and sustained chronic post-injury responses (Hefendehl et al. 2014). The phagocytic capacity of aged microglia is reduced as is their endocytosis (Orre et al. 2014). During their lifetimes, microglia respond to various stimuli and produce ROS, which can induce DNA damage with age and thus the accumulation of mutations that produce dysregulation (Perry and Holmes 2014). These mutations can trigger a substantial loss of microglia (through cell death), and the renewal of this cell population may be impaired as a result of telomere shortening, which in turn is caused by a decrease in telomerase activity. Taken together, these observations suggest that aged microglia decline in homeostatic function and become susceptible to deterioration (Lourbopoulos et al. 2015).
Several lines of evidence from humans and mice support the notion of microglial senescence, which leads to neurodegeneration. Microglia from healthy aging human brains show an increased expression of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-12), and similar findings were obtained using a mouse model (Lee et al. 2013). Conditioned media from bone marrow-derived macrophages of aged rats show an increase in the expression of pro-inflammatory mediators in glial cells. Also, there is an age-related increase in macrophage infiltration into the brain, and this is combined with enhanced expression of IFN-γ and the TLR 4 agonist (Barrett et al. 2015). All these features are likely contribute to damaging the cascade, thus impairing neuronal function. Higher expression of these cytokines produces tissue degeneration. Therefore, the increased levels of these molecules in senescent microglia may contribute to brain damage during aging and even to the onset of neurodegenerative diseases.
The efficiency of central nervous system remyelination declines with age. This decline is in part due to an age-associated decrease in the phagocytic removal of myelin debris, which contains inhibitors of oligodendrocyte progenitor cell differentiation. It has been shown that the expression of genes involved in the retinoid X receptor (RXR) pathway decreases with age in both myelin-phagocytosing human monocytes and mouse macrophages. Macrophage-specific RXRa (Rxra) knockout mice revealed that loss of function in young mice causes delayed myelin debris uptake and slowed remyelination. In contrast, RXR agonists partially restore myelin debris phagocytosis in aged macrophages (Natrajan et al. 2015). These results reveal the RXR pathway as a positive regulator of myelin debris clearance and a key player in the age-related impairment of remyelination.
The K+ channel expression pattern of microglia strongly depends on the cellular microenvironment, and it has been recognized as a sensitive marker of the functional state of this cell population. Although age-related changes in the microglial phenotype are accompanied by alterations in the expression of voltage-activated microglia, there are no modifications in Ca2+-activated or K+ channels (Schilling and Eder 2015).
Alternative activation of microglia by IL-4 is claimed to support growth and repair processes after central nervous system injury. Aged mice show reduced functional recovery after spinal cord injury. This reduction is associated with the impaired induction of IL-4 receptor α (IL-4Rα) in microglia. The failure to successfully promote an IL-4/IL-4Rα response in aged mice results in attenuated expression of arginase, IL-1β, and chemokine ligand 2 and diminished recruitment of IL-4Rα+ macrophages to the injured spinal cord. The expression of this key signaling cascade is reduced with age, thus correlating with impaired functional recovery after spinal cord damage (Fenn et al. 2014).
There are few data on how aging affects the function of osteoclasts. Bone mass is maintained by a delicate balance between formation and resorption. At the cell level, the rates of bone formation and resorption reflect the number and activity of stromal/osteoblastic cells and osteoclasts – both of macrophage origin. The former regulate the number and activity of osteoclasts through expression of the soluble receptor activator of NF-кB ligand (RANKL), M-CSF, and osteoprotegerin (OPG). With advancing age, RANKL expression in whole bone and in cultured bone marrow cells from both humans and animals gradually increases, while the expression of OPG either decreases or remains unchanged. RANKL expression is also increased in early stromal/osteoblastic cells from aged mice. Furthermore, the osteoclast progenitor pool increases with advancing age in these animals. It has been demonstrated that aging significantly enhances stromal/osteoblastic cell-induced osteoclastogenesis, promotes the expansion of the osteoclast precursor pool, and alters the relationship between osteoblasts and osteoclasts. Consistent with these changes, the efficacy of osteoclasts to form bone is also impaired (Cao et al. 2005). All these modifications may contribute to the osteoporosis associated with aging.
Human skeletal aging is characterized by a gradual loss of bone mass as a result of excess bone resorption that is not counteracted by new bone formation. Human bone marrow cells show an increased expression of the M-CSF receptor and RANK with age. The generation of osteoclasts in vitro is also augmented with age (Chung et al. 2014). These findings support the hypothesis that human bone marrow cells and their products contribute to skeletal aging by increasing the generation of bone-resorbing osteoclasts.
It was recently shown that Wnt4 attenuates bone loss in mouse models of osteoporosis and skeletal aging by inhibiting NF-кB via noncanonical Wnt signaling. In addition to promoting bone formation, Wnt4 inhibits osteoclast formation and bone resorption. Mechanistically, Wnt4 impedes NF-кB activation mediated by Tak1 in macrophages and osteoclast precursors independently of β-catenin (Yu et al. 2014).
In summary, the aging process impairs the function of macrophages and tissue-specific macrophages, thus leading not only to a deficient immune response but also to the development of several pathologies in the tissues in which these cells reside.
Molecular Mechanisms Involved in Macrophage Aging
The data presented so far indicate an age-associated malfunction of macrophages. Most of the aforementioned studies describe this dysfunction but do not shed light on its origin. Many theories have been formulated to explain the aging process. Given that immunosenescence is a hallmark of aging, these theories may also explain the changes that occur in the immune system as a result of maturation (Fig. 3).

