
Abstract
Dimethylsulfoniopropionate (DMSP) is an organosulfur zwitterion produced by various marine algae and Bacteria as an osmolyte, cryoprotectant, defense molecule, and antioxidant. In the marine environment in particular, it can be degraded by the Bacteria in various ways. In this chapter we cover the biochemistry and physiology of the various pathways of DMSP catabolism, including the three core enzymes DMSP dethiomethylase (EC 4.4.1.3, the so-called DMSP lyase), DMSP demethylase (EC 2.1.1.269), and DMSP CoA transferase/lyase (EC 2.3.1.x). Six isoenzyme classes of DMSP dethiomethylase have been purified and confirmed in marine Bacteria thus far, with a further isoenzyme found in algae that may also occur in Bacteria – these are all discussed in detail. Methodologies for enzyme assays and the synthesis of DMSP hydrochloride are given, including those for radio- and stable-isotope labelling.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others

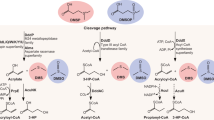
1 Introduction
Dimethylsulfoniopropionate (DMSP, also termed 3-(dimethylsulfaniumyl)propanoate or dimethyl-β-propiothetin) is a compound that has gained significant interest from biologists, ocean scientists, and climate scientists alike in the last 70 years since its discovery by Challenger and Simpson (1948), following the earlier observation by Haas (1935) that red algae of the Rhodophyta such as Polysiphonia lanosa (L.) Tandy and Polysiphonia fucoides (Hudson) Greville evolved dimethylsulfide (DMS) as a gas when exposed to air. Challenger and Simpson (1948) further examined P. lanosa, confirming Haas’s (1935) observation of DMS emission in air, tap water, and distilled water, but not if kept in jars of seawater. They found that the addition of sodium hydroxide caused DMS emission – a hydrolysis reaction still familiar to many today as the basis of the common DMSP gas chromatography assays. To identify the DMS precursor compound, specimens were laboriously extracted in ethanol, concentrated under reduced pressure, and treated with tetrathiocyanatodiamminechromate(III) precipitating the DMS precursor, which was then converted to the hydrochloride over many steps via the sulfate and the chloroplatinate. The compound they were left with is what we now know as DMSP hydrochloride (DMSP-HCl). The precise role of DMSP in vivo has been debated but is now widely considered to be a multifunction compound used as an osmolyte (primarily), antioxidant, cryoprotectant, and defense compound (Bullock et al. 2017). DMSP biosynthesis is found across many taxa in the Eukarya, viz., the Anthozoa, Chlorophyta, Rhodophyta , Phaeophyta, Chrysophyta, Dinoflagellata, Haptophyta, Viridiplantae, Cryptophyta, and Ochrophyta, and in some members of the Bacteria from the Alphaproteobacteria, such as Labrenzia aggregata LZB033 from the family Rhodobacteraceae (Otte et al. 2004; Curson et al. 2017), and in very small concentrations in members of the “Cyanobacteria.” The role of DMSP in the latter is not fully understood at this time. Bacteria that degrade DMSP have been isolated mostly from marine environments, but there is no doubt that they will also live in association with larger Rhodophyta, etc., just as DMS-degrading Bacteria such as Methylophaga spp. do (Li et al. 2007).
The environmental significance of DMSP and its daughter compounds DMS and acrylate have been reviewed many times, and there is an enormity of environmental data from oceans all over the world – much of this is not relevant to the biologist, but good reviews on the subject for background information on the marine sulfur cycle are Kelly (1996), Keine (1993, 1996), Wood (1996), and Kelly et al. (1993), all of which are short reviews and perspectives that, while over 20 years old, are very useful; for a longer and more up-to-date review on the subject, Schäfer et al. (2010) gives much more depth. Obviously, the reader is directed to Charlson et al. (1987) for the overarching significance of DMS and DMSP in the environment.
The bacterial lysis of DMSP into DMS or other organosulfur species was classically thought to be catalyzed by a DMSP lyase that ultimately catalyzed the lysis of DMSP into DMS and acrylate:
In the last 15 or so years, a significant canon of work emanating from the Johnston and Todd laboratories at the University of East Anglia in the United Kingdom has changed this picture, and a variety of new enzymes and pathways have been identified in a range of organisms. Intensive protein structural studies from the Zhang group at the State Key Laboratory of Microbial Technology, Shandong University, China, have given key evolutionary, functional, and mechanistic detail, and considerable work by the Moran and Whitman laboratories at the University of Georgia, USA, have identified downstream catabolic pathways and given structures of many core enzymes. This is a testament not only to the hard work and dedication of these groups but also of the enormous international priority of our understanding of sources and sinks of DMSP in the oceans.
Herein we review the biochemical and physiological literature on DMSP catabolism in aerobic Bacteria and give some information on the distribution of the genes encoding each enzyme; though the ecology of these systems is not within the scope of this chapter, we have given the basics to underpin the biochemistry. To aid the reader, we have also curated methods for enzyme assays and the synthesis of DMSP, including stable- and radio-isotope-labelled versions.
2 Synthesis, Storage, and Determination of DMSP
DMSP is not widely commercially available and must usually be made by the end user. Several synthetic methods have been published, which we give here in a format more accessible to most biologists who do not have chemistry skills. Research Plus Inc. of New Jersey, USA, does sell DMSP-HCl for around US$3900 per gram (£2990, €3350), but it can be made much more economically in-house, for around US$0.10–0.30 per gram (£0.08–0.24, €0.09–0.27). Hypothetically, one could extract DMSP-HCl from Rhodophyta as Challenger and Simpson (1948) did, and while this would be too inconvenient for most purposes, it could be of use for production of some radiolabelling combinations that are not possible chemically owing to the cost of the precursors, whereas sodium [14C]-bicarbonate and sodium [35S]-sulfate, for example, are very economical and could be used to “spike” a living organism easily enough, though the work-up of the compound is very long-winded and there will be much loss to the bench – Aronoff (1956) covers all the core methodologies for labelling of plants and algae and extraction of labelled metabolites.
2.1 Synthesis of Dimethylsulfoniopropionate
Analytical grade reagents should be used throughout to ensure high yields and higher purity products, and the work conducted in a ducted fume hood. Only glass-distilled deionized water (ddH2O) should be used. DMSP is most commonly prepared by the Michael addition (Michael 1886) of dimethylsulfide (the Michael donor) and acrylic acid (the Michael acceptor) and then recovered as the hydrochloride salt (DMSP-HCl), which should be stored in non-actinic glass under argon, with the jar held within a box containing dried silica gel at −20 °C to ensure longevity. Solution preparation is covered elsewhere in this chapter. Acrylic acid is supplied with either hydroquinone or 4-methoxyphenol as a stabilizer – this does not impact use in these syntheses and there is no need to attempt to purify the acid further, which will only result in polymerization! We have given methods for sulfur-34 and sulfur-35 labelling as well as carbon-12 and carbon-13 labelling, which can be applied to the methyl groups or to the propionate moiety – it is possible to synthesize mixed labelled compounds, e.g., [14C1,2]dimethyl-[35S]-sulfonio-[13C1,2,3]-propionate hydrochloride, should one wish, which may be useful in tracing fates of each functional group in physiological work or for use in combined stable-isotope probing (SIP) and microautoradiography (MAR) methods in ecology without needing to run two sets of incubations, for example. Other labels such as tritium or deuterium could be used by obtaining the appropriate labelled starting material but as hydrogen atoms are wont to exchange in solution, they have relatively little practical use in physiological work, so we have not included them here. As detailed in Boden and Hutt (2018a) in this handbook, carbon-11 labelling is very useful for short-term labelling in biological systems as it has a half-life of c. 20 min – we discuss [11CU]-dimethylsulfide synthesis in that chapter (though it is by no means straightforward and requires a skilled radiochemist) which could be used to make [11C1,2]-dimethylsulfoniopropionate if a sufficiently rapid methodology was used.
In all methods given, a slight molar excess of acrylic acid can be used, which affords the convenience that the smell of unreacted DMS will not be a problem during work-up, though we recommend the whole process is conducted in a fume hood in any case.
2.1.1 DMSP-HCl from Dimethylsulfide, Acrylic Acid, and Hydrogen Chloride Gas
This is a somewhat unnecessary method as hydrochloric acid (see below) can be used as the solvent in place of benzaldehyde very easily, though it is much faster and does not require refluxing or cooling overnight. A1, 500 mL QuickFit 3-neck flask is held in an ice bath over a magnetic stirrer, with a thermometer in one of the side necks, and a gas-inlet tube in the other, reaching to the bottom of the vessel, leaving the central neck open. A large glass stirring “flea” is added, followed by 250 mL dimethylsulfide (3.40 mol), which is cooled to 0 °C with stirring. 250 mL acrylic acid (3.65 mol) is then added, followed by 250 mL benzaldehyde. A cylinder or lecture bottle of hydrogen chloride gas is then connected to the gas-inlet tube and bubbled through the mixture at a rapid pace, with stirring, for 30 min. Alternatively, air from a pump can be forced through a train of three 500 mL glass Drechsel bottles, one filled with ddH2O, the others both filled with concentrated hydrochloric acid, and the outflow from this train used, but that will take significantly longer to react with the reactants. A third alternative is to generate hydrogen chloride using CaCl2 or NaCl and concentrated sulfuric acid (per Arnáiz (1995), then washed through concentrated hydrochloric acid in a Drechsel bottle). A white precipitate of DMSP-HCl will form, which is recovered by filtration at the pump onto a course glass fiber filter, which is then washed with 200 mL benzaldehyde and lyophilized for 24–48 h to remove the solvent. It is then dissolved in boiling ethanol before recrystallizing by cooling slowly to −20 °C by standing the beaker in a domestic freezer. The DMSP-HCl is then recovered by filtration at the pump and recrystallized a second time. Theoretical yield is around 580 g, for around US$165 (£125, €140). This method was modified from Steinke et al. (1998).
