
Abstract
The human gut microbiota, comprising many hundreds of different microbial species, has closely co-evolved with its human host over the millennia. Diet has been a major driver of this co-evolution, in particular dietary non-digestible carbohydrates. This dietary fraction reaches the colon and becomes available for microbial fermentation, and it is in the colon that the great diversity of gut microorganisms resides. For the vast majority of our evolutionary history humans followed hunter-gatherer life-styles and consumed diets with many times more non-digestible carbohydrates, fiber and whole plant polyphenol rich foods than typical Western style diets today.
*Corresponding author.
Access provided by Autonomous University of Puebla. Download reference work entry PDF
Similar content being viewed by others


Keywords:
3.1 3.1 Introduction
The human gut microbiota, comprising many hundreds of different microbial species, has closely co-evolved with its human host over the millennia. Diet has been a major driver of this co-evolution, in particular dietary non-digestible carbohydrates. This dietary fraction reaches the colon and becomes available for microbial fermentation, and it is in the colon that the great diversity of gut microorganisms resides. For the vast majority of our evolutionary history humans followed hunter-gatherer life-styles and consumed diets with many times more non-digestible carbohydrates, fiber and whole plant polyphenol rich foods than typical Western style diets today. Adaption of the Western-style diet over the past 50 years may thus have resulted in a significant down-shift or adverse modulation in the fermentative and metabolic output of the colonic microbiota. Short chain fatty acids (SCFA), the major end-products of bacterial carbohydrate fermentation, impact on many critical physiological processes including energy metabolism, lipid and cholesterol levels, carcinogenesis and gene expression. Additionally, the close inter-relationship between bacteria within our gut and the gastrointestinal lymphatic tissue is critical to the optimal functioning of our immune system. Modulation of these microbiota related processes as a result of our modern dietary environment may have played a role in the growing incidence of allergic diseases and chronic diseases like cardiovascular disease, obesity and cancer, often called the diseases of affluence. Recent advances in functional foods, particularly prebiotics, offer a means of modulating the gut microbiota and potentially redressing some of the deleterious consequences of modern diet on our gut microbiota and health, both locally within the gut and systemically. There is a growing body of evidence, mainly from animal studies, but also from notable human interventions, that prebiotic dietary supplementation can improve host health. Similarly, prebiotics have been repeatedly shown in human and animal interventions to modulate the relative abundance of bacteria within the gut microbiota, particularly bringing about an increased abundance of bifidobacteria, seen as beneficial microbiota constituents. However, in many cases the underlying mechanisms of effect remain to be determined. Until recently, studies examining the impact of the gut microbiota on human health or the efficacy of microbiota targeted functional foods were greatly limited by the fact that up to 70% of the gut microbiota are resistant to culture under laboratory conditions. Additionally, many gut bacteria are new to science, lacking even closely related representatives within microbial culture collections. This made assigning function to particular intestinal bacteria difficult if not impossible in most cases. Recent advances in post-genomics technologies such as genomics, metagenomics and metabonomics, are allowing researchers to derive the metabolic and microbiological consequences of microbiota modulation in a culture independent manner and link these to physiological biomarkers. These culture independent approaches allow the determination of not just the metabolic potential of the gut microbiota, in terms of the genes comprising the microbiota metagenome, but the metabolic kinetics of the gut microbiota in terms of metabolic fluxes or changes in metabolite profiles upon microbiota modulation. In this chapter, we will discuss how metagenomics and metabonomics are being applied to study the consequences of diet:microbe interactions in the gut and particularly, the microbiological and metabolic consequences of prebiotic ingestion. These technologies hold great promise in elucidating the underlying molecular basis of prebiotic induced health effects.
3.2 3.2 From Genomics to Metagenomics
The Human Genome Project (HGP) (Abdellah et al., 2004; Venter et al., 2001) provided a human genetic blueprint which has proved an extremely useful tool in identifying determinants of heritable diseases and providing the possibility of linking particular genotypes to disease risk (Desiere, 2004). During its completion the HGP also gave rise to many of the bioinformatic tools necessary for the emergence of other high resolution, data rich molecular technologies such as transcriptomics, proteomics and metabolomics. Additionally, it created the necessary scientific and commercial interest which has lead to technological leaps in terms of DNA sequencing capabilities. However, it has become apparent that this genetic blueprint does not tell the whole story of our individuality and other factors, environmental factors, play an important role in human health and disease susceptibility. These extra-genomic environmental factors which include diet, xenobiotic compounds (e.g., cooked food mutagens, drugs, environmental chemicals) and our intestinal microbiota closely interact with an individuals genome to regulate gene expression, metabolic pathways and epigenetics, and consequently, impact on an individuals health and disease susceptibility (Johnson et al., 2008; Martin et al., 2007). The advent of high through-put sequencing has enabled the rapid genetic characterization of microbial genomes and whole microbial community genomes or “metagenomes” derived directly from microbial consortia in a culture independent manner (Goldberg et al., 2006). Metagenomics is defined as “the application of modern genomics techniques to the study of communities of microbial organisms directly in their natural environments, bypassing the need for isolation and lab cultivation of individual species” and has provided unparalleled insight into the composition and metabolic potential of microorganisms comprising important communities in the terrestrial and marine environments (Chen and Pachter, 2005; McHardy and Rigoutsos, 2007; Rusch et al., 2007). This approach has now been adopted by a large scale international sequencing project, the Human Microbiome Project, run by the NIH in the United States (see Turnbaugh et al., 2007). This multi-disciplinary project aims to sequence and characterize the gut microbiota giving a global view of the metabolic potential encoded by the gut microbiota. The collaborative sequencing effort will result in the completion of an estimated 1,000 microbial genome sequences as well as metagenomic analysis of microbial communities resident in different regions of the body including the gastrointestinal and urogenital tract, oral and nasal cavities and the skin. This genomic encyclopaedia will revolutionize both our understanding of the human associated microbiota and the way in which human microbiology is carried out in the future, given the enormous reference database of sequence information on both culturable and un-culturable microbiota members that will be generated.
3.3 3.3 The Human Gut Microbiota as an Extension of the Human Genome
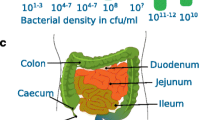
The human gut microbiota is estimated to comprise up to 1,000 different microbial species which colonize to various degrees and in different ecological niches along the length of the alimentary canal, from mouth to anus. The composition and activity of the gut microbiota differs between the different geographical regions of the gut. Host physiology plays an important role in nurturing these gastrointestinal microbial communities and can also play a regulatory role in bacterial colonization. Gastric acid, rapid flow of digesta, enzymatic and bile secretions all limit the growth of bacteria in the upper gut and it is not until the colon that sizable bacterial populations develop (Conway, 1995; Hayashi et al., 2005; Wang et al., 2005). In the healthy adult, a climax microbial consortium resides within the colon and this microbiota displays a remarkable compositional stability and a high degree of homeostasis and self-regulation which greatly impedes colonization by allochthonous microorganisms ingested by the host (Marchesi and Shanahan, 2007). Additionally, many bacteria, even distantly related species share phenotypic traits and there is a degree of functional redundancy shared between some microbiota components, ensuring for the host a steady supply of metabolites of microbial origin including vitamins and SCFAs for example. This homeostasis also ensures essential functions within the gut microbiota are maintained irrespective of fluctuations in the relative abundance of individual bacterial strains e.g., upon bacteriophage attack (Kurokawa et al., 2007). These activities include fermentative and enzymatic transformation of plant storage and structural carbohydrates, many of which are recalcitrant to degradation and digestion in the upper gut, allowing the host to derive energy from otherwise inaccessible substrates. The gut microbiota also enables host access to plant derived polyphenolic compounds most of which escape digestion in the upper gut of which some possess strong biological activities including antioxidant, phytoeostrogenic, vaso-dilation activities. Important co-metabolic processes occur between host and microbiota, explemified by the entero-hepatic circulation of bile acids (Ley et al., 2008; Nicholson et al., 2005). The gut microbiota is largely fermentative, converting un-digested carbohydrate and protein into short chain fatty acids and there is also a complex array of mutualistic, symbiotic and cross-feeding interactions between bacteria within the gut microbiota in SCFA transformation and metabolism (Duncan et al., 2004; Morrison et al., 2006).
3.4 3.4 Limitations of the Microbiological Culture Based Approaches
The extent to which the gut microbiota contributes towards human health is only becoming clear now and until relatively recently the colon was viewed solely as a retention tank for faeces, where water absorption was considered its most important contribution to systemic biological function. A major contributor towards this lack of understanding and a major hurdle to studying the gut microbiota is the fact that the vast majority of intestinal bacteria are recalcitrant to growth under laboratory conditions and up to 30% of bacterial species within an individual’s gut microbiota may be novel phylotypes and new to science (Eckburg et al., 2005; Suau et al., 1999). Therefore there are major limitations to the traditional culture based microbiological approach for studying the gut microbiota. These include;
-
Traditional culture based approaches are limited to monitoring only the culturable minority of the gut microbiota. About 70–80% of intestinal bacteria are considered recalcitrant to cultivation in pure culture, many requiring thus far unidentified growth factors or co-culture with other intestinal microorganisms to fulfil their mutualistic or symbiotic life strategies.