Molecular view of macrophage aging. Altered gene expression caused by the accumulation of DNA damage and by epigenetic changes may, in part, explain the dysfunctions observed in aged macrophages
Aging and Altered Gene Expression
Aging is associated with changes in gene expression of several cell types. Immune cells lose the expression of several genes. As discussed above, aged macrophages also show altered expression of many genes (TLRs, pro-inflammatory cytokines, chemokines, MHC class II molecules, signal transduction molecules, transcription factors, etc.), which may explain the loss of some functional activities. The molecular basis of this altered expression is related to impaired signal transduction (MAPK, protein kinase C, etc.) (Chelvarajan et al. 2006). In other cases, changes in gene expression result from age-related modifications of one or more transcriptional factors. For example, it was demonstrated that loss of MHC class II expression in aged macrophages is caused by lower levels of transcription factors that bind to the promoters of these genes, thereby reducing binding efficiency (Herrero et al. 2001). Moreover, changes in gene expression may be due to epigenetic mechanisms. In this regard, methylation of CpG islands decreases during cellular senescence, and the activity of the DNA-methyl transferase is also lower in aged cells. Furthermore, the acetylation and deacetylation of histones are involved in cell senescence. The histone acetyl transferase activity of p300/CBP is reduced in several tissues in aged mice (Li et al. 2002). Moreover, the histone deacetylase silent information regulator 2 (Sir2) and its homologs in mammals sirtuin (SIRT)1 and SIRT6 are involved in regulating genomic stability and aging in yeast, worms, and mice (Mostoslavsky et al. 2006).
Although many functions of innate immune cells decline with age, the elderly show a chronic low level of inflammation, which is a major contributor to age-associated frailty and morbidity, as well as increased mortality. This low level is caused by increased basal inflammatory cytokine production by innate cells such as macrophages – a process known as inflammaging. It is currently unclear how healthy macrophages are maintained throughout life and how inflammation is linked to myeloid dysfunction during aging. It has been shown that autophagy, an intracellular degradation mechanism, regulates the acquisition of major aging features in macrophages (Stranks et al. 2015). In the absence of the essential autophagy gene Atg7, macrophage populations are increased, and key functions such as phagocytosis and nitrite burst are reduced, while the inflammatory cytokine response is increased – a phenotype also observed in aged macrophages. Macrophages from aged mice exhibit a significantly reduced autophagic flux compared to that of young mice. Interestingly, in aged macrophages, the promoter regions of Atg5 and LC3B, two genes involved in autophagy, are hyper-methylated, and this is accompanied by low gene expression. Treatment of aged mice and their derived macrophages with methyltransferase inhibitor or specific DNA methyltransferase siRNA restores the expression of Atg5 and LC3 in vivo and in vitro (Khalil et al. 2016). On the basis of these observations, it is pertinent to study the epigenetic regulation of gene expression during aging in macrophages.
Another mechanism that could explain the major susceptibility of aged macrophages to apoptosis is related to ER stress. When macrophages phagocyte debris or pathogens, ER stress is induced through activation of three transducers located on the ER membrane, namely, inositol-requiring enzyme-1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA-like ER kinase (PERK). This activation either relieves ER stress or induces cellular apoptosis. IRE1 triggers apoptosis through TRAF and Jun amino-terminal kinase (JNK) or through IRE1-dependent decay (RIDD), which is dependent on the ribonucleolytic function of IRE1. Normally, IRE1 targets specific mRNAs, such as x-box binding protein 1 (XBP1), to exert its ribonucleolytic function and induce splicing of XBP1. However, prolonged ER stress induces RIDD and leads to indiscriminate degradation of membrane-associated mRNAs regardless of their sequences. In response to tunicamycin (TM), a known ER stress inducer in macrophages, peritoneal macrophages of aged mice are more susceptible to apoptosis than macrophages from young mice. Also, aged macrophages express less phosphorylated IRE1α (p-IRE1α) than young ones. Knocking down XBP1 using small interference RNA-targeted XBP1 (si-XBP1) increases the protein levels of p-IRE1α and reduces apoptosis in aged macrophages but not in young ones. Moreover, concurrently knocking down the gene expression of both IRE1α and XBP1 abrogates the apoptosis-reducing effects of si-XBP1 in aged macrophages (Song et al. 2013). These results suggest that the IRE1α-XBP1 axis contributes to age-associated apoptosis induced by ER stress, and they identify a novel interaction by which aging enhances ER stress-induced apoptosis in macrophages.
Aging is the single biggest risk factor for malignant transformation. Among the most common age-associated malignancies in humans are non-melanoma skin cancers. Mutant H-Ras activation in mouse epidermis, a frequent event in cutaneous squamous cell carcinoma (SCC), causes a differential outcome in aged versus young mice. While H-Ras activation in the young skin results in hyperplasia that is accompanied mainly by rapid hair growth, activation in aged skin leads to more dysplasia and gradual progression to in situ SCC. The progression of SCC is associated with increased inflammation; pronounced accumulation of immune cells, including T cells, macrophages, and mast cells; and also excessive cell senescence. A strong age-dependent anti-inflammatory response involving enhanced IL4/IL10 expression was reported. Furthermore, upon switching off oncogenic H-Ras activity, young but not aged skin regenerates successfully. This observation would suggest a failure of the aged epidermal stem cell to restore damaged tissue. These findings support an age-dependent link between the accumulation of senescent cells, immune infiltration, and cancer progression, which together may contribute to the increased risk of cancer associated with old age (Golomb et al. 2015).
Telomere Shortening
Telomeres are chromatin structures that cap and protect the ends of chromosomes. In vertebrates, they are formed by tandem repeats of hexamer sequences (TTAGGG) that are associated with various specific proteins involved in the maintenance and regulation of telomere length (Zhang et al. 2016). With self-replication, telomeres lose TTAGGG repeats because conventional DNA polymerases are not able to completely replicate linear chromosomes (Lansdorp 2005). Progressive telomere shortening has detrimental effects; chromosome caps are unprotected, leading to genomic instability and cell death. However, in healthy cells, telomere erosion initiates a cell senescence program that prevents further divisions, thereby protecting cells from excessive telomere loss and cell death. Telomere shortening is involved in the aging process and in the regulation of replicative lifespan. Late generations of the telomerase KO mice (Terc−/−) show severe telomere dysfunction characterized by critically short telomeres and end-to-end fusions. These mice suffer from various age-related diseases that affect highly proliferative tissues (Blasco 2002). Among these tissues, the generation and function of immune cells are impaired by telomere attrition. Numerous studies have confirmed that loss of telomeric DNA with progressive telomere shortening occurs in cells of the hematopoietic system as a result of a function of normal replicative aging. Terc−/− mice have been reported to show reduced proliferative capacity of T and B cells (Blasco 2002).
