
Abstract
Monogenic diabetes, accounting for 1–3% of diabetes cases, results from mutations that impair pancreatic β-cell function. Monogenic forms of diabetes are often misdiagnosed as either type 1 or type 2 diabetes. A molecular diagnosis based on an emerging genetic classification enables personalized treatment, better prediction of disease progression, as well as screening, early diagnosis, and genetic counseling of family members. Historically, monogenic forms of diabetes were termed maturity-onset diabetes of the young (MODY). The different MODY subtypes differ in age of onset, manifestation of hyperglycemia, patterns of glucose-stimulated insulin secretion, and response to treatments. Furthermore, several monogenic forms of childhood and adolescent diabetes are associated with extrapancreatic manifestations and can feature a range of genetic syndromes. In this chapter, monogenic β-cell diabetes subtypes will be described according to their molecular etiologies and categorized based on their clinical implications.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
Differentiating Monogenic Diabetes from Type 1 and Type 2 Diabetes
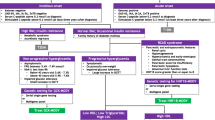
Type 1 and 2 diabetes account for the majority (≥95%) of all diabetes. Identifying rare monogenic forms of β-cell diabetes among the vast majority of type 1 and 2 diabetes patients can be challenging. A diagnosis of monogenic diabetes should be suspected if a patient with a clinical diagnosis of type 1 diabetes also has a family history of diabetes including individuals with noninsulin dependence. Furthermore, the detection of measurable C-peptide and a lack of autoantibodies against pancreatic antigens 5 years after diagnosis is uncommon in type 1 diabetes and increases the probability that a patient has monogenic diabetes [1].
Differentiation of monogenic diabetes from young-onset type 2 diabetes can usually be made clinically and should be suspected when hyperglycemia is observed in the absence of obesity, acanthosis nigricans, or polycystic ovarian syndrome and when plasma HDL levels are normal (or high) and triacylglycerol is in the normal to low range.
Phenotypic Categorization of Monogenic Diabetes
Powerful molecular genetic technologies have allowed the identification of gene mutations in patients with diabetes resulting primarily in pancreatic β-cell dysfunction. This has allowed to group monogenetic diabetes forms such as maturity-onset diabetes of the young, permanent neonatal diabetes mellitus (PNDM) or transient neonatal diabetes mellitus (TNDM), which can now usually be assigned to a specific genetic subgroup. Definition of precise genetic subgroups has allowed for personalized management of affected patients, leading to more appropriate treatment, prognostic information, and genetic counseling.
In this chapter, the description of monogenic forms of diabetes will be based on phenotypic categories aiming for the best clinical identification of and differentiation between distinct genetic subtypes. Monogenic diabetes subtypes will be phenotypically categorized into four groups: (1) diabetes diagnosed before 6 months of age, (2) mild familial fasting hyperglycemia, (3) familial young-onset diabetes, and (4) diabetes with extrapancreatic features.
Diabetes Diagnosed Before 6 Months of Age
KATP Channel Gene Mutations (KCNJ11 and ABCC8)
Diabetes diagnosed before 6 months of age likely has a genetic monogenic etiology of neonatal diabetes and is not caused by an autoimmune pathology. Neonatal diabetes is a rare condition and is defined as a disease onset before 6 months of age. Clinically, neonatal diabetes has two subdivisions, termed transient (TNDM) and permanent (PNDM) neonatal diabetes mellitus [2]. Neonatal diabetes resolves in ≈ 50% of all patients and in the majority of TNDM. Over ten distinct genetic anomalies or mutations have been identified causing the disease (Table 1). The majority of cases of TNDM have a mutation that maps to a locus on the long arm of chromosome 6, and mutations in two overlapping genes, ZAC and HYMA1, have been identified as the predominant cause of transient neonatal diabetes [3]. Mutations in the genes encoding the β-cell ATP-sensitive potassium channel, a key regulator of nutrient-induced insulin secretion in pancreatic β-cells, have been shown to cause TNDM. Activating mutations in the KCNJ11 and ABCC8, genes encoding two ATP-sensitive K− channel (KATP channel) subunits Kir6.2 and SUR1, which prevent closure of the KATP channel and thus inhibit insulin secretion, are now known to cause permanent neonatal diabetes [4, 5]. Mutations in the KATP channel genes are found in ≈ 50% of patients with PNDM; however, they can also cause TNDM. The majority of patients with PNDM have isolated diabetes, and PNDM should also be suspected if the parents do not have diabetes. The patients are usually diagnosed at birth or in the first week of life. They may constitute a syndrome of developmental delay with or without epilepsy. Identification and genetic diagnosis of patients with KCNJ11 and ABCC8 gene mutations is particularly important because, despite often having low or no insulin secretion and undetectable C-peptide and being insulin dependent, oral sulfonylurea treatment provides the most effective therapy and should be tried [5, 6]. Treatment is usually with glibenclamide at higher doses than type 2 diabetic patients (0.4–0.8 mg/kg/day). In addition to pancreatic β-cells, glibenclamide also binds to SUR subunits of the KATP channel in the nerves, muscle, and brain, where it enables, respectively, improvement of the diabetes and associated neurological symptoms. Permanent neonatal diabetes can also be caused by mutations in β-cell transcription factors leading to abnormal pancreatic development and is often associated with other developmental anomalies, defects in the glucose sensing, insulin secretory defects, and accelerated β-cell decompensation [6]. Approximately 10% of cases of permanent diabetes have not been assigned to a specific gene defect. About 10% of neonatal diabetes is caused by syndromes that frequently are associated with pancreatic aplasia and that can be caused by mutations in several transcription factors (Table 1).