2.1.2 DMSP-HCl from Dimethylsulfide, Acrylic Acid, and Hydrochloric Acid
A 5000 mL QuickFit 3-neck flask is held in a sand bath on top of a magnetic stirring hot plate, with a thermometer in one of the side necks and a glass stopper in the other. 250 mL dimethylsulfide (3.40 mol) is added to 1620 mL 2 M hydrochloric acid and the mixture stirred with a glass stirring “flea.” 233 mL acrylic acid (3.40 mol) is added and an Allihn or Dimroth condenser of 50 cm length added to the central neck, with cold tap water entering from the bottom olive. More sand is added to the bath if necessary to bring it level with the liquid in the flask and is then heated to bring the flask contents to 95 °C and is then adjusted to maintain the temperature of the flask contents at 95 °C for 2 h under reflux – check the position of the vapor ring in the condenser from time to time, ensuring it does not escape the column. Heating is ceased after 2 h and the flask contents allowed to cool to room temperature – ideally overnight – with the reflux condenser still in place. The flask contents (c. 2.1 L) are divided into 4 or 5 500 mL round-bottomed flasks and the liquid is removed under reduced pressure using a rotary evaporator to concentrate it down until an oily liquid or white solid remains. The contents of each flask are pooled in a 1-L beaker and a 1:1 mixture of sodium-dried diethyl ether and absolute ethanol added (c. 100–200 mL) to resuspend it – if an oily liquid was present, it will at this stage usually turn into a white solid, at which point the solid is recovered by filtration at the pump on Whatman No. 1 filter paper. 500 mL absolute ethanol is poured through the solid on the filter paper in 2–3 batches, followed by 1000 mL sodium-dried diethyl ether. The solid material is then dissolved in boiling absolute ethanol and cooled to −20 °C slowly to bring about recrystallization using a domestic freezer before recovering the crystals of DMSP-HCl at the pump. A second round of recrystallization is recommended. The theoretical yield is about 580 g, for around US$70 (£55, €62). This method was adapted from Alcolombri et al. (2017).
2.1.3 [14C1,2] or [13C1,2]–Dimethylsulfoniopropionate Hydrochloride Synthesis from [14CU]– Or [13CU]–Dimethylsulfide, Acrylic Acid, and Hydrochloric Acid
The production of carbon-14 or carbon-13 labelled DMSP-HCl with the labels on the two S-methyl groups is easily achieved using the appropriately labelled dimethylsulfide synthesized according to Boden and Hutt (2018a), also in this handbook. The method given above should be scaled down and microscale reflux used (some microscale methodology is given in Boden and Hutt (2018a), but Aronoff (1956) is an excellent resource for radiobiochemical methods) for the carbon-14 version and standard semi-micro methods for carbon-13.
2.1.4 Dimethyl-[34S] or [35S]-Sulfoniopropionate Hydrochloride from [34S]- or [35S]-Dimethylsulfide, Acrylic Acid, and Hydrochloric Acid
Preparation of sulfur-34 or sulfur-35 labelled dimethylsulfide is given in Boden and Hutt (2018a). Synthesis should proceed as though carbon-13 or carbon-14, respectively, scaled appropriately.
2.1.5 Dimethylsulfonio-[13C1 or 1,2,3] or [14C1]-Propionate from Dimethylsulfide, [13C1 or 1,2,3]- or [14C1]-Acrylic Acid, and Hydrochloric Acid
Carbon-13 or carbon-14 labelling of the propionate moiety can be achieved using commercially available from Sigma-Aldrich (carbon-13, at 99% isotopic purity) American Radiolabelled Chemicals Ltd. (carbon-14, at specific activities of up to 5 mCi/mmol, taking care to procure the neat liquid and not a solution in ethanol or base). Multiple carbon-13 labels are available: [13C1]-acrylic acid or [13C1,2,3]-acrylic acid – the former is around US$3900 per gram (£3000, €3400); the latter is a custom product.
2.2 Preparation, Storage, and Use of DMSP Solutions for Culture Work
Solid DMSP-HCl should be stored at −20 °C in non-actinic glass, under argon for maximum stability, in a box of silica gel. Stock solutions (>0.5 M are possible) are made in glass-distilled deionized water (ddH2O) and are then sterilized into sterile, acid-washed glass serum bottles (120 mL) using 0.22 μm polycarbonate syringe filters or 0.22 μm polyethersulfone or polyvinylidene fluoride Sterivex™ filter units (MilliporeSigma, Burlington, Massachusetts, USA) if larger volumes are required. Bottles are wrapped in foil (or non-actinic glass can be used) and the solutions flushed with filter-sterilized argon at 10 mL/min for c.30 min to remove air, and bottles are then sealed with PTFE-coated butyl rubber septa and are stored at 4 °C – solutions should be monitored for initial concentration and reexamined at intervals to check for degradation, but this should be negligible if stored correctly and using very pure DMSP-HCl.
For batch cultures in shake flasks, DMSP is added from stock solutions to 5–20 mM typically – in organisms that can only consume the acrylate moiety, that is 15–60 mmol C per liter, which would usually support growth to 1–4 g dry biomass/L. For those only consuming the DMS moiety, that is 10–40 mmol C per liter, which would usually support 0.7–3.0 g dry biomass/L. Since DMS is toxic to many organisms >5 mM, it may be prudent to keep the concentration low for organisms that lyse it and consume only that group. For continuous-flow chemostat cultures, DMSP should be treated as a labile substrate and the “fractional flow” method of Boden and Hutt (2018b) used.
2.3 Determination of DMSP in Microbial Cultures
The usual means of DMSP determination is by alkaline lysis into DMS and quantification by gas chromatography (GC) – in very low concentration (nM) samples, such as seawater, samples are then purged with a carrier gas and the DMS cryotrapped then released by heating in boiling water. For the 1–20 mM range commonly used for bacterial growth, this is not necessary. Since DMSP is formed by cultures, this should either be measured first or driven off. Culture samples (8 mL) are harvested by centrifugation to remove biomass (13,000 g, 30 min at 4 °C), and 5.00 mL of the supernate is transferred into a 20 mL serum bottle, sealed with a PTFE-coated butyl rubber septum. Two needles are inserted, one just below the septum (“output”) and one right to the bottom of the bottle (“input”). A stream of water-saturated (by passage through two Drechsel bottles filled with Ballotini beads and ddH2O) argon or oxygen-free nitrogen is fed into the input needle at about 5 mL/min, for about 10 min, which should remove most if not all DMS – this can be checked using methods from Boden and Hutt (2018a). 5 mL of a freshly made solution of 10 M NaOH is drawn into a syringe and injected into the “input” needle, and the “output” needle then rapidly removed to avoid loss of liberated DMS. After removing both needles, the bottle is incubated in the dark for >6 h and then equilibrated to the room temperature of the room in which DMS will now be quantified. DMS will now be partitioned over the gas and liquid phases; thus determination should either be against a calibration curve based on DMS solutions treated with NaOH and incubated for the same time period, or the partitioning should be determined and accounted for (Przyjazny et al. 1983) – DMS can then be quantified in the headspace by GC or liquid phase by colorimetric means (cf. Boden and Hutt 2018a) and the total DMS then determined, which is equal to the total DMSP. If samples cannot be analyzed immediately and centrifugation is not possible (e.g., collecting a time course from a DMSP-limited chemostat), samples can be strongly acidulated to pH <1.0 using 0.1 mL concentrated sulfuric acid per 10 mL. It is worth noting that del Valle et al. (2011) examined sample acidulation methods for specimens containing Phaeocystis spp. from the Haptophyta of the Eukarya and found that acidulated samples still suffered DMSP lysis; thus it is key to test the stability of DMSP in an acidulated sample, e.g., 7 days before attempting use in a key experiment. Kinsey and Kieber (2016) have developed a microwave-based method, in which 4 mL aliquots in serum bottles were boiled in a 900 W domestic microwave (<10 s) and then cooled in ice – this could give reasonably high throughput when preserving samples.
3 Enzymology of DMSP Degradation by the Bacteria
3.1 Dimethylsulfoniopropionate Dethiomethylases (EC 4.4.1.3)
Otherwise known – somewhat imprecisely – as “DMSP lyases”, this group of isoenzymes (i.e., identical in reaction catalyzed but different in structure) catalyze the canonical cleavage of DMSP into DMS, acrylate, and a proton:
Herein we use “DMSP dethiomethylase” in line with the Enzyme Commission (viz., official) nomenclature, which is a more accurate description of the enzyme and gives a precise location of lytic activity. Owing to many of the different DMSP dethiomethylases isolated having gene codes beginning with ddd, it can be somewhat confusing to the uninitiated – as such, we have herein assigned each isoenzyme an arbitrary class number for the sake of clarity herein, but it may be of use to workers in the future. It is worth noting that a similarly wide array of isoenzymes or paralogs may also be present in the Eukarya, viz., Emiliania huxleyi (Steinke et al. 1998).