-
Many phylogenetically diverse bacteria share phenotypic traits and can grow under the same growth conditions and on the same nutrient media, even in the presence of selective supplements (Tuohy and McCartney, 2006). This necessitates sub-culturing followed by biochemical and genetic characterization before the microorganisms may be phylogenetically positioned; a time consuming and costly process.
-
Bacteria show a high degree of genetic plasticity, acquiring genetic determinants through horizontal DNA transfer from even distantly related species and genome duplication followed by divergent evolution within bacterial species is common. This genomic plasticity is driven by environmental selective pressure and often involves transfer of environmentally important genetic traits. Repeated passage of bacterial cultures under the selective pressure of pure culture growth conditions in the laboratory will thus result in loss of traits relevant to the natural environment from which these organisms were isolated (Lee et al., 2008). Environmentally driven genomic instability in bacteria should thus be taken into consideration when conducting functional studies on bacteria, such as probiotics, isolated from gastrointestinal ecosystems.
Thus, the reasons why we can not culture the majority of gut bacteria include the following factors; the unknown growth requirements of the bacteria, the selectivity of the media that are used, the stress imposed by the cultivation procedures, the necessity of strictly anoxic conditions, and difficulties with simulating the interactions of bacteria with other microbes and host cells. The traditional microbiological culture approach is therefore is not well suited to the enumeration of intestinal bacteria or species diversity measures especially for complex microbial consortia, such as the human gut microbiota. Advances in microbial genetics, and the adoption of the 16S rRNA gene as a universal microbial molecular chronometer, have allowed the development of tools which greatly facilitate culture independent microbial ecology studies (Frank and Pace, 2008; Handelsman, 2004; Tuohy and McCartney, 2006). Ribosomal RNAs are excellent molecules for measuring evolutionary relationships among organisms (Olsen et al., 1986). In contrast to traditional taxonomy, which is based on phenotypic traits, this kind of taxonomy reflects natural evolutionary relationships among organisms (Woese et al., 1990). Prokaryotic ribosomes contain two subunits, the sizes being 50S and 30S. The 50S subunit contains about thirty-four proteins as well as 5S rRNA (120 bases), and 23S rRNA (about 2,900 bases), and the 30S subunit contains about twenty-one proteins and 16S rRNA (about 1,500 bases). The 16S rRNA has been the most widely employed molecule to develop the phylogeny of prokaryotes. Within the various regions of 16S rRNA, the degree of conservation differs considerably. Analyses of rRNA sequences have revealed signature sequences, short stretches of rRNA, that are unique to a certain group or groups of organisms enabling the phylogenetic placement and identification of bacteria (Blaut et al., 2002).
Recent large scale sequencing projects have significantly increased the number of 16S rRNA genes archived in public databases such as GenBank, enhancing our understanding of how bacteria are related to one another at the phylogenetic level (Frank and Pace, 2008). However, many of these 16S rRNA gene sequences were derived from direct cloning experiments where environmental DNA is extracted, purified and sequenced either directly or upon generation of clone libraries and as such do not correspond to previously cultured bacteria. Indeed, many phylotypes identified within the gut microbiota are only distantly related to previously cultured bacteria. Where close relatives have been cultivated, comparisons have been drawn between these known and cultured bacteria and close un-culturable novel relatives in terms of putative physiology and ecological function. However, such functional inferences are limited since many core metabolic functions appear to be shared between distantly related bacteria whilst traits enabling occupation of a particular ecological niche may be strain specific and may not be shared with close relatives. The gut microbiota in particular appears to be a hot bed of bacterial promiscuity, with many intestinal bacteria showing evidence of high-frequency heterologous DNA transfer. Many of the genes carried by highly transmissible genetic elements within the gut microbiota encode ecologically important functional genes, involved in substrate metabolism, drug resistance or antimicrobial production (Salyers et al., 2004; Tuohy et al., 2002, 2004). Recent comparative genomic studies on the intestinal microorganism Bifidobacterium longum, clearly illustrated the significance of DNA transfer within the gut microbiota. This important member of what may be considered the beneficial gut microbiota, and close relative of common probiotic microorganisms, proved to be highly susceptible to deletion and loss of genetic traits upon repeated passage through pure culture. Upon comparative genomic analysis, several DNA regions, encoding intestinally important traits including oligosaccharide and polyol utilization, arsenic resistance and lantibiotic production, unique to the intestinally isolated B. longum strain DJO10A appeared to have been deleted from the culture collection strain, B. longum NCC2705. The authors were able to demonstrate this loss of intestinally relevant traits upon culturing B. lonugm for more than 1,000 generations under laboratory conditions (Lee et al., 2008). Similarly, comparative genomics studies with the important gastrointestinal and nosocomial pathogen, Clostridium difficile showed that a core of only 19.7% of genes were shared between 75 clinically relevant strains compared to a C. difficile 630 whole genome DNA microarray (Stabler et al., 2006). Thus despite the growing number of novel 16S rRNA species identified, we know little about how intestinal bacteria behave in their own specific ecological niche within the gut.
3.5 3.5 Gut Microbiota Community Level Phylogenetic Analysis
Studies on the composition and species richness of the human gut microbiota have been greatly facilitated by the development of molecular techniques and tools based around the phylogenetic information encoded by the bacterial 16S rRNA gene (Tuohy and McCartney, 2006; Zoetendal et al., 2004). The gut microbiota species composition may be determined by directly isolating bacterial DNA from intestinal samples followed by separation and DNA sequencing of the 16S rRNA genes present. Upon phylogenetic positioning, these 16S rRNA gene sequences can be used to generate a picture of the relative abundance of different bacterial species or phylotypes present in the original environmental sample. Using this and similar approaches a detailed picture of the composition of the human gut microbiota has now emerged. Suau et al. (1999) showed that 95% of 16S rRNA gene sequences recovered from a single fecal sample of a healthy adult volunteer fell within one of three phylogenetic groupings: the Bacteroides group, the C. coccoides-E. rectale group, and the C. leptum group. The Clostridium groups, C. leptum and C. coccoides-E. rectale include many bacteria previously described as important members of the gut microbiota including species of Eubacterium, Ruminococcus, Butyrivibrio and Faecalibacterium prausnitzii. More recently the divisional structure of the gut microbiota at the phylum level has been described using a similar approach with the two most abundant bacterial phyla being the Gram negative Bacteroidetes and the Gram positive, low GC% Firmicutes (Eckburg et al., 2005; Louis et al., 2007; Wang et al., 2005). Seventy two per cent of the 395 phylotypes detected by Eckburg et al. (2005) belonged to the clostridial group of Firmicute bacteria, most in clusters XIVa (also referred to as the Clostridium coccoides group) or IV (Clostridium leptum) and the majority of these were either novel, or unrelated to species held in culture collections. Other important though less abundant phyla include the Proteobacteria, Actinobacteria, Fusobacteria and the Verrucomicrobia phyla (Eckburg et al., 2005; Wang et al., 2005). These lesser phyla none-the-less contain many bacteria important for human health and disease including the enterobacteria and bifidobacteria. Moreover, the bacterial community in the stomach and jejunum has been shown to differ from that in the distal ileum, ascending colon and rectum, with the major phylogenetic groups being similar in the distal ileum and rectum (Bik et al., 2006; Suau et al., 1999; Wang et al., 2005) also showed that a major proportion, up to 70% of the 16S rRNA species present in this fecal microbiota belonged to novel phylogenetic lineages all be it within the three dominant groupings. Only 24% of clones corresponded to previously identified bacterial phylotypes. More recently, Gill et al. (2006) using a direct high throughput sequencing of metagenomic DNA present in fecal samples collected from two American individuals found that 22.2% of phylotypes were novel and 83.3% of phylotypes corresponded to previously uncultured bacterial species. It is likely that as more metagenomic-level studies add 16S rRNA gene sequences onto the databases, the proportion of un-recognized phylotypes within an individual’s gut microbiota will fall considerably. However, as discussed above, full community phylogeny does not necessarily translate into an understanding of bacterial function or community function and other approaches are needed to link bacterial identity and ecological functioning.
3.6 3.6 Community Finger-Printing Techniques (e.g., DGGE)
Fingerprinting techniques have been used to study bacterial communities and appear to be ideal for monitoring community shifts and comparing communities between GI sites and among animals.