Insulin (INS) Gene Mutations
Heterozygous mutations in the insulin gene (INS) account for 15–20% of cases of PNDM [7]. Patients with PNDM caused by an INS mutation have permanent diabetes without extrapancreatic features, except a low birth weight, which is a feature of all subtypes of neonatal diabetes. Mutations in the INS gene that result in the synthesis of abnormal insulin proteins have been found in humans to result in an early-onset diabetes-like phenotype [8]. These abnormal insulin proteins have altered metabolic properties and usually present with inappropriately high serum insulin levels and high insulin/C-peptide ratios due to abnormal posttranslational processing and an increased half-life. In many cases, diabetes develops only in individuals with underlying insulin resistance or other risk factors for diabetes. Some mutations in the insulin gene have been reported to segregate with early-onset diabetes with incomplete penetrance and are inherited in an autosomal dominant manner [6].
Mild Familial Fasting Hyperglycemia
Glucokinase (GCK) Gene Mutations
Heterozygous mutations in the glucokinase (GCK, MODY2) gene, encoding the β-cell hexokinase IV, should be suspected in patients with mild fasting hyperglycemia (5.5–8.0 mmol/l) that show no or little deterioration with age [9, 10]. The phosphorylation of glucose at the sixth carbon position is the first step in glycolysis and is catalyzed by a family of enzymes called hexokinases. Glucokinase is expressed mainly in the liver and endocrine pancreas and is a unique member of this family. In contrast to hexokinases I, II, and III, glucokinase is characterized by a high substrate specificity for glucose, a high KM of about 10 mM (versus 0.1–0.001 mM for the other hexokinases), and a lack of inhibition by metabolites, such as glucose 6-phosphate or glucose 1,6-bisphosphate. These unique biochemical properties allow glucokinase to serve as the glucose sensor of the pancreatic β-cell by integrating glucose metabolism and insulin secretion [11].
Genetic linkage between DNA polymorphisms in the glucokinase gene (Fig. 1) on the short arm of chromosome 7 (7p15-p14) and MODY was initially reported in families of French origin. More than 80 different GCK mutations have been identified since then and, depending on the population, may represent from 11% to 63% of all MODY. Impairment in the enzymatic activity of mutant GCK leads to decreased glycolytic flux in pancreatic β-cells [11, 12]. This translates in vivo into a rightward shift in the dose-response curve relating blood glucose and insulin secretion rates (ISRs) obtained during a graded intravenous glucose infusion. Average ISRs over a glucose range between 5 and 9 mM are 61% lower in MODY2 subjects than in control subjects [12]. Complete loss of glucokinase activity in subjects with homozygous mutations in the GCK gene (T228M and M210K) causes neonatal diabetes, a rare form of diabetes that requires insulin therapy within the first weeks of life [13]. In contrast, individuals with activating glucokinase mutations (e.g., HNF4αV455M) develop an autosomal-dominant form of familial hyperinsulinism due to a leftward shift of the dose-response curve relating blood glucose and insulin secretion rates [14]. These genetic findings highlight the importance of glucokinase as a glucose sensor and critical regulator for insulin secretion in pancreatic β-cells.

Mutations in the glucokinase gene
Glucokinase-deficient mice have been shown to be an excellent animal model for the genetic defect in humans. Mice that lack glucokinase activity die perinatally with severe hyperglycemia and phenotypically resemble rare forms of neonatal diabetes. Heterozygous mice have elevated blood glucose levels and reduced insulin secretion. Expression of GCK in β-cells in the absence of expression in the liver can prevent perinatal death of GCK null mice, providing strong evidence for the need of β-cell GCK in glucose sensing and for maintaining normal glucose levels [15].
13C nuclear magnetic spectroscopy studies have revealed that a hepatic glucose cycling defect also contributes to the molecular etiologies of GCK mutation phenotype. Patients with GCK mutations have decreased net accumulation of hepatic glycogen and augmented hepatic gluconeogenesis after a meal [16]. These results suggest that, in addition to β-cell dysfunction, abnormalities in liver glycogen metabolism contribute to the hyperglycemia in patients with glucokinase-deficient diabetes [16].
Fetal insulin secretion in response to maternal glycemia is an important determinant for intrauterine growth. Glucose-sensing defects in pancreatic β-cells, caused by a heterozygous mutation in the glucokinase gene, can reduce fetal growth and birth weight in addition to causing hyperglycemia after birth [17]. Fetuses that have inherited a glucokinase mutation from the mother or father have a reduced birth weight of 521 g (p = 0.0002) compared to unaffected siblings [17]. It is likely that these changes in birth weight reflect changes in fetal insulin secretion that are influenced directly by the fetal genotype and indirectly through maternal hyperglycemia, by the maternal GCK genotype [17].
Hyperglycemia in subjects with GCK mutations frequently manifests in the neonatal period and invariably develops before adolescence [18, 19]. Most MODY2 subjects exhibit an increased fasting glucose set point; however, glucose metabolism can be regulated at this new level, thereby producing adequate insulin responses with only small increments of plasma glucose during an oral glucose tolerance test and hemoglobin A1c levels rarely exceeding 7.5%. The release of insulin in response to arginine in MODY2 is preserved. Glucokinase deficiency is not associated with an increased incidence of diabetic complications, including proliferative retinopathy, neuropathy, or proteinuria, and other manifestations of the metabolic syndrome such as hypertension, obesity, or dyslipidemia [19]. This finding is also consistent with the low frequency of coronary heart disease in MODY2 patients. Hypoglycemic medication is not appropriate for most patients with heterozygous GCK mutations because their hyperglycemia is invariably mild, their glycemic regulation is maintained, and medication has a minimal effect. Pregnancy is the one exception in which hypoglycemic therapy might be considered, in particular when excess fetal growth can be documented. Clinical features of GCK mutations are summarized in Table 2.