3.1.1 Quantitative Enzyme Assays
DMSP dethiomethylases can be assayed in cell-free extracts (CFEs) using discontinuous analytical or radiometric enzyme assay. 0.1 M Tricine-HCl or Tris-HCl (pH 8.0) containing 20 mM DMSP-HCl is dispensed in 5 mL volumes into 30 mL glass serum bottles, which are then stoppered with PTFE-coated butyl rubber septa. Headspaces of bottles are flushed with argon for a few minutes to exclude oxygen-mediated enzyme activities. Cell-free extract (0.1–0.5 mL) is injected after preheating the bottles to the optimal growth temperature of the organism. Vials are incubated at the same temperature, with 0.5 mL removed every 30 s into sterile serum bottles similarly stoppered and charged with 0.5 mL 50% (v/v) perchloric acid. The concentration of DMS (cf. Boden and Hutt (2018a) for methods) and/or acrylate are then determined in each vial. Acrylate is best determined by chromatographic methods as colorimetric assays are not specific enough – it can be determined by HPLC using a C18 column with a water-acetonitrile gradient and a photodiode array detector (Shintani 1995) – extracting the acrylate into a small volume of toluene from each vial by shaking for 10 min, removing the organic layer, and drying over anhydrous sodium sulfate before injecting into the instrument may give cleaner peaks and can be a useful means of concentrating the analyte. The enzyme activity is expressed in nmol DMS or nmol acrylate formed min−1 (mg protein)−1. Alternatively, carbon-14 labelled DMSP (see previous section for synthetic methods) labelled at the methyl or propionate ends can be used in the same assay – it should be diluted in “cold” DMSP to give a specific activity of 10 mCi/mol, i.e., 1 μCi per assay vial – and after extinction of enzyme activity with perchloric acid, aliquots are added to an acid-safe liquid scintillation cocktail (e.g., Ultima Gold by Perkin-Elmer, not to be confused with Ultima Gold XR, etc., Ultima Gold AB is also suitable), left overnight in the dark and then counted in a calibrated scintillation counter against suitable quench curves for the cocktail used, with DMS or acrylate concentrations then determined, and thus enzyme activity.
3.1.2 Rapid Screening Qualitative Enzyme Assays
For rapid screening of, e.g., protein purification fractions for acrylate production, thin-layer chromatography run rapidly or overnight can be a highly convenient tool for qualitative or semiquantitative screening. Acrylate is converted to the free acid and Heck coupled (Heck and Nolley 1972) to iodobenzene in an alkaline solution over a palladium catalyst (e.g., FibreCat 1001 or nanopalladium), which yields (E)-cinnamic acid, eliminating iodine. This reaction can be completed in 30 min at 60 °C if acrylate is in the 0.01–10.00 mM range – typical for DMSP dethiomethylase assays – and is stoichiometric (yield 96%). The reacted fractions are extracted into ethyl acetate and spotted onto silica gel plates and developed with 4:1:1 toluene-acetic acid-ethanol (<1 h). Fully dried plates (fume hood, overnight) are sprayed with 0.12 M ferric chloride in 2 M HCl, (E)-cinnamic acid giving a bright yellow spot (Rf 0.74, Bobbio et al. 1987). Alternatively F254 plates can be used and the spot visualized using 254 nm ultraviolet light. Acrylate can be determined by densitometry if necessary (Mei et al. 2015; Bobbio et al. 1987) – the more modern TLC rapid screening systems could make this procedure even quicker.
3.1.3 Differentiation of Isoenzymes Using Artificial DMSP Homologues
A useful differentiation between Class III (DddP), Class IV (DddQ), and Class V (DddW) DMSP dethiomethylase isoenzymes was given by Burkhardt et al. (2017), which showed that Class V isoenzymes had the widest substrate diversity for synthetic DMSP homologues, whereas Class III isoenzymes could not degrade trimethylammonium propionate (TMAP), and Class IV isoenzymes could not degrade TMAP, dimethyltelluriopropionate (DMTeP), nor racemic, (S)-, or (R)-2-methyl-dimethylsulfoniopropionate (2-Me-DMSP). These data could potentially provide a useful means to differentiate enzymes in cell-free extracts by using these substrates (synthetic methods are given in Burkhardt et al. (2017)), but one should take caution that this has thus far only been demonstrated with isoenzymes from Ruegeria pomeroyi DSM 15171T and Phaeobacter inhibens DSM 17395 and only expressed in E. coli rather than in the native cellular milieu, which may contain species that bind to these homologues and alter activities, etc.
Lei et al. (2018) have undertaken important phylogenetic studies of DMSP dethiomethylase isoenzymes of the cupin type and have attempted to understand the superfamily to which they belong. They also identified that N,N,N′,N′-tetrakis(2-pyridylmethyl)ethane-1,2-diamine (TPEN), a d-block metal chelator, inhibited all known cupin-type DMSP dethiomethylase isoenzymes, which will better enable both physiological and environmental distinguishing between these isoenzymes and other modes of DMSP catabolism. They also suggest that only Class I (DddL), Class II (DddY), and Class VII (Alma1) DMSP dethiomethylase isoenzymes are genuine DMSP dethiomethylases, since Class IV (DddQ) has higher specific activity for 3-methyl-DMSP, and Class V (DddW) and Class VI (DddK) isoenzymes have high substrate promiscuity – they did not consider Class III (DddP) isoenzymes as they are not cupin family proteins. Class IV and Class V isoenzymes were phylogenetically distant from Classes I and II, but Class IV was close to Class I. Clearly further work to assess the in vivo in situ (i.e., in the native organism in the natural environment) role of all proposed DMSP dethiomethylase isoenzymes is needed.
Comparative properties of the seven known classes of DMSP dethiomethylase are curated in Table 1.
3.1.4 Class I (DddL) Isoenzymes
The DddL isoenzyme (Class I) was first identified by Curson et al. (2008) in Rhodobacter sphaeroides ATCC 17023T (origin unknown, probably marine) and Sulfitobacter pontiacus EE-36 (salt marsh, Georgia, USA, Buchan et al. (2000)). It comprises a single subunit of c. 25 kDa and contained no known functional domains at the time of discovery, pace a single transmembrane motif.
We ran a homology model using the S. pontiacus EE-36 DddL amino acyl sequence using SWISS-MODEL in July 2018, which found a 100% amino acyl sequence match to the crystal structure of DddL by Hehemann et al. that is as yet unpublished (SMTL ID: 4b29.1), which indicates a cupin structure (cf. Class III, DddP), with Zn(II) and Ni(II) ions bound.
Zeng et al. (2016) investigated the diversity of DMSP dethiomethylase genes in surface water of Kongsfjorden, Svalbard (Bokmål, “the king’s fjord”), finding all of the dddL clones were allied to the genus Sulfitobacter. The dddL gene was also identified in Bacteria communities associated to corals (Raina et al. 2009).
3.1.5 Class II (DddY) Isoenzymes and the Curson–Johnston Pathway
In Alcaligenes faecalis subsp. faecalis M3A, the dddY gene was identified as being responsible for the same DMSP lysis to DMS ; the encoded periplasmic DddY protein of c. 46 kDa had no known functional domains. The dddY gene was not found in major marine metagenome databases (Reisch et al. 2011a) but is probably important in DMSP cycling in marine sediments (Curson et al. 2011). In this organism, DMS is emitted without further metabolism, but the acrylate group is assimilated into biomass. Acrylate is first bound to coenzyme A (CoA) by acrylate CoA transferase (AcuN, EC 2.3.1.x), yielding acryloyl-CoA:
The acryloyl-CoA is then double-hydrated into 3-hydroxypropionate (3HPA) by acryloyl-CoA hydratase (AcuK, EC 4.2.1.54):
3HPA can then be oxidized to 3-oxopropionate (malonate semialdehyde) by 3-hydroxypropionate dehydrogenase (DddA, EC 1.1.1.59), yielding NADH:
3-oxopropionate is ligated to CoA by 3-oxopropionate dehydrogenase (DddC, EC 1.2.1.18), forming acetyl-CoA, carbon dioxide, and NAD(P)H – the former can be assimilated via Krebs’ cycle:
The DddY isoenzyme structure was solved from a recombinant protein in Escherichia coli expressing the dddY gene of Acinetobacter bereziniae NIPH 3 with the addition of a C-terminal His tag (Li et al. 2017). DddY comprises two domains – a cap domain and a catalytic domain. The former is at the N-terminus and is composed of α-helices, “capping” the catalytic domain. The C-terminal catalytic domain is a beta barrel comprised of eight antiparallel β-strands – this barrel contains the catalytic site. Like all the other DMSP dethiomethylases known, it contains a cupin motif. All cupin superfamily proteins use a divalent cation in their catalytic site – in DddY this is Zn(II), bound sufficiently to not be removed by 10 mM EDTA. The catalytic mechanism has been resolved: when the active site is “empty,” the Zn(II) ion is chelated by two histidine residues (His265 and His338) and a glutamic acid residue (Glu269) and is bound to water. The other feature of the active site is a tyrosine residue (Tyr271), which has an –O− charge in the “empty” state. The carboxyl head of DMSP coordinates with the Zn(II) ion via the hydroxyl oxygen, the water leaving. An electron is then transferred from Tyr271 to the hydrogen on the α-carbon of DMSP, which is displaced, transferring an electron to the β-carbon, forming a double bond between the carbons, and then on to the sulfur, cleaving the C-S bond and liberating DMS and acrylate, leaving Tyr271 with a –OH group. It is believed (Li et al. 2017) that all other DMSP dethiomethylases are cupin superfamily proteins, based on homology analyses.
