
Abstract
This review presents insights on the suppression of specific factors of host defense mechanisms with an emphasis on the effects of exogenous AGEs. The data are derived from studies of humans and mice. We propose that the loss of these defenses is the driving force behind the increased oxidative stress and the pathogenesis of both T1DM and T2DM and their complications. Two components of a complex and powerful homeostasis system that provide cell-protective liaisons between cellular AGE receptors (AGER1) and the NAD + -dependent deacetylase sirtuin 1 (SIRT1) are highlighted. An imbalance between host defenses and increased oxidant challenges from the environment appear to form the basis of cell injury that underlies diabetes mellitus. We introduce the concept that reduced levels of AGEs, either by restriction in the diet or by the use of agents block the action(s) of uptake of AGEs as novel cost-efficient strategies in the prevention and treatment of the current diabetes epidemic.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
Introduction
Background
AGEs are prooxidant molecules that were initially thought to arise primarily from endogenous sources. Their presence in excess amounts was only thought to be seen in patients with diabetes mellitus or in aging [1, 2]. It is now clear that the diet is a principal source of AGEs in normal subjects [3], as well as those with diabetes mellitus [4–6]. AGEs are present in the body as a part of normal metabolism, but their levels are tightly controlled. It is only when their levels become high and remain chronically elevated, as in diabetes and aging, that they are associated with organ damage.
Relationship Between AGEs and Diabetes
It has long been recognized that patients with diabetes (both type 1 and type 2) have high circulating and tissue levels of AGEs [7, 8]. Furthermore, the levels of AGEs have been shown to be associated with both the development of complications and mortality in experimental models [9] and possibly in humans. A large study of patients in Canada and Great Britain showed that the number of individuals with diabetes increased by approximately 50%, and when they compared mortality in a population with and without diabetes, there was an excessive risk of mortality in those with diabetes, but over a period of 13 years that risk had decreased [10]. This was interpreted to be partly due to earlier detection of diabetes that contributed to higher prevalence of prediabetes and to improvements in the management of diabetes. Increased prevalence of diabetes has also been found in the USA and other parts of the world [11], and parallels the increase in obesity (Fig. 1). With respect to AGEs, it should also be noted that the increase in obesity is associated with the change in food habits in the developed world. Namely, there has been an increase in the consumption of foods that are high in AGES: red meat, “fast food,” and heat-processed foods [12]. In fact, AGEs may be a significant factor underlying both the risk of developing both T1 and T2 diabetes, and their complications [12], as will be emphasized below. One particularly disturbing fact is that T2D is becoming more frequent in the young, where it has been found to be much more aggressive, and the early loss of beta cell function appears to be more rapid [11]. The fact that AGEs have been shown to directly injure beta cells [13] potentially gives them a key role in the induction of both T1D and T2D. Also, the fact that AGEs can be controlled by dietary modifications or drugs makes the control of AGEs a high priority across the age spectrum.

Evolution of Man and Changes in Body Habitus
The appearance of insulin-dependent T1D (and latent T1D) in aging, while not as frequent as T2D, is now a recognized phenomenon [14] and may be related to a loss of beta cell function. While the mechanisms are surely complex, the fact that AGEs are directly toxic to the beta cells and that there is a documented increase in AGEs with aging suggests that AGEs may be one underlying contributing factor.
For instance, insulin resistance in patients with T2D can be reduced by the restriction of AGE intake [15]. Finally, the excess of AGEs in ingested food may play a role in changes in the gut microbiome, which may influence the development of beta cell injury. Namely, AGE restriction reduces the incidence of T1D in NOD mice [16–18] and, although the gut microbiome was not investigated in these studies, the importance of both the microbiome and gender was recently explored [17].
In this review, we present insights from studies of AGEs in humans and mice. While we will emphasize the effects of exogenous AGEs and the suppression of specific host defense mechanisms, it should be noted that AGEs are also formed intracellularly, where they are critical for several normal intracellular functions. It is only when the overall levels of AGEs in the extracellular and the intracellular spaces exceed the ability of the native antioxidant (and AGE) defenses that they pose a problem. This outcome is most evident in chronic disease conditions in which high levels of oxidative stress (OS) are sustained.
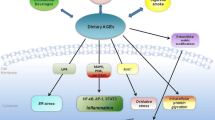
Insights from studies of humans and mice are therefore discussed with an emphasis on the effects of chronic AGEs and the chronicity as the major factor underlying the suppression of specific host defense mechanisms and factors (Fig. 2). Loss in defenses is very likely a driving force behind the increased oxidative stress and the pathogenesis of both T1DM and T2DM and their complications [3]. We have found new links between cellular AGE receptors (AGER1) and the NAD+-dependent deacetylase sirtuin 1 (SIRT1), two components of a complex and powerful homeostasis system that has cell-protective effects. Thus, one potential cause of the “epidemic” nature of diabetes and obesity may be the imbalance between depleted host defenses and overt exposure to oxidants (AGEs) from the environment, mainly the diet [12] (Fig. 3). The results include beta cell injury that predisposes to the clinical syndrome, i.e., diabetes mellitus. Therefore, restricting or blocking the effects of sustained exposure to AGEs in the diet could be a novel and cost-efficient strategy in the prevention and treatment of diabetes.

Relationship between AGEs, Host Defenses and Inflammation Responses

Contribution of AGEs to Obesity, Diabetes and Their Complications
Over the past decade, it has become apparent that the interactions between AGEs, advanced lipoxidation endproducts (ALEs), and oxidized lipids are far more prevalent in vivo than previously estimated. Substantial amounts of oxidized lipids are generated by AGE precursors [19]. Oxidized lipids were studied in T2D patients, in whom consumption of a test meal containing either a low or high oxidized fatty acid content showed that the levels of conjugated dienes in serum chylomicrons were increased in those with poor glycemic control and remained elevated for longer periods of time, compared to those with better glycemic control (HbA1c <10). Interestingly, the levels in T2D with good control did not differ from nondiabetic controls. While unsaturated fatty acids from exogenous sources can act as major donors of reactive carbonyls and are more efficient catalysts of AGE or ALE production than is glucose, this fact appears to have escaped serious attention. However, Staprans et al. [20] showed that oxidized cholesterol in the diet can accelerate atherosclerosis by increasing oxidized cholesterol levels in circulating LDL and chylomicron remnants. Oxidized fatty acids were not found to play a role in the formation of oxidized cholesterol fractions in this study. It is important to note, however, that since ALEs can also interact with AGE receptors, these compounds could underlie processes currently attributed to free fatty acids, such as beta cell injury, insulin resistance, and atherosclerosis [21]. Also while fatty acids like AGEs are thought to play a major part in atherosclerosis [19, 22], the fact that fatty acids have a very low affinity for certain receptors (including toll-like receptor 4 [TLR4]) suggests that free fatty acids at circulating levels may play a lesser role, whereas TLR4 could directly interact with AGEs [21, 23]. Intracellular AGE formation is usually tightly controlled, partly by the balance between nascent oxidants and antioxidants, and by the glyoxalase system and other enzymes that reduce OS and inhibit AGE formation [24]. Extracellular AGE-modified proteins, including those liberated from tissues, are sequestered by AGE receptors [25], internalized, and degraded by proteolytic digestion. The resulting products are normally excreted by the kidneys [26, 27]. Therefore, the levels of AGEs in tissues and cells are increased when renal function is decreased. Another reason for delayed AGE detoxification is that proteins and lipids modified by AGEs are resistant to degradation, which delays their turnover and interferes with tissue repair.
We will focus on methylglyoxal (a reactive AGE) and two new aspects of the cellular anti-OS host (innate) defense system: AGE receptor-1 (AGER1) and the NAD + -dependent deacetylase sirtuin 1 (SIRT1). These two components are part of a complex and powerful cellular antioxidant defense system that controls cellular oxidative stress at physiological levels. The major prooxidant AGE receptor (RAGE) mediates increased cellular inflammation and oxidative stress, and a secreted form circulates and is able to bind circulating AGEs [28]. As host defenses are breached by a chronic excess of oxidants from the environment and AGEs are allowed to accumulate in tissues and cells, the result is a sustained increase of oxidative stress, leading to cell injury. This sustained change in homeostasis could underlie the increased susceptibility to diabetes mellitus and its complications.
Thus, AGEs may play a central role in the induction of diabetes and its complications (Fig. 3), and their control may need to be reassessed in the management of both T1 and T2 diabetes mellitus.
Brief Definition of Bioreactive AGEs
General Comments
The term AGEs is given to a series of prooxidant metabolic derivatives of nonenzymatic reactions between reducing sugars and free amines of proteins, largely α-NH2 or ε-NH2 groups, as well as of aminolipids and nucleic acids [29–31]. When AGEs are initially formed, they have high oxidant potential; however, as they progressively decay, they become less active. Extracellular and intracellular reactive carbonyl precursors (i.e., glyoxal, 3-deoxyglucosone, or methylglyoxal) generate AGEs orglycoxidants; including; (N-; epsilon-carboxymethyl-lysine, CML), MG-imidazolone-H1 (MG-H1 or crosslink-forming endproducts such as pentosidine [32, 33]. The chemical process of glycation, initially identified by Prof. Maillard (1912), is sensitive to pH, high temperature, hydration, type of sugar, and acid or base buffering conditions [34]. This reaction is slow and strictly regulated in vivo. However, under supraphysiological conditions, AGE formation may occur at vastly accelerated rates. It is important to note that the amount of AGEs formed depends on the substrate source (animal or plant), temperature applied, the amount of available water (hydration), and the time of exposure to the increased temperature [35].