Familial Young-Onset Diabetes
Patients in whom diabetes is diagnosed before age 25, who have a strong family history of diabetes, and who do not exhibit phenotypic characteristics of type 1 and 2 patients should be evaluated for mutations in the genes encoding for transcription factors hepatocyte nuclear factor-1α (HNF1A), hepatocyte nuclear factor-4α (HNF4A), pancreatic and duodenal homeobox gene-1 (PDX1, formerly termed IPF1), and neurogenic differentiation 1 gene (NEUROD1) [20].
Hepatocyte Nuclear Factor-1α (HNF1A, MODY3)
Mutations in the HNF1A gene are the most common monogenic forms of transcription factor in young-onset diabetes, accounting for 1–2% of all diabetes. HNF1α is a homeodomain transcription factor composed of an N-terminal dimerization domain, a POU-homeobox DNA-binding domain, and a C-terminal transactivation domain. HNF1α is expressed in the liver, kidney, intestine, and pancreatic islets where it directs tissue-specific gene expression. The gene encoding HNF1α is located on the long arm of chromosome 12 (12q24.2) and was identified as the MODY3 gene through a combination of genetic linkage analysis and positional cloning [21]. Depending on the population, HNF1A mutations account for 21–73% of all monogenic early-onset diabetes. More than 190 different HNF1α mutations have been found to co-segregate with diabetes in UK, German, French, Danish, Italian, Finnish, North American, and Japanese families (Fig. 2). They include missense, nonsense, deletion, insertion, and frame shift mutations. Most HNF1α mutations can be predicted to result in loss of function. However, mutant HNF1α proteins with an intact dimerization domain may impair pancreatic β-cell function by forming nonproductive dimers with wild-type protein, thereby exhibiting dominant negative activity. This mechanism has been shown for frameshift mutation HNF1α-P291fsinsC. Overexpression of HNF1α-P291fsinsC in MIN6 cells, a murine β-cell line, resulted in 40% inhibition of the endogenous HNF1α activity in a dose-dependent manner [22]. Furthermore, the formation of heterodimers between wild-type and HNF1α-P291fsinsC mutant proteins has been observed, indicating that this mutant protein has dominant negative activity [22]. Codon 291, in the poly-C tract of exon 4, is a frequent site for mutations in the HNF1α gene. This is likely due to slipped mispairing during DNA replication, thereby causing this region to be a mutation hotspot [23].

Functional domains and mutations in HNF1α
Hypomorphic HNF1α mutations may also contribute to the development of type 2 diabetes in some populations. The HNF1α(G319S) variant is associated with type 2 diabetes in the Canadian Oji-Cree population with odd ratios of 4.0 and 1.97 in individuals with homozygous and heterozygous G319S mutations, respectively [24]. This mutation is located in the proline-rich transactivation domain and substitutes a conserved glycine residue. Clinical studies indicate that the G319S variant in the Canadian Oji-Cree population is associated with earlier onset of diabetes in women, lower body mass index, and higher plasma glucose after oral glucose challenge [24].
HNF1α mutations lead to β-cell dysfunction and result in elevated fasting glycemia and impaired glucose-stimulated insulin secretion. Patients with HNF1α mutations have decreased serum levels of highly sensitive C-reactive protein (hsCRP) and altered patterns of plasma protein fucosylation [25]. Other clinical features of HNF1A diabetes (Table 3) include increased proinsulin-to-insulin ratios, increased responsiveness to sulfonylureas, and lower body mass index (BMI) [21–26]. HNF1A mutations are highly penetrant, with 63% of MODY3 diagnosed by the age of 25 years, 78.6% by 35 years, and 95.5% by 55 years. Subjects with HNF1α mutations have a more rapid deterioration in β-cell function than GCK diabetes subjects [26].
Heterozygous HNF1α patients frequently require treatment with oral hypoglycemic agents or insulin [26]. A genetic diagnosis of patients with HNF1A diabetes is important because this genetic subgroup exhibits a high sensitivity to sulfonylurea drugs and patients should initially be treated with very low doses. Sulfonylurea therapy is highly effective since it can bypass glycolytic metabolism by directly binding to KATP channels and stimulating insulin release – which is beneficial since HNF1α activates many metabolic genes and HNF1A diabetes results from defective glycolysis and ATP production required for normal insulin synthesis and secretion [27]. During pregnancy, insulin may be required and remains the most common treatment for this patient group.
Hepatocyte Nuclear Factor-4α (HNF4A, MODY1)
HNF4α is an orphan member of the superfamily of ligand-dependent transcription factors. It contains a zinc finger region (amino acids 48–128) and binds DNA as a homodimer. Two transcriptional activation domains, designated AF-1 and AF-2, flank the DNA-binding domain. AF-1 consists of the first 24 amino acids and functions as a constitutive autonomous activator of transcription. The AF-2 transactivation domain of HNF4α, spanning amino acid residues 128–366, includes the dimerization interface and ligand-binding domain.