It is worth noting that a DMSP dethiomethylase was purified in the mid-1990s (van der Maarel 1996a, b) from Desulfovibrio desulfuricans subsp. aestuarii DSM 10141 (previously “Desulfovibrio acrylicus, ” which is a later heterotypic synonym of D. desulfuricans subsp. aestuarii, Shivani et al. 2016) – this has since been shown to be a Class II isoenzyme (DddY) by Curson et al. (2011).
3.1.6 Class III (DddP) Isoenzymes and the Reisch–Whitman Pathway
Kirkwood et al. (2010) identified the DddP isoenzyme in Roseovarius nubinhibens DSM 15170T , which has a subunit mass of c. 48 kDa, existing as a homodimer in vivo at about 95 kDa. The dddP gene was expressed in E. coli and cleaved DMSP into DMS and acrylate per the canonical DMSP dethiomethylase reaction. The recombinant enzyme was not inhibited by 2,2′-bipyridyl or EDTA at 2.5 or 25 mM, respectively, which was taken to indicate that it was not a metalloenzyme. ICP-OES analysis of purified enzyme samples did not detect any common metals found in metalloenzymes above background levels, based on digestion of the enzyme in 2.5% (v/v) nitric acid – though the detection limit was c. 5–10 nmol for most metals. The isoenzyme has a low affinity for DMSP and is slow in comparison with the other isoenzymes (Km 13.8 mM, VMAX 300 nmol min−1 (mg protein)−1).
Wang et al. (2015) have resolved the structure of the DddP isoenzyme from Ruegeria lacuscaerulensis DSM 11314T expressed in E. coli with a C-terminal His tag. It was found to belong to the M24 peptidase superfamily, which also includes bona fide M24B peptidases, M24B prolidases, M24B aminopeptidases, and creatinases (EC 3.5.3.3), all from the Bacteria. Unlike the biochemical studies of Kirkwood et al. (2010), the enzyme was found to contain two Fe(III) ions per homodimer, which was also observed by Hehemann et al. (2014) in Roseobacter denitrificans ISM DddP expressed in E. coli using ICP-MS and TRXF to determine metals. Each DddP monomer comprises an N-terminal and C-terminal domain and adopts a “pita bread” fold of 15 β-strands and 16 α-helices, as observed in M24 peptidases (Bazan et al. 1994). The dimer has two active centers, each formed of four β-strands and three loops from the C-terminal domain of one monomer and the α3 helix from the N-terminal domain of the other monomer. The active site is complex in terms of amino acyl residues required for function, and the Fe(III) ions are bridged and coordinated by Glu421. The sulfur of DMSP is bound by Trp95 and Tyr117, forming strong π-interactions with the S+ moiety. Asp337 acts as a nucleophile, attacking the H on the β-carbon of the carboxyl end of DMSP, which is bound by the oxygens to Tyr366 and an Fe(III). Electrons are donated by Tyr366 to the carbonyl oxygen and to the carbonyl α and β-carbons in sequence, before finally onto the sulfur, cleaving off DMS and forming acrylate, releasing a proton to Asp377. Evolutionary analyses indicate that DddP ancestors are functional peptidases and that a mutation of the active site resulted in loss of peptidase activity and “conversion” to a lyase (Wang et al. 2015).
The dddP gene was found in the genomes of various Alphaproteobacteria and in members of the Ascomycota of the Eukarya, in which functional DMSP dethiomethylase activity was observed (Todd et al. 2009). In the Zeng et al. (2016) investigation of the diversity of DMSP dethiomethylase genes in surface water of Kongsfjorden, Svalbard, dddP genes were mostly allied to the Rhodobacteraceae , with no specific affiliation to known genera.
The downstream catabolism of acrylate in Ruegeria pomeroyi proceeds via a different route to the Curson-Johnston pathway – it should be noted that while we have grouped this with Class III (DddP) isoenzymes, both Class IV (DddQ) and Class V (DddW) isoenzymes can also catalyze the first step, with DMS being catabolized or released, as discussed in Sect. 3.1.6. In the first step of the Reisch-Whitman pathway (Reisch et al. 2013), acrylate is bound to CoA by propionate-CoA ligase (PrpE, EC 6.2.1.17) that can also use acrylate:
Acryloyl-CoA can either proceed downstream or be moved out of the pathway into a “storage pool” by hydroxylation by the acrylate utilization hydratase (AcuH), which yields 3-hydroxypropionyl-CoA:
This reaction can proceed in either direction, and given the toxicity of acryloyl-CoA, AcuH likely serves to ensure it does not reach a hazardous concentration within the cell, by storing it as a pool of 3-hydroxylpropionyl-CoA when the concentration reaches a threshold related to the Km of the AcuH (Cao et al. 2017; Wang et al. 2017), and pooled 3-hydroxylpropionyl-CoA can be converted back if levels of acryloyl-CoA in the cell decrease, draining the pool again.
Acryloyl-CoA proceeding through catabolism is reduced by an NADPH-dependent acryloyl-CoA reductase (AcuI or YdhH, EC 1.3.1.64), EC to form propionyl-CoA:
The propionyl-CoA is then carboxylated by propionyl-CoA carboxylase (PccAB, EC 6.4.1.3) to form (S)-methylmalonyl-CoA :
(S)-methylmalonyl-CoA is converted to succinyl-CoA by methylmalonyl-CoA mutase (McmA, EC 5.4.99.2):
Succinyl-CoA then enters Krebs’ cycle at the point of oxidation to succinate by succinyl-CoA synthetase (SucCD, EC 6.2.1.5):
3.1.7 Class IV (DddQ) Isoenzymes
In Ruegeria pomeroyi DSM 15171T , in addition to the dddP gene, that encoding a second DMSP degrading enzyme was also identified – dddQ, encoding DddQ, of c. 22 kDa (Todd et al. 2011). We ran homology models using SWISS-MODEL, based on the DddQ sequence from R. pomeroyi DSM 15171T , in July 2018 and found 100% amino acyl match to an “unpublished” crystal structure by Chong et al. (SMTL ID: 5cu1.1), which shows a cupin family structure with a non-covalently bound Fe(III) ion in the catalytic domain. Li et al. (2014a) reported the structure and mechanism of DddQ, which was rebutted by Alcolombri et al. (2014a) on the basis of very low kinetic parameters, indicating that the enzyme was perhaps not a bona fide DMSP dethiomethylase. Li et al. (2014b) responded noting that the enzyme has a KM of 21.5 mM and giving robust evidence to support it being a true DMSP dethiomethylase. Lei et al. (2018) argue that DddQ is only an opportunistic DMSP dethiomethylase owing to substrate promiscuity and shows much higher specific activities with 3-methyl-dimethylsulfoniopropionate (3-Me-DMSP) than with DMSP itself. Unlike Class I (DddL) and Class II (DddY) DMSP dethiomethylases, it shows a wide range of activities, also lysing ethylmethylsulfoniopropionate (EMSP), dimethylsulfoniopropionate (DESP), and tetramethylenesulfoniopropionate (TMSP).
The dddQ gene from Ruegeria lacuscaerulensis DSM 11314T was cloned into E. coli with a C-terminal His tag and the recombinant protein was used for structural work. The enzyme was a homodimer, with a monomer of 22 kDa, and had optimal activity at pH 8.0 and 30 °C, and Mn(II) and Co(II) ions increased the specific activity, though only Zn(II) was actually present in the native enzyme as purified, and excess Zn(II) inhibited the enzyme – which is in common with many other zinc metalloenzymes in which a second Zn(II) ion binds, blocking the active site.
The mechanism of DddQ was resolved by Li et al. (2014a). The Zn(II) in the active site was coordinated by His125, His163, Glu129, and Tyr131, which stabilized the structure of the enzyme. When DMSP entered the active site, the carboxyl carbon of the propionate “head” of the molecule became coordinated to the Zn(II) ion, which released Tyr131, which then deviated by 25°, aligning its –O− moiety with a hydrogen on the α-carbon, to which it transferred an electron, which then went onto the β-carbon and finally onto the sulfur, eliminating DMS and forming a double bond between the α and β carbons to yield acrylate and donating a proton to the negatively charged Tyr131. Interestingly, Brummett and Dey (2016) demonstrated a violet DddQ from R. lacuscaerulensis DSM 11314T, which had two Fe(III) ions coordinated per homodimer – very different from the Zn(II)-dependent version that Li et al. (2014) identified in the same organism, even though both were done in the same gene expressed into E. coli.
3.1.8 Class V (DddW) Isoenzymes
The DddW DMSP dethiomethylase was identified in Ruegeria pomeroyi DSM 15171T by Todd et al. (2012) and further characterization done by Brummett et al. (2015). It is another cupin family protein, with a 17 kDa monomer that is found as a homodimer in vivo. When the dddW gene is expressed in E. coli in minimal medium without added metal ions, an insoluble protein is formed (Brummett et al. 2015), but adding d-block divalent cations to the medium resulted in the formation of a soluble and active enzyme, with Fe(II) being preferentially uptaken and which was required for activity, though Mn(II) was also used to some degree.