HSCs display telomere shortening during in vitro culture and in vivo aging (Zimmermann et al. 2004). HSCs derived from humans and mice lose telomeric DNA with age despite the presence of detectable telomerase activity (Allsopp et al. 2001). Moreover, telomere shortening occurs during serial transplantation of HSCs, coinciding with impaired function (Allsopp et al. 2001). These observations suggest that telomere attrition alters the capacity of HSCs to generate blood cells. In support of this notion, HSCs from telomerase-deficient mice characterized by short telomeres show a reduced ability to repopulate irradiated mice (Allsopp et al. 2001).
To determine the effect of aging on macrophages, bone marrow-derived macrophages was produced in vitro. In this cellular model, the effect of aging on this cell population without the influence of other cell types that may be affected by this process was analyzed. It was showed that telomeres shorten as macrophages age, leading to decreased GM-CSF- but not M-CSF-dependent proliferation of these cells as a result of reduced phosphorylation of signal transducer and activator of transcription (STAT)5a. Macrophages from aged mice showed shortened telomeres, increased susceptibility to oxidants, and an accumulation of intracellular ROS. In these macrophages STAT5a oxidation was reduced, which led to the decreased phosphorylation observed. Interestingly, the same cellular defects were found in macrophages from Terc−/− mice, thereby suggesting that telomere loss is responsible for enhanced oxidative stress, for reduced Stat5a oxidation and phosphorylation, and, ultimately, for impaired GM-CSF-dependent macrophage proliferation (Sebastian et al. 2009). The microglia of these animals also show deficient capacity to proliferate (Raj et al. 2015).
DNA Damage
Accumulation of DNA damage may also explain the aging process. Increasing experimental data suggest that somatic mutations accumulate and increase exponentially during aging (Tucker et al. 1999). This may be due to a greater number of mutations or to deficient DNA repair activity. DNA damage produced by these mutations may alter gene expression patterns, the generation of modified proteins, and some cellular functions. To repair this damage, cells have developed a DNA damage response, which includes the detection of the lesion and the activation of cell cycle checkpoints and several repair mechanisms to repair the damage. Deficiencies in some of the components of the DNA damage response lead to senescence and premature aging, thus supporting the notion that accumulated DNA damage contributes to aging.
Advanced age is accompanied by impaired mitochondrial functions, such as lowered oxidative capacity, reduced oxidative phosphorylation, decreased ATP production, a significant increase in ROS generation, and a diminished antioxidant defense. Mitochondrial biogenesis declines with age due to alterations in mitochondrial dynamics and the inhibition of mitophagy – an autophagy process that removes dysfunctional mitochondria (Sobenin et al. 2015). Mitochondrial aging is characterized by structural alterations to this organelle and by mitochondrial DNA (mtDNA) damage (Yao et al. 2013). Somatic mtDNA mutations during aging are due to the high mutation rate of the mitochondrial genome, which is at least 5- to 15-fold higher than that in the nuclear genome. Mitochondrial dysfunction contributes to the development of oxidative stress. The electron transport chain constantly produces superoxide radical anions, which, in the case of mitochondrial dysfunction, cause electron leakage. These electrons go on to form hydroxyl radicals and hydrogen peroxide from superoxide and ROS associated with lipid and protein oxidation. ROS production induces macrophages nonresponsive to growth factors such as GM-CSF (Sebastian et al. 2009). ROS formation may trigger a cascade of events such as inflammation, cellular apoptosis, and DNA damage.
Alterations in the maintenance of telomere length and in the nucleotide excision repair (NER) and nonhomologous end-joining (NHEJ) repair pathways limit stem cell function in an age-dependent manner by intrinsically diminishing the self-renewal and proliferative capacity of HSCs. Moreover, elevated levels of ROS contribute to impaired HSC function. Studies in mice deficient for the ataxia-telangiectasia mutated (Atm) gene show that the self-renewal capacity of HSCs depends on Atm-mediated inhibition of oxidative stress. Atm-deficient mice show progressive bone marrow failure, which results from a defect in HSC function that is associated with elevated ROS (Ito et al. 2004). Therefore, DNA damage- and ROS-dependent HSC failure may lead to an impaired production of blood cells, among these macrophages, during aging.
Few reports have assessed the direct influence of DNA damage and ROS on macrophage biology. The activation of macrophages leads to an increase in ROS and NO production, as well as many pro-inflammatory cytokines that result in the clearance of the invading pathogen. However, this prooxidant environment may also cause DNA damage in macrophages themselves, including the induction of apoptosis (Xaus et al. 2000). This observation suggests that it is crucial for these cells to be supported by highly efficient antioxidant defenses. In this regard, it has been shown that the levels of antioxidant defenses , such as superoxide dismutase activity, decrease with aging in macrophages, although no data about DNA damage in these cells has been reported.
In addition, elevated levels of ROS modulate some redox-sensitive transcription factors. Among these, NF-κB is highly relevant because it is a key regulator of macrophage biology. It is thought that the phosphorylation of IκB, the inhibitory subunit of NF-κB, is the key step in NF-κB redox activation. ROS-mediated phosphorylation of IκB, leading to its ubiquitination and degradation, allows the NF-κB complex to be translocated to the nucleus and act as a transcriptional activator. In contrast, direct oxidation of critical cysteine residues in the p50 subunit of NF-κB decreases its DNA-binding activity (Piette et al. 1997). It has been described that macrophages undergo oxidative stress with aging, as reflected by an increase in the oxidized glutathione/reduced glutathione ratio (De La Fuente et al. 2004). Thus, alteration of the redox status in macrophages during aging may alter the activity of NF-κB and the expression of its target genes, which in turn may lead to the loss of some functional activities.