AGEs are formed by the nonenzymatic interaction of hydroxyl groups (typically from sugars) and amino groups, preferably lysine or arginine. These intermediates are unstable and spontaneously degrade or undergo redox cyclization reactions, releasing reactive oxygen species which can modify proteins, lipids, or nucleic acids either in the intracellular or extracellular spaces, as well as in mixtures of nutrients, under high temperature. This review will focus on one of the active AGE precursors (methylglyoxal, MG) which readily modifies proteins containing arginine residues to form derivatives MG-imidazolone-H1 (MG-H1). The modification of proteins by MG is particularly important since it is directed to arginine residues, which have a high probability of location at functional sites [36]. Unlike some of the earlier AGE precursors, MG derivatives are relatively stable, but still quite reactive. For these reasons, MG-H1 modification of proteins and lipids may be quantitatively one of the more important modifications in the pathogenesis of diseases, especially diabetes and diabetes-associated complications.
AGE Targets
Rather than consider each individual organ/cell type targeted by AGEs, we here will provide representative examples and direct the reader to the literature. The examples serve mainly to familiarize the reader with the broad clinical significance of AGEs and the importance of keeping their levels as low as possible.
Metabolism of Intracellular Methylglyoxal
General Comments
There are two glyoxalase enzymes, glyoxalase-1 (Glo-1) and glyoxalase 2. Since methylglyoxal is the major substrate for Glo-1, if Glo-1 levels are reduced, the intracellular levels of methylglyoxal increase to cytotoxic levels. Thus, Glo-1 is an important part of the intracellular antioxidant system, especially glutathione and the control of MG-derived AGEs [4]. Glo-1 has an antioxidant response element in its promoter region, which binds Nrf2 [37]. Therefore when Nrf2 is upregulated acutely by increased OS, due to MG, it could bind to the Glo-1 promoter and induce the translation of Glo-1. The result would be reduced intracellular MG levels. While this feedback mechanism may control acute changes in MG levels, we have found that Nrf2 levels are decreased in chronic high OS conditions, such as diabetes. The net result of this downregulation of an important regulator of intracellular MG levels promotes cell injury and eventually cell death.
Cell Surface AGE Receptors
General Comments
There are two general types of AGE receptors. One class serves to bind, internalize, and degrade AGEs. AGER1 is the best example in this class [12]. This receptor also serves to control excessive intracellular OS and is therefore a major part of the cellular antioxidant defense system. There is a second group of receptors that also bind AGEs, but instead of detoxifying AGEs, these receptors increase OS and inflammation [12]. Thus, as a group they are classified as prooxidant receptors. RAGE is the classic example of this class.
A complete description of these receptors is beyond the scope of this review, but suffice it to say that elucidation of AGE interactions with AGE receptors has proven to be complicated. This is because the prooxidant receptors, i.e., those other than AGER1, have a relatively low AGE affinity. They bind molecules other than AGEs, and their primary structure is quite varied. The fact that AGEs, like other oxidant species, can signal through non-AGE receptors, such as scavenger receptors, G-protein-coupled receptors, pattern-recognition receptors, and toll-like receptors, as well as via receptor-independent pathways, is underappreciated and has led to considerable confusion in the field. For these reasons, we will focus on AGER1 and RAGE.
Receptors that bind AGEs are differentially regulated by ambient levels of both AGEs and OS. For instance, AGER1 is upregulated by acute rises in AGEs, but is depleted, suppressed, or unresponsive in the presence of chronically high levels of OS (as in diabetes or chronic kidney disease). Importantly, intracellular antioxidant systems and some of the extracellular host defenses, such as lysozyme and defensins are also depleted under high OS. On the other hand, RAGE, which is also upregulated by AGEs, remains upregulated in the continued presence of high AGE levels. Thus, it perpetuates high oxidative stress states.
AGER1 – Defense Against AGE Toxicity
AGER1, which is encoded by the gene DDOST, is an evolutionarily conserved type 1 transmembrane protein present on the cell surface, the inner membrane of the endoplasmic membranes, and in mitochondria [12]. AGE-specific receptors were first recognized on peripheral monocytes [38–40]. AGER1 expression increases after acute exposure to increased levels of AGEs, like many other receptors. Thus, under normal conditions, AGER1 levels inversely correlate with intracellular AGEs and directly with serum AGEs. However, AGER1 becomes downregulated when exposed to persistently elevated levels of AGEs, arising largely from ingestion of food containing large amounts of AGEs and/or the presence of diabetes. Since the kidneys are the major site of AGE disposal and AGER1 removes AGEs from the blood, AGER1 levels usually directly correlate with the amount of AGEs present in the urine. The removal of circulating AGEs promotes cell stability and protects the entire body against overt OS. The mechanisms of AGER1 action include inhibition of the activation of NADPH oxidases p47phox (also known as neutrophil cytosol factor 1) and gp91 by suppression of Tyr311 and Tyr332 phosphorylation of PKCδ [41]. These actions prevent NFκB p65 97 activation and nuclear translocation, two processes that are promoted by AGE binding to RAGE. AGER1 also prevents AGE-initiated transactivation of EGFR due to high oxidative stress [42]. Thus, AGER1 may also restrict hyperactivity of other G-protein-coupled receptor kinases. Since increased levels of p66Shc are linked to diabetes mellitus, atherosclerosis, and kidney disease, it should be noted that AGER1 inhibits AGE-induced Ser36 phosphorylation of the p66Shc isoform of SHC-transforming protein 1, a major oxidative stress and apoptosis-promoting adaptor protein. AGER1 also inhibits AGE-mediated suppression of the antioxidant effect of FOXO3 on superoxide dismutase 2 (SOD2) expression, perhaps because of its role in the negative regulation of p66Shc activity. These in vitro data were confirmed in mice transgenic for AGER1 which had decreased formation of occlusive atheromas caused by wire-injury, a high fat diet or T2DM [43]. If these findings in mice apply to humans, the significance of reduced levels of AGER1 in diabetes mellitus may explain the increased incidence of atherosclerosis in these patients.
Very recently, a potentially important protective synergism between AGER1 and SIRT1 has been identified [86]. SIRT1 is thought to play a major part in insulin signaling and secretion, insulin resistance, inflammation, lifespan, as well as tissue fibrosis, affecting the cardiovascular system and kidneys in diabetes. Unfortunately, both SIRT1 and AGER1 are suppressed in patients with diabetes mellitus, especially those with complications characterized by high levels of AGEs and oxidative stress, such as diabetes. We recently found that AGER1 overexpression blocks AGE-induced suppression of SIRT1 [44], thereby inhibiting NFκB p65 hyperacetylation and inflammatory events. AGER1 also prevents AGE-induced impaired signaling via the insulin receptor and insulin receptor substrate 1 (IRS1) in adipocytes, and results in prevention of an AGE-induced decrease in glucose uptake [45]. These data indicate that AGER1 provides SIRT1 with a shield against a high external oxidant load. This may mitigate inflammation while preserving the metabolic actions of insulin. This conclusion is reinforced by the observation that AGER1 protein levels in peripheral blood mononuclear cells of healthy humans correlate with circulating AGE levels [3].
Thus, since the level of AGER1 expression normally correlates directly with those of intracellular antioxidant systems (e.g.,SIRT1, NAMPT, SOD2, and GSH) and negatively with prooxidant pathways (e.g., RAGE, NADPH oxidase, and p66shc), AGER1 may be important in the maintenance of normal homeostasis. An obvious corollary is that reduced AGER1 expression levels may be a marker of compromised host defenses in patients.
RAGE – Propagation of AGE Toxicity
In contrast to AGER1, RAGE activation promotes both ROS and inflammation in acute and chronic diseases. Whereas, AGER1 is relatively specific for AGEs, RAGE binds multiple ligands, including high mobility group protein B1 (HMGB1), amyloid β protein, and members of the calcium-binding S100 protein group. RAGE is a prominent member of a family of low-affinity, pattern-recognition receptors that function at the interface of innate and adaptive immunity [46]. While the binding characteristics for AGEs by AGER1 are well-understood, those for multiligand RAGE are not as clear. Activation of full-length, cell-associated RAGE induces an array of signaling events, including MAPK p38–JNK and JAK–STAT, CDC42–RAC and others, many of which may act as both the result and the cause of ROS. Whereas full-length RAGE does not contribute to the endocytosis or removal of AGEs, the extracellular domains of RAGE may be shed as soluble variants possibly contributing to AGE clearance [47]. Even though an association between RAGE and diabetic complications has been reported, it has been difficult to assign a primary role to this receptor other than that of an ROS transducer. RAGE may be principally modulated by ambient OS. The best evidence for this supposition is that low OS, as after restricting external AGEs, markedly suppresses RAGE mRNA and protein levels in diabetic mice and after sevelamer carbonate in T2DM with kidney disease [9, 48]. Similarly, AGE restriction in either healthy humans or those with diabetes mellitus to levels markedly below their baseline (>60%) reduces RAGE levels in peripheral blood mononuclear cells, indicating that RAGE is regulated by AGEs entering from the external environment. In fact, RAGE mRNA and protein levels, in peripheral blood mononuclear cells from healthy individuals, directly correlate with serum levels of AGEs and oxidative stress, as well as with ingested AGE levels [3]. It is important to note that RAGE levels are only modestly elevated in patients with diabetes mellitus without complications. One can conclude that both AGER1 and RAGE respond to the presence of AGEs in the environment, but in discordant directions. Findings in both animals and humans offer new perspectives on the role of RAGE in diabetes mellitus. As with other signal transduction molecules that regulate proinflammatory events, the often noted upregulation of RAGE may constitute the result rather than a cause of increased OS. When host defenses are compromised and basal OS increases, RAGE may be upregulated and amplify OS. Other prooxidant scavenger receptors that bind AGEs, as well as galectin-3 also seem to function in this manner [49].