The HNF4A gene is located on chromosome 20q (20q12-q13.1) and plays a critical role in the normal function of the liver, intestine, kidney, and pancreatic islets [28, 29]. Clinical studies demonstrated that loss-of-function mutations in HNF4α (Fig. 3) cause diabetes by compromising β-cell function. Prediabetic subjects with HNF4α mutations have normal sensitivity to insulin and first-phase insulin responses to intravenous glucose [30]. However, compared with normal subjects, mutant HNF4A patients exhibit a decrease in plasma C-peptide concentration, decrease in absolute amplitude of insulin secretory oscillations, and reduced insulin secretion rates in response to intravenous glucose infusions as blood glucose levels increase above 7 mmol/l [30–32]. Furthermore, HNF4A haploinsufficiency leads to diminished glucagon secretory responses to arginine, suggesting a role of the HNF4A gene in α-cell function [33].

Functional domains and mutations in HNF4α. DBD: DNA binding domain, LBD: Ligand binding domain, Pro: Proline-rich repressor domain
Clinically, HNF4A resembles HNF1A diabetes. Patients have a progressive pancreatic β-cell defect and frequently develop severe diabetes and complications, including micro- and macrovascular angiopathy and peripheral neuropathy (Table 4). About 30% of cases with MODY1 require insulin therapy, and the majority of the remainder are treated with oral antidiabetic drugs. Molecular studies indicate that the mechanism by which HNF4α deficiency causes an impairment in insulin secretion is because of abnormal pancreatic islet gene expression. Several genes of the glucose-stimulated insulin secretion pathway in pancreatic β-cells are regulated by HNF4α. They include the glucose transporter-2 (GLUT-2) and enzymes of glycolysis, including aldolase B, glyceraldehyde-3-phosphate dehydrogenase, and l-pyruvate kinase [29]. HNF4α also regulates the expression of other transcription factors, such as HNF1α, which itself is a transcriptional activator of the insulin gene [29]. Together, these observations suggest that diminished HNF4α activity can impair glucose-stimulated insulin secretion by decreasing the expression of genes involved in glucose entry and metabolism in pancreatic β-cells as well as insulin gene transcription [29]. Since HNF4α proteins are not only expressed in pancreatic β-cells but also play a key role in hepatocyte differentiation, mutations in this gene could be expected to result in pleiotropic phenotypes. Indeed, subjects with HNF4A haploinsufficiency have diminished serum apolipoprotein (Apo)A2, apoC3, Lp(a), and triglyceride levels compared to normal controls or patients, reflecting the reduced HDL cholesterol and increased LDL cholesterol levels [34].
The first HNF4α mutation was found in R-W pedigree, a family of German ancestry. The affected members of the R-W family have a nonsense mutation, Q268X, in the HNF4α gene [35]. This mutation generates a truncated protein that contains an intact DNA-binding domain but lacks part of the AF-2 region. Functional studies of this mutation revealed that the mutant protein lacks transcriptional activity and does not interact with the wild-type HNF4α in a dominant negative fashion [29].
Additional HNF4α variants associated with MODY1 have since then been identified and include F75fsdelT, K99fsdelAA, R154X, R127W, V255M, E276Q, V393I, and G115S [29, 30]. F75fsdelT and K99fsdelAA are frameshift mutations that lead to truncated HNF4α proteins [35–38]. HNF4α(R154X) produces a truncated protein containing only the DNA-binding domain and the AF-1 transactivation domain. This mutant protein lacks transactivation potential and may exert a mild dominant-negative effect on the activity of wild-type HNF4α in β-cells. In contrast to the frameshift or nonsense mutants, the functional properties of HNF4α missense mutants are more varied. HNF4α(V393I), located in the AF-2 domain, leads to a twofold decrease in transactivation potential [31]. Other sequence variants, such as HNF4α(R127W) and HNF4α(V255M), have a modest reduction in transcriptional activation [36]. Only one missense mutation that is located in the DNA-binding domain of HNF4α has been reported. This mutation, HNF4α(G115S), leads to an impairment in the ability of the mutant protein to bind to HNF4 consensus binding sites, thereby reducing its transactivation activity.
Heterozygous HNF4A carriers have increased birth weight (by ≈ 800 g) and macrosomia and may also cause transient or persistent hyperinsulinemic hypoglycemia. This marked increase in birth weight reflects increased insulin secretion in the fetus, with some neonates exhibiting transient or prolonged hypoglycemia [31]. The most likely explanation is that increased insulin secretion in the fetus progresses to reduced insulin secretion and diabetes in adolescence or early adulthood. Clinical management is usually by long-term treatment with low-dose sulfonylureas (for the same reason than HNF1A diabetes), which seem to be effective. In light of the abnormal lipoprotein profile, the cardiovascular risk factors should be monitored and appropriately treated.
Pancreatic and Duodenal Homeobox Gene-1 (PDX1, MODY4)
Heterozygous mutations in PDX1 gene are rare causes of familial young-onset diabetes. From limited affected patients with this monogenic form, it seems that the diabetes phenotype, penetrance, and pathophysiology is similar to HNF1A and HNF4A diabetes. Insulin promoter factor-1 or pancreatic and duodenal homeobox gene-1 (PDX1, IPF1) (Fig. 4) is a homeodomain transcription factor that is required for endocrine and exocrine pancreas development as well as insulin gene expression in the adult islet. PDX1 binds to promoters of target genes as a heterodimer with the ubiquitously expressed homeodomain protein PBX [39]. PDX1 is an essential gene for early pancreas development. During development in mice, PDX1 is initially expressed at 8.5 days postcoitum (dpc) in the dorsal and ventral gut epithelia that will later develop into a pancreas. At 9.5 dpc, PDX1 expression marks the dorsal and ventral pancreatic buds of the gut and later is restricted to differentiating insulin-producing β-cells and somatostatin-producing δ-cell [40]. Targeted disruption of the PDX1 gene results in a failure of the pancreas to develop [41]. Furthermore, β-cell-specific inactivation of the mouse PDX1 gene leads to β-cell dedifferentiation, loss of proper glucose sensing, insulin processing, and the development of diabetes [42]. Thus, PDX1 appears to be a key regulator in early pancreas formation and later in maintaining islet pattern of hormone expression and normoglycemia.