Denaturing gradient gel electrophoresis (DGGE) and similar techniques (e.g., temperature gradient gel electrophoresis, TGGE and temporal temperature gradient gel electrophoresis, TTGE) profile or finger-print of the gut microbiota by generating snap-shots of bacterial richness encoded by 16S rRNA gene fragments within environmental samples (Muyzer et al., 1993). Two additional microbial community fingerprinting techniques are single strand conformation polymorphism (SSCP) and terminal-restriction fragment length polymorphism (TRFLP) analyses. DGGE, TGGE, and TTGE are based on sequence-specific melting behavior of amplicons, SSCP on the secondary structure of single stranded DNA, and T-RFLP on specific target sites for restriction enzymes. Double stranded DNA (ds-DNA) melts into single-stranded DNA at different rates across a gradient of denaturant (e.g., urea, or temperature) depending on its DNA sequence. DGGE takes advantage of this differential ds-DNA melting to separate 16S rRNA gene fragments of equal length amplified from microbial communities across a denaturing urea and formamide gradient on a polyacrylamide gel. The resultant pattern of DNA bands on the DGGE gel constitutes a fingerprint of the different 16S rRNA gene fragments amplified from the original sample and thus the species make-up of the sampled microbial community. This approach generates a snap-shot that can then be used to assess changes in species richness over time or in response to different environmental stimuli, such as the presence of growth substrate, antibiotics or xenobiotics. The phylogenetic identity of differential bands can be determined following band excision and sequencing. It has been reported that DGGE or TGGE are sensitive enough to detect bacteria that constitute up to 1% of the total bacterial community. This means that only the most dominant bacteria will be represented. Theoretically, each DGGE band corresponds to a single operational taxonomic unit (OTU), where the total banding pattern is reflective of a community’s species richness and diversity (Muyzer et al., 1993). Earlier workers have excised and directly PCR-amplified and sequenced (Ampe et al., 1999; Ovreas et al., 1997) or PCR cloned and sequenced DGGE bands to successfully identify the taxonomic units of interest (Iwamoto et al., 2000; Zwart et al., 1998). Conversely, recent investigators (Abecia et al., 2007; Ercolini, 2004; Ercolini et al., 2003; Kowalchuk et al., 1997; van Beek and Priest, 2002) have reported that band excision and sequencing of DGGE bands might not provide unequivocal identifications as a result of the co-migration of DNA fragments from different taxa to the same positions within DGGE gels.
This approach has been employed to show that individuals harbor unique collections of intestinal bacteria, and that the species composition of the adult gut microbiota is remarkably stable over time (Zoetendal et al., 1998). DGGE has recently been employed to follow the compositional changes that occur within the human gut microbiota upon ingestion of prebiotic functional foods (Tannock et al., 2004); to characterize the mucosa associated microbiota in Crohn’s disease patients (Martinez-Medina et al., 2006); to monitor microbiota changes within the infant gut upon dietary intervention (Nielsen et al., 2007); to monitor the impact of antibiotics and probiotic intervention on the gut microbiota (Saarela et al., 2007; Yap et al., 2008) and has shown that the gut microbiota profile presented by the patient before pharmaceutical intervention can predict individual susceptibility to diarrhoea (De La Cochetière et al., 2008).
3.7 3.7 Limitations of PCR Based Techniques
Many of molecular microbiology tools rely on efficient extraction of community DNA, PCR and cloning to characterize community 16S rRNA. This approach has a number of limitations, and bias towards recovery of particular 16S rRNA species may be introduced at different stages. Firstly, the method used to isolate bacterial DNA from faeces or mucosal specimens may select for a certain bacteria based on cell wall conformations; Gram positive bacteria by and large are more difficult to disrupt than Gram negative species. PCR is a competitive reaction governed by melting and renaturation efficiencies of the target DNA sequences and therefore PCR based approaches may select for particular pools of 16S rRNA based on PCR amplification efficiency rather than the relative abundance of bacteria and their 16S rRNA genes in the test sample. High GC, Gram positive bacteria, such as the bifidobacteria, consequently are often under-represented in 16S rRNA gene libraries derived from competitive PCR reactions (Farris and Olson, 2007). Primer sequences to amplify bacterial DNA are based on highly conserved regions within the 16S rRNA gene. Even slight differences in the DNA sequence in these primer binding regions might result in bacteria not being detected and information on the highly conserved regions within the 16S rRNA gene derives only from already sequenced species. Finally, cloning of 16S rRNA gene fragments may introduce bias since different cloning vectors and host strains favor certain DNA sequences (Bonnet et al., 2002; Sipos et al., 2007). The limitations of these approaches must be recognized particularly when considering the relative abundance of bacteria within an ecosystem and methods which enable direct visualization and enumeration of phylogenetically related groups of bacteria within environmental samples may be more suited to this task. Hongoh et al. (2003) detected a significant increase in the expected number of phylotypes by lowering the annealing temperature, and a significant decrease in the proportion of clones belonging to the predominant group by raising the number of PCR cycles.
3.8 3.8 Molecular Characterization of the Gut Microbiota in situ
Fluorescence in situ hybridization (FISH) using 16S rRNA targeted oligonucleotide probes provides a direct means of enumerating specific bacterial populations in environmental samples without the need for microbiological culture and in a phylogenetically relevant manner (Amann et al., 1992). Here, bacterial cells are visualized either by epifluorescence microscopy or flow cytometry. The advantage of this approach is that phylogenetically related bacteria ranging from kingdom or phylum levels to species level, depending on probe design, may be enumerated directly in situ. This approach is now commonly applied to enumerate changes in relative abundance of bacteria within the human gut microbiota. As with other molecular approaches it is important to realize the limitations of FISH, which include: variability in permeabilization of target cells and the need for prior 16S rRNA gene information for probe design. FISH does however, allow the estimation of relative bacterial population levels within mixed microbial consortia without the bias introduced by DNA recovery and subsequent PCR and cloning (Amann and Fuchs, 2008). FISH gives an accurate picture of relative abundance of bacteria in mixed consortia because the bacteria are enumerated directly without selective amplification or culture (Amann and Fuchs, 2008). Table 3.1 shows the relative abundances of bacteria within the human gut microbiota as determined by FISH.
Use of flow cytometry allows fluorescence activated cell sorting of bacterial cells. Where 16S rRNA targeted FISH is employed to label bacteria, FACS can be used for the physical separation or sorting of microorganisms according to their phylogenetic groupings from mixed microbial consortia (Ben-Amor et al., 2005; Kalyuzhnaya et al., 2006; Lay et al., 2007). The cell sorted bacterial groups can then be subjected to other molecular procedures to identify bacteria enumerated by the particular FISH probe employed or to investigate the genetic potential of cell sorted bacteria following cloning and DNA sequencing in a culture independent manner. This approach has been used to identify the species make up of viable, injured and dead fractions in human faeces (Ben-Amor et al., 2005; Lay et al., 2007) also used this approach to monitor which bacterial species were enumerated by C. leptum group specific FISH probes by carrying out PCR-TTGE (a DGGE type method) on cell sorts of fecal bacteria labeled with the C. leptum probes (Clep866-CY5/cp or Fprau645-CY5). The C. leptum group, which include important butyrate producing bacteria, constitute one of the dominant groupings within the gut microbiota and covers many different phylogenetic clades. Since the majority of bacteria within the gut are unculturable such culture independent techniques which have the potential for separating out bacterial species without the need for microbiological culture may prove invaluable in assigning ecological function to these dominant and prevalent unculturable moieties.
The prebiotic concept, defined as “non-digestible (by the host) food ingredients that have a beneficial effect through their selective metabolism in the intestine” requires that certain dietary fibers are selectively metabolized by the gut microbiota leading to a concomitant improvement in host health (Gibson and Roberfroid, 1995). The FISH technique has recently been employed to demonstrate that the ability of various foodstuffs and ingredients to bring about a change within the relative abundance of gut bacterial populations seen as beneficial towards human health. The fructans, inulin and oligofructose, galactooligosaccharides, lactulose, resistant starch, whole grain wheat have all been shown to elicit a significant relative increase in numbers of bifidobacteria within the human gastrointestinal tract upon dietary intervention using FISH (Costabile et al., 2008; Depeint et al., 2008). Indeed, true prebiotics can reproducibly be shown to bring about a selective modulation of the gut microbiota, whereby relative abundance of what are termed the beneficial microbiota (currently including bifidobacteria and/or lactobacilli) using either FISH or more traditional culture based approaches. In parallel, as outlined elsewhere in this volume, there is a growing body of evidence from both animal and human studies that prebiotics can bring about improvements in mineral absorption and bone health, improved immune function and relief of allergic and inflammatory disease; protection from gastrointestinal infections including infectious diarrhea and travellers diarrhea; reduced risk of colon cancer and through modulation of blood lipid profiles and energy metabolism, reduce the risk of the metabolic syndrome, type 2 diabetes and coronary vascular disease. However, there is a lack of mechanistic data linking changes within the gut microbiota with biological mechanisms underpinning the observed health effects. The challenge for the prebiotics field of research in the coming years is to bridge this gap and link prebiotic induced modulations within the gut microbiota with specific physiological processes responsible for improved health or protection from disease.
3.9 3.9 Culture-Independent Functional Characterization of the Gut Microbiota’s Metabolic Potential
The 16S rRNA based molecular tools mentioned above have contributed to a picture of the gut microbiota as being made up of mainly unculturable bacteria, with many novel species. This approach has also contributed greatly to the extensive 16S rRNA gene database used for bacterial phylogeny and taxonomy and has proved invaluable in characterizing species make up and composition of the gut microbiota. However, knowing the phylogeny of a microorganism may not necessarily shed light on its ecological function. These issues highlight an important problem, how to assign ecological function to novel or unculturable bacteria? Recent advances in high throughput sequencing allow the rapid genome sequencing and more over rapid and accurate compilation of community metagenomes (Committee on Metagenomics, 2007). Sequence based metagenomics applies shot-gun sequencing of total DNA extracted from an environmental sample and was originally pioneered in marine and terrestrial environments (Gill et al., 2006; Kurokawa et al., 2007; Turnbaugh et al., 2007). This approach has also recently been applied to study the human gut microbiota, e.g., the Human Microbiome Project described above. The functional or metabolic potential of the human gut microbiota may thus be accessed through these large scale sequencing projects targeting not just community 16S rRNA genes, but potentially all bacterial genes present.