Examples of Conditions in Which AGEs May Play a Pathogenic Role
AGEs and the Induction of T1D in Children
A rising incidence of type 1 diabetes in children has been noted throughout the world prompting calls for new prognostic indicators [50]. In a large study, islet cell antibody-positive children were evaluated for predictors of T1D [51]. An assay for an AGE (CML) was included, based on evidence implicating the environment in the development of T1D in twins [51]. The authors studied 7,287 unselected school children, of whom 115 were ICA positive and 32 monozygotic and 32 dizygotic twins discordant for diabetes, and followed them for 7 years. They found that CML was increased in ICA+ and prediabetic children as well as in diabetic and nondiabetic twins and that elevated levels of CML were a persistent and independent predictor of diabetes progression. Thus, AGEs are another risk factor, in addition to ICA and HLA risk. The familial environment explained 75% of the CML variance, confirming their previous data. Thus, it could be concluded that CML is a potential therapeutic target in ICA+ children.
AGEs and the Induction of T1D in Adults
Recently, it has been recognized that autoimmune diabetes also occurs in adults [14]. A study of 6,156 adults attending adult diabetes clinics in Europe revealed that 541 had auto antibodies, of which most recognized GADA (~90%). Of the total population, ~10% did not require insulin (latent autoimmune diabetes). The majority of those with high GADA levels (403/541) were female, lean, and treated with insulin. Overall, LADA is much more frequent than adult-onset autoimmune type diabetes. While AGEs were not examined in these patients, the data from the autoimmune diabetes in children would suggest that presence of high AGEs is a reasonable possibility and should be examined in the LADA population, since this is a modifiable risk factor.
AGEs and Pancreatic Beta Cells
The role of β-cell responses to AGEs was examined in mice treated with AGE-BSA (bovine serum albumin modified by AGEs) [13]. The investigators found that treated mice had higher glucose levels and lower insulin levels in response to a glucose challenge than controls, despite normal insulin sensitivity and normal islet morphology. Isolated islets from these mice also had lower glucose-stimulated insulin secretion. In addition, ATP production in isolated islet cells was reduced by AGEs, while the glucose-stimulated insulin secretion was restored by a sulfonylurea derivative. AGEs also inhibited nitric oxide activity by activating inducible nitric oxide synthase (iNOS) activity. Aminoguanidine reversed the inhibitory effects on ATP production and insulin secretion, leading the authors to conclude that AGEs inhibit cytochrome c oxidase and ATP production, which leads to impaired glucose-stimulated insulin secretion, mediated by iNOS-dependent nitric oxide production.
AGEs as Initiators of Insulin Resistance and T2D
This question was studied in several different models of T2DM (db/db, or C57B6 mice fed a high fat diet as well as C57B6 mice with age-related T2DM) [12]. The role of AGEs was evaluated using AGE restriction. The results show that there was a decrease in oxidative stress and an improvement in insulin resistance (IR), despite persistent hyperglycemia or obesity, high fat intake, or advanced age. The direct role of food AGEs in IR was further supported by study in which a low-AGE diet was supplemented with methylglyoxal-modified albumin (MG+) and compared to the low AGE diet (MG−). Mice fed an MG+ diet, as well as mice fed regular mouse chow, but not pair-fed, age-matched MG− mice, had an early onset of age-associated insulin resistance [52], increased adiposity, and inflammatory changes, which could not be attributed to advanced age or overnutrition. MG+ mice, in addition to impaired insulin receptor signaling and low insulin-stimulated glucose uptake, had suppressed tissue expression of key defense factors such as SIRT1, AGER1, and plasma adiponectin [52]. The marked acceleration of T2DM onset in MG+ mice over five generations cannot be attributed to genetic effects, neither can the doubling of obesity in humans in the last generation.
However, the loss of anti-AGE and OS-regulatory genes function across generations could reflect epigenetic changes, because of the gradual increase in oxidant levels over several generations in both mice and humans. Although further investigation is required, impaired host defenses could gradually result in hyper-responsiveness to inflammatory stimuli and, thus, increased susceptibility to disease. For instance, offspring of obese or diabetic parents are at higher risk of developing these phenotypes as adults.
That AGEs can influence insulin sensitivity was also explored in human subjects with T2DM and insulin resistance, who were exposed to AGE restriction for 4 months [15]. Compared to a control group, there were substantial reductions in plasma insulin, leptin, and pro-inflammatory TNF and RAGE in patients exposed to AGE restriction. In contrast, AGER1, SIRT1, and adiponectin were increased. These responses were accentuated in peripheral monocytes by NFκB p65 hyperacetylation, which was likely due to SIRT1 suppression. Further, gene transfer and silencing studies showed that SIRT1 actions were under the control of AGER1 in monocytes/macrophages, where SIRT1 exerts anti-inflammatory functions, or in adipocytes, where it regulates glucose utilization [53, 54].
Further studies exploring this new link between AGEs and diabetes may help explain how the modern environment depletes host defenses and contributes to the metabolic syndrome and diabetes type 2.
Brain
Diabetes is associated with increased risk of clinically verified Alzheimer’s disease, especially if diagnosed in mid-life [55]. This has led to the search for modifiable factors in diabetic and prediabetic individuals. Insulin resistance in an asymptomatic, late middle-aged cohort was found to be associated with progressive atrophy in brain regions associated with Alzheimer’s disease [55, 56]. Since insulin resistance is responsive to lowering AGEs, this could be an interesting area to explore as a novel therapeutic approach [15]. After initially proposing that AGEs could be involved in the pathogenesis of Alzheimer’s disease, it was then hypothesized that methylglyoxal could be a major contributor to this disease [56]. This postulate was supported by the observation that higher levels of methylglyoxal in 267 serially followed older adults with normal cognitive function at baseline were associated with a faster rate of cognitive decline [57]. Since methylglyoxal levels can be modulated by diet and/or drugs, this result could have pathogenetic implications and a potential therapeutic strategy. A study of the toxicity of methylglyoxal in neural cell types revealed that neurons are sixfold more susceptible to methylglyoxal injury compared to astrocytes, which could be due to the fact that the methylglyoxal degrading enzyme (Glo-1) had a ninefold higher expression level in astrocytes compared to neurons [58]. Finally, methylglyoxal led to glycation of occludin in cerebral microvessels, making them more permeable, which could contribute to dysfunction of the blood-brain barrier [59].
Recent transgenerational studies from our group mice fed an MG-supplemented diet (MG+) showed that age-related brain dysfunction and SIRT1 deficiency are associated with elevated brain MG [60]. Cognitive decline was promoted by AGEs, which also promoted the metabolic syndrome, via SIRT1 deficiency. AGEs and Aβ, which are known to be toxic to the brain were increased to levels similar to those found in old mice. These changes, however, could not be attributed to aging, since they were absent in pair-fed, genetically identical, and age-matched MG- mice. These findings were consistent with, and supported, new clinical findings, which suggested that abnormally high levels of circulating MG was a determinant of cognitive decline in older humans, as well as young elderly, who were cognitively intact at baseline [57, 61]. Serum levels of MG, a marker associated positively with high dietary AGE intake and negatively with SIRT1 levels, also predicted impaired insulin sensitivity over time in this population. In summary, these findings point to AGEs as a new environmental risk factor for the new complex of dementia and the metabolic syndrome in older adults.
Brain dysfunction, as well as IR, is associated with dietary factors, especially since modern diets are replete with prooxidant AGEs in addition to calories or specific nutrients. Furthermore, it has been suggested that the benefits derived from calorie restriction on cognition are related to the increase in SIRT1 expression in the brain and a decrease in the consumption of AGEs, because of the restriction of food intake. This postulate is supported by observations on our recent study in mice, where we found that the MG+ diet reproduced age-related metabolic changes, insulin receptor defects, and inflammation [52]. These changes, however, were absent in mice fed the low MG diet (MG−), despite identical caloric intake by both groups. These data provide evidence that age-related changes, which had previously been attributed to calorie restriction, could be partly due to AGE restriction. We have shown that changes in the SIRT1 pathway are linked to AGE receptor levels and that AGE receptors are expressed in brain neurons, microglia, and endothelium. In the current experiments AGER1, an anti-AGE receptor, was downregulated in the brain of MG+ mice [60]. However, RAGE, a signaling receptor linked to neurotoxicity, was enhanced. We reasoned that since AGER1 can prevent the suppression of systemic SIRT1, it could have a similar effect in the brain. This was supported by the finding that low intracellular AGEs and ROS in neurons isolated from MG- mice were associated with higher AGER1 and SIRT1 levels, compared to neurons from regular diet and MG+ mice. Furthermore, it is possible that AGER1 depletion could delay the clearance of AGE-modified molecules. This may be the cause of higher AGE deposits, such as AGE-Aβ, SIRT1 suppression, and glial activation in the brains of MG+ but not MG− mice. The chronic and sustained nature of these effects was reflected in behavioral changes in MG+ mice, which mirrored the early cognitive changes seen in older humans. Importantly, these changes were absent in mice on the low-AGE diet (MG− mice). Together with the animal data, the clinical findings indicate that chronic exposure to exogenous AGEs can result in a gradual depletion of host defenses before clinical evidence of cognitive or metabolic disturbances appear. A critical and novel finding afforded by the animal studies is that a reduced exposure to oral AGEs preserved key brain gene function and also averted cognitive decline, as well as metabolic derangements and changes in motor function. These data in mice must be examined in clinical trials.