Functional domains and mutations in PDX1
The PDX1 gene maps to human chromosome 13 (13q12.1) and is involved in several human disorders including pancreatic agenesis and diabetes [43, 44]. A single nucleotide deletion within codon 63 (Pro63fsdelC) of the human PDX1 gene has been reported to cause pancreatic agenesis. This patient inherited the mutant allele from his parents who were heterozygous for the same mutation [43]. Heterozygous family members have early-onset diabetes (range 17–67 years) (Table 5). The point deletion leads to an out-of-frame protein downstream of the PDX1 transactivation domain, resulting in a nonfunctional protein lacking the homeodomain that is essential for DNA binding [45]. Expression studies of the mutant PDX1(Pro63fsdelC) protein in eukaryotic cells revealed a second PDX1 isoform that resulted from an internal translation initiation at an out-of-frame AUG. The reading frame crosses over to the wild-type IPF1 reading frame at the site of the point deletion just carboxy-proximal to the transactivation domain, resulting in a second PDX1 isoform that contains the COOH-terminal DNA-binding domain but lacks the amino-terminal transactivation domain. This terminal domain PDX1 isoform may inhibit the transactivation functions of wild-type PDX1, suggesting that a dominant-negative mechanism may contribute to the development of diabetes in individuals with this mutation. Six of eight affected heterozygotes in this pedigree were treated with diet or oral hypoglycemic agents. None of the family members carrying the PDX1(Pro63fsdelC) mutation showed ketosis or other indications of severe insulin deficiency [45].
Other PDX1 mutations that predispose carriers to diabetes include D76N, C18R, R197H, Q59L, and InsCCG243 [46, 47]. The PDX1(InsCCG243) mutation is linked in two French families with a late-onset form of type 2 diabetes and autosomal inheritance, in which insulin secretion becomes progressively impaired over time. The nondiabetic carriers have lower than normal insulin levels at high glucose levels. The InsCCG243 mutation occurs at the COOH-terminal border of PDX1 homeodomain required for transactivation. Three PDX1 missense mutations (C18R, D76N, and R197H) were found in diabetic subjects from Great Britain. Functional analysis of these mutations suggest that they exhibit decreased binding activity to the human insulin gene promoter and reduced activation of the insulin gene in response to hyperglycemia [41]. These mutations are estimated to have a frequency of 1% in the English population and may predispose to type 2 diabetes (relative risk of 3.0). The PDX1 mutations (D76N) and (Q59L) were also found in French, late-onset type 2 diabetic families with a relative risk of 12.6 for diabetes and with decreased glucose-stimulated insulin secretion in nondiabetic individuals. These mutations are located in the amino-terminal transactivation region that mediates insulin transcription. In summary, hypomorphic PDX1 variants may lead to a progressive impairment of β-cell function and glucose homeostasis in concert with other inherited metabolic abnormalities and risk factors such as age, obesity-related insulin resistance, and physical inactivity. Therefore, PDX1 mutations may also be involved in the polygenic basis of late-onset type 2 diabetes [46, 47].
Neurogenic Differentiation Factor 1 (NEUROD1, MODY6)
Similar to PDX1 mutations, heterozygous NEUROD1 gene carriers are rare and, based on information of few patients, exhibit similar clinical features than the above transcription factor forms of monogenic diabetes. NEUROD1/Beta2 (Fig. 5) belongs to the basic helix-loop-helix (bHLH) family of transcription factors that is involved in determining cell type during development. NEUROD1 is composed of a bHLH DNA-binding domain and a C-terminal transactivation domain that interacts with the cellular coactivators p300 and CBP. NEUROD1 is expressed in the pancreatic islets, intestine, and brain [48]. Mice deficient for NEUROD1 function have abnormal islet morphology and overt diabetes and die after birth [49].

Functional domains and mutations in NEUROD1/Beta2
Mutations in the NEUROD1 gene have been reported as being associated with diabetes in two families with autosomal-dominant inheritance [49]. One of the families had a G to T substitution in codon 111, causing a substitution of Arg to Leu (R111L) in the proximal bHLH domain. In vitro studies suggest that NEUROD1(R111L) has lost its DNA-binding activity and is less effective in transactivating the insulin promoter. Clinical features of subjects with this mutation are similar to type 2 diabetes with high fasting serum insulin levels, elevated levels of insulin 2 h after oral glucose, and an average age of diagnosis of 40 (range 30–59 years) [49].
The second mutation in the NEUROD1 gene consists of an insertion of a cytosine residue in a poly-C tract in codon 206 (206 + C) [49]. NeuroD1(H206fsinsC) gives rise to a truncated polypeptide lacking the C-terminal transactivation domain, a region that associates with the coactivators CBP and p300. This mutant retains its ability to bind to DNA; however, it has lost its ability to activate transcription through the deletion of the protein domain that interacts with coactivator p300 [49]. The clinical profile of patients with this truncated protein is more severe and shares clinical features of monogenic early-onset diabetes such as low endogenous insulin secretion and early age of onset (range 17–56 years) [49].