Gill et al. (2006) examined the fecal metagenome of two unrelated healthy American adults. They found that these metagenomes were enriched for genes involved in the metabolism of carbohydrates, amino acids and xenobiotics, methanogenesis and the biosynthesis of vitamins and isoprenoids, compared to the human genome itself and other bacterial communities. They also pointed out that the fecal metagenome encoded many functions not represented in the human genome, in particular, genes involved in the metabolism of major plant structural and storage polysaccharides such as xylans, pectins, arabino-containing carbohydrates and fructans. The abundance of such genes within the gut microbiome and their rarity within the human genome is of key importance to the overall metabolic capacity encoded in the combined human-microbiota metagenome. Some 81 different glycosyl hydrolase families as well as key genes involved in the production of short chain fatty acids (SCFA, acetate, butyrate, propionate and succinate) could be assigned to the gut microbiota highlighting the fact that carbohydrate fermentation appears to be a major energy source in the colon, driving the energy economy within the microbiota, building cross-feeding interrelationships and providing about 50% of the daily energy requirements of the gut wall. In a second metagenomics study, Kurokawa et al. (2007) compared the metagenomes of 13 healthy Japanese individuals of different age, with two American fecal metagenomes (Gill et al., 2006). They found that genes involved in carbohydrate metabolism and transport were enriched in all 13 metagenomes, compared to a database constructed from COG (Cluster of Orthologous Groups) assigned genome sequences of 243 different bacteria not commonly found within human gut microbiota. Genes involved in lipid metabolism however, were under-represented in the gut metagenomes compared to this reference database. These observations support the notion that the human gut microbiota has evolved along-side its human host to complement human encoded functions and allows the host access to dietary nutrients not digested or absorbed in the upper gut like complex plant storage polysaccharides. SCFA derived from microbial fermentation in the colon contribute about 10% of our daily energy intake, and play important physiological roles including regulating cellular differentiation and proliferation in the gut wall, cholesterol synthesis and de novo lipogenesis in the liver, and may act as an energy source systemically (Macfarlane and Gibson, 1997). Similarly, 90% of plant derived polyphenolic compounds reach the colon where they are transformed into biologically active and available intermediates by the resident microbiota (Spencer et al., 2001). This interaction between our intestinal microbiota and plant foods, both carbohydrate and polyphenolic moieties has important implications for human health and disease and in devising optimal human diet as well as functional food design. For much of our evolutionary history, humans followed a hunter-gatherer life strategy and consumed diets rich in whole plant foods particularly high in dietary fiber and polyphenols. Whole plant foods (fruits, vegetables, whole grain cereals) and dietary fiber are recognized as beneficial for human health and in epidemiological studies these foods are often inversely related to risk of chronic human diseases including coronary vascular disease and cancer. Both for present day humans and for our prehistoric ancestors the maintenance of a stable, fermentative gut microbiota would have been essential to maximize energy and nutrient recovery as well as non-nutrient functional benefit from our diet (Cassidy, 2006; Pompei et al., 2007). However, our modern, Western style diets, low in fiber and whole plant foods, and high in refined sugars, protein and saturated fat, appear to be out of step with our co-evolved hunter-gatherer intestinal microbiota. It is not surprising that there appears to be a strong link between adoption of this Western style diet and increased incidence of certain cancers, coronary vascular disease, obesity and the metabolic syndrome, and immunological diseases, like inflammatory bowel disease and allergy, in which the gut microbiota have been proposed to play an aetiological role and for which certain whole plant foods are proving protective. Modulating our modern gut microbiota through dietary supplementation with Prebiotics in particular can mediate positive health effects on a number of important physiological functions including de novo lipogenesis Beylot (2005), mineral absorption (Abrams et al., 2005; Holloway et al., 2007), regulation of satiety and body fat deposition (Delzenne et al., 2007; So et al., 2007) and importantly, providing butyrate as an energy supply to the colonic mucosa (Pool-Zobel and Sauer, 2007).
One of the more surprising insights provided by recent metagonomic studies has been the observation that the gut microbiota may be involved in the aetiology of obesity and that the composition of the gut microbiota is fundamentally different in the obese compared to the lean individuals at the phylum level. There is currently an epidemic of obesity which appears to develop at the population level upon adoption of a Western-style diet. In the UK there has been a sharp increase in the incidence of obesity over the past 15 years and it has been estimated that by 2,050 60% men in the UK will be obese Foresight (2007). There is a genetic component to obesity with over 600 genes reported to play a role in energy metabolism and body weight, and there is a sub-population genetically predisposed to obesity (Wardle et al., 2008). However, the sudden increase in the incidence of obesity since the 1980s has occurred at a rate which far outstrips that of human genomic evolution, showing that the obesogenic environment has a major role to play. This obesogenic environment impacts on the quantity of food we eat, the types of foods we eat, satiety, energy recovery from the diet, epigenetic programming, mental state and exercise, all of which play important roles in determining the risk of obesity. There are strong epidemiological data linking diets high in fat and refined carbohydrates with obesity (Johnson et al., 2008). Conversely, diets rich in fiber and whole plant foods are inversely associated with obesity (Astrup et al., 2008). Gordon and co-workers have recently shown that the gut microbiota differs between obese and lean individuals at the phylum level in both animal models of obesity and in human subjects (Ley et al., 2005, 2006; Turnbaugh et al., 2006). These authors report that obesity is associated with a reduced abundance of intestinal Bacteroidetes and increased abundance of Firmicutes in the genetic ob/ob mouse model of obesity and in humans. This altered microbiota is associated with increased fermentation end products in caecal and fecal samples, and differences in the metabolic potential of the gut microbiota with an increased energy harvest from the diet (Ley et al., 2005, 2006). Interestingly, obesity appears to be transferred concomitantly with this obese type microbiota and when germ-free animals are associated with caecal contents of obese mice they too become obese despite despite eating less food (Turnbaugh et al., 2006, 2008). However, it also appears that diet has a strong influence on the composition and activity of the obese-associated microbiota. It appears that the gut microbiota of obese humans who lose weight after one year on either a low carbohydrate or a low fat diet returns to a lean type profile (Ley et al., 2006). Similarly, there is some evidence from animal studies that certain fibers, particularly prebiotics may reduce the risk of obesity itself and its associated pathologies. Cani et al. (2007a) showed that when conventional mice are placed on a high fat diet obesity and insulin resistance may be induced by increased plasma levels of the highly inflammatory Gram negative bacterial wall fragment, lipopolysaccharide. They observed a die-off in the intestinal microbiota in high fat fed animals. This high-fat diet with low fermentable carbohydrate may have contributed towards increased availability or though depleted mucosal barrier function have contributed to increased uptake of intestinal LPS thus providing the immunological trigger for the chronic low grade inflammation characteristic of obesity and predisposing to insulin resistance. The same authors later found that dietary supplementation with the prebiotic oligofructose, in these same high-fat fed animals, resulted in reduced plasma LPS, reduced inflammation and improved insulin sensitivity. These physiological changes were strongly associated with an increased abundance of intestinal bifidobacteria in these animals (Cani et al., 2007b). Further studies are required to investigate in more depth any apparent aetiological role of the obese-type microbiota in body weight gain and conversely, in the ability of different diets to induce an obese-type microbiota or reduce the risk of becoming obese by modulating gut microbiota composition and activity through dietary interventions for example with prebiotics.
The sequence based metagonomics is a powerful tool for measuring the metabolic potential of the gut microbiota and recent high level investment in metagenomic studies of the gut microbiota will generate a wealth of sequence data for data-mining and the design of future gut microbial ecology studies. Identifying the genetically encoded functions which are enriched or under-represented within a microbial community can shed light on the ecological role of that community as a whole (Turnbaugh et al., 2007). Additionally, identifying ecologically sensitive genes or sets of genes which enable a particular strain to successfully colonize an ecological niche within the gut will greatly enrich our understanding of how bacteria interact with each other, our diet and their human host to mediate either beneficial health effects or induce disease. A good place to start may be to characterize the composition and commonality of ecologically important genes constituting the gut microbiota mobile metagenome, encoded by transmissible plasmids, transposons and bacteriophages, which is becoming recognized as an important determinant of strain identity and ecological function within the gut (Lee et al., 2008). However, as observed by Kurokawa et al. (2007) not all open reading frames (ORFs) identified in metagenomic studies can be annotated to known function by comparisons with reference databases. In the existing gut microbiota metagenomes between 45% and 80% of protein-coding genes observed in the 13 Japanese (Kurokawa et al., 2007) and two American (Gill et al., 2006) fecal metagenomes respectively could not be assigned a metabolic function at the 90% threshold identity upon BLASTP analysis against their reference database of 243 non-gut bacterial genomes. Another limitation of the metagonomic approach is that it generates data on the metabolic potential of a microbial ecosystem, the potential encoded by the genotype of the organisms present which is only translated into metabolic kinetics or phenotype in response to particular environmental stimuli. Another post-genomics approach has recently emerged with the potential to measure the metabolic kinetics or community phenotype of the gut microbiota and possibly relate these changes in microbial derived metabolites to particular intestinal bacteria irrespective of whether they can be cultured under laboratory conditions or whether they are new to science.