AGEs are also involved in abnormalities of peripheral nerves in diabetes; macrophages were found to ingest glycated myelin from the peripheral nerves of patients with T2D [21], which may contribute to the neuropathy in diabetes. This is also an area deserving of further investigation, especially since AGEs are a modifiable risk factor.
Estrogen has been shown to have protective effects against the development of Alzheimer’s disease in both men and women [62]. Studies in mice reveal that estradiol protects against postischemic hippocampal neurodegeneration [63]. Since estrogen is derived from testosterone by aromatase action, several investigators have examined the levels and expression of aromatase in brain and brain injury [62, 64]. While aromatase is not expressed in neurons, in the presence of OS, aromatase is expressed in astrocytes and this is associated with neuroprotection in animals [64]. Studies in the hippocampus of both men and women have shown estradiol-mediated neuroprotection [65], and a recent study of single nucleotide polymorphism in the gene coding for aromatase shows that the genotype associated with the greatest levels of estradiol was associated with greater hippocampal gray matter in males [62]. Thus, estrogen has neuroprotective effects in both men and women, and could be important in the development of Alzheimer’s disease.
In summary, AGE-induced reductions in estrogen receptor function and estradiol production may be associated with cognitive and peripheral nerve dysfunction. In this context, both methylglyoxal and Glo-1 have both been shown to play significant roles in the function of brain cell types and the blood-brain barrier.
Kidney
AGE EXCRETION
The kidneys are a major site for the disposal of oxidants from the circulation, especially AGEs. The kidneys receive 10% of the cardiac output. In addition, other than the brain and heart, they are the only organs in which blood flow is determined by cardiac output, rather than vasocontraction. Low molecular weight AGE-modified peptides may pass into the glomerular filtrate and be presented to the cells lining the tubules. After passing the glomerulus, this large amount of blood enters a rich capillary bed around the tubules. Thus, the tubules are directly exposed to higher molecular weight AGE modified molecules in the blood on both their luminal and abluminal sides. Therefore, they are exposed to AGE-modified molecules of varied molecular weight. Because of their large exposure to circulating AGEs and their role in the removal of AGEs, the kidneys are a target for AGE-induced oxidative stress. The exact mechanism for the disposal of AGEs in the kidneys remains incompletely understood, but since very reactive AGEs (such as methylglyoxal) are highly charged, they are unlikely to be filtered in large amounts through a normal glomerular barrier; however, an AGE-modified barrier might be more permeable. The fact that albuminuria is a sign of diabetic kidney disease suggests that the glomerular filter may be altered before there are other changes in kidney function [66]. However, if large amounts of AGEs are to be removed by the kidneys, they must be actively excreted. This conclusion is supported by the observation that when the levels of AGEs in the circulation are reduced by either dietary restriction or drugs, the kidneys excrete an increased amount of AGEs [6, 15]. This suggests that the kidneys are injured by high circulating levels of oxidants, such as those present in T2D. On the other hand, relatively inactive AGEs (such as CML modified peptides) are not highly charged and may more easily pass the glomerular filter and would be less dependent on intact glomerular or tubular function.
AGE Renal Toxicity (Animal Studies)
Direct evidence of the toxicity of AGEs for normal kidney structure and function was obtained in normal mice who received AGEs intravenously for 4 weeks [67]. These animals developed deposits of AGEs in the kidneys, in association with both glomerular and interstitial fibrosis, suggesting that circulating AGEs can be deposited in the kidneys and cause kidney disease. Specifically, AGEs induced an increase in glomerular extracellular matrix alpha 1(IV) collagen, laminin B1, and transforming growth factor beta 1 mRNA levels, as well as glomerular hypertrophy. The AGE response was specific because the coadministration of an AGE inhibitor, aminoguanidine, reduced all these changes. Recently, it was found that TGFβ-induced changes in mir-192 and p53 that resulted in a fibrotic response in glomerular mesangial cells [68].
AGE Removal (Clinical Studies of Normal Subjects and Diabetic Patients with Kidney Disease)
The importance of the excretion of circulating AGEs could be inferred from a study of nearly 1,500 participants without cardiovascular disease or type 2 diabetes mellitus. They found that increased cystatin C (a marker of decreased kidney function) carried a threefold increased risk of progression from normoglycemia to prediabetes [69]. This suggests that abnormalities in kidney function, possibly due to an ability to excrete excess amounts of ingested oxidants (AGEs, ALEs) result in the initiation of a series of events that carry a high risk of developing frank diabetes in an otherwise normal population.
Two recent studies of adult T2DM patients with established kidney disease revealed that reduction of AGEs, using a drug that binds AGEs in the intestine and places them in the stool, showed that there was a reduction of HbA1c and albuminuria (in women and blacks), in concert with reduced prooxidants and increased antioxidant mechanisms [70]. Namely, serum and cellular AGEs, TNFR1, and RAGE were reduced. Adiponectin, AGER1, Nrf2, SIRT1, and ERα are among the antioxidants there were increased.
AGEs and the Induction Diabetic Kidney Disease (DKD)
High levels of AGEs and chronic inflammation in T2D may predispose to the development of DKD. AGEs induce inflammation (especially TNFα) in patients and in animal models [4, 15, 71, 72]. The association between high methylglyoxal levels and progression of DKD was confirmed in type 1 diabetes in a longitudinal study of T1D children whose follow-up included a kidney biopsy [73]. Another study of long-term survivors of T1D followed at the Joslin clinic revealed that DKD was present only in those with high levels of AGEs [74]. Finally, another group of investigators at the Joslin clinic found that increased TNF receptors were a better predictor of progressive CKD in both T1D and T2D in the USA [75, 76]. These data were confirmed in a study of 106 nonobese Japanese with T2D, where it was found that circulating TNF receptor 2 was associated with the development of stage 3 CKD (GFR ~30) [77]. Since AGEs enhance the expression of TNF receptors [78] and AGEs are increased in T2D, these studies suggest that AGEs could be a significant contributor to the progression of DKD in patients with either T1 or T2 diabetes. In this regard, as noted above, TNF receptors can be lowered by currently available drugs, as well as by reducing the intake of amount of AGE-rich food.
Finally, a recent review suggests that AGEs are uremic toxins and proposes both nonpharmacologic and pharmacologic interventions to reduce AGEs in patients with CKD [79].
AGEs Toxicity in Hemodialysis Patients
AGEs levels are often several folds higher in hemodialysis patients than in normal controls [80]. These patients have a very high incidence of complications and a high mortality rate, especially those who also have diabetes (USRDS). Reduction of AGEs by a drug that sequesters AGEs in the gut, thereby preventing them from being taken into the body, led to a substantial reduction of both AGEs and other risk factors for CVD [63]. These changes were apparent after only 3 weeks of treatment. In a cross-sectional study of 189 dialysis patients [64], 139 hemodialysis and 50 peritoneal dialysis patients showed that serum CML level correlated significantly with dietary AGE intake, based on 3-day food records (P = 0.003). While no correlation was observed with protein, fat, saturated fat, or carbohydrate intake, both serum CML and MG levels correlated with blood urea nitrogen (P = 0.03 and P = 0.02, respectively) and serum albumin levels (P = 0.04 and P = 0.02, respectively). The authors confirmed the fact that dietary AGE content, independently of other diet constituents, is an important contributor to the high serum AGE levels in patients with renal failure. Moreover, the lack of correlation between serum AGE levels and dietary protein, fat, and carbohydrate intake indicates that a reduction in dietary AGE content can be obtained safely without compromising the content of obligatory nutrients in these very ill patients who are generally malnourished.