Diabetes with Extrapancreatic Features
A number of very rare diabetes-related disorders have been identified where diabetes subtypes are associated with extra pancreatic features. They are frequently underdiagnosed but can in theory be easily recognized because of their comorbidities.
Hepatocyte Nuclear Factor-1β (HNF1B, MODY5)
HNF1α and HNF1β are homologous proteins belonging to a large superfamily of homeodomain-containing transcription factors. As such, HNF1β is structurally similar to HNF1α with an N-terminal dimerization domain, a POU-homeobox DNA-binding domain, and a C-terminal transactivation domain. HNF1α and 1β bind to DNA as homo- and/or heterodimers. The HNF1 genes have an overlapping tissue distribution, but HNF1α/HNF1β ratios differ from one organ to another with HNF1α being the predominant form in the liver and HNF1β the major form in the kidney. Inactivation of the HNF1β gene in mice results in early embryonic lethality by day 7.5 of development. HNF1β-deficient embryos exhibit an abnormal extraembryonic region, poorly organized ectoderm, and no discernible visceral endoderm [50, 51].
The gene encoding HNF1B (Fig. 6) maps to chromosome 17q (17cen-q21.3), and genetic variation in this gene is responsible for several human disorders, including early-onset diabetes (Table 6), familial hypoplastic glomerulocystic kidney disease, and Müllerian aplasia [52–54]. HNF1β mutations are rare causes of diabetes, and only a few HNF1B families have been identified and studied. HNF1B diabetes develops early in life (10–25 years) and ultimately requires insulin replacement therapy to control hyperglycemia. The first HNF1β mutation found to be associated with early-onset monogenic diabetes was HNF1β(R177X)2 [51]. Nephropathy, in addition to diabetes, was found in this pedigree suggesting that decreased expression levels of HNF1β in the kidney contribute to renal dysfunction [52]. This loss-of-function mutation generated a truncated protein lacking the C-terminal transactivation domain.

Functional domains and mutations in HNF1β
Diabetes, renal dysfunction progressing to end-stage renal disease, and Müllerian aplasia have also been described in patients with HNF1B mutations [53]. Birth weight is frequently reduced by ≈ 800 g, due to reduced insulin secretion in the developing fetus. The diabetes phenotype of HNF1B carriers resembles HNF1A diabetes except that they are more insulin resistant. In contrast to other familial, early-onset diabetes subtypes, patients with mutations in the HNF1B gene have early and rapidly progressing familial hypoplastic glomerulocystic kidney disease that can be associated with nephron agenesis and is distinct from the diabetic nephropathy in type 1 or type 2 diabetes [53–55]. In addition, they may have increased risk to develop prostate cancer [56]. They may also exhibit other clinical features such as uterine and genital abnormalities, gout, hyperuricemia, exocrine pancreatic dysfunction, and abnormal liver function tests. In summary, there is increasing evidence that normal expression levels and activity of HNF1β are critical for β-cell function and pancreas and kidney development and that both loss-of-function and gain-of-function mutations can lead to disease in these organs [57].
Maternally Inherited Diabetes and Deafness (MIDD)
MIDD accounts for ≈ 1% of patients with diabetes. The vast majority (>85%) of MIDD patients carry a mutation in the mitochondrial DNA at position 3243 (A to G) [58]. The prevalence of this mutation seems to be higher in Japanese compared to the Caucasian populations. The average age when MIDD is diagnosed is 37 years old. The main extrapancreatic manifestation that patients with MIDD experience is sensorineuronal hearing loss [59]. Hearing loss usually precedes the onset of diabetes and is marked by a decrease in perception of high tone frequencies which progressively declines over the years, to severe hearing loss at all frequencies. MIDD has also been associated with a number of other issues and at the most severe end of the spectrum may include renal dysfunction, gastrointestinal problems, and cardiomyopathy manifestations. The penetrance of MIDD has been estimated to be more than 80% by the age of 70 years.
The A3243G mutation in mitochondrial DNA is heteroplasmic and can be present in any tissue. However, it is more commonly present in tissues with lower replication rates such as muscle, neurons, and pancreatic β-cells. The presence of this mutation can lead to decreased glucose metabolism, reduced function of the respiratory chain, and a decrease in oxidative phosphorylation, which ultimately could result in a decrease of ATP production. This decrease in ATP has been suggested to impact on high-energy demanding processes such as insulin secretion by pancreatic β-cells, muscle contraction, or neuronal neurotransmitter release [58].
Clinical management of MIDD is initially by dietary modification and hypoglycemic agents; however, insulin is usually required by 2 years after diagnosis. Because metformin can interfere with mitochondrial function, this hypoglycemic agent should be used with caution or be avoided, since there is a theoretical risk of inducing or exacerbating lactic acidosis. Since MIDD is a maternally transmitted disease, affected fathers should be reassured in genetic counseling that they will not transmit the disease to their children (Table 7).
Carboxyl Ester Lipase (CEL) Gene Mutations
Mutations in the variable number of tandem repeats (VNTR) of the carboxyl ester lipase (CEL) gene can cause β-cell dysfunction and early-onset diabetes (mean age of diagnosis, 36 years). CEL is expressed in the exocrine acinar cells. Affected individuals have phenotypes consistent with reduced CEL activity. Furthermore, they develop glucose intolerance and often exhibit asymptomatic exocrine failure and altered serum lipids [60]. The mechanism by which carboxyl ester lipase deficiency in the acinar cells causes progressive failure of the β-cells is unknown. The pancreas of subjects with CEL mutations shows atrophy and possible fat infiltration on imaging and marked fibrosis at autopsy [61].