Homology modelling (June 2018) of the R. pomeroyi DddW amino acyl sequence using SWISS-MODEL gave close sequence matches to an unpublished homodimeric cupin protein from Halorhodospira halophila by Pakskovsky et al. (SMTL ID: 3ibm.1) bound to Zn(II), as well as the Schnicker et al. (2017) DddW structure.
Lei et al. (2018) demonstrate some substrate promiscuity in DddW, with almost the same specific activity for DMSP as for tetramethylenesulfoniopropionate (TMSP), as well as lower specific activities for ethylmethylsulfoniopropionate (EMSP), dimethylsulfoniopropionate (DESP), and 2-methyl-DMSP (as had been observed by Burkhardt et al. (2017) previously).
3.1.9 Class VI (DddK) Isoenzymes
Many organisms from the “SAR11” clade of the Roseobacterales contain the dddK gene. In Candidatus Pelagibacter ubique, the Km of DddK is 82 mM, versus 13 mM for DmdA, suggesting that demethylation would be the preferential pathway at low levels of DMSP and that dethiomethylation would only be used at high levels (Sun et al. 2016). It is worth noting that demethylation yields MT, which is required by Ca. P. ubique as a sulfur source, whereas dethiomethylation yields DMS, which cannot be used, and thus the sulfur is wasted when the DddK is used at high DMSP levels – Sun et al. (2016) predicted that 59% of DMSP would be catabolized to DMS rather than MT in Ca. P. ubique, which makes sense given the relatively low levels of sulfur assimilated into biomass in general, but Schnicker et al. (2017) noted that when DddK used Ni(II) in the active site, the enzyme was almost as efficient as DmdA, and this may not be the case in situ in the marine environment.
Lei et al. (2018) demonstrate some substrate promiscuity in DddK, with reasonably strong specific activities for ethylmethylsulfoniopropionate (EMSP), dimethylsulfoniopropionate (DESP), and tetramethylenesulfoniopropionate (TESP).
The Ca. P. ubique DddK is 16.5 kDa and has a promiscuity for d-block divalent cations, showing the highest specific activities with Ni(II), Mn(II), and Fe(II) in that order (Schnicker et al. 2017). The crystal structure of DddK was resolved, with a single cupin domain. The coordination of metal ions varied between crystals of the enzyme containing nickel or iron with zinc. The DMSP cleavage mechanism comprised the binding of DMSP, eliminating water from the active site – the carboxylate group of DMSP binding the complexed metal ion by both oxygens, which were also both H-bonded by Tyr64 and Tyr122, the former donating an electron to one of the hydrogens on the α-carbon of DMSP, which in turn donated to the α-β carbon bond, and finally to the sulfur, liberating DMS. Water then “reset” the active site when acrylate left.
3.1.10 Class VII (Alma1) Isoenzymes
Algal DMSP dethiomethylases were studied by Steinke et al. (1998), who identified a number of “isoenzymes.” Alcolombri et al. (2015) have recently purified a DMSP dethiomethylase (Alma1) from Emiliania huxleyi , which was found to be a homotetramer of 160 kDa, with a subunit size of about 40 kDa. Seven paralogs of Alma1 were found in E. huxleyi and Isochrysis Parke spp. ( Haptophyta ) – Alma2, Alma3, Alma4, Alma5, Alma6, and Alma7 – six of which presumably correspond to the six “isoenzymes,” or more correctly, paralogs, of Steinke et al. (1998).
Phylogenetic analysis of the Alma1 gene found homologues in the Bacteria, though functionality of a bacterial Class VII DMSP dethiomethylase has yet to be demonstrated – as such we have tentatively included it herein, since it is possibly a bacterial isoenzyme also.
3.2 Dimethylsulfoniopropionate CoA–Transferase/Lyase(DddD) and the Alcolombri–Tawfik Pathway
In a Marinomonas pontica MWYL1, the dddD gene was identified as being involved in the breakdown of DMSP and the liberation of DMS (Todd et al. 2007), but unlike the DMSP dethiomethylase isoenzymes, DddD did not yield acrylate. The gene was expressed in E. coli with an N-terminal His tag and was purified by affinity chromatography and anion exchange, yielding a protein of 90 kDa. In purified form, it did not liberate DMS from DMSP using the standard assay methods for DMSP dethiomethylases (Alcolombri et al. 2014b), but when acetyl-CoA was added, a specific activity of 5000 nmol DMS released min−1 (mg protein)−1 was recorded. Vmax was about 60,000 nmol DMS released min−1 (mg protein)−1, and kcat was 10.8 s, with optimum activity at pH 8.0 and 30 °C. Acryloyl-CoA (used in the Reisch-Whitman pathway) and DMSP-CoA did not act as cofactors for the enzyme. The DddD sequence resembled that of a carnitine CoA-transferase, but carnitine was not used as a substrate by the purified enzyme. The dddD gene was also found in Pseudomonas sp. J465 and Psychrobacter sp. J466 , both obtained from the gut of Clupea harengus L. (Atlantic herring, from the Actinopterygii of the Eukarya), which both grew on DMSP as a sole carbon and energy source, liberating DMS (Curson et al. 2010). In both of these species, it was found in an operon with dddT (DMSP transporter), dddB (possible aldehyde dehydrogenase), dddC (3-oxopropionate dehydrogenase), iclR (repressor), and dddR (regulator). The dddD gene was also found in Bacteria associated to corals (Raina et al. 2009).
The reaction that the DMSP-CoA transferase-lyase (DddD, EC 2.3.1.x) catalyzes releases acetate as well as DMS and proceeds via a two-step transfer of CoA from acetyl-CoA to DMSP, forming 3-hydroxypropionate-CoA:
Some CoA is then recycled, which liberates free 3-hydroxypropionate (HPA), but most is broken down to acetyl-CoA, which can be recycled by DddD, or can enter Krebs’ cycle, etc.
3HPA-CoA is lysed by DddD to release 3HPA which is oxidized to 3-oxopropionate (malonate semialdehyde) by 3-hydroxypropionate dehydrogenase (DddA, EC 1.1.1.59), yielding NADH:
3-oxopropionate is ligated to CoA by 3-oxopropionate dehydrogenase (DddC, EC 1.2.1.18), forming acetyl-CoA, carbon dioxide, and NAD(P)H – the former can be assimilated.
DddA and DddC are also found in the Curson-Johnston pathway.
The structure of DddD comprises a C-terminal domain similar to L-carnitine CoA-transferase (CaiB, EC 2.8.3.21) and an N-terminal domain similar to α-(2S)-2-methylacryl-CoA racemase (Mcr, EC 5.1.99.4). Where CaiB is a homodimer of two intertwined subunits, DddD’s predicted structure features the same structure but with the subunits fused into a single domain. The crystal structure of DddD has not yet been resolved, and homology modelling (May 2018) using SWISS-MODEL brings only close hits to individual domains or subunits of other enzymes with resolved structures.
The DMSP CoA-transferase/lyase enzyme assay should be demonstrated as distinct from DMSP dethiomethylases by quantifying the acetate produced. To serum bottles (30 mL), 5 mL volumes of 50 mM HEPPS-NaOH buffer at pH 8.0, containing 2 mM DMSP-HCl and 0.5 mM acetyl-CoA, are added. The septa are added and bottles flushed with argon to preclude any downstream oxidative reactions (details of this method are given above – cf. Sect. 3.1). Bottles are incubated at 30 °C for 20 min to equilibrate temperature, and then 0.1 mL cell-free extract is added. Bottles without acetyl-CoA should be assayed for DMS production per the DMSP dethiomethylase assays given previously to preclude this enzyme also being present. Bottles are incubated at 30 °C with very gentle agitation. At 1 min intervals for 10 min, bottles are sacrificed by injecting 0.5 mL 50% (v/v) perchloric acid and incubating in an ice-salt slush. While headspace DMS could be measured, it is the production of acetate that distinguishes DMSP CoA-transferase/lyase. Various colorimetric acetate assay kits are available, e.g., MegaZyme (Dublin, Éire) and Sigma-Aldrich (Poole, UK), but bottles will need to be neutralized with aqueous potassium hydroxide first. After neutralizing, HPLC can be used on a C18 column using 25 mM sulfuric acid as the mobile phase at 0.3 mL/min will elute acetate at around 3–4 min – a refractive index detector or a diode array or UV detector at 200–210 nm is suitable – acetate determination methods by HPLC are widely published. GC can be used to measure acetic acid directly at low pH after quenching with perchloric acid, e.g., using a phosphoric acid-coated Porapak Q column and flame ionization detector (White and Leenheer 1975). Following the determination of acetate at each time point, the specific enzyme activity is given in nmol acetate produced min−1 (mg protein)−1.
3.3 Dimethylsulfoniopropionate Demethylase (DmdA, EC 2.1.1.269) and the Reisch–Moran Pathway
This pathway is found in marine heterotrophs, particularly those adapted to oligotrophy. DMSP is demethylated and ultimately dissimilated to acetate (which is assimilated into biomass), methanethiol (MT, which can be used as a sulfur source or emitted into the environment), and carbon dioxide. Through this pathway, ATP is consumed but reducing equivalents ([H]) are generated in the form of FADH2 and NADH, which are used to generate proton motive force (Δp) and thus ATP. It is important to note that Ca. P. ubique uses this pathway and is known to reduce reduced forms of sulfur (Tripp et al. 2008) such as DMSP or methionine and cannot use sulfate, which is unusual given concentrations in the oceans (cf. Boden and Hutt (2018a) for further discussion on this). Reisch et al. (2011a) postulate that the energetic cost of sulfate reduction during assimilation is too much for an oligotrophic organism in an environment where electron donors are scarce; thus the ancestor that lost this ability may have survived given it could not “accidentally” waste [H] assimilating sulfate, killing itself in the process.