3.10 3.10 Measuring the Metabolic Kinetics of the Human Gut Microbiota Through Metabonomics
Metabonomics is defined as “a systems approach to examining the changes in hundreds or thousands of low-molecular-weight metabolites in an intact tissue or biofluid” (Nicholson et al., 2005). The human metabonome thus comprises; the metabolites derived from human genome encoded determinants, metabolites of derived from the human microbiota, and the flux in these combined metabolite profiles under different environmental perturbations e.g., interactions with diet, pharmaceuticals, carriage of parasites, and chronic disorders like cardiovascular disease and cancer. This technology employs 1H-nuclear magnetic resonance (NMR) spectrometry and mass spectrometry-based techniques to profile metabolites in biofluids like urine, plasma and fecal water, generating a picture of the metabolic kinetics of an organism at a particular point in time. By applying image analysis followed by multivariate statistics, metabonomics is currently being used to track changes in these metabolite profiles in response to dietary modulation, including prebiotic, or pharmaceutical interventions, and to generate distinctive metabolite profiles in disease states such as inflammatory bowel disease, bowel cancer and cardiovascular disease. Such studies offer the possibility of developing new diagnostic tools or novel therapeutic targets and importantly deliver tools with the breath and resolution to derive functional data on the behavior of the gut microbiota in situ and in response to dietary change (Marchesi et al., 2007; Martin et al., 2008; Holmes et al., 2008; Solanky et al., 2005). The metagenomic studies described above illustrate clearly the degree of co-operation between the human genome and the gut microbiota and that the gut microbiota has co-evolved alongside mammals over time (Ley et al., 2008; Nicholson et al., 2005) also put forward the concept of the gut microbiota co-evolving and metabolically complementing the human genome resulting in a close symbiotic relationship involved in co-metabolism of a range of dietary and xenobiotic compounds. These authors suggested that gut microbiota structure and composition reflects this symbiotic relationship with only the bacterial populations beneficial to the host predominating in the human microbiota in health and contributing to co-metabolic activities. Of course this symbiotic relationship is impacted by other life-style influences such as diet, exercise, stress and drug or xenobiotic intake. Recognizing the contribution of microbiota derived compounds observable in metabolite profiles of human biofluids to metabolic processes at the whole organism level, Nicholson et al. (2005) combined high resolution analytical techniques with image analysis and multivariate statistics to establish the metabonomics concept.
Recent metabonomics studies have highlighted the potential contribution of microbiotal derived metabolites or co-metabolites in the aetiology of chronic human diseases. Dumas et al. (2006) found that in mice genetically predisposed to impaired glucose homeostasis and non-alcoholic fatty liver disease (NAFLD) maintained on a high fat diet, microbial activities within the gut lead to reduced choline bioavailability. This mimics choline-deficient diets already known to induce NAFLD and insulin resistance (IR), key initial steps in the development of the metabolic syndrome and obesity. 1H-NMR metabolite profiling using principle component analysis was able to identify low circulating plasma phosphatidylcholine and high urinary excretion levels of methylamines (dimethylamine, trimethylamine and trimethylamine-N-oxide), gut microbe:host co-metabolites, as characteristic of high-fat fed animals with NAFLD and impaired glucose metabolism. Holmes et al. (2008) later illustrated the power of the metabonomics approach in grouping individuals according to their urine metabolite profiles at the population level, and relating these profiles to geography, diet and disease risk. These authors showed that urinary formate (and, to a lesser extent, hippurate) was inversely related to blood pressure (BP) and coronary vascular disease (CVD) risk. Again 1H-NMR was used to generate urinary metabolite profiles of 4,630 human volunteers in the UK, the USA, China and Japan. Individuals could be separated in a blind manner according to geography with East Asians separating from UK/USA populations; Japanese living in Japan separating from Japanese living in the USA, and populations of northern (Guangxi) and Southern (Beijing and Shanxi) China showing distinct urinary metabolite profiles. The authors reported that urinary profiles of people in the UK and USA were similar. The main differentiating metabolites between the populations were of dietary origin, including amino acids, creatine and trimethylamine-N-oxide; acetylcarnitine, tricarboxylic acid cycle intermediates involved in energy metabolism and dicarboxylic acids like suberate. Holmes et al. (2008) also identified a group of microbial compounds or which derive from the gut microbiota:host co-metabolic processing including hippurate, phenylacetylglutamine, methylamines and formate. Formate can either be formed from endogenous one-carbon metabolism or upon fermentation of non-digestible carbohydrates in the colon by diverse bacteria including certain clostridia (mainly belonging to clusters XVI, IV, XIV), the Actinobacteria including Bifidobacterium species, and to a lesser extent, the Proteobacteria, see Table 3.1 . Hippurate, an end product of aromatic (benzoic acid) co-metabolism and a likely end product of polyphenolic metabolism by the host and gut microbiota, was also inversely related to BP and positively correlated with fiber intake, while high BP was associated with diets high in animal protein and the urinary metabolite, aniline (Table 3.2 ).
The same group have also recently shown that the close relationship between mammals and their intestinal microbiota extends through-out the life-span. Distinct 1H-HMR urinary metabolite profiles were found in dogs in response to dietary change (a calorie restricted diet compared to normal chow) and at different ages in this longitudinal study. Urinary metabolite profiles shifted rapidly before age 1 year (early life) after which the metabolic signature stabilized between 1 and 2 years of age. This change in metabolite profiles in to first 12 months of life corresponds to the emergence and successive development of the gut microbiota and the dietary change upon weaning. A second metabolic shift was observed in middle-age (years 5–9) before profiles again underwent a metabolic transformation in old age after about 10 years. Many of the differentiative metabolites had their origins in the microbiota:host co-metabolism highlighting the role of the gut microbiota in the ageing process, from successive development of the gut microbiota in puppies to modulation of the gut microbiota in middle and old age, times often associated with the onset of chronic disease (Wang et al., 2007).
3.11 3.11 Metabonomics and Disease States (IBD and Colon Cancer)
Metabonomics has been applied to investigate changes in metabolic profiles in order to identify mechanisms involved in certain diseases. Metabonomic analysis of either fecal extracts (Marchesi et al., 2007) or colonic mucosa (Balasubramanian et al., 2008) in patients with active inflammatory bowel disease (IBD) both showed a reduction in SCFA in these patients compared to control individuals, in particular in acetate but also in butyrate. Balasubramanian et al. found however, that in patients in remission the values for these metabolites were similar to control. They also reported that the concentration of formate was significantly lower in patients with active ulcerative colitis (UC) compared to patients with active Crohn’s disease (CD) and that this difference may serve as a biomarker for the distinction between active UC and CD. It is important to accurately diagnose IBD at an early stage as a correct differentiation between CD and UC defines treatment and prognosis.
The above metabolites differentiating UC and CD include microbial metabolites originating from the fermentation of carbohydrates by the gut microbiota highlighting the importance of the microbiota in the aetiology of IBD. Similar findings have been observed in studies looking at blood metabolites in different mouse models of colitis (Chen et al., 2008). The metabonomics analysis revealed increased levels of stearoyl lysophosphatidylcholine and lower levels of oleoyl lysophosphatidylcholine in blood which the authors traced to an inhibition of stearoyl-CoA desaturase 1 (SCD1) expression in the liver. As this inhibition did not only occur in a dextran sulphate sodium (DSS)-induced colitis model but also in Citrobacter rodentium-induced colitis Chen et al. concluded that the observed inhibition of SCD1 is highly likely to be due to the disruption of the intestinal microbiota and the resulting inflammation. Furthermore Marchesi et al. reported higher quantities of amino acids lysine, leucine, isoleucine, valine and alanine – products of bacterial protein metabolism – in faeces of CD patients compared to controls.
Although these studies identified metabolites of microbial origin as playing important physiological roles at the whole body or system level, they did not attempt to link these metabolite profiles with specific bacteria within the gut microbiota.
A study in colon cancer and polypectomized patients attempted to attribute changes in metabolic profiles to changes in bacterial diversity by combining DGGE and metabonomics analysis of fecal water (Scanlan et al., 2008). The authors reported a significantly increased diversity in the Clostridium leptum and the Clostridium coccoides subgroups as well as relatively higher levels of amino acids such as valine, leucine, isoleucine, glutamate and tyrosine and lower levels of methylamine in fecal water of colon cancer and polypectomized patients compared to control individuals. The altered amino acid profile together with the increased diversity may suggest a higher incidence of potentially detrimental species of clostridia.
The modulating effect of microorganisms on systemic metabolite profiles (blood, jejunal wall and longitudinal mysenteric muscle tissue) was also confirmed by infection with Trichinella spiralis in NIH Swiss mice which subsequently caused post-infective irritable bowel syndrome (IBS) – an intestinal disorder characterized by abdominal pain, vomiting and either diarrhoea or constipation (Martin et al., 2006). The metabonomic signature of the T. spiralis-infected mice revealed an increased energy metabolism, fat mobilisation and a disruption of amino acid metabolism as well as muscular hypertrophy. The treatment of the infected mice with probiotic Lactobacillus paracasei resulted in metabolic profiles closer to those of uninfected mice indicating a partial normalisation of the muscular activity and the disordered energy metabolism.