Summary
Monogenic forms of diabetes result from mutations that are essential for normal pancreatic beta cell function. They are rare and frequently misdiagnosed as type 1 and 2 diabetes. Clinical classifications of monogenic diabetes subtypes based on genetic etiologies help aid the diagnosis and differentiation of these subtypes and should in many cases be followed up by genetic diagnosis. Precise knowledge of the genetic molecular etiology of monogenic diabetes allows personalized treatment, better prediction of disease progression, screening of family members, and genetic counseling.
References
Patel KA, Oram RA, Flanagan SE, et al. Type 1 diabetes genetic risk score: a novel tool to discriminate monogenic and type 1 diabetes. Diabetes. 2016. pii: db151690.
Deeb A, Habeb A, Kaplan W, et al. Genetic characteristics, clinical spectrum, and incidence of neonatal diabetes in the Emirate of AbuDhabi, United Arab Emirates. Am J Med Genet. 2016;170:602–9.
Docherty LE, Kabwama S, Lehmann A, et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype-phenotype correlation in an international cohort of patients. Diabetologia. 2013;56:758–62.
Babenko AP, et al. Activating mutations in the ABC C8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355:456–66.
Babiker T, Vedovato N, Patel K, et al. Successful transfer to sulfonylureas in KCNJ11 neonatal diabetes is determined by the mutation and duration of diabetes. Diabetologia. 2016;59:1162–6.
Greeley ASW, Naylor RN, Philipson LH. Bell, GI Neonatal diabetes: an expanding list of genes allows for improved diagnosis and treatment. Curr Diab Rep. 2011;11:519–32.
Støy J, Steiner DF, Park SY, Ye H, et al. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord. 2010;11:205–15.
Liu M, Sun J, Cui J, et al. INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med. 2015;42:3–18.
Cuesta-Muñoz AL, Tuomi T, Cobo-Vuilleumier N, et al. Clinical heterogeneity in monogenic diabetes caused by mutations in the glucokinase gene (GCK-MODY). Diabetes Care. 2010;33:290–2.
Osbak KK, Colclough K, Saint-Martin C, Beer NL, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30:1512–26.
Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39:647–52.
Byrne MM, Sturis J, Clement K, et al. Insulin secretory abnormalities in subjects with hyperglycemia due to glucokinase mutations. J Clin Invest. 1994;93:1120–30.
Njolstad PR, Sovik O, Cuesta-Munoz A, et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N Engl J Med. 2001;344:1588–92.
Glaser B, Kesavan P, Heyman M, et al. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226–30.
Grupe A, Hultgren B, Ryan A, Ma YH, Bauer M, Stewart TA. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell. 1995;83:69–78.
Velho G, Petersen KF, Perseghin G, et al. Impaired hepatic glycogen synthesis in glucokinase-deficient (MODY-2) subjects. J Clin Invest. 1996;98:1755–61.
Hattersley AT, Beards F, Ballantyne E, Appleton M, Harvey R, Ellard S. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet. 1998;19:268–70. [see comments].
Froguel P, Zouali H, Vionnet N, et al. Familial hyperglycemia due to mutations in glucokinase. Definition of a subtype of diabetes mellitus. N Engl J Med. 1993;328:697–702. [see comments].
Pearson ER, Velho G, Clark P, et al. Beta-cell genes and diabetes: quantitative and qualitative differences in the pathophysiology of hepatic nuclear factor-1alpha and glucokinase mutations. Diabetes. 2001;50 Suppl 1:S101–7.
Frayling TM, Evans JC, Bulman MP, et al. Beta-cell genes and diabetes: molecular and clinical characterization of mutations in transcription factors. Diabetes. 2001;50 Suppl 1:S94–100.
Yamagata K, Oda N, Kaisaki PJ, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. 1996;384:455–8. [see comments].
Yamagata K, Yang Q, Yamamoto K, et al. Mutation P291fsinsC in the transcription factor hepatocyte nuclear factor-1alpha is dominant negative. Diabetes. 1998;47:1231–5.
Kaisaki PJ, Menzel S, Lindner T, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in MODY and early-onset NIDDM: evidence for a mutational hotspot in exon 4. Diabetes. 1997;46:528–35. [erratum appears in Diabetes 1997 Jul;46(7):1239].
Hegele RA, Cao H, Harris SB, Hanley AJ, Zinman B. The hepatic nuclear factor-1alpha G319S variant is associated with early-onset type 2 diabetes in Canadian Oji-Cree. J Clin Endocrinol Metab. 1999;84:1077–82.
Owen KR. RD Lawrence lecture 2012: assessing aetiology in diabetes: how C-peptide, CRP and fucosylation came to the party! Diabet Med. 2013;30:260–6.
McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem. 2013;50:403–15.
Pearson ER, Liddell WG, Shepherd M, Corrall RJ, Hattersley AT. Sensitivity to sulphonylureas in patients with hepatocyte nuclear factor-1alpha gene mutations: evidence for pharmacogenetics in diabetes. Diabet Med. 2000;17:543–5.
Chen WS, Manova K, Weinstein DC, et al. Disruption of the HNF-4 gene, expressed in visceral endoderm, leads to cell death in embryonic ectoderm and impaired gastrulation of mouse embryos. Genes Dev. 1994;8:2466–77.