The loss of DNA damage repair mechanisms contributes to the aging associated with DNA damage. One example is the metabolic syndrome that occurs in DNA polymerase η (pol η) deficiency. Mice depleted of pol η (pol η−/−) are characterized by obesity, with visceral fat accumulation, hepatic steatosis, hyperleptinemia, hyperinsulinemia, and glucose intolerance. The infiltration of macrophages into adipose tissue is apparent in these animals. Furthermore, the plasma and adipocytes of this mouse model show increased expression of inflammatory cytokines. In comparison to that of wild-type mice, the adipose tissue of pol η−/− mice exhibits increased DNA damage and a greater DNA damage response, as reflected by upregulation and/or phosphorylation of ATM, phosphorylated H2AX (γH2AX), and poly[ADP-ribose] polymerase 1 (PARP-1). Concomitantly, the adipose tissue of this mouse shows increased cellular senescence as early as 4 weeks of age, as indicated by the upregulation of senescence markers, including p53, p16Ink4a , and p21, and senescence-associated (SA) β-gal activity. Treatment of pol η−/− mice with a p53 inhibitor, pifithrin-α, reduces adipocyte senescence and attenuates the metabolic abnormalities (Chen et al. 2015). These studies indicate that elevated DNA damage is a root cause of adipocyte senescence, which in turn plays a determining role in the development of obesity and insulin resistance.
Nijmegen breakage syndrome 1 (NBS1) is a component of the MRE11 complex, which is a sensor of DNA double-strand breaks and plays a crucial role in the DNA damage response. Given that activated macrophages produce large amounts of ROS that can cause DNA lesions, the role of NBS1 in the functional activity of macrophages was examined. In mice expressing a hypomorphic allele of Nbs1 (Nbs1ΔB/ΔB), ROS induced by macrophage activation caused increased levels of DNA damage that were associated with defects in proliferation, delayed differentiation, and increased senescence accompanied by telomere shortening. Furthermore, upon stimulation, Nbs1ΔB/ΔB macrophages exhibited increased expression of pro-inflammatory cytokines. Macrophage proliferation was also drastically decreased (Pereira-Lopes et al. 2015). These results suggest that macrophages require mechanisms of protection against the DNA damage induced by the ROS produced during pro-inflammatory activation.
Conclusions and Future Directions
Macrophages are a key component of both innate and adaptive immunity and are of utmost importance in the elimination of invading pathogens, the initiation of an immune response by activating T cells, and in the resolution of inflammation and tissue repair. The deterioration of the immune system, called immunosenescence, is a hallmark of the aging process, and it contributes to the increased mortality and major incidence of immune diseases and cancer observed in the elderly. Given the relevance of macrophages in the immune system, the alterations that these cells undergo during aging may play a key role in immunosenescence.
Here we have summarized increasing experimental data about how aging affects macrophage functions. We, and many others, have described that most of these functions are altered in aged humans, rats, and mice. These observations suggest that dysfunctional macrophages are involved in the deterioration of the immune system with advanced age. However, most of these studies used peritoneal macrophages or blood monocytes, which may be influenced by their interaction with other cell types that are also affected by aging, thus providing a limited view of this process in macrophages. In contrast, bone marrow-derived macrophages provide an extraordinary model through which to study the effect of aging exclusively on the genomic expression of these phagocytic cells, without the influence of other cell types. However, this model does not reflect the precise function of macrophages in tissues in vivo. Therefore, the integration of data from all macrophage models offers the best strategy to assess how aging affects macrophage function and the molecular mechanisms involved in this process.
Many theories have been postulated to explain the aging process. Although these theories have been demonstrated in many cell types, very few data are available regarding the cellular and molecular mechanisms involved specifically in macrophage aging. It was demonstrated that these cells undergo telomere shortening with aging and also showed the influence of the accumulation of ROS and DNA damage in these cells with aging. These studies could shed light on the origin of macrophage dysfunction with age. As an integrative model, we propose that the shortening of telomeres leads to an increase in ROS. This increase in turn induces DNA breakage, which would result in the mutations that can explain most of the alterations described in aged macrophages (Fig. 4). This hypothesis is validated by two recent studies. In the first, chimeric mice with hyper-long telomeres were generated. These mice accumulated fewer cells with short telomeres and less DNA damage with age and expressed lower levels of p53. In highly renewing compartments, such as the blood, cells with hyper-long telomeres were longitudinally maintained or enriched with age (Varela et al. 2016). In the second one, blocking the oxidative damage by means of a transgenic mouse model with moderate overexpression of human G6PD induced lower levels of ROS-derived damage and greater protection from aging-associated functional decline, including extended median lifespan in females (Nobrega-Pereira et al. 2016).