Toxicity of AGEs in Peritoneal Dialysis Patients
The fact that reduction of AGEs by dialysis is independent of dialysis method was shown in an intervention trial conducted in 26 renal failure patients on maintenance peritoneal dialysis who were randomized to either a high or a low AGE diet for 4 weeks. Those on the low dietary AGE intake had decreased serum CML (P < 0.002) and serum MG (P < 0.008) and lowered levels of two glycated lipid molecules which induce inflammatory responses in several cell types: CML-LDL (P < 0.011) and CML-apoB (P < 0.028) [81]. On the other hand, patients on the regular diet were found to have increased serum CML (P < 0.028), serum MG (P < 0.09), CML-LDL (P < 0.011), and CML-apoB (P < 0.028). Other findings related to metabolic changes that are directly related with cardiovascular risk were serum AGE correlated with BUN (CML, P < 0.002, MG, P < 0.05), serum creatinine (CML, P < 0.05; MG, P < 0.004), total serum protein (CML, P < 0.05, MG, P < 0.05 for MG), serum albumin (P < 0.02 for CML; r = 0.4; P < 0.05 for MG), and serum phosphorus (CML; P < 0.006; MG, P < 0.01). The authors concluded that dietary glycotoxins contribute significantly to the elevated AGE levels in renal failure patients, and that dietary AGE restriction is an effective and feasible method to reduce excess toxic AGEs, and possibly associated cardiovascular mortality, and favorably alter several metabolic parameters.
Mortality in T2D Patients Admitted to the ICU, and the Effects of the Levels of AGEs and Diabetic Kidney Disease on Outcome in Acute Trauma Patients
The presence of T2D has been associated with increased 1-year mortality in patients who had been admitted to an ICU (36%) compared to 29.1% in nondiabetic patients [82]. However, the presence of kidney disease was associated with a 1-year mortality of 54.6%, again emphasizing the importance of kidney function in controlling oxidative stress. Another study of AGEs in acute trauma patients admitted to the ICU revealed that those with the most severe trauma had elevated levels of AGEs that persisted, whereas the levels defervesced within one week in those with less severe injuries.
These data show that restricted exposure to AGEs is important to prevent diabetic kidney disease, progression of established nephropathy, and to better control responses to acute injury.
AGEs in Cardiovascular Disease
General Comments
While there have been many experimental animal studies showing that chronic, high levels of AGEs promote the induction and progression of cardiovascular disease [12], a recent study of 7,447 Mediterranean region subjects showed that a low-AGE diet reduced the incidence of major cardiovascular events in persons at high cardiovascular risk [83]. This study was a randomized prospective trial of three diets, two Mediterranean diets (one supplemented with virgin olive oil and one supplemented with nuts) and a regular diet. This study confirms a number of previous trials and suggests that a low-AGE diet is cardio-protective in a European population. Small studies in more diverse populations [3] suggest that these data may apply more broadly. The latter study points out the fact that it is not only the amount and type of food ingested, but that the method of food preparation plays a major role. Namely, food that is rich in animal protein and is cooked at high temperature for prolonged periods contains a large amount of cytotoxic AGEs.
Studies in rat models of hypertension revealed that methylglyoxal-mediated changes in arterial wall medial/intimal thickness were associated with hypertension [11]. Importantly, these changes were attenuated by metformin.
The question of the effects of AGE-crosslinking on large blood vessels was studied in rats with streptozotocin-induced diabetes treated with the AGE-breaker, alagebrium for 1–3 weeks [63]. The diabetes-induced increase of large artery stiffness was reversed as measured by systemic arterial compliance, aortic impedance, and carotid artery compliance and distensibility. These findings could be important in the treatment of patients with diabetes-related complications.
Estrogen in Men with Diabetes and the Risk of Peripheral Vascular Disease
Our recent unpublished data show that monocytes of aged men with T2D have suppressed levels of ERα mRNA, which is reversed by removal of AGEs. Thus AGEs may play a role in the effects of estradiol on peripheral vascular disease. While there have been few studies of estrogen and the risk of aging-related diseases in men, a recent cross-sectional study of Framingham Heart Study participants revealed that there was a 62% increased risk of incident diabetes in per cross-sectional doubling of estradiol levels [84]. This risk was 40% for estrone, which associated with impaired fasting glucose at visit 7. In follow-up over a period of 6.8 years, there was an increasing risk of diabetes with increasing quartiles of both estradiol and estrone and an increased incidence of diabetes with increasing quartiles of estrone. Thus, at the last visit, in those with a twofold increase in total estrone at visit 7, there was a 77% increase in the risk of incident diabetes. An analysis of the same study subjects showed that the age-related increase in total estrone was greater than that in total estradiol. Estrone was positively associated with smoking, BMI, and testosterone.
Total and free estradiol were associated with diabetes, BMI, testosterone, and comorbid conditions. Additionally, free estradiol was associated negatively with smoking. Finally, an investigation of a middle-aged community-based sample suggests that lower free testosterone and higher estrone concentrations may be associated with peripheral vascular disease and ankle-brachial index change in men, but sex hormones did not affect these parameters in women. Finally, peripheral vascular disease may also be related to AGEs and their influence on estrogen levels.
In summary, there is now considerable evidence that the amount of AGEs provided in the food environment can have a profound effect on cardiovascular disease. However, AGEs can be controlled by modifying the diet or by introducing drugs that modify uptake or inhibit AGEs.
Liver
Nonalcoholic Fatty Liver Disease
Visceral fat is a risk factor for both T2D and nonalcoholic fatty liver disease (NAFLD) and the two diseases are often seen together [14]. Both diseases may be preceded by the metabolic syndrome, and both have been associated with elevated AGE levels (JCEM/Plos1). A recent study in animals to address the relationship between high AGE intake and NAFLD [85] showed that chronically increased levels of dietary AGEs were associated with the initiation of inflammation, in the absence of steatosis. The authors concluded that high levels of dietary AGEs could be a precipitating factor for the initiation and progression of NAFLD.
Estrogen and Liver Disease
Since estrogen receptor alpha (the major ER isoform in the liver) is downregulated in high OS conditions, such as T2D, and estrogen has been shown to be protective against the induction of inflammation in the liver, an investigation of models of steatosis in mice was conducted [86]. Both estradiol and tamoxiphen (which mimics estradiol effects in the liver) had hepatoprotective effects against steatosis and NAFLD. These data were confirmed in men with obesity and NAFLD [87]. Finally, in a study of 251 postmenopausal women, of whom 37% had NAFLD, those with hormone replacement therapy had a low frequency of metabolic syndrome and insulin resistance, compared to those who did not have hormone replacement therapy [88]. The authors suggested that hormone replacement therapy could be a protective factor against liver disease, but cautioned that this remains as an area of investigation.
These studies in mice and humans suggest that AGEs can be hepatotoxic and that restriction of AGEs can have positive effects on NAFLD.
AGEs at Different Chronological Ages
Gestation and Infants
Since maternal diabetes has been associated with an elevated risk of diabetes and obesity in their offspring, and the transmission of AGEs from mother to fetus was unknown, AGEs were examined in sera of healthy mothers in labor (n = 60), their infants, and in infant foods [89]. Significant correlations were found between newborn and maternal serum CML (sCML) (P = 0.001), serum methylglyoxal derivatives (sMGs) (P = 0.001), and 8-isoprostanes (P = 0.001). High maternal sMGs predicted higher infant insulin or homeostasis model assessment (P = 0.027) and CML and adiponectin levels in infants negatively correlated with maternal sCML (P = 0.011). Importantly, the levels of sAGEs significantly increased with the initiation of processed infant food intake, raising daily AGE consumption by ~7.5-fold in a year, suggesting that the high content of AGEs in processed food could not be handled by the infants’ kidneys. These data are consistent with the observations that processed food commercially available for infants often has a very high content of AGEs and suggest that AGE exposure in infants may predispose to the later development of diabetes [90].
Adults
While it has generally been believed that oxidative stress (OS) inevitably increases with aging and underlies the increased incidence of cardiovascular (CVD) or kidney (CKD) disease in this period, it is now a subject of active debate as to whether unopposed OS is an obligatory process of normal aging in humans. Furthermore, with the rapid rise in the number of aging persons, it is of concern whether OS is principally the cause or the result of chronic diseases of late adulthood and whether it can be modified. Importantly, it is critical to understand if increased OS is an inevitable component of normal aging, the age of onset of such an increase, whether it can be reduced in healthy adults, and whether increased OS in patients with chronic diseases can be reduced. AGEs play a significant role in the pathogenesis of many chronic diseases in middle age and the aged, including CVD, CKD, and diabetes. In a recent study, the levels of AGEs and OS in 345 healthy urban adults 18–45 years old or older than 60 years were examined to determine if they were correlated with dietary AGEs, if AGEs and OS could be modified by restricting the amount of AGEs in the diet, and if the levels of AGEs correlated with changes in AGER1 levels [3]. Both serum (s) CML and sMG were higher on average in healthy participants older than 60 years than in 18–45-year-olds and as a group independently correlated with dietary AGEs (P = 0.0001). Somewhat surprisingly, some of the 18–45-year-old participants had sCML values in the range found in participants older than 60 years. These findings clearly established that the intake of dietary AGEs strongly affects the levels of circulating AGEs, OS, and proinflammatory markers at all ages. In addition, the levels of AGER1 in PMNC positively correlated with serum and urine AGEs and oxidant stress markers in healthy participants. One of the most important findings in this study was that reducing dietary AGE intake significantly decreased OS in healthy adults. This suggests that increased OS is not an obligate correlate of aging, that reduction of AGEs in the diet could be a safe and efficient intervention in normal aging, and that it may improve outcomes in age-related diseases.