From Ca. Pelagibacter ubique and Ruegeria pomeroyi DSM 15171T , a DMSP demethylase (DmdA) was identified by Reisch et al. (2008), which uses tetrahydrofolate (THF) as its cofactor and produces 3-(methylthio)propanoate (MMPA):
The MMPA produced is bound to coenzyme A (CoA) and is then further oxidized in a manner reminiscent of the β-oxidation of fatty acids (Reisch et al. 2011b):
This is catalyzed by 3-(methylsulfanyl)propionyl-coenzyme A ligase (DmdB , EC 6.2.1.44), and the MMPA-CoA produced is then dehydrogenated by the flavoprotein 3-(methylthio)propionyl-coenzyme A dehydrogenase (DmdC , EC 1.3.99.B13), into (2E)-3-(methylthio)prop-2-enoyl-CoA (MTA-CoA) – the dehydrogenation occurring between the α and β carbons:
(2E)-3-(methylthio)prop-2-enoyl-CoA hydratase (DmdD , EC 4.2.1.155) hydrolyses this into acetaldehyde, methanethiol (MT), carbon dioxide, and CoA, in a reaction similar to that of crotonase (EC 4.2.1.17). It has been proposed (Reisch et al. 2011a) that DmdD is not essential for this reaction and that other – as yet unknown – enzymes may be able to catalyze this step, since organism without dmdD can still carry it out, such as R. pomeroyi:
The MT is either assimilated as a sulfur source or further dissimilated by methanethiol oxidase (MtoX, EC 1.8.3.4) as discussed in Boden and Hutt (2018a) or released into the environment, and the acetaldehyde oxidized to acetate(by an NADP-dependent acetaldehyde dehydrogenase (AldB, EC 1.2.1.4) – DddB may be an aldehyde dehydrogenase, Fowler (2015)), which can then be assimilated into biomass, either via the glyoxylate shunt across Krebs’ cycle or via the Erb pathway (Erb et al. 2009).
Keine and Taylor (1988) demonstrated that DMSP can be converted to mercaptopropionate (MPA) in anoxic sediments and was shown under air by a member of the Bacteria by Visscher and Taylor (1994). This could occur via a second demethylation of MMPA, and has not been demonstrated conclusively as a metabolic step, and may represent only a dead-end catabolite (Reisch et al. 2011a).
The crystal structure of DmdA was resolved by Schuller et al. (2012) and was found to be related to the T-protein of the glycine cleavage system (EC 2.1.2.10), which is an aminomethyltransferase. DmdA was found as a homodimer, with each DmdA subunit containing two domains – a Greek key surrounded by three α-helices and a 5-strand antiparallel β-sheet with α-helices on each side, respectively. The THF binding site in other THF-dependent methyltransferases has been determined as a cloverleaf-like confluence of domains (Leys et al. 2003; Scrutton and Leys 2005). THF is bound by Tyr206 and Glu204, with Tyr95 ring stacked with one of the rings of THF. DMSP was bound by the carboxyl group to Leu37, while Tyr206 is H-bonded to the primary amino group on THF. Electrons from the lone pair on a secondary amino group on the folate ring of THF are donated to one of the S-methyl carbons of DMSP, forming an SN2-type intermediate, with water molecules in the active site mediating proton transfer, and one of the S-methyl groups of DMSP ending up bridging the remaining MMPA and THF as the latter methylates and the former is eliminated simultaneously. DMSP probably polarizes to some degree, lowering the activation energy required for the SN2 methyl transfer reaction. In a bona fide, in vitro SN2 nucleophilic substitution, the rate-limiting (slower) step would be the donation of electrons from the lone pair to the S-methyl group of DMSP, so it is likely the case in vivo here.
In the analysis of DMSP degradation gene diversity by Zeng et al. (2016) in surface water of Kongsfjorden, Svalbard, dmdA genes were mostly affiliated to the Rhodobacteraceae , specifically Sulfitobacter , Ruegeria , Roseobacter , Octadecabacter, and Thalassobius .
The DMSP demethylase enzyme assay is conducted under anoxia to prevent oxidation of THF – this can be done in an anoxic chamber per Reisch et al. (2008) or in a glove bag filled with argon or by more skilled workers, using Hungate technique in Warburg flasks – not easy to get hold of anymore, though many laboratories still have them. A two-side-arm Thunberg cell could also be used – sold in 2018 as “anaerobic cell with glass pouches” – or a 5 mL serum bottle with two all-glass 0.2 mL gas-tight syringes to take the place of the side arms, plus two needles with stop-cocks for flushing the bottle). We have given both methodologies here – the preparative steps with flushing of bottles, etc. can be done as normal if working in anoxia, of course – for laboratories without anoxic chambers, the Hungate method is a very suitable alternative; however, it does require a degree of skill to perform properly and practice of handling an air-sensitive electrochrome (e.g., methyl viologen solution reduced with dithionite) with this method is useful training – they will change color if oxygen exposure occurs. Methods given here are adapted from Reisch et al. (2008), using the single-time point determination of products. For a more reliable approach, a discontinuous assay sacrificing flasks every 1 min for 10 min should be used such that the initial rate can be precisely determined – in a 10 min single-time point assay (per Reisch et al. 2008), all of the DMSP could have been consumed within the first 1 min, for example, or not consumed at all until 8 min – vastly under- or overestimating the true specific activity.
All water, solutions/buffers, etc. needed to be deoxygenated by sparging with oxygen-free nitrogen or argon that has been saturated with water using a Drechsel bottle of Ballotini beads filled with ddH2O (this prevents loss of water volume). To a 250 mL QuickFit conical flask, 200 mL water is added using a volumetric pipette or volumetric flask and the meniscus marked on the outside carefully. The flask is emptied and dried in an oven. 10.23 g DMSP-HCl is added and the flask flushed with argon. 100 mL 0.4 M HEPES supplemented with 1 mM disodium edetate is added, along with a glass stirring “flea.” A SubaSeal vaccine stopper is added and the contents stirred to dissolve the DMSP, at which point the stopper is removed, and, under a stream of argon, 0.5 M NaOH is added dropwise until the solution is pH 6.5 (c. 20 mL). The solution is then diluted to the previously marked “200 mL” mark by adding 0.4 M HEPES-NaOH pH 7.5, supplemented with 1 mM disodium EDTAate. This solution will contain c. 1 mM acrylate (Reisch et al. 2008) owing to alkaline hydrolysis of DMSP during the neutralization step, and thus the concentration of DMSP will be slightly below 300 mM (“buffered DMSP solution”). A 6.85 mM solution of tetrahydrofolic acid is made in thoroughly deoxygenated water in a small serum bottle filled with argon. A 0.2 M solution of dithiothreitol (DTT) is prepared similarly.
Warburg flask method : a two-side-arm Warburg flask is stoppered at the main neck and gas-in/out stoppers connected to each side arm. The flask is flushed with argon for 10 min and then 0.9 mL buffered 300 mM DMSP, 0.1 mL 0.2 M DTT and 0.1 mL 6.85 mM THF solution are added to the main chamber. The stopper is replaced and gas flushing continued for 5 min. The stopper on the “gas out” side arm is removed, with the gas still flowing, and 0.1 mL cell-free extract (CFE) is added and the stopper replaced. The stopper on the “gas in” side arm is removed and pointed such that gas still flows into the flask – 0.2 mL 7.6 M orthophosphoric acid is added to this side arm and the stopper replaced. After another 1–2 min of gas flow, the supply is turned off and the side-arm stoppers twisted into their “closed” position, isolating the contents of the flask from the atmosphere. The flask is incubated at 30 °C for 15 min to equilibrate the contents and the reaction initiated by flicking the CFE into the main chamber and returning to the incubator. After 10 min incubation with gentle agitation, the reaction is stopped by flicking the acid into the main chamber to quench the reaction. The flask is then opened and 0.5 mL of the contents removed using a Gilson and precipitated protein removed by brief centrifugation (13,000 g, 15 min, 4 °C). 5-methyl-THF or MMPA can be determined in the supernate by HPLC – a reverse-phase SB-AQ column (Agilent) using 25 mM sodium dihydrogen phosphate and 120 mM orthophosphoric acid in 1.18 M acetonitrile at 0.75 mL/min. Absorption at 214 nm is used for MMPA and 280 nm for 5-methyl-THF, and the specific activity given in nmol MMPA or 5-methyl-THF formed min−1 (mg protein)−1.
Anoxic chamber method : The same set-up methodology is observed, but the reaction is conducted in a serum bottle of argon (or chamber gas – usually 5% (v/v) hydrogen in nitrogen), the reactants added and a septum applied, at which point it can be removed from the chamber. The CFE is added by injection and the quench acid similar.
4 Research Needs
The twenty-first-century canon of research on “DMSP lyases” and other pathways of DMSP catabolism have made enormous advances in this field, which has grown massively, with protein crystal structures to molecular ecology all completed in little over a decade. That said, there are still gaps in our understanding and further work is needed:
-
1.