Integrative model of the effect of aging on macrophages. The shortening of telomeres leads to an increase in ROS. This increase in turn induces DNA breakage, which would result in the mutations that can explain most of the alterations described in aged macrophages
Abbreviations
- ABCA:
-
ATP-binding cassette transporter
- AMD:
-
Age-related macular degeneration
- AML1/CFB:
-
Acute myeloid leukemia/core-binding factor
- ATF6:
-
Activating transcription factor-6
- Atm:
-
Ataxia-telangiectasia mutated
- C/EBP:
-
CCAAT/enhancer-binding protein
- CNV:
-
Choroidal neovascularization
- COX:
-
Cyclooxygenase
- CSF1R:
-
M-CSF receptor
- DAMP:
-
Damage-associated molecular pattern
- ds:
-
Double-stranded
- DTH:
-
Delayed-type hypersensitivity
- EGF:
-
Epithelial growth factor
- ER:
-
Endoplasmic reticulum
- ERK:
-
External-regulated kinase
- FcγRI:
-
Fc-γ receptor I
- GH-R:
-
Growth hormone receptor
- γH2AX:
-
Phosphorylated H2AX
- GHS-R:
-
Growth hormone secretagogue receptor
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HSC:
-
Hematopoietic stem cell
- IFN-γ:
-
Interferon gamma
- IκB:
-
Inhibitor of NF-κB
- IL:
-
Interleukin
- IL-4Rα:
-
IL-4 receptor α
- IRE1:
-
Inositol-requiring enzyme-1
- JNK:
-
Jun amino-terminal kinase
- LC:
-
Langerhans cells
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MCAF:
-
Macrophage chemotactic and activating factor
- M-CFU:
-
Macrophage colony-forming unit
- M-CSF:
-
Macrophage colony-stimulating factor
- M-CSF-R:
-
M-CSF receptor
- MHC:
-
Major histocompatibility complex
- MIP-1:
-
Macrophage inflammatory protein-1
- MR:
-
Mannose receptor
- mtROS:
-
Mitochondrial reactive oxygen species
- MZ:
-
Marginal zone
- MZM:
-
Macrophage of MZ
- NBS1:
-
Nijmegen breakage syndrome 1
- Nbs1ΔB/ΔB:
-
Mice expressing a hypomorphic allele of Nbs1
- NER:
-
Nucleotide excision repair
- NF-κB:
-
Nuclear factor-kappa B
- NHEJ:
-
Non-homologous end-joining
- NO:
-
Nitric oxide
- NOS2:
-
Inducible nitric oxide synthase
- OPG:
-
Osteoprotegerin
- PARP-1:
-
Poly[ADP-ribose] polymerase 1
- PPRγ:
-
Peroxisome proliferator-activated receptor-γ
- PERK:
-
Protein kinase RNA-like ER kinase
- PGE2:
-
Prostaglandin E2
- pol η:
-
Polymerase η
- SCC:
-
Squamous cell carcinoma
- pi3k-AKT:
-
Phosphoinositide-3-kinase-Akt
- p-IRE1α:
-
Phosphorylated IRE1α
- si-XBP1:
-
Small interference RNA-targeted XBP1
- PKC:
-
Protein kinase C
- PRR:
-
Pattern recognition receptors
- RANKL:
-
Receptor activator of NF-κB ligand
- RIDD:
-
IRE1-dependent decay
- ROS:
-
Reactive oxygen species
- STING:
-
Stimulator of IFN genes
- RXR:
-
Retinoid X receptor
- SA:
-
Senescence-associated
- Sir2:
-
Silent information regulator 2
- SIRT:
-
Sirtuin
- STAT:
-
Signal transducer and activator of transcription
- Tak1:
-
Transforming growth factor-β-activated kinase-1
- Terc−/−:
-
Telomerase KO mice
- TGF:
-
Tumor growth factor
- TLR:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor alpha
- TRAF6:
-
TNF-receptor-associated factor 6
- VEGF:
-
Vascular endothelial growth factor
References
Agius E, Lacy KE, Vukmanovic-Stejic M, Jagger AL, Papageorgiou AP, Hall S, Reed JR, Curnow SJ, Fuentes-Duculan J, Buckley CD, Salmon M, Taams LS, Krueger J, Greenwood J, Klein N, Rustin MH, Akbar AN (2009) Decreased TNF-alpha synthesis by macrophages restricts cutaneous immunosurveillance by memory CD4+ T cells during aging. J Exp Med 206:1929–1940
Allsopp RC, Cheshier S, Weissman IL (2001) Telomere shortening accompanies increased cell cycle activity during serial transplantation of hematopoietic stem cells. J Exp Med 193:917–924
Angelovich TA, Shi MD, Zhou J, Maisa A, Hearps AC, Jaworowski A (2016) Ex vivo foam cell formation is enhanced in monocytes from older individuals by both extrinsic and intrinsic mechanisms. Exp Gerontol 80:17–26
Arnold L, Henry A, Poron F, Baba-Amer Y, Van Rooijen N, Plonquet A, Gherardi RK, Chazaud B (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069
Ashcroft GS, Horan MA, Ferguson MW (1998) Aging alters the inflammatory and endothelial cell adhesion molecule profiles during human cutaneous wound healing. Lab Investig 78:47–58
Barrett JP, Costello DA, O’Sullivan J, Cowley TR, Lynch MA (2015) Bone marrow-derived macrophages from aged rats are more responsive to inflammatory stimuli. J Neuroinflammation 12:67
Birjandi SZ, Ippolito JA, Ramadorai AK, Witte PL (2011) Alterations in marginal zone macrophages and marginal zone B cells in old mice. J Immunol 186:3441–3451
Blasco MA (2002) Immunosenescence phenotypes in the telomerase knockout mouse. Springer Semin Immunopathol 24:75–85
Bouchlaka MN, Sckisel GD, Chen M, Mirsoian A, Zamora AE, Maverakis E, Wilkins DE, Alderson KL, Hsiao HH, Weiss JM, Monjazeb AM, Hesdorffer C, Ferrucci L, Longo DL, Blazar BR, Wiltrout RH, Redelman D, Taub D, Murphy WJ (2013) Aging predisposes to acute inflammatory induced pathology after tumor immunotherapy. J Exp Med 210:2223–2237
Canan CH, Gokhale NS, Carruthers B, Lafuse WP, Schlesinger LS, Torrelles JB, Turner J (2014) Characterization of lung inflammation and its impact on macrophage function in aging. J Leukoc Biol 96:473–480
Cao JJ, Wronski TJ, Iwaniec U, Phleger L, Kurimoto P, Boudignon B, Halloran BP (2005) Aging increases stromal/osteoblastic cell-induced osteoclastogenesis and alters the osteoclast precursor pool in the mouse. J Bone Miner Res 20:1659–1668
Chelvarajan RL, Liu Y, Popa D, Getchell ML, Getchell TV, Stromberg AJ, Bondada S (2006) Molecular basis of age-associated cytokine dysregulation in LPS-stimulated macrophages. J Leukoc Biol 79:1314–1327