3.12 3.12 Measuring the Impact of Microbiota Modulation Using Metabonomics
As described above, the gut microbiota interacts intimately with host metabolism to mediate health and disease. These interactions are complex, often involving multiple and interconnected metabolic pathways, which makes a classical approach whereby one or a few metabolites are monitored impractical for investigating microbe:host metabolic interactions. Recently, metabonomics has been employed to measure the consequences of microbiota modulation. Yap et al. (2008) investigated the impact of the broad spectrum glycopeptide antibiotic, vancomycin, on the gut microbiota and the metabonome of female mice. Vancomycin was chosen because it is active against Gram positive bacteria and is poorly absorbed across the gut wall which means it will reach the large bowel. Vancomycin induced changes in the composition of the gut microbiota as determined by 16S rRNA targeted PCR-DGGE was reflected in changes in metabolite profiles in faeces and urine. Vancomycin intervention had a dramatic impact on phenolic regions of the NMR spectrum, with reduced levels of urinary hippurate and phenylacetylglycine which are produced through microbiota:host co-metabolic pathways. Although these changes in urine metabolites appeared to be transitory, particular metabolites took longer to return to pre-treatment levels, with hippurate in particular only returning to pre-vancomycin levels 19 days after the vancomycin intervention. Microbiotal choline metabolism also appeared to be disrupted by vancomycin treatment. Reduced concentrations of trimethylamine (TMA) and trimethylamine-N-oxide (TMAO), gut microbial and hepatic detoxification end products of choline metabolism respectively, were observed in urine post-vancomycin treatment. Vancomycin intervention had a dramatic effect on carbohydrate fermentation, a key functional activity of the gut microbiota, with reduced levels of acetate, propionate and n-butyrate and elevated fecal oligosaccharide concentrations. Considering the important and diverse biological roles of these SCFA in the host, antibiotic disruption of this key microbiotal function may have a significant impact on host health. Such an impact on carbohydrate fermentation and SCFA production may have been expected considering the key roles played by Gram positive bacteria like the bifidobacteria and species belonging to the C. leptum and C. coccoides groups in polysaccharide and oligosaccharide fermentation and in cross-feeding on SCFA. Interestingly, although alterations in fermentation end products lasted 13 days post-vancomycin treatment, populations of C. leptum and C. coccoides appeared to recover from day 2 onwards, indicating that disruption of other bacterial groups present may have had a more dramatic impact on SCFA production than bacteria belonging to these clostridial groupings. Vancomycin intervention also disrupted protein handling by the gut microbiota with elevated levels of amino acids in faeces and reduced levels of creatine and α-ketoisocaproate in urine.
Using a defined microbiota animal model of the infant gut microbiota, Martin et al. (2008) recently described the impact of probiotics (Lactobacillus paracasei and L. rhamnosus) and prebiotics (two different GOS preparations) on the gut microbiota and host metabonome. This simplified animal model of the human infant gut microbota comprised ex-germ-free animals colonized with strains of E. coli, B. breve, B. longum, Staphylococcus epidermis, S. aureus, C. perfringens and Bacteroides distasonis isolated from a healthy twenty day old breast fed human infant. The authors showed distinct metabolite profiles in urine, plasma, fecal extracts and intact liver tissue upon prebiotic induced microbiota modulation using NMR based metabonomics. Supplementation with either prebiotic resulted in elevated population levels of the bifidobacterial strains present, B. breve and B. longum and reduced levels of C. perfringens. When given in combination with L. paracasei, reduced numbers of Bacteroides distasonis were observed, while reduced numbers of fecal E. coli were observed in animals dosed with prebiotic and L. rhamnosus. Prebiotic treatment appeared to reduce bacterial proteolysis, with lower concentrations of lysine observed in faeces, isobutyrate in the caecum and N-acetyl-glycoproteins in urine. Changes were also observed in choline metabolism upon prebiotic intervention. Co-metabolic processes in the metabolism of dietary choline have been previously shown to impact on insulin resistance, non-alcoholic fatty liver disease and type 2 diabetes in animal models. Both TMA and TMAO concentrations were altered in the urine and liver respectively, indicating prebiotic induced changes in choline metabolism by the gut microbiota. Prebiotic intervention impacted significantly on levels of lipids stored in the liver, with reduced triglycerides and increased concentrations of polyunsaturated fatty acids. Similarly, Prebiotic intervention resulted in increased hepatic glutamate, gutamine, branched-chain amine acids and alanine, and when mice were dosed with prebiotic plus L. paracasei, increased hepatic glycogen was observed indicating a stimulation of gluconeogenesis and glycogenesis. Prebiotic intervention also appeared to stimulate animal energy expenditure as indicated by increased levels of taurine and creatine in urine post-prebiotic supplementation which derive from increased muscular activity. Changes in SCFA within the gastrointestinal tract were measured by gas chromatography and correlated with prebiotic induced alterations in microbiota population levels. In general there was a negative association between SCFA concentrations and population levels of C. perfringens and E. coli and a positive association between SCFA and bifidobacteria, the lactobacilli and Bacteroides diastasonis, corresponding with perceived prebiotic modes of action within the gut microbiota. However, it is difficult to extrapolate these data to the human situation since there are a number of major differences between these model microbiota systems and humans. Principally, in the lack of microbiota complexity, differences in gut physiology (rodents are copiophagious, and their upper gut are colonized by large populations of bacteria unlike healthy humans) and bacterial species may have different biological roles in different animals.
Recently, Li et al. (2008) in an attempt facilitate direct human microbial ecology studies at the “omics” level, combined both metagenomics (16S rRNA targeted PCR-DGGE) and metabonomics (metabolite profiling by NMR) to generate a matrix of differential urinary metabolites and unique bacterial genotypes present in fecal samples collected from four generations of a single Chinese family. These authors were thus able to correlate individual bacterial species identified within the fecal microbiota of the human volunteers by 16S rRNA gene targeted PCR-DGGE with particular profiles of metabolites present in urine. This powerful approach offers for the first time a real insight into the in vivo functioning of even unculturable and previously uncharacterized members of the gut microbiota directly without the need for bacterial cultivation and in biological samples which can be collected in a non-invasive manner.
3.13 3.13 Conclusion
Recent insights into the composition and make up of the human gut microbiota and the evolution of powerful and high resolution data rich analytical techniques are revolutionizing the way we view the human intestinal microbiota. It is clear that our resident microbiota, which has co-evolved with us over the millennia, impacts on a range of human metabolic processes and appears to be particularly effected by recent population level changes in human diet, particularly the reduction in fiber and whole plant food ingestion and adoption of the Western-style diet which as occurred with growing affluence over the past 50 years. Nowhere is this more clearly illustrated than in the fact that the gut microbiota of the obese people appears to be different to lean people and that this obese-type microbiota returns to a lean type profile upon weight loss. The application of metagenomics approaches, particularly the recent Human Microbiome Project which aims to genome sequence up to 1,000 gastrointestinal bacteria as well as directly sequence functional communities in different body sites will provide a valuable encyclopaedia of genetic information mapping out the metabolic potential of bacteria residing on or in the human body. Metabonomics, on the other hand offers the possibility of tracking changes in metabolite profiles at the systems level allowing direct measurement of metabolic kinetic or metabolite flux over experimental time courses such as before and after dietary intervention or in the presence or absence of disease. A key recent development has been the combining of these two approaches (Li et al., 2008) offering a powerful tool for direct study of the human gut microbiota in vivo and upon dietary modulating. These omics based approaches are thus providing tools of sufficient resolution to allow researchers to realistically address one of the most fundamental and tantalizing questions in the area of functional foods research, “how do probiotics and prebiotics really work.”