Stoffel M, Duncan SA. The maturity-onset diabetes of the young (MODY1) transcription factor HNF4alpha regulates expression of genes required for glucose transport and metabolism. Proc Natl Acad Sci U S A. 1997;94:13209–14.
Malecki MT, Yang Y, Antonellis A, Curtis S, Warram JH, Krolewski AS. Identification of new mutations in the hepatocyte nuclear factor 4alpha gene among families with early onset Type 2 diabetes mellitus. Diabet Med. 1999;16:193–200.
Pearson ER, Pruhova S, Tack CJ, et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte.nuclear factor 4alpha mutations in a large European collection. Diabetologia. 2005;48:878–85.
Byrne MM, Sturis J, Fajans SS, et al. Altered insulin secretory responses to glucose in subjects with a mutation in the MODY1 gene on chromosome 20. Diabetes. 1995;44:699–704.
Herman WH, Fajans SS, Smith MJ, Polonsky KS, Bell GI, Halter JB. Diminished insulin and glucagon secretory responses to arginine in nondiabetic subjects with a mutation in the hepatocyte nuclear factor-4alpha/MODY1 gene. Diabetes. 1997;46:1749–54.
Shih DQ, Dansky HM, Fleisher M, Assmann G, Fajans SS, Stoffel M. Genotype/phenotype relationships in HNF-4alpha/MODY1: haploinsufficiency is associated with reduced apolipoprotein (AII), apolipoprotein (CIII), lipoprotein(a), and triglyceride levels. Diabetes. 2000;49:832–7.
Yamagata K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature. 1996;384:458–60.
Navas MA, Munoz-Elias EJ, Kim J, Shih D, Stoffel M. Functional characterization of the MODY1 gene mutations HNF4(R127W), HNF4(V255M), and HNF4(E276Q). Diabetes. 1999;48:1459–65.
Moller AM, Urhammer SA, Dalgaard LT, et al. Studies of the genetic variability of the coding region of the hepatocyte nuclear factor-4alpha in Caucasians with maturity onset NIDDM. Diabetologia. 1997;40:980–3.
Hani EH, Suaud L, Boutin P, et al. A missense mutation in hepatocyte nuclear factor-4 alpha, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. J Clin Invest. 1998;101:521–6.
Dutta S, Gannon M, Peers B, Wright C, Bonner-Weir S, Montminy M. PDX:PBX complexes are required for normal proliferation of pancreatic cells during development. Proc Natl Acad Sci U S A. 2001;98:1065–70.
Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993;12:4251–9.
Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–9.
Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. Beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–8.
Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15:106–10.
Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. 1997;17:138–9.
Stoffers DA, Stanojevic V, Habener JF. Insulin promoter factor-1 gene mutation linked to early-onset type 2 diabetes mellitus directs expression of a dominant negative isoprotein. J Clin Invest. 1998;102:232–41.
Hani EH, Stoffers DA, Chevre JC, et al. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin Invest. 1999;104:R41–8.
Macfarlane WM, Frayling TM, Ellard S, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest. 1999;104:R33–9.
Naya FJ, Huang HP, Qiu Y, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–34.
Malecki MT, Jhala US, Antonellis A, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–8.
Coffinier C, Thepot D, Babinet C, Yaniv M, Barra J. Essential role for the homeoprotein vHNF1/HNF1beta in visceral endoderm differentiation. Development. 1999;126:4785–94.
Barbacci E, Reber M, Ott MO, Breillat C, Huetz F, Cereghini S. Variant hepatocyte nuclear factor 1 is required for visceral endoderm specification. Development. 1999;126:4795–805.
Horikawa Y, Iwasaki N, Hara M, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–5.
Lindner TH, Njolstad PR, Horikawa Y, Bostad L, Bell GI, Sovik O. A novel syndrome of diabetes mellitus, renal dysfunction and genital malformation associated with a partial deletion of the pseudo-POU domain of hepatocyte nuclear factor-1beta. Hum Mol Genet. 1999;8:2001–8.
Bingham C, Bulman MP, Ellard S, et al. Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet. 2001;68:219–24.
Bingham C, Ellard S, Allen L, et al. Abnormal nephron development associated with a frameshift mutation in the transcription factor hepatocyte nuclear factor-1 beta. Kidney Int. 2000;57:898–907. [see comments].
Gudmundsson J, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–83.
Pearson ER, et al. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor 1a and -1b mutations. Diabetes Care. 2004;27:1102–7.
Murphy R, Turnbull DM, Walker M, Hattersley AT. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A > G mitochondrial point mutation. Diabet Med. 2008;25:383–99.
Kokotas H, Petersen MB, Willems PJ. Mitochondrial deafness. Clin Genet. 2007;71:379–91.
Raeder H, Johansson S, Holm PI, et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. 2006;38:54–62.
Raeder H, et al. Pancreatic lipomatosis is a structural marker in nondiabetic children with mutations in carboxyl-ester lipase. Diabetes. 2007;56:444–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this entry
Cite this entry
Stoffel, M. (2017). Maturity-Onset Diabetes of the Young: Molecular Genetics, Clinical Manifestations, and Therapy. In: Poretsky, L. (eds) Principles of Diabetes Mellitus. Springer, Cham. https://doi.org/10.1007/978-3-319-18741-9_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-18741-9_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-18740-2
Online ISBN: 978-3-319-18741-9
eBook Packages: MedicineReference Module Medicine