Drugs That Influence AGEs
General Comments
There are at least three classes of drugs that may reduce the amount of AGEs presented to the GI tract. One approach is to bind AGEs in the gut and eliminate them in the stool. Sevelamer carbonate fits into this category [63]. This effectively reduces circulating and cellular AGEs in patients with diabetic kidney disease, increases antioxidant defenses, and decreases prooxidant molecules. The second group of oral drugs include those that bind or chelate AGEs in both food and after they have been absorbed into the body. They are soluble and pass both the intestinal barrier and enter most cells. There are a number of drugs in this category (listed in general order of efficacy of binding AGEs): metformin, vitamin B analogues such as pyridoxamine and benfotiamine, and aminoguanidine. In addition to these drugs, a drug that breaks formed AGE complexes in tissues makes them more available for degradation. Finally, a third category is an injectable form of soluble RAGE which has become available. There are animal study reports on this drug , but none in humans.
Oral Drugs Directly Influencing the Degradation of AGEs
-
1.
Metformin: In a study of 57 subjects with T2D, of whom 30 were treated with metformin , methylglyoxal levels were elevated in all T2D, compared to 28 controls [10]. Methylglyoxal levels correlated with rising HbA1c levels in the nonmetformin treated patients and low-dose metformin patients (<1,000 mg/day), but not in the high dosage (1,500–2,000 mg/day). The authors concluded that metformin reduces methylglyoxal levels in a dose-dependent fashion and that this effect is independent of its effects on glycemic levels.
-
2.
Aminoguanidine: There is a rich literature showing that aminoguanidine decreases nephropathy, cardiac hypertrophy, and aortic lesions in various animal models. Age-related cardiac hypertrophy has been prevented in both Sprague-Dawley and Fischer 344 rats by aminoguanidine treatment. While AGE clearance declined in untreated aged rats, this was blunted by aminoguanidine treatment, and loss of renal mass with aging was prevented in Sprague-Dawley rats. Aminoguanidine treatment was also shown to reduce aortic accumulation of injected AGEs, reduce mononuclear cell infiltration, and improve vasodilatory responses to acetylcholine and nitroglycerin. The metabolic turnover of food-derived reactive orally absorbed advanced glycation endproducts (AGEs) or glycotoxins may be delayed in patients with diabetes and kidney disease. Another study asked whether pharmacologic inhibition of dietary AGE bioreactivity by aminoguanidine (AG) improved the turnover and renal excretion of radio-labeled AGEs in normal Sprague-Dawley rats. The radio-labeled AGE diet produced serum absorption and urinary excretion peaks kinetically distinct from those of free [14C]glucose or [125I]ovalbumin. Namely, 26% of the orally absorbed AGE ovalbumin was excreted in the urine, whereas after AG treatment, urinary excretion of dietary AGEs increased markedly (to >50% of that which was absorbed). More than 60% of tissue-bound radioactivity was found to be covalently deposited in kidneys and liver, whereas after treatment with AG, tissue AGE deposits were reduced to <15% of the amount found in untreated AGE-fed controls. Thus, reduction of dietary bioreactive AGEs by aminoguanidine improves their renal elimination and prevents tissue deposition of AGEs derived from the food. The authors concluded that this may protect against excessive tissue AGE toxicity in diabetic patients, especially those with renal disease [6]. A clinical trial was conducted, using two doses of aminoguanidine, in 691 T1D subjects with nephropathy and retinopathy. The primary end point was doubling of serum creatinine, and secondary end points included proteinuria, retinopathy, and kidney function. After a follow-up of 2–4 years, the primary end point was not reached. However, in the aminoguanidine group, the estimated GFR fell more slowly, proteinuria was decreased, and fewer subjects reached a three-step or greater progression of retinopathy.
-
3.
Pyridoxamine: Hudson and colleagues have shown that pyridoxamine scavenges methylglyoxal and related pathogenic reactive carbonyl species, which keeps them from interacting with proteins [91]. Since pyridoxamine is an oral drug with a good safety profile, it has been the subject of clinical trials in diabetic kidney disease, of which none reached the study goal of reducing the doubling of serum creatinine. However, the study showed that those with the lowest serum creatinine at entrance appeared to have more benefit than either the middle or upper third suggesting there might be some benefit in early stages of DKD. A third trial is now in progress and will be reported in the future.
-
4.
Benfotiamine: In a study of 20 inpatients with T2DM in a randomized crossover design, the effects of a low-AGE (LAGE) and high-AGE (HAGE) meal on flow-mediated dilatation (FMD) and laser-Doppler flowmetry, serum E-selectin, intracellular adhesion molecule 1, and vascular cell adhesion molecule 1, oxidative stress, and serum AGE were assessed at baseline and 2, 4, and 6 h after each meal. While the meals had identical ingredients, they had different amounts of AGEs (15,100 compared with 2.750 kU AGE or the HAGE and LAGE meals, respectively). The differences were obtained by varying the cooking temperature and time. Flow-mediated dilation decreased by 36.2% (P < 0.01 for all compared with baseline) after the HAGE meal. After the LAGE meal, FMD decreased by 20.9% (P < 0.01) (P < 0.001 for all compared with the HAGE meal) [92]. After the HAGE meal, both macrovascular function and microvascular function were impaired (−67.2%), and serum AGE and markers of endothelial dysfunction and oxidative stress were increased. The authors concluded that a HAGE meal in patients with T2DM can induce a more pronounced acute impairment of vascular function than a LAGE meal that is otherwise identical. They suggested that chemical modifications of food during cooking may play a major role in influencing the extent of acute postprandial vascular dysfunction. This study was followed by an analysis of the effect of benfotiamine on the same parameters in this population in 13 type 2 diabetes patients given a meal with a high AGE content before and after a 3-day therapy with benfotiamine (1,050 mg/day). The same measures as in the first study were repeated [93]. They found that the effects of HAGE on both FMD and reactive hyperemia were completely prevented by benfotiamine. While serum markers of endothelial dysfunction and oxidative stress, as well as AGE, increased after HAGE, these effects were significantly reduced by benfotiamine treatment.
-
5.
Alagebrium: Alagebrium was developed as a compound that had the capability of breaking preformed AGE links with proteins. A large number of animal studies have been published, but there are no clinical trials showing efficacy.
Summary
The current diabetes epidemic coincides with a series of environmental changes. A major shift over the past half century is the enrichment of nutrients by AGEs. Importantly, AGEs are highly palatable and appetite-enhancing, prooxidant substances that simultaneously drive overnutrition and oxidant overload. The result of this sustained oxidant overload is that host defenses are overwhelmed and basal levels of oxidative stress are increased, factors recognized as “chronic inflammation. ” These changes can impair both insulin production and insulin sensitivity and lead to diabetes mellitus. Evidence from transgenerational animal studies and human trials indicates that high basal oxidative stress precedes both T1DM and T2DM. These changes are the result of increased levels of AGEs that are both externally derived as well as endogenously derived and present themselves as altered host defenses. However, they can be reduced or even eradicated by relatively straightforward changes in the way food is prepared (as well as the amount and types of foods) and can be supplemented with currently available drugs that are also quite inexpensive. Thus, the techniques and approaches are currently available to manage the burgeoning population of diabetes mellitus and the complications that accompany this disease. There is sufficient basic science information and results from clinical trials to conclude that this is a manageable health issue that can and should become a part of everyday clinical care.
References
Ward RA, McLeish KR. Methylglyoxal: a stimulus to neutrophil oxygen radical production in chronic renal failure? Nephrol Dial Transplant. 2004;19(7):1702–7.
Kim KM, Kim YS, Jung DH, Lee J, Kim JS. Increased glyoxalase I levels inhibit accumulation of oxidative stress and an advanced glycation end product in mouse mesangial cells cultured in high glucose. Exp Cell Res. 2012;318(2):152–9.
Vlassara H, Cai W, Goodman S, Pyzik R, Yong A, Chen X, Zhu L, Neade T, Beeri M, Silverman JM, Ferrucci L, Tansman L, Striker GE, Uribarri J. Protection against loss of innate defenses in adulthood by low advanced glycation end products (AGE) intake: role of the antiinflammatory AGE receptor-1. J Clin Endocrinol Metab. 2009;94(11):4483–91.
Vlassara H, Cai W, Crandall J, Goldberg T, Oberstein R, Dardaine V, Peppa M, Rayfield EJ. Inflammatory mediators are induced by dietary glycotoxins, a major risk factor for diabetic angiopathy. Proc Natl Acad Sci U S A. 2002;99(24):15596–601.
Koschinsky T, He CJ, Mitsuhashi T, Bucala R, Liu C, Buenting C, Heitmann K, Vlassara H. Orally absorbed reactive glycation products (glycotoxins): an environmental risk factor in diabetic nephropathy. Proc Natl Acad Sci U S A. 1997;94(12):6474–9.
He C, Sabol J, Mitsuhashi T, Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes. 1999;48(6):1308–15.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–20.
Vlassara H, Brownlee M, Cerami A. Nonenzymatic glycosylation: role in the pathogenesis of diabetic complications. Clin Chem. 1986;32(10 Suppl):B37–41.
Cai W, He JC, Zhu L, Chen X, Zheng F, Striker GE, Vlassara H. Oral glycotoxins determine the effects of calorie restriction on oxidant stress, age-related diseases, and lifespan. Am J Pathol. 2008;173(2):327–36.
Lind M, Garcia-Rodriguez LA, Booth GL, Cea-Soriano L, Shah BR, Ekeroth G, Lipscombe LL. Mortality trends in patients with and without diabetes in Ontario, Canada and the UK from 1996 to 2009: a population-based study. Diabetologia. 2013;56:2601–8.