Protein structural work – Crystal structures of the class I (DddL) DMSP dethiomethylase isoenzyme and the DMSP CoA-transferase/lyase (DddD) are needed, as are further determinations of the metals associated with the cupin-type isoenzymes. This probably needs to be done from enzyme purified from the native organism rather than by recombinant expression to remove possibilities of, e.g., nickel contamination from affinity resins (or Co(II)-charged Talon® (GE Lifesciences) resin could be used instead). The class VII (Alma1) putative isoenzyme from the Bacteria remains to be purified and activity demonstrated.
-
2.
Kinetic work – Core kinetic parameters for class I (DddL) and class II (DddY) isoenzymes are missing and determination of these – And those of other isoenzymes – From a wider range of organisms is needed.
-
3.
Ecological work – Now that all seven isoenzyme genes have been identified, plus those of other enzymes of DMSP catabolism, a full screen of transcriptomes from marine samples from around the world are needed – Bacterioneuston, surface water, deep water, sediments, fish GI tracts etc., to explore the whole diversity of all of these isoenzymes.
-
4.
Inhibitors – Screening in particular of the DMSP dethiomethylase isoenzymes for specific inhibitors would better enable DMSP degradation rate studies by the marine chemists, as they would be able to partition their data into the different isoenzyme routes.
References
Alcolombri U, Elias M, Vardi A, Tawfik DS (2014a) Ambiguous evidence for assigning DddQ as a dimethylsulfoniopropionate lyase and oceanic dimethylsulfide producer. Proc Natl Acad Sci 111:E2078–E2079
Alcolombri U, Laurino P, Lara-Astiaso P, Vardi A, Tawfik DS (2014b) DddD is a co-A-transferase/lyase producing dimethyl sulfide in the marine environment. Biochemistry 53:5473–5475
Alcolombri U, Ben-Dor S, Feldmesser E, Levin Y, Tawfik DS, Vardi A (2015) Identification of the algal dimethyl sulfide-releasing enzyme: a missing link in the marine sulfur cycle. Science 348:1466–1469
Alcolombri U, Lei L, Meltzer D, Vardi A, Tawfik DS (2017) Assigning the algal source of dimethylsulfide using a selective lyase inhibitor. ACS Chem Biol 12:41–46
Arnáiz FJ (1995) A convenient way to generate hydrogen chloride in the freshman lab. J Chem Educ 72:1139
Aronoff S (1956) Techniques of radiobiochemistry. Iowa State College Press, Ames
Bazan JF, Weaver LH, Roderick SL, Huber R, Matthews BW (1994) Sequence and structure comparison suggest that methionine aminopeptidase, prolidase, aminopeptidase p, and creatinase share a common fold. Proc Natl Acad Sci 91:2473–2477
Bobbio FO, Bobbio PA, de Souza SC (1987) Separation and identification of cinnamic acids by TLC. J Chem Educ 64:182–182
Boden R, Hutt LP (2018a) Bacterial metabolism of C1 organosulfur compounds. In: Rojo F (ed) aerobic utilization of hydrocarbons, oils and lipids. Handbook of hydrocarbon and lipid metabolism. Springer, Cham
Boden R, Hutt LP (2018b) Determination of kinetic parameters and metabolic modes using the chemostat. In: Steffan R (ed) Consequences of microbial interaction with hydrocarbons, oils and lipids: biodegradation and bioremediation. Handbook of hydrocarbon and lipid microbiology. Springer, Cham
Brummett AE, Schincker NJ, Crider A, Todd JD, Dey M (2015) Biochemical, kinetic and spectroscopic characterization of Ruegeria pomeroyi DddW – a mononuclear iron-dependent DMSP lyase. PLoS One 10:e0127288
Brummett AE, Dey M (2016) New mechanistic insight from substrate- and product-bound structures of the metal-dependent dimethylsulfoniopropionate lyase DddQ. Biochemistry 55:6162–6174
Buchan A, Collier LS, Neidle EL, Moran MA (2000) Key aromatic-ring-cleaving enzyme, protocatechuate 3,4-dioxygenase, in the ecologically important Roseobacter lineage. Appl Environ Microbiol 66:4662–4672
Bullock HA, Luo H, Whitman WB (2017) Evolution of dimethylsulfoniopropionate metabolism in marine phytoplankton and bacteria. Front Microbiol 8:637
Burkhardt I, Lauterbach L, Brock NL, Dickschat JS (2017) Chemical differentiation of three DMSP lyases from the marine Roseobacter group. Org Biomol Chem 15:4432–4439
Cao HY, Wang P, Xu F, Li PY, Xie BB, Qin QL, Zhang YZ, Li CY, Chen XL (2017) Molecular insight into the acryloyl-CoA hydration by AcuH for acrylate detoxification in dimethylsulfoniopropionate-catabolizing bacteria. Front Microbiol 8:2034
Challenger F, Simpson MI (1948) Studies on biological methylation. Part XII. A precursor of dimethyl sulfide evolved by Polysyphonia fastigiata.Dimethyl-2-carboxyethylsulfonium hydroxide and its salts. J Chem Soc 0:1591–1597
Charlson RJ, Lovelock JE, Andreae MO, Warren SG (1987) Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate. Nature 326:655–661.
Curson AR, Rogers R, Todd JD, Brearley CA, Johnston AW (2008) Molecular genetic analysis of a dimethylsulfoniopropionate lyase that liberates the climate-changing gas dimethylsulfide in several marine α-proteobacteria and Rhodobacter sphaeroides. Environ Microbiol 10:757–767
Curson AR, Sullivan MK, Todd JD, Johnston AW (2010) Identification of genes for dimethyl sulfide production in bacteria in the gut of Atlantic herring (Culpea hargengus). ISME J 4:144–146
Curson AR, Sullivan MJ, Todd JD, Johnston AW (2011) DddY, a periplasmic dimethylsulfoniopropionate lyase found in taxonomically diverse species of Proteobacteria. ISME J 5:1191–1200
Curson ARJ, Liu J, Bermejo Martínez A, Green RT, Chan Y, Carrión O, Williams BT, Zhang S-H, Yang G-P, Bulman Page PC, Zhang X-H, Todd JD (2017) Dimethylsulfoniopropionate biosynthesis in marine bacteria and identification of the key gene in this process. Nat Microbiol 2:17009
del Valle DA, Slezak D, Smith CM, Rellinger AN, Kieber DJ, Kiene RP (2011) Effect of acidification on preservation of DMSP in seawater and phytoplankton cultures: evidence for rapid loss and cleavage of DMSP in samples containing Phaeocystis sp. Mar Chem 124:57–67
Erb TJ, Fuchs G, Alber BE (2009) (2S)-methylsuccinyl-CoA dehydrogenase closes the ethylmalonyl-CoA pathway for acetyl-CoA assimilation. Mol Microbiol 73:922–1008
Fowler EK (2015) On the molecular diversity of dimethylsulphoniopropionate catabolism by marine bacteria. Ph.D Thesis. University of East Anglia, UK
Haas P (1935) The liberation of methyl sulphide by seaweed. Biochem J 29:1297–1299
Heck RF, Nolley JP (1972) Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl and styryl halides. J Org Chem 37:2320–2322
Hehemann J-H, Law A, Redecke L, Boraston AB (2014) The structure of RdDddP from Roseobacter denitrificans reveals that DMSP lyases in the DddP-family are metalloenzymes. PLoS One 9:e103128
Keine RP (1993) Microbial sources and sinks for methylated sulfur compounds in the marine environment. In: Murrell JC, Kelly DP (eds) Microbial growth on C1 compounds. Intercept Ltd, Andover, pp 15–34
Keine RP (1996) Microbiological controls on dimethylsulfide emissions from wetlands and the ocean. In: Murrell JC, Kelly DP (eds) NATO ASI series vol 39. Microbiology of atmospheric trace gases. Springer-Verlag, Berlin, pp 205–225
Keine RP, Taylor BF (1988) Biotransformations of organosulfur compounds in sediments via 3-mercaptopropionate. Nature 322:148–150
Kelly DP (1996) A global perspective on sources and sinks of biogenic trace gases: an atmospheric system driven by microbiology. In: Murrell JC, Kelly DP (eds) NATO ASI series vol 39. Microbiology of atmospheric trace gases. Springer-Verlag, Berlin, pp 1–16
Kelly DP, Malin G, Wood AP (1993) Microbial transformations and biogeochemical cycling of one-carbon substrates containing sulphur, nitrogen or halogens. In: Murrell JC, Kelly DP (eds) Microbial growth on C1 compounds. Intercept Ltd, Andover, pp 47–63
Kinsey JD, Kieber DJ (2016) Microwave preservation method for DMSP, DMSO, and acrylate in unfiltered seawater and phytoplankton culture samples. Limnol Oceanogr Methods 14:196–209