Chen H, Ma F, Hu X, Jin T, Xiong C, Teng X (2013) Elevated COX2 expression and PGE2 production by downregulation of RXRalpha in senescent macrophages. Biochem Biophys Res Commun 440:157–162
Chen YW, Harris RA, Hatahet Z, Chou KM (2015) Ablation of XP-V gene causes adipose tissue senescence and metabolic abnormalities. Proc Natl Acad Sci USA 112:E4556–E4564
Chung PL, Zhou S, Eslami B, Shen L, Leboff MS, Glowacki J (2014) Effect of age on regulation of human osteoclast differentiation. J Cell Biochem 115:1412–1419
Danon D, Kowatch MA, Roth GS (1989) Promotion of wound repair in old mice by local injection of macrophages. Proc Natl Acad Sci USA 86:2018–2020
De La Fuente M, Hernanz A, Guayerbas N, Alvarez P, Alvarado C (2004) Changes with age in peritoneal macrophage functions. Implication of leukocytes in the oxidative stress of senescence. Cell Mol Biol (Noisy-le-grand) 50:OL683–OL690. Online Pub
Denkinger MD, Leins H, Schirmbeck R, Florian MC, Geiger H (2015) HSC aging and senescent immune remodeling. Trends Immunol 36:815–824
Epelman S, Lavine KJ, Randolph GJ (2014) Origin and functions of tissue macrophages. Immunity 41:21–35
Fallah MP, Chelvarajan RL, Garvy BA, Bondada S (2011) Role of phosphoinositide 3-kinase-Akt signaling pathway in the age-related cytokine dysregulation in splenic macrophages stimulated via TLR-2 or TLR-4 receptors. Mech Ageing Dev 132:274–286
Fenn AM, Hall JC, Gensel JC, Popovich PG, Godbout JP (2014) IL-4 signaling drives a unique arginase+/IL-1beta+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Ralpha after traumatic spinal cord injury. J Neurosci 34:8904–8917
Ghosh AK, Garg SK, Mau T, O’Brien M, Liu J, Yung R (2015) Elevated endoplasmic reticulum stress response contributes to adipose tissue inflammation in aging. J Gerontol A Biol Sci Med Sci 70:1320–1329
Golomb L, Sagiv A, Pateras IS, Maly A, Krizhanovsky V, Gorgoulis VG, Oren M, Ben-Yehuda A (2015) Age-associated inflammation connects RAS-induced senescence to stem cell dysfunction and epidermal malignancy. Cell Death Differ 22:1764–1774
Gomez CR, Karavitis J, Palmer JL, Faunce DE, Ramirez L, Nomellini V, Kovacs EJ (2010) Interleukin-6 contributes to age-related alteration of cytokine production by macrophages. Mediat Inflamm 2010:475139
Gordon S (2016) Phagocytosis: an immunobiologic process. Immunity 44:463–475
Gowers IR, Walters K, Kiss-Toth E, Read RC, Duff GW, Wilson AG (2011) Age-related loss of CpG methylation in the tumour necrosis factor promoter. Cytokine 56:792–797
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14:571–578
Harry GJ (2013) Microglia during development and aging. Pharmacol Ther 139:313–326
Hefendehl J, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M (2014) Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13:60–69
Herrero C, Marques L, Lloberas J, Celada A (2001) IFN-gamma-dependent transcription of MHC class II IA is impaired in macrophages from aged mice. J Clin Invest 107:485–493
Hinojosa CA, Akula Suresh Babu R, Rahman MM, Fernandes G, Boyd AR, Orihuela CJ (2014) Elevated A20 contributes to age-dependent macrophage dysfunction in the lungs. Exp Gerontol 54:58–66
Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak W, Suda T (2004) Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431:997–1002
Jackaman C, Radley-Crabb HG, Soffe Z, Shavlakadze T, Grounds MD, Nelson DJ (2013) Targeting macrophages rescues age-related immune deficiencies in C57BL/6J geriatric mice. Aging Cell 12:345–357
Jackaman C, Dye DE, Nelson DJ (2014) IL-2/CD40-activated macrophages rescue age and tumor-induced T cell dysfunction in elderly mice. Age (Dordr) 36:9655
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, Mccarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL (2014) Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371:2488–2498
Jiang M, Xiang Y, Wang D, Gao J, Liu D, Liu Y, Liu S, Zheng D (2012) Dysregulated expression of miR-146a contributes to age-related dysfunction of macrophages. Aging Cell 11:29–40
Khalil H, Tazi M, Caution K, Ahmed A, Kanneganti A, Assani K, Kopp B, Marsh C, Dakhlallah D, Amer AO (2016) Aging is associated with hypermethylation of autophagy genes in macrophages. Epigenetics 11:381–388
Lansdorp PM (2005) Major cutbacks at chromosome ends. Trends Biochem Sci 30:388–395
Lee DC, Ruiz CR, Lebson L, Selenica ML, Rizer J, Hunt JB Jr, Rojiani R, Reid P, Kammath S, Nash K, Dickey CA, Gordon M, Morgan D (2013) Aging enhances classical activation but mitigates alternative activation in the central nervous system. Neurobiol Aging 34:1610–1620
Li Q, Xiao H, Isobe K (2002) Histone acetyltransferase activities of cAMP-regulated enhancer-binding protein and p300 in tissues of fetal, young, and old mice. J Gerontol A Biol Sci Med Sci 57:B93–B98
Lin L, Lee JH, Buras ED, Yu K, Wang R, Smith CW, Wu H, Sheikh-Hamad D, Sun Y (2016) Ghrelin receptor regulates adipose tissue inflammation in aging. Aging (Albany NY) 8:178–191
Linehan E, Dombrowski Y, Snoddy R, Fallon PG, Kissenpfennig A, Fitzgerald DC (2014) Aging impairs peritoneal but not bone marrow-derived macrophage phagocytosis. Aging Cell 13:699–708
Liscovsky MV, Ranocchia RP, Alignani DO, Gorlino CV, Moron G, Maletto BA, Pistoresi-Palencia MC (2011) CpG-ODN+IFN-gamma confer pro- and anti-inflammatory properties to peritoneal macrophages in aged mice. Exp Gerontol 46:462–467
Locati M, Mantovani A, Sica A (2013) Macrophage activation and polarization as an adaptive component of innate immunity. Adv Immunol 120:163–184
Lourbopoulos A, Erturk A, Hellal F (2015) Microglia in action: how aging and injury can change the brain’s guardians. Front Cell Neurosci 9:54