Linder BL, Fradkin JE, Rodgers GP. The TODAY study: an NIH perspective on its implications for research. Diabetes Care. 2013;36(6):1775–6.
Vlassara H, Striker GE. AGE restriction in diabetes mellitus: a paradigm shift. Nat Rev Endocrinol. 2011;7(9):526–39.
Zhao Z, Zhao C, Zhang XH, Zheng F, Cai W, Vlassara H, Ma ZA. Advanced glycation end products inhibit glucose-stimulated insulin secretion through nitric oxide-dependent inhibition of cytochrome c oxidase and adenosine triphosphate synthesis. Endocrinology. 2009;150(6):2569–76.
Zimmerman GA, Meistrell 3rd M, Bloom O, Cockroft KM, Bianchi M, Risucci D, Broome J, Farmer P, Cerami A, Vlassara H, et al. Neurotoxicity of advanced glycation endproducts during focal stroke and neuroprotective effects of aminoguanidine. Proc Natl Acad Sci U S A. 1995;92(9):3744–8.
Uribarri J, Cai W, Ramdas M, Goodman S, Pyzik R, Chen X, Zhu L, Striker GE, Vlassara H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: potential role of AGER1 and SIRT1. Diabetes Care. 2011;34(7):1610–6.
Peppa M, Brem H, Cai W, Zhang JG, Basgen J, Li Z, Vlassara H, Uribarri J. Prevention and reversal of diabetic nephropathy in db/db mice treated with alagebrium (ALT-711). Am J Nephrol. 2006;26(5):430–6.
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110(22):9066–71.
Karlsson FH, Tremaroli V, Nookaew I, Bergstrom G, Behre CJ, Fagerberg B, Nielsen J, Backhed F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99–103.
Cai W, He JC, Zhu L, Peppa M, Lu C, Uribarri J, Vlassara H. High levels of dietary advanced glycation end products transform low-density lipoprotein into a potent redox-sensitive mitogen-activated protein kinase stimulant in diabetic patients. Circulation. 2004;110(3):285–91.
Staprans I, Pan XM, Rapp JH, Feingold KR. Oxidized cholesterol in the diet is a source of oxidized lipoproteins in human serum. J Lipid Res. 2003;44(4):705–15.
Vlassara H, Brownlee M, Cerami A. Accumulation of diabetic rat peripheral nerve myelin by macrophages increases with the presence of advanced glycosylation endproducts. J Exp Med. 1984;160(1):197–207.
Hodgkinson CP, Laxton RC, Patel K, Ye S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28(12):2275–81.
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–25.
Thornalley PJ. Glyoxalase Istructure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. 2003;31(Pt 6):1343–8.
Vlassara H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev. 2001;17(6):436–43.
Makita Z, Bucala R, Rayfield EJ, Friedman EA, Kaufman AM, Korbet SM, Barth RH, Winston JA, Fuh H, Manogue KR, et al. Reactive glycosylation endproducts in diabetic uraemia and treatment of renal failure. Lancet. 1994;343(8912):1519–22.
Makita Z, Radoff S, Rayfield EJ, Yang Z, Skolnik E, Delaney V, Friedman EA, Cerami A, Vlassara H. Advanced glycosylation end products in patients with diabetic nephropathy. N Engl J Med. 1991;325(12):836–42.
Schmidt AM, Stern DM. Receptor for age (RAGE) is a gene within the major histocompatibility class III region: implications for host response mechanisms in homeostasis and chronic disease. Front Biosci. 2001;6:D1151–60.
Brownlee M, Cerami A, Vlassara H. Advanced glycosylation end products in tissue and the biochemical basis of diabetic complications. N Engl J Med. 1988;318(20):1315–21.
Bucala R, Makita Z, Koschinsky T, Cerami A, Vlassara H. Lipid advanced glycosylation: pathway for lipid oxidation in vivo. Proc Natl Acad Sci U S A. 1993;90(14):6434–8.
Baynes JW, Thorpe SR. Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes. 1999;48(1):1–9.
Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem. 1996;271(17):9982–6.
Monnier VM, Bautista O, Kenny D, Sell DR, Fogarty J, Dahms W, Cleary PA, Lachin J, Genuth S. Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes. 1999;48(4):870–80.
Finot PA. The absorption and metabolism of modified amino acids in processed foods. J AOAC Int. 2005;88(3):894–903.
Brands CMJ, Alink G, van Boekel M, Jongen W. Mutagenicity of heated sugar-casein systems: effect of the maillard reaction. L Agric Food Chem. 2000;48:2271–5.
Ahmed N, Thornalley PJ. Advanced glycation endproducts: what is their relevance to diabetic complications? Diabetes Obes Metab. 2007;9(3):233–45.
Vlassara H. Advanced glycosylation in nephropathy of diabetes and aging. Adv Nephrol Necker Hosp. 1996;25:303–15.
Vlassara H, Brownlee M, Cerami A. High-affinity-receptor-mediated uptake and degradation of glucose-modified proteins: a potential mechanism for the removal of senescent macromolecules. Proc Natl Acad Sci U S A. 1985;82(17):5588–92.
Li G, Yan Q, Oen HO, Lennarz WJ. A specific segment of the transmembrane domain of Wbp1p is essential for its incorporation into the oligosaccharyl transferase complex. Biochemistry. 2003;42(37):11032–9.
He C, Koschinsky T, Sabol J, Buenting C, Liu C, Vlassara H. Mononuclear (MN) cell AGE receptor-1 (AGE-R1) mRNA expression and its relationship to diabetic complications. Diabetes (Suppl). 1997;46:48A (Abstract).
Cai W, Torreggiani M, Zhu L, Chen X, He JC, Striker GE, Vlassara H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-delta: implications for vascular disease. Am J Physiol Cell Physiol. 2009;298(3):C624–34.
Cai W, He JC, Zhu L, Lu C, Vlassara H. Advanced glycation end product (AGE) receptor 1 suppresses cell oxidant stress and activation signaling via EGF receptor. Proc Natl Acad Sci U S A. 2006;103(37):13801–6.
Torreggiani M, Liu H, Wu J, Zheng F, Cai W, Striker G, Vlassara H. Advanced glycation end product receptor-1 transgenic mice are resistant to inflammation, oxidative stress, and post-injury intimal hyperplasia. Am J Pathol. 2009;175(4):1722–32.
Cai W, Uribarri J, Zhu L, Chen X, Swamy S, Zhao Z, Grosjean F, Simonaro C, Kuchel GA, Schnaider-Beeri M, Woodward M, Striker GE, Vlassara H. Oral glycotoxins are a modifiable cause of dementia and the metabolic syndrome in mice and humans. Proc Natl Acad Sci U S A. 2014;111:4910–5.
Vlassara H, Striker GE. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinol Metab Clin North Am. 2013;42(4):697–719.
Schmidt AM, Hasu M, Popov D, Zhang JH, Chen J, Yan SD, Brett J, Cao R, Kuwabara K, Costache G, et al. Receptor for advanced glycation end products (AGEs) has a central role in vessel wall interactions and gene activation in response to circulating AGE proteins. Proc Natl Acad Sci U S A. 1994;91(19):8807–11.
Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–55.
Cai W, He JC, Zhu L, Chen X, Wallenstein S, Striker GE, Vlassara H. Reduced oxidant stress and extended lifespan in mice exposed to a low glycotoxin diet: association with increased AGER1 expression. Am J Pathol. 2007;170(6):1893–902.
Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, Cerami A. Identification of galectin-3 as a high-affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med. 1995;1(6):634–46.
Gale EA. The rise of childhood type 1 diabetes in the 20th century. Diabetes. 2002;51(12):3353–61.
Beyan H, Riese H, Hawa MI, Beretta G, Davidson HW, Hutton JC, Burger H, Schlosser M, Snieder H, Boehm BO, Leslie RD. Glycotoxin and autoantibodies are additive environmentally determined predictors of type 1 diabetes: a twin and population study. Diabetes. 2012;61(5):1192–8.
Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci U S A. 2012;109(39):15888–93.
Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nat Rev Endocrinol. 2009;5(7):367–73.
Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46.
Willette AA, Xu G, Johnson SC, Birdsill AC, Jonaitis EM, Sager MA, Hermann BP, La Rue A, Asthana S, Bendlin BB. Insulin resistance, brain atrophy, and cognitive performance in late middle-aged adults. Diabetes Care. 2013;36(2):443–9.
Vlassara H, Brownlee M, Cerami A. Excessive nonenzymatic glycosylation of peripheral and central nervous system myelin components in diabetic rats. Diabetes. 1983;32(7):670–4.
Beeri MS, Moshier E, Schmeidler J, Godbold J, Uribarri J, Reddy S, Sano M, Grossman HT, Cai W, Vlassara H, Silverman JM. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech Ageing Dev. 2011;132(11–12):583–7.
Belanger M, Yang J, Petit JM, Laroche T, Magistretti PJ, Allaman I. Role of the glyoxalase system in astrocyte-mediated neuroprotection. J Neurosci. 2011;31(50):18338–52.
Tellez-Nagel I, Korthals JK, Vlassara HV, Cerami A. An ultrastructural study of chronic sodium cyanate-indiuced neuropathy. J Neuropathol Exp Neurol. 1977;36(2):352–63.