References
Abdellah Z, Ahmadi A, Ahmed S, Aimable M, Ainscough R, Almeida J et al. (2004) International human genome sequencing consortium. Nature 409:860–921
Abecia L, Fondevila M, Balcells J, Edwards JE, Newbold CJ McEwan NR (2007) Effect of antibiotics on the bacterial population of the rabbit caecum FEMS Microbiol. Lett 272:144–153
Abrams SA, Griffin IJ, Hawthorne KM, Liang L, Gunn SK, Darlington G et al. (2005) A combination of prebiotic short- and long-chain inulin-type fructans enhances calcium absorption and bone mineralization in young adolescents. Am J Clin Nutr 82:471–476
Amann R, Fuchs BM (2008) Single-cell identification in microbial communities by improved fluorescence in situ hybridization techniques. Nat Rev Microbiol 6:339–348
Amann RI, Zarda B, Stahl DA, Schleifer KH (1992) Identification of individual prokaryotic cells by using enzyme-labeled, rRNA-targeted oligonucleotide probes. Appl Environ Microbiol 58:3007–3011
Ampe F, ben Omar N, Moizan C, Wacher C, Guyot JP (1999) Polyphasic study of the spatial distribution of microorganisms in Mexican pozol, a fermented maize dough, demonstrates the need for cultivation-independent methods to investigate traditional fermentations. Appl Environ Microbiol 65:5464–5473
Astrup A, Dyerberg J, Selleck M, Stender S (2008) Nutrition transition and its relationship to the development of obesity and related chronic diseases. Obes Rev 9:Suppl 1:48–52
Balasubramanian K, Kumar S, Singh RR, Sharma U, Ahuja V, Makharia GK, Jagannathan NR (2008) Metabolism of the colonic mucosa in patients with inflammatory bowel diseases: an in vitro proton magnetic resonance spectroscopy study. Magnon Reson Imaging 2. [Epub ahead of print]
Ben-Amor K, Heilig H, Smidt H, Vaughan EE, Abee T, de Vos WM (2005) Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis. Appl Environ Microbiol 71:4679–4689
Beylot M (2005) Effects of inulin-type fructans on lipid metabolism in man and in animal models. Br J Nutr 93:S163–S168
Bik EM, Eckburg PB, Gill SR, Nelson KE, Purdom EA, Francois F et al. (2006) Molecular analysis of the bacterial microbiota in the human stomach. Proc Natl Acad Sci USA 103:732–737
Blaut M, Collins MD, Welling GW, Doré J, van Loo J, de Vos W (2002) Molecular biological methods for studying the gut microbiota: the EU human gut flora project. Br J Nutr 87(Suppl. 2):S203–S211
Bonnet R, Suau A, Doré J, Gibson GR, Collins MD (2002) Differences in rDNA libraries of faecal bacteria derived from 10- and 25-cycle PCRs. Int J Syst Evol Microbiol 52:757–763
British Nutrition Foundation (2004) http://www.nutrition.org.uk/home.asp?siteId=43§ionId=609&parentSection=324&which=1
Campbell TC, Chen J (1994) Diet and chronic degenerative diseases: perspectives from China. Am J Clin Nutr 59:1153S–1161S
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, FavaF, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R (2007a) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56(7):1761–1772
Cani PD, Neyrinck AM, Fava F, Knauf C, Burcelin RG, Tuohy KM et al. (2007b) Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 50:2374–2383
Cassidy A (2006) Factors affecting the bioavailability of soy isoflavones in humans. J AOAC Int 89:1182–1188
Chen K, Pachter L (2005) Bioinformatics for whole-genome shotgun sequencing of microbial communities. PLoS Comp Biol 1:24
Chen C, Shah YM, Morimura K, Krausz KW, Miyazaki M, Richardson TA, Morgan ET, Ntambi JM, Idle JR, Gonzalez FJ (2008) Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab 7:135–147
Committee on Metagenomics (2007) Challenges and Functional Applications The new science of metagenomics: revealing the secrets of our microbial planet. National Research Council, The National Academies Press, Washington, DC, USA http://www.nap.edu/catalog/11902.html accessed on 27 March 2007.
Conway PL (1995) Human colonic bacteria: role in nutrition. In: Gibson GR, Macfarlane GT (eds) Physiology and Pathology, CRC Press, Boca Raton, FL, pp. 1–18
Costabile A, Klinder A, Fava F, Napolitano A, Fogliano V, Leonard C et al. (2008) Whole-grain wheat breakfast cereal has a prebiotic effect on the human gut microbiota: a double-blind, placebo-controlled, crossover study. Br J Nutr 99:110–120
De La Cochetière MF, Durand T, Lalande V, Petit JC, Potel G, Beaugerie L (2008) Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb Ecol 56:395–402
Delzenne NM, Cani PD, Neyrinck AM (2007) Modulation of glucagon-like peptide 1 and energy metabolism by inulin and oligofructose: experimental data. J Nutr 137:2547S–2551S
Depeint F, Tzortzis G, Vulevic J, I’anson K, Gibson GR (2008) Prebiotic evaluation of a novel galactooligosaccharide mixture produced by the enzymatic activity of Bifidobacterium bifidum NCIMB 41171: in healthy humans: a randomized, double-blind, crossover, placebo-controlled intervention study. Am J Clin Nutr 87:785–789
Desiere F (2004) Towards a systems biology understanding of human health: interplay between genotype, environment and nutrition. Biotechnol Annu Rev 10:51–84
Duncan SH, Louis P, Flint HJ (2004) Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl Environ Microbiol 70:5810–5817
Dumas ME, Barton RH, Toye A, Cloarec O, Blancher C, Rothwell A et al. (2006) Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci USA 103:12511–12516
Dunitz M (1983) In: Bukitt D (ed) Don’t forget fiber in your diet. Singapore. Arco, p. 32
Eaton SB, Eaton SB III, Konner MJ (1997) Paleolithic nutrition revisited: a twelve year retrospective. Euro J Clin Nut 51:207–216
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M et al. (2005) Diversity of the human intestinal microbial flora. Science 308:1635–1638
Ercolini D (2004) PCR-DGGE fingerprinting: novel strategies for detection of microbes in food. J Microbiol Methods 56:297–314
Ercolini D, Hill PJ, Dodd CE (2003) Bacterial community structure and location in Stilton cheese. Appl Environ Microbiol 69:3540–3548
Farris MH, Olson JB (2007) Detection of Actinobacteria cultivated from environmental samples reveals bias in universal primers. Lett Appl Microbiol 45:376–381
Frank DN, Pace NR (2008) Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol 24:4–10
“Forsight: tackling obesity” document www.foresight.gov.uk (2007)
Flint HJ (2006) In: Logan NA, Lappin-Scott HM, Oyston PCF (eds) Prokaryote diversity: mechanisms and significance. The significance of prokaryote diversity in the human gastrointestinal tract. SGM Symposium, UK, pp. 65–90
Goldberg SM, Johnson J, Busam D, Feldblyum T, Ferriera S, Friedman R et al. (2006) A Sanger/pyrosequencing hybrid approach for the generation of high-quality draft assemblies of marine microbial genomes. Proc Natl Acad Sci USA 103:11240–11245
Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312:1355–1359
Gibson GR, Roberfroid MB (1995) Dietary modulation of the human colonic microbiota: introducing the concept of prebiotics. J Nutr 125:1401–1412
Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685
Hayashi H, Takahashi R, Nishi T, Sakamoto M, Benno Y (2005) Molecular analysis of jejunal, ileal, caecal and recto-sigmoidal human colonic microbiota using 16S rRNA gene libraries and terminal restriction fragment length polymorphism. J Med Microbiol 54:1093–1101
Hongoh Y, Yuzawa H, Ohkuma M, Kudo T (2003) Evaluation of primers and PCR conditions for the analysis of 16S rRNA genes from a natural environment. FEMS Microbiol Lett 221(2):299–304
Holloway L, Moynihan S, Abrams SA, Kent K, Hsu AR, Friedlander AL (2007) Effects of oligofructose-enriched inulin on intestinal absorption of calcium and magnesium and bone turnover markers in postmenopausal women. Br J Nutr 97:365–372
Holmes E, Loo RL, Stamler J, Bictash M, Yap IK, Chan Q et al. (2008) Human metabolic phenotype diversity and its association with diet and blood pressure. Nature 453:396–400
Institute of Medicine (2002) Dietary reference intakes. energy, carbohydrate, fiber, fat, fatty acids, cholesterol, protein, and amino acids. Washington, DC: National Academy Press
Iwamoto T, Tani K, Nakamura K, Suzuki Y, Kitagawa M, Eguchi M, Nasu M (2000) Monitoring impact of in situ biostimulation treatment on groundwater bacterial community by DGGE. FEMS Microbiol Ecol 32:129–141
Johnson IT, Belshaw NJ (2008) Environment, diet and CpG island methylation: epigenetic signals in gastrointestinal neoplasia. Food Chem Toxicol 46:1346–1359
Johnson L, Mander AP, Jones LR, Emmett PM, Jebb SA (2008) Energy-dense, low-fiber, high-fat dietary pattern is associated with increased fatness in childhood. Am J Clin Nutr 87:846–854
Kalyuzhnaya MG, Zabinsky R, Bowerman S, Baker DR, Lidstrom ME, Chistoserdova L (2006) Fluorescence in situ hybridization-flow cytometry-cell sorting-based method for separation and enrichment of type I and type II methanotroph populations. Appl Environ Microbiol 72:4293–4301
Kurokawa K, Itoh T, Kuwahara T, Oshima K, Toh H, Toyoda A (2007) Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res 14:169–181
Kowalchuk GA, Stephen JR, De Boer W, Prosser JI, Embley TM, Woldendorp JW (1997) Analysis of ammonia-oxidizing bacteria of the beta subdivision of the class Proteobacteria in coastal sand dunes by denaturing gradient gel electrophoresis and sequencing of PCR-amplified 16S ribosomal DNA fragments. Appl Environ Microbiol 63:1489–1497
Lay C, Doré J, Rigottier-Gois L (2007) Separation of bacteria of the Clostridium leptum subgroup from the human colonic microbiota by fluorescence-activated cell sorting or group-specific PCR using 16S rRNA gene oligonucleotides. FEMS Microbiol Ecol 60:513–520
Lay C, Rigottier-Gois L, Holmstrøm K, Rajilic M, Vaughan EE, de Vos WM et al. (2005) Colonic microbiota signatures across five northern European countries. Appl Environ Microbiol 71:4153–4155
Lee JH, Karamychev VN, Kozyavkin SA, Mills D, Pavlov NV, Polpuchine NN, Richardson PM, Shakhova VV, Slesarev AI, Weimer B, O’Sullivan DJ (2008) Comparative gneomic analysis of the gut bacterium Bifidobacterium longum reveals loci susceptible to deletion during pure culture growth. BMC Genomics 27:9:247
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102:11070–11075
Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS et al. (2008) Evolution of Mammals and Their Gut Microbes. Science 320:1647–1651
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444:1022–1023
Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H et al. (2008) Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci USA 105:2117–2122
Louis P, Scott KP, Duncan SH, Flint HJ (2007) Understanding the effects of diet on bacterial metabolism in the large intestine. J Appl Microbiol. 102:1197–208
Macfarlane GT, Gibson GR (1997) In Mackei RI, White BA (eds) Gastrointestinal microbiology: gastrointestinal ecosystems and fermentations, Carbohydrate Fermentation, Energy Transduction and Gas Metabolism in the Human Large Intestine. vol 1. International Thomson Publishing, UK p. 269
Marchesi J, Shanahan F (2007) The normal intestinal microbiota. Curr Opin Infect Dis 20:508–513
Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F et al. (2007) Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. Proteome Res 6:546–551
Martin FP, Dumas ME, Wang Y, Legido-Quigley C, Yap IK, Tang H et al. (2007) A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol Syst Biol 3:112
Martin FP, Wang Y, Sprenger N, Yap IK, Lundstedt T, Lek P et al. (2008) Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol 4:157
Martinez-Medina M, Aldeguer X, Gonzalez-Huix F, Acero D, Garcia-Gil LJ (2006) Abnormal microbiota composition in the ileocolonic mucosa of Crohn’s disease patients as revealed by polymerase chain reaction-denaturing gradient gel electrophoresis. Inflamm Bowel Dis 12:1136–1145
McHardy AC, Rigoutsos I (2007) What’s in the mix: phylogenetic classification of metagenome sequence samples. Curr Opin Microbiol 10:499–503
Morrison DJ, Mackay WG, Edwards CA, Preston T, Dodson B, Weaver LT (2006) Butyrate production from oligofructose fermentation by the human faecal flora: what is the contribution of extracellular acetate and lactate? Br J Nutr 96:570–577
Mueller S, Saunier K, Hanisch C, Norin E, Alm L, Midtvedt T et al. (2006) Differences in fecal microbiota in different European study populations in relation to age, gender, and country: a cross-sectional study. Appl Environ Microbiol 72:1027–1033
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Nielsen S, Nielsen DS, Lauritzen L, Jakobsen M, Michaelsen KF (2007) Impact of diet on the intestinal microbiota in 10-month-old infants. J Pediatr Gastroenteorl Nutr 44:613–618
Nicholson JK, Holmes E, Wilson ID (2005) Gut microorganisms, mammalian metabolism and personalized health care. Nat Rev Microbiol 3:431–438
Olsen GJ, Lane DJ, Giovannoni SJ, Pace NR & Stahl DA (1986) Microbial ecology and evolution: a ribosomal RNA approach. Annual Reviews of Microbiology 40:337–365
Ovreas L, Forney L, Daae FL, Torsvik V (1997) Distribution of bacterioplankton in meromictic Lake Saelenvannet, as determined by denaturing gradient gel electrophoresis of PCR-amplified gene fragments coding for 16S rRNA. Appl Environ Microbiol 63:3367–3373
Pool-Zobel BL, Sauer J (2007) Overview of experimental data on reduction of colorectal cancer risk by inulin-type fructans. J Nutr 137:2580S–2584S
Pompei A, Cordisco L, Amaretti A, Zanoni S, Matteuzzi D, Rossi M (2007) Folate production by bifidobacteria as a potential probiotic property. Appl Environ Microbiol 73:179–185
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S et al. (2007) The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 53:77
Saarela M, Maukonen J, von Wright A, Vilpponen-Salmela T, Patterson AJ, Scott KP, Hämynen H, Mättö J (2007) Tetracycline susceptibility of the ingested Lactobacillus acidophilus LaCH-5 and Bifidobacterium animalis subsp. lactis Bb-12 strains during antibiotic/probiotic intervention. Int J Antimicor Agenet 29(3):271–280
Salyers AA, Gupta A, Wang Y (2004) Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends Microbiol 12:412–416
Sipos R, Székely AJ, Palatinszky M, Révész S, Márialigeti K, Nikolausz M (2007) Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol Ecol 60:341–350
So PW, Yu WS, Kuo YT, Wasserfall C, Goldstone AP, Bell JD et al. (2007) Impact of resistant starch on body fat patterning and central appetite regulation. PLoS ONE 2:e1309
Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD et al. (1999) Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol 65:4799–4807
Stabler RA, Gerding DN, Songer JG, Drudy D, Brazier JS, Trinh HT, Witney AA, Hinds J, Wren BW (2006) Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteroil 188:7297–7305
Spencer JP, Schroeter H, Rechner AR, Rice-Evans C (2001) Bioavailability of flavan-3-ols and procyanidins: gastrointestinal tract influences and their relevance to bioactive forms in vivo. Antioxid Redox Signal 3:1023–1039
Solanky KS, Bailey NJ, Beckwith-Hall BM, Bingham S, Davis A, Holmes E et al. (2005) Biofluid 1H NMR-based metabonomic techniques in nutrition research - metabolic effects of dietary isoflavones in humans. J Nutr Biochem 16:236–244
Stewart JA, Chadwick VS, Murray A (2006) Carriage, quantification, and predominance of methanogens and sulfate-reducing bacteria in faecal samples. Lett Appl Microbiol 43:58–63
Tannock GW, Munro K, Bibiloni R, Simon MA, Hargreaves P, Gopal P et al. (2004) Impact of consumption of oligosaccharide-containing biscuits on the fecal microbiota of humans. Appl Environ Microbiol 70:2129–2136
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI (2008) Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3:213–223
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI (2007) The human microbiome project. Nature 449:804–810
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031
Tuohy K, Rowland IR, Rumsby PC (2002) In: Atherton KT (ed) Genetically modified crops assessing safety. Taylor and Francis, London, 110–137
Tuohy KM, McCartney AL (2006) Molecular microbial ecology of the human gut. In: Gibson GR, Rastall RA (eds) Prebiotics: development and application. Wiley, London, 135–155
van Beek S, Priest FG (2002) Evolution of the lactic acid bacterial community during malt whisky fermentation: a polyphasic study. Appl Environ Microbiol 68:297–305
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG et al. (2001) The sequence of the human genome. Science 291:1304–1351
Wardle J, Carnell S, Haworth CM, Plomin R (2008) Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr 87:398–404
Wang M, Ahrné S, Jeppsson B, Molin G (2005) Comparison of bacterial diversity along the human intestinal tract by direct cloning and sequencing of 16S rRNA genes. FEMS Microbiol Ecol 54:219–231
Wang Y, Lawler D, Larson B, Ramadan Z, Kochhar S, Holmes E et al. (2007) Metabonomic investigations of aging and caloric restriction in a life-long dog study. J Proteome Res 6:1846–1854
Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea, Bacteria and Eucarya. Proc Natl Acad Sci USA 87:4576–4579
Yap IK, Li JV, Saric J, Martin FP, Davies H, Wang Y, Wilson ID, Nicholson JK, Utzinger J, Marchesi JR, Holmes E (2008) Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J Proteome Res 7:3718–3728
Zoetendal EG, Cheng B, Koike S, Mackie RI (2004) Molecular microbial ecology of the gastrointestinal tract: from phylogeny to function. Curr Issues Intest Microbiol 5:31–47
Zoetendal EG, Akkermans AD, De Vos WM (1998) Temperature gradient gel electrophoresis analysis of 16S rRNA from human fecal samples reveals stable and host-specific communities of active bacteria. Appl Environ Microbiol 64:3854–3859
Zwart G, Huismans R, van Agterveld MP, Van de Peer Y, De Rijk P, Eenhoorn H, Muyzer G, van Hannen EJ, Gons HJ, Laanbroek HJ (1998) Divergent members of the bacterial division Verrucomicrobiales in a temperate freshwater lake1. FEMS Microbiol Ecol 25:159–169
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Springer Science+Business Media, LLC
About this entry
Cite this entry
Tuohy, K.M., Abecia, L., Deaville, E.R., Fava, F., Klinder, A., Shen, Q. (2009). Post-Genomics Approaches towards Monitoring Changes within the Microbial Ecology of the Gut. In: Charalampopoulos, D., Rastall, R.A. (eds) Prebiotics and Probiotics Science and Technology. Springer, New York, NY. https://doi.org/10.1007/978-0-387-79058-9_3
Download citation
DOI: https://doi.org/10.1007/978-0-387-79058-9_3
Publisher Name: Springer, New York, NY
Print ISBN: 978-0-387-79057-2
Online ISBN: 978-0-387-79058-9
eBook Packages: Chemistry and Materials ScienceReference Module Physical and Materials ScienceReference Module Chemistry, Materials and Physics