
Abstract
High-throughput methods for screening of physical and functional interactions now provide the means to study virus–host interactions on a genome scale. The limited coverage of these methods and the large size and uncertain quality of the identified interaction sets, however, require sophisticated computational approaches to obtain novel insights and hypotheses on virus infection processes from these interactions. Here, we describe the central steps of bioinformatics methods applied most commonly for this task and highlight important aspects that need to be considered and potential pitfalls that should be avoided.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others
Key words
- Virus–host interactions
- Yeast two-hybrid
- RNA interference
- Computational analysis
- Databases
- Functional enrichment analysis
- Clustering
- Interaction prediction
1 Introduction
Large-scale screens of virus–host interactions using either yeast two-hybrid (Y2H) or RNA interference (RNAi) now provide substantial resources for the computational analysis and modeling of processes involved in virus infection and proliferation [1, 2]. Following the first genome-wide Y2H screen of virus–host interactions in EBV [3], similar screens have been performed for hepatitis C virus (HCV) [4], vaccinia virus [5], H1N1 and H3N2 influenza virus [6], and HIV-1 [7]. An overview of these studies is provided in our recently published review [1]. Since then, numerous additional virus–host Y2H screens have been published including dengue virus [8], influenza virus polymerase [9], flavivirus NS3 and NS5 proteins [10], murine γ-herpesvirus 68 [11], SARS [12], chikungunya virus [13], papaya ringspot virus NIa-Pro protein [14] and human T-cell leukemia virus type 1 and type 2 [15].
In contrast to Y2H, which detects binary physical virus–host interactions, RNAi can also identify functional interactions of the virus with so-called host factors (HF), which are involved in protein complexes, signaling pathways, or cellular processes relevant for infection, as well as HFs binding to viral nonprotein components (e.g., nucleic acids) [16, 17]. Genome-wide RNAi screens of viral HFs were first performed in Drosophila systems for an insect picornavirus [18] and dengue [19] and influenza viruses [20]. Subsequently, genome-scale screens in human cells were published for HIV-1 [7, 21–23], West Nile virus (WNV) [24], HCV [25–27], and influenza virus [28–31] (see Table 2 in [1]). Most recently, a genome-scale study was published performing RNAi for 17 different viruses [32].
Not surprising for high-throughput methods, reproducibility of both Y2H and RNAi large-scale screens is extremely low resulting in very small overlaps between independent screens of virus–host interactions. This can be best assessed for RNAi screens as here several independent screens of the same viruses have been performed including HIV-1, HCV, and influenza virus [7, 21, 23, 25, 27, 28, 30, 31]. In all cases, overlaps were modest ranging from 3 to 6 % for HIV-1 [33], 3–16 % for HCV and 1–12 % for influenza virus. Overlaps are similarly low when comparing different Y2H screens of the same virus or between Y2H and RNAi screens as is exemplified by the case of dengue virus. In the most recent dengue-human Y2H screen by Khadka et al. [8], 188 interactions were identified involving 105 proteins. Only three of these had been identified as HFs in previous RNAi screens (<3 %) and only 1 of 20 (5 %) previously published interactions was detected. Reasons that have been suggested for these discrepancies are differences in the experimental setup, such as differences in cell culture systems, virus isolates, or siRNA pools, as well as different criteria to determine the final set of published interactions. These differences may lead to different subsets of targets and HFs identified such that a large number of interactions is missed in each screen (false negatives). In addition, many of the detected interactions may be false positives, i.e., wrongly detected, due to unspecific interactions of “sticky” proteins in case of Y2H and off-target effects in case of RNAi.
Both the large size and varying quality of the high-throughput screens make it difficult to directly obtain insights on virus infection processes from the screening results. Accordingly, computational and systems biology approaches are necessary to integrate results from different screens and additional data sources as well as identify general trends and connections among the targeted proteins such as common pathways and biological processes they are involved in. An overview of computational approaches used for these purposes was recently published [1]. In this chapter, the corresponding methods are described in more detail and potential pitfalls are highlighted.
2 Resources and Databases
The first step in the analysis of virus–host interactions generally is the compilation of both virus–host and cellular interactions from previously published studies. Although for large-scale screens these data are commonly provided as supplementary material, it is cumbersome to trawl the available literature and download all required supplementary tables. Furthermore, for small-scale studies, interaction data is mostly provided within the main text. To alleviate this problem, many interaction databases have been developed that collect and store interaction data. A number of such databases focus specifically on virus–host interactions, notably the HIV-1, human protein interaction database at NCBI [34], VirHostNet [35], VirusMINT [36], HPIDB [37], and ViPR[38]. In most of these cases, interactions were obtained based on extensive literature curation. While this increases the quality of the data, it requires a continuous curation effort to keep the data up-to-date. Unfortunately, most of these databases are no longer actively updated and currently none of them can be considered as a standard repository for virus–host interactions.
Alternatively, virus–host interactions can be obtained from protein interaction repositories with a more general focus, such as BioGRID [39] and BIND [40]. Both rely on a combination of manual curation and high-throughput submission. However, only BioGRID is still actively maintained as the most recent updates to BIND occurred in mid-2006. Despite its active status, BioGRID by far does not provide a complete picture of either viral or cellular interactomes as it depends on authors submitting their interaction data to BioGRID or the availability and the area of interest of manual curators. Unfortunately, virus–host interactions appear to be included only to a limited degree in BioGRID with many of the most recent studies not covered. In contrast, cellular interactions, in particular human interactions, are much better covered by BioGRID and other actively maintained protein interaction databases such as MINT [41], IntAct [42], or DIP [43]. Furthermore, the human protein reference database (HPRD) provides a large collection of manually curated human interactions but no new release has been published since 2010 [44].
In summary, none of the resources available on viral and cellular protein interactions likely covers all known interactions. Thus, the best strategy to capture as much information as possible is to combine data from all of these resources as they are often to a large degree complementary. For virus–host interactions an additional literature screen is generally necessary as little up-to-date information is contained in available databases. Furthermore, when compiling virus–host and cellular interaction networks, annotations with regard to the type of interaction—which are generally available in all discussed databases—should be taken into account. In particular, protein–gene interactions should be distinguished from protein–protein interactions and for the latter type of interactions, binary and indirect (via other proteins) physical interactions should be treated separately from functional interactions. In most cases, this is best done based on the annotated experimental methods as the interaction type annotation of most databases is not sufficiently fine-grained.
3 Virus–Host Interactions in the Context of the Cellular Interactome
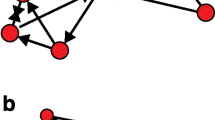
One of the most commonly used approaches for the analysis of virus–host interactomes focuses on evaluating general characteristics of viral targets or HFs within the host networks. Despite their incompleteness and uncertain quality, networks of cellular interactions compiled from public databases as described in the previous section are generally used as an approximation of the true host interactome. Many previous studies have found interesting trends for viral (and also bacterial) targets and HFs mostly with regard to centrality and interconnectedness of these proteins [3, 4, 6, 28, 45–47]. Centrality measures aim to quantify the importance of a protein within the host interactome (Fig. 1a). The most well-known and most easily computable centrality measure is the degree of a protein, i.e., the number of its interactions. The motivation behind this centrality concept is that high-degree proteins (so-called hubs) likely interact with and influence many different pathways and processes and, thus, are important for the cellular system. Indeed, degree has been reported to be correlated to essentiality of a protein for cell survival [48, 49]. Several studies on virus–host interactions indicated a tendency for viruses to interact with or depend on highly connected host proteins [3, 4, 28, 45–47], suggesting that virus tend to target essential proteins or proteins involved in many different pathways.

Network centrality measures and network randomization. (a) Various network centrality measures were calculated for an example network (degree, betweenness, and distance centrality, annotated next to the proteins). The most central protein in terms of degree and distance is F, whereas betweenness centrality is highest for protein E, which has only two interactions. The reason for this is that any shortest path from one protein on the left (A–D) to any protein on the right (F–I), has to pass E. In contrast, proteins G–I have betweenness 0, as there is no shortest path going through them. Removal of any of the high betweenness-proteins (C–F) disconnects the networks, but not removal of the other proteins. (b) Calculation of distances (d ) in an unweighted network using breadth-first search. In this example, all distances from A to any of the other proteins are calculated. The gray arrows indicate the order in which the interactions are traversed. In a breadth-first search, all interaction partners of a protein are visited before their interactions are traversed. (c) To determine centrality measures for viral targets and HFs, first centrality measures of all proteins in the cellular network are determined using existing tools (e.g., cytoHubba) and then values for the relevant proteins are selected. (d) Randomization of networks by rewiring of interactions. Afterwards, each protein has the same number of interactions as in the original network
Alternative centrality measures include distance and betweenness centrality, which focus on more global aspects. Both were found to be significantly increased for viral targets and HFs, mostly independent of the correlation between degree and distance or betweenness centrality [4, 45]. In case of distance centrality, proteins are considered central if the average distance, i.e., the length of the shortest path, to any other protein in the network is small. Distance centrality of a protein is then calculated as the sum of the reciprocals of the distances to the other proteins. For this purpose, shortest path lengths between any pair of proteins have to be calculated. In case of unweighted interaction networks, this can be done most easily using breadth-first searches starting from each protein in the network (Fig. 1b). Betweenness centrality of a protein P, on the other hand, sums up the fraction of shortest paths between any pair of proteins that pass P. Thus, proteins are central if most shortest paths between many pairs of proteins go through them. Such proteins are called bottlenecks and the most extreme case of a bottleneck would be a protein whose removal disconnects the network (Fig. 1a). Betweenness centrality for unweighted interaction networks can be calculated most efficiently using Brandes’