Kirkwood M, Le Brun NE, Todd JD, Johnston AWB (2010) ThedddP gene of Roseovarius nubinhibens encodes a novel lyase that cleaves dimethylsulfoniopropionate into acrylate plus dimethyl sulfide. Microbiology (UK) 156:1900–1906
Lei L, Phaneendra Cherukuri K, Alcolombri U, Meltzer D, Tawfik DS (2018) The dimethylsulfoniopropionate (DMSP) lyase and lyase-like cupin family consists of bona fide DMSP lyases as well as other enzymes with unknown function. Biochemistry 57:3364–3377
Leys D, Brasan J, Scrutton NS (2003) Channelling and formation of ‘active’ formaldehyde in dimethylglycine oxidase. EMBO J 22:4038–4048
Li TD, Doronina NV, Ivanova EG, IuA T (2007) Vitamin B12-independent strains of Methylophaga marina isolated from Red Sea algae. Mikrobiologiia 76:88–94
Li C-Y, Wei T-D, Zhang S-H, Chen X-L, Gao X, Wang P, Xie B-B, Su H-N, Qin Q-L, Zhang X-Y, Yu J, Zhang H-H, Zhou B-C, Yang G-P, Zhang Y-Z (2014a) Molecular insight into bacterial cleavage of oceanic dimethylsulfoniopropionate into dimethyl sulfide. Proc Natl Acad Sci 111:1026–1031
Li C-Y, Chen X-L, Xie B-B, Su H-N, Qin Q-L, Zhang Y-Z (2014b) Reply to Tawfik et al.: DddQ is a dimethylsulfoniopropionate lyase involved in dimethylsulfoniopropionate catabolism in marine bacterial cells. Proc Natl Acad Sci 111:E2080
Li C-Y, Zhang D, Chen X-L, Wang P, Shi W-L, Li P-L, Zhang X-Y, Qin Q-L, Todd JD, Zhang Y-Z (2017) Mechanistic insights into dimethylsulfoniopropionate lyase DddY, a new member of the cupin superfamily. J Mol Biol 429:3850–3862
Mei S, Taejun C, Tetsuya T, Hitomi O, Yuji A (2015) A simple TLC-densitometric method for the quantification of acrylic acid in aqueous solutions. J Plan Chromatgr Mod TLC 28:12–16
Michael A (1886) Ueber die Addition von Natriumacetissig- und Natriummalonsäureäthern zu den Aethern ungesättigter Säuren. J Prakt Chem 35:349–356
Otte ML, Wilson G, Morris JT, Moran BM (2004) Dimethylsulfoniopropionate (DMSP) and related compounds in higher plants. J Exp Bot 55:1919–1925
Przyjazny A, Janicki W, Chrzanowski W, Staszewski R (1983) Headspace gas chromatographic determinations of distribution coefficients of selected organosulphur compounds and their dependence on some parameters. J Chromatogr 280:249–260
Raina JB, Tapiolas D, Willis BL, Bourne DG (2009) Coral-associated bacteria and their role in the biogeochemical cycling of sulfur. Appl Environ Microbiol 75:3492–3501
Reisch CR, Moran MA, Whitman WB (2008) Dimethylsulfoniopropionate-dependent demethylase (DmdA) from Pelagibacter ubique and Silicibacter pomeroyi. J Bacteriol 190:8018–8024
Reisch CR, Moran MA, Whitman WB (2011a) Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front Microbiol 2:172
Reisch CR, Stoudemayer MJ, Varaljay VA, Amster IJ, Moran MA, Whitman WB (2011b) Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473:208–211
Reisch CR, Crabb WM, Gifford SM, Teng Q, Stoudemayer MJ, Moran MA, Whitman WB (2013) Metabolism of dimethylsulfoniopropionate by Ruegeria pomeroyi DSS-3. Mol Microbiol 89:774–791
Schäfer H, Myronova N, Boden R (2010) Microbial degradation of dimethylsulphide and related C1-Sulphur compounds: organisms and pathways controlling fluxes of Sulphur in the biosphere. J Exp Bot 61:315–334
Schnicker NJ, de Silva SM, Todd JD, Dey M (2017) Structural and biochemical insights into dimethylsulfoniopropionate cleavage by cofactor-bound DddK from the prolific marine bacterium Pelagibacter. Biochemistry 56:2873–2885
Schuller DK, Reisch CR, Moran MA, Whitman WB, Lanzilotta WN (2012) Structures of dimethylsulfoniopropionate-dependent demethylase from the marine organism Pelagabacter ubique. Protein Sci 21:289–298
Scrutton NS, Leys D (2005) Crystal structure of DMGO provides a prototype for a new tetrahydrofolate-binding fold. Biochem Soc Trans 33:776–779
Shintani H (1995) HPLC analysis of toxic additives and residual monomer from dental plate. J Liq Chromatogr 18:613–626
Shivani Y, Subhash Y, Ch S, ChV R (2016) Halodesulfovibrio spirochaetisodalis gen. nov., sp. nov. and reclassification of four Desulfovibrio spp. Int J Syst Evol Microbiol 67:87–93
Steinke M, Wolfe GV, Kirst GO (1998) Partial characterisation of dimethylsulfoniopropionate (DMSP) lyase isozymes in 6 strains of Emiliania huxleyi. Mar Ecol Prog Ser 175:215–225
Sun J, Todd JD, Thrash JC, Qian Y, Qian MC, Temperton B, Guo J, Fowler EK, Aldrich JT, Nicora CD, Lipton MS, Smith RD, De Leenheer P, Payne SH, Johnston AW, Davie-Martin CL, Halsey KH, Giovannoni SJ (2016) The abundant marine bacterium Pelagibacter symultaneously catabolizes dimethylsulfoniopropionate to the gases dimethyl sulfide and methanethiol. Nat Microbiol 1:16065
Todd JD, Rogers R, Li YG, Wexler M, Bond PL, Sun L, Curson ARJ, Malin G, Steinke M, Johnston AWB (2007) Structural and regulatory genes required to make the gas dimethyl sulfide in bacteria. Science 315:666–669
Todd JD, Curson AR, Dupont CL, Nicholson P, Johnston AW (2009) The dddP gene, encoding a novel enzyme that converts dimethylsulfoniopropionate into dimethyl sulfide, is widespread in ocean metagenomes and marine bacteria and also occurs in some ascomycete fungi. Environ Microbiol 11:1376–1385
Todd JD, Curson ARJ, Kirkwood M, Sullivan MJ, Green RT, Johnston AWB (2011) DddQ, a novel, cupin-containing dimethylsulfoniopropionate lyase in marine roseobacters and in uncultured marine bacteria. Environ Microbiol 13:427–438
Todd JD, Kirkwood M, Newton-Payne S, Johnston AW (2012) DddW, a third DMSP lyase in a model Roseobacter marine bacterium, Ruegeria pomeroyi DSS-3. ISME J 6:223–226
Tripp HJ, Kitner JB, Schwalbach MS, Dacey JW, Wilhelm LJ, Giovannoni SJ (2008) SAR11 marine bacteria require exogenous reduced Sulphur for growth. Nature 452:741–744
van der Maarel MJEC, Aukema W, Hansen TA (1996a) Purification and characterization of a dimethylsulfoniopropionate cleaving enzyme from Desulfovibrio acrylicus. FEMS Microbiol Lett 143:3241–3245
van der Maarel MJEC, van Bergeijk S, van Werkhoven AF, Laverman AM, Meijer WG, Stam WT, Hansen TA (1996b) Cleavage of dimethylsulfoniopropionate and reduction of acrylate by Desulfovibrio acrylicus sp. Arch Microbiol 166:109–115
Visscher PT, Taylor BF (1994) Demethylation of dimethylsulfoniopropionate to 3-mercaptopropionate by an aerobic marine bacterium. Appl Environ Microbiol 60:4617–4619
Wang P, Chen X-L, Li C-Y, Gao X, Zhu D-Y, Xie B-B, Qin Q-L, Zhang X-Y, Su H-N, Zhou B-C, Xun L-Y, Zhang Y-Z (2015) Structural and molecular basis for the novel catalytic mechanism and evolution of DddP, an abundant peptidase-like bacterial dimethylsulfoniopropionate lyase: a new enzyme from an old fold. Mol Microbiol 98:289–301
Wang P, Cao HY, Chen XL, Li CY, Zhang XY, Qin QL, Todd JD, Zhang YZ (2017) Mechanistic insight into acrylate metabolism and detoxification in marine dimethylsulfoniopropionate-catabolizing bacteria. Mol Microbiol 105:674–688
White WR, Leenheer JA (1975) Determination of free formic and acetic acids by gas chromatography using the flame ionization detector. J Chromatogr Sci 13:386–389
Wood AP (1996) Sulfur, carbon and nitrogen interactions. In: Murrell JC, Kelly DP (eds) NATO ASI series Vol. 39. Microbiology of atmospheric trace gases. Springer-Verlag, Berlin, pp 281–295
Zeng Y-X, Qiao Z-Y, Yu Y, Li H-R, Luo W (2016) Diversity of bacterial dimethylsulfoniopropionate degradation genes in surface seawater of the. Arctic Kongsfjorden Sci Rep 6:33031
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this entry

Cite this entry
Boden, R., Hutt, L.P. (2019). Aerobic Bacterial Catabolism of Dimethylsulfoniopropionate. In: Rojo, F. (eds) Aerobic Utilization of Hydrocarbons, Oils, and Lipids. Handbook of Hydrocarbon and Lipid Microbiology . Springer, Cham. https://doi.org/10.1007/978-3-319-50418-6_52
Download citation
DOI: https://doi.org/10.1007/978-3-319-50418-6_52
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-50417-9
Online ISBN: 978-3-319-50418-6
eBook Packages: Biomedical and Life SciencesReference Module Biomedical and Life Sciences