Cai W, Uribarri J, Zhu L, Chen X, Swamy S, Zhao Z, Grosjean F, Simonaro C, Kuchel GA, Schnaider-Beeri M, Woodward M, Striker GE, Vlassara H. Oral glycotoxins are a modifiable cause of dementia and the metabolic syndrome in mice and humans. Proc Natl Acad Sci U S A. 2014;111(13):4940–5.
West RK, Moshier E, Lubitz I, Schmeidler J, Godbold J, Cai W, Uribarri J, Vlassara H, Silverman JM, Beeri MS. Dietary advanced glycation end products are associated with decline in memory in young elderly. Mech Ageing Dev. 2014;140:10–2.
Bayer J, Rune G, Kutsche K, Schwarze U, Kalisch R, Buchel C, Sommer T. Estrogen and the male hippocampus: genetic variation in the aromatase gene predicting serum estrogen is associated with hippocampal gray matter volume in men. Hippocampus. 2013;23(2):117–21.
Vlassara H, Uribarri J, Cai W, Goodman S, Pyzik R, Post J, Grosjean F, Woodward M, Striker GE. Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol. 2012;7(6):934–42.
Vlassara H, Cai W, Chen X, Serrano EJ, Shobha MS, Uribarri J, Woodward M, Striker GE. Managing chronic inflammation in the aging diabetic patient with CKD by diet or sevelamer carbonate: a modern paradigm shift. J Gerontol A Biol Sci Med Sci. 2012;67(12):1410–6.
Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. 2001;47(4):318–29.
Waheed S, Matsushita K, Astor BC, Hoogeveen RC, Ballantyne C, Coresh J. Combined association of creatinine, albuminuria, and cystatin C with all-cause mortality and cardiovascular and kidney outcomes. Clin J Am Soc Nephrol. 2013;8(3):434–42.
Vlassara H, Striker LJ, Teichberg S, Fuh H, Li YM, Steffes M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc Natl Acad Sci U S A. 1994;91(24):11704–8.
Kato M, Dang V, Wang M, Park JT, Deshpande S, Kadam S, Mardiros A, Zhan Y, Oettgen P, Putta S, Yuan H, Lanting L, Natarajan R. TGF-beta induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci Signal. 2013;6(278):ra43.
Donahue RP, Stranges S, Rejman K, Rafalson LB, Dmochowski J, Trevisan M. Elevated cystatin C concentration and progression to pre-diabetes: the Western New York study. Diabetes Care. 2007;30(7):1724–9.
Yubero-Serrano EM, Woodward M, Poretsky L, Vlassara H, Striker GE. Effects of sevelamer carbonate on advanced glycation end products and antioxidant/pro-oxidant status in patients with diabetic kidney disease. Clin J Am Soc Nephrol CJASN. 2015;10(5):759–66.
Zheng F, He C, Cai W, Hattori M, Steffes M, Vlassara H. Prevention of diabetic nephropathy in mice by a diet low in glycoxidation products. Diabetes Metab Res Rev. 2002;18(3):224–37.
Wu J, Guan TJ, Zheng S, Grosjean F, Liu W, Xiong H, Gordon R, Vlassara H, Striker GE, Zheng F. Inhibition of inflammation by pentosan polysulfate impedes the development and progression of severe diabetic nephropathy in aging C57B6 mice. Lab Invest. 2011;91(10):1459–71.
Beisswenger PJ, Howell SK, Russell GB, Miller ME, Rich SS, Mauer M. Early progression of diabetic nephropathy correlates with methylglyoxal-derived advanced glycation end products. Diabetes Care. 2013;36:3234–9.
Schwedler SB, Bobadilla N, Striker LJ, Vaamonde CA, Herrera-Acosta J, Striker GE. Pentosan polysulfate treatment reduces cyclosporine-induced nephropathy in salt-depleted rats. Transplantation. 1999;68(10):1583–8.
Gohda T, Niewczas MA, Ficociello LH, Walker WH, Skupien J, Rosetti F, Cullere X, Johnson AC, Crabtree G, Smiles AM, Mayadas TN, Warram JH, Krolewski AS. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol. 2012;23(3):516–24.
Schwedler SB, Gilbert T, Moreau E, Striker LJ, Merlet-Benichou C, Striker GE. Nephrotoxin exposure in utero reduces glomerular number in sclerosis-prone but not sclerosis-resistant mice. Kidney Int. 1999;56(5):1683–90.
Izumi Y, Yabe D, Taniguchi A, Fukushima M, Nakai Y, Hosokawa M, Okumura T, Nin K, Matsumoto K, Nishimura F, Nagasaka S, Seino Y. Circulating TNF receptor 2 is associated with the development of chronic kidney disease in non-obese Japanese patients with type 2 diabetes. Diabetes Res Clin Pract. 2013;99(2):145–50.
Wu J, Zhang R, Torreggiani M, Ting A, Xiong H, Striker GE, Vlassara H, Zheng F. Induction of diabetes in aged C57B6 mice results in severe nephropathy: an association with oxidative stress, endoplasmic reticulum stress, and inflammation. Am J Pathol. 2010;176(5):2163–76.
Stinghen AE, Massy ZA, Vlassara H, Striker GE, Boullier A. Uremic toxicity of advanced glycation end products in CKD. J Am Soc Nephrol JASN. 2016;27:354–70.
Peppa M, Uribarri J, Cai W, Lu M, Vlassara H. Glycoxidation and inflammation in renal failure patients. Am J Kidney Dis. 2004;43(4):690–5.
Uribarri J, Peppa M, Cai W, Goldberg T, Lu M, He C, Vlassara H. Restriction of dietary glycotoxins reduces excessive advanced glycation end products in renal failure patients. J Am Soc Nephrol JASN. 2003;14(3):728–31.
Christiansen CF, Johansen MB, Christensen S, O’Brien JM, Tonnesen E, Sorensen HT. Type 2 diabetes and 1-year mortality in intensive care unit patients. Eur J Clin Invest. 2013;43(3):238–47.
Striker GE, Klahr S. Clinical trials in progression of chronic renal failure. Adv Intern Med. 1997;42:555–95.
Jasuja GK, Travison TG, Davda M, Rose AJ, Zhang A, Kushnir MM, Rockwood AL, Meikle W, Coviello AD, D’Agostino R, Vasan RS, Bhasin S. Circulating estrone levels are associated prospectively with diabetes risk in men of the framingham heart study. Diabetes Care. 2013;36:2591–6.
Striker LJ, Striker GE. Administration of AGEs in vivo induces extracellular matrix gene expression. Nephrol Dial Transplant. 1996;11 Suppl 5:62–5.
Miyashita T, Toyoda Y, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Hepatoprotective effect of tamoxifen on steatosis and non-alcoholic steatohepatitis in mouse models. J Toxicol Sci. 2012;37(5):931–42.
Lupia E, Zheng F, Grosjean F, Tack I, Doublier S, Elliot SJ, Vlassara H, Striker GE. Pentosan polysulfate inhibits atherosclerosis in Watanabe heritable hyperlipidemic rabbits: differential modulation of metalloproteinase-2 and -9. Lab Invest. 2012;92(2):236–45.
Florentino G, Cotrim HP, Florentino A, Padilha C, Medeiros-Neto M, Bragagnol G, Schwingel P. Hormone replacement therapy in menopausal women: risk factor or protection to nonalcoholic fatty liver disease? Ann Hepatol. 2012;11(1):147–9.
Mericq V, Piccardo C, Cai W, Chen X, Zhu L, Striker GE, Vlassara H, Uribarri J. Maternally transmitted and food-derived glycotoxins: a factor preconditioning the young to diabetes? Diabetes Care. 2010;33(10):2232–7.
Vlassara H, Striker G. Glycotoxins in the diet promote diabetes and diabetic complications. Curr Diab Rep. 2007;7(3):235–41.
Chetyrkin SV, Zhang W, Hudson BG, Serianni AS, Voziyan PA. Pyridoxamine protects proteins from functional damage by 3-deoxyglucosone: mechanism of action of pyridoxamine. Biochemistry. 2008;47(3):997–1006.
Negrean M, Stirban A, Stratmann B, Gawlowski T, Horstmann T, Gotting C, Kleesiek K, Mueller-Roesel M, Koschinsky T, Uribarri J, Vlassara H, Tschoepe D. Effects of low- and high-advanced glycation endproduct meals on macro- and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am J Clin Nutr. 2007;85(5):1236–43.
Uribarri J, Stirban A, Sander D, Cai W, Negrean M, Buenting CE, Koschinsky T, Vlassara H. Single oral challenge by advanced glycation end products acutely impairs endothelial function in diabetic and nondiabetic subjects. Diabetes Care. 2007;30(10):2579–82.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this entry
Cite this entry
Vlassara, H., Striker, G.E. (2017). Advanced Glycation Endproducts (AGEs) and Chronic Complications in Diabetes. In: Poretsky, L. (eds) Principles of Diabetes Mellitus. Springer, Cham. https://doi.org/10.1007/978-3-319-18741-9_20
Download citation
DOI: https://doi.org/10.1007/978-3-319-18741-9_20
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18740-2
Online ISBN: 978-3-319-18741-9
eBook Packages: MedicineReference Module Medicine