Lumeng CN, Liu J, Geletka L, Delaney C, Delproposto J, Desai A, Oatmen K, Martinez-Santibanez G, Julius A, Garg S, Yung RL (2011) Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J Immunol 187:6208–6216
Ly LV, Baghat A, Versluis M, Jordanova ES, Luyten GP, Van Rooijen N, Van Hall T, Van Der Velden PA, Jager MJ (2010) Aged mice, outgrowth of intraocular melanoma depends on proangiogenic M2-type macrophages. J Immunol 185:3481–3488
Mahbub S, Deburghgraeve CR, Kovacs EJ (2012) Advanced age impairs macrophage polarization. J Interf Cytokine Res 32:18–26
Metchnikoff E, Mitchell PC (1908) The prolongation of life: optimistic studies. New York/London, G. P. Putnam’s sons
Mirsoian A, Bouchlaka MN, Sckisel GD, Chen M, Pai CC, Maverakis E, Spencer RG, Fishbein KW, Siddiqui S, Monjazeb AM, Martin B, Maudsley S, Hesdorffer C, Ferrucci L, Longo DL, Blazar BR, Wiltrout RH, Taub DD, Murphy WJ (2014) Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J Exp Med 211:2373–2383
Mitzel DN, Lowry V, Shirali AC, Liu Y, Stout-Delgado HW (2014) Age-enhanced endoplasmic reticulum stress contributes to increased Atg9A inhibition of STING-mediated IFN-beta production during Streptococcus pneumoniae infection. J Immunol 192:4273–4283
Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, Mills D, Patel P, Hsu JT, Hong AL, Ford E, Cheng HL, Kennedy C, Nunez N, Bronson R, Frendewey D, Auerbach W, Valenzuela D, Karow M, Hottiger MO, Hursting S, Barrett JC, Guarente L, Mulligan R, Demple B, Yancopoulos GD, Alt FW (2006) Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124:315–329
Nakamura R, Sene A, Santeford A, Gdoura A, Kubota S, Zapata N, Apte RS (2015) IL10-driven STAT3 signalling in senescent macrophages promotes pathological eye angiogenesis. Nat Commun 6:7847
Natrajan MS, De La Fuente AG, Crawford AH, Linehan E, Nunez V, Johnson KR, Wu T, Fitzgerald DC, Ricote M, Bielekova B, Franklin RJ (2015) Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain 138:3581–3597
Nobrega-Pereira S, Fernandez-Marcos PJ, Brioche T, Gomez-Cabrera MC, Salvador-Pascual A, Flores JM, Vina J, Serrano M (2016) G6PD protects from oxidative damage and improves healthspan in mice. Nat Commun 7:10894
Nyugen J, Agrawal S, Gollapudi S, Gupta S (2010) Impaired functions of peripheral blood monocyte subpopulations in aged humans. J Clin Immunol 30:806–813
Ogawa T, Kitagawa M, Hirokawa K (2000) Age-related changes of human bone marrow: a histometric estimation of proliferative cells, apoptotic cells, T cells, B cells and macrophages. Mech Ageing Dev 117:57–68
Orre M, Kamphuis W, Osborn LM, Jansen AH, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging 35:2746–2760
Pereira-Lopes S, Tur J, Calatayud-Subias JA, Lloberas J, Stracker TH, Celada A (2015) NBS1 is required for macrophage homeostasis and functional activity in mice. Blood 126:2502–2510
Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224
Piette J, Piret B, Bonizzi G, Schoonbroodt S, Merville MP, Legrand-Poels S, Bours V (1997) Multiple redox regulation in NF-kappaB transcription factor activation. Biol Chem 378: 1237–1245
Pinto AR, Godwin JW, Chandran A, Hersey L, Ilinykh A, Debuque R, Wang L, Rosenthal NA (2014) Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (Albany NY) 6:399–413
Raj DD, Moser J, Van Der Pol SM, Van Os RP, Holtman IR, Brouwer N, Oeseburg H, Schaafsma W, Wesseling EM, Den Dunnen W, Biber KP, De Vries HE, Eggen BJ, Boddeke HW (2015) Enhanced microglial pro-inflammatory response to lipopolysaccharide correlates with brain infiltration and blood-brain barrier dysregulation in a mouse model of telomere shortening. Aging Cell 14:1003–1013
Rodriguez-Menocal L, Faridi MH, Martinez L, Shehadeh LA, Duque JC, Wei Y, Mesa A, Pena A, Gupta V, Pham SM, Vazquez-Padron RI (2014) Macrophage-derived IL-18 and increased fibrinogen deposition are age-related inflammatory signatures of vascular remodeling. Am J Physiol Heart Circ Physiol 306:H641–H653
Rossi DJ, Bryder D, Seita J, Nussenzweig A, Hoeijmakers J, Weissman IL (2007) Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447:725–729
Sans-Fons MG, Yeramian A, Pereira-Lopes S, Santamaria-Babi LF, Modolell M, Lloberas J, Celada A (2013) Arginine transport is impaired in C57Bl/6 mouse macrophages as a result of a deletion in the promoter of Slc7a2 (CAT2), and susceptibility to Leishmania infection is reduced. J Infect Dis 207:1684–1693
Schilling T, Eder C (2015) Microglial K(+) channel expression in young adult and aged mice. Glia 63:664–672
Sebastian C, Herrero C, Serra M, Lloberas J, Blasco MA, Celada A (2009) Telomere shortening and oxidative stress in aged macrophages results in impaired STAT5a phosphorylation. J Immunol 183:2356–2364
Seidler S, Zimmermann HW, Bartneck M, Trautwein C, Tacke F (2010) Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol 11:30
Sene A, Khan AA, Cox D, Nakamura RE, Santeford A, Kim BM, Sidhu R, Onken MD, Harbour JW, Hagbi-Levi S, Chowers I, Edwards PA, Baldan A, Parks JS, Ory DS, Apte RS (2013) Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab 17:549–561
Sharma R, Kapila R, Haq MR, Salingat V, Kapasiya M, Kapila S (2014) Age-associated aberrations in mouse cellular and humoral immune responses. Aging Clin Exp Res 26:353–362
Shaw AC, Panda A, Joshi SR, Qian F, Allore HG, Montgomery RR (2011) Dysregulation of human toll-like receptor function in aging. Ageing Res Rev 10:346–353
Sheng J, Ruedl C, Karjalainen K (2015) Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity 43:382–393
Smallwood HS, Lopez-Ferrer D, Squier TC (2011) Aging enhances the production of reactive oxygen species and bactericidal activity in peritoneal macrophages by upregulating classical activation pathways. Biochemistry 50:9911–9922
Sobenin IA, Zhelankin AV, Sinyov VV, Bobryshev YV, Orekhov AN (2015) Mitochondrial aging: focus on mitochondrial DNA damage in atherosclerosis – a mini-review. Gerontology 61: 343–349
Song Y, Shen H, Du W, Goldstein DR (2013) Inhibition of x-box binding protein 1 reduces tunicamycin-induced apoptosis in aged murine macrophages. Aging Cell 12:794–801
Spadaro O, Goldberg EL, Camell CD, Youm YH, Kopchick JJ, Nguyen KY, Bartke A, Sun LY, Dixit VD (2016) Growth hormone receptor deficiency protects against age-related NLRP3 inflammasome activation and immune senescence. Cell Rep 14:1571–1580
Stout-Delgado HW, Cho SJ, Chu SG, Mitzel DN, Villalba J, El-Chemaly S, Ryter SW, Choi AM, Rosas IO (2016) Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am J Respir Cell Mol Biol 55:252
Stranks AJ, Hansen AL, Panse I, Mortensen M, Ferguson DJ, Puleston DJ, Shenderov K, Watson AS, Veldhoen M, Phadwal K, Cerundolo V, Simon AK (2015) Autophagy controls acquisition of aging features in macrophages. J Innate Immun 7:375–391
Suchy D, Labuzek K, Buldak L, Szkudlapski D, Okopien B (2014) Comparison of chosen activation markers of human monocytes/macrophages isolated from the peripheral blood of young and elderly volunteers. Pharmacol Rep 66:759–765
Sugawara Y, Azuma N, Onodera S, Tsunoka Y, Morimoto M (2011) Th2 immune responses and alternatively activated macrophages (AAMacs) in helminth infection in aged mice. J Vet Med Sci 73:511–516
Takahashi R, Totsuka S, Ishigami A, Kobayashi Y, Nagata K (2015) Attenuated phagocytosis of secondary necrotic neutrophils by macrophages in aged and SMP30 knockout mice. Geriatr Gerontol Int 16:135–142
Tucker JD, Spruill MD, Ramsey MJ, Director AD, Nath J (1999) Frequency of spontaneous chromosome aberrations in mice: effects of age. Mutat Res 425:135–141
Valledor AF, Borras FE, Cullell-Young M, Celada A (1998) Transcription factors that regulate monocyte/macrophage differentiation. J Leukoc Biol 63:405–417
Varela E, Munoz-Lorente MA, Tejera AM, Ortega S, Blasco MA (2016) Generation of mice with longer and better preserved telomeres in the absence of genetic manipulations. Nat Commun 7:11739
Verschoor CP, Johnstone J, Loeb M, Bramson JL, Bowdish DM (2014) Anti-pneumococcal deficits of monocyte-derived macrophages from the advanced-age, frail elderly and related impairments in PI3K-AKT signaling. Hum Immunol 75:1192–1196
Wang Y, Wehling-Henricks M, Samengo G, Tidball JG (2015) Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 14:678–688
Wynn TA, Vannella KM (2016) Macrophages in tissue repair, regeneration, and fibrosis. Immunity 44:450–462
Xaus J, Cardo M, Valledor AF, Soler C, Lloberas J, Celada A (1999) Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity 11:103–113
Xaus J, Comalada M, Valledor AF, Lloberas J, Lopez-Soriano F, Argiles JM, Bogdan C, Celada A (2000) LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 95:3823–3831
Yao YG, Kajigaya S, Feng X, Samsel L, Mccoy JP Jr, Torelli G, Young NS (2013) Accumulation of mtDNA variations in human single CD34+ cells from maternally related individuals: effects of aging and family genetic background. Stem Cell Res 10:361–370
Yeramian A, Martin L, Serrat N, Arpa L, Soler C, Bertran J, Mcleod C, Palacin M, Modolell M, Lloberas J, Celada A (2006) Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J Immunol 176:5918–5924
Youm YH, Grant RW, Mccabe LR, Albarado DC, Nguyen KY, Ravussin A, Pistell P, Newman S, Carter R, Laque A, Munzberg H, Rosen CJ, Ingram DK, Salbaum JM, Dixit VD (2013) Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18:519–532
Yu B, Chang J, Liu Y, Li J, Kevork K, Al-Hezaimi K, Graves DT, Park NH, Wang CY (2014) Wnt4 signaling prevents skeletal aging and inflammation by inhibiting nuclear factor-kappaB. Nat Med 20:1009–1017
Zandi S, Nakao S, Chun KH, Fiorina P, Sun D, Arita R, Zhao M, Kim E, Schueller O, Campbell S, Taher M, Melhorn MI, Schering A, Gatti F, Tezza S, Xie F, Vergani A, Yoshida S, Ishikawa K, Yamaguchi M, Sasaki F, Schmidt-Ullrich R, Hata Y, Enaida H, Yuzawa M, Yokomizo T, Kim YB, Sweetnam P, Ishibashi T, Hafezi-Moghadam A (2015) ROCK-isoform-specific polarization of macrophages associated with age-related macular degeneration. Cell Rep 10:1173–1186
Zhang J, Rane G, Dai X, Shanmugam MK, Arfuso F, Samy RP, Lai MK, Kappei D, Kumar AP, Sethi G (2016) Ageing and the telomere connection: an intimate relationship with inflammation. Ageing Res Rev 25:55–69
Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, Macpherson G, Randolph GJ, Scherberich J, Schmitz J, Shortman K, Sozzani S, Strobl H, Zembala M, Austyn JM, Lutz MB (2010) Nomenclature of monocytes and dendritic cells in blood. Blood 116:e74–e80
Zimmermann S, Glaser S, Ketteler R, Waller CF, Klingmuller U, Martens UM (2004) Effects of telomerase modulation in human hematopoietic progenitor cells. Stem Cells 22:741–749
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Section Editor information
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this entry

Cite this entry
Lloberas, J., Tur, J., Vico, T., Celada, A. (2019). Molecular and Cellular Aspects of Macrophage Aging. In: Fulop, T., Franceschi, C., Hirokawa, K., Pawelec, G. (eds) Handbook of Immunosenescence. Springer, Cham. https://doi.org/10.1007/978-3-319-99375-1_46
Download citation
DOI: https://doi.org/10.1007/978-3-319-99375-1_46
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-99373-7
Online ISBN: 978-3-319-99375-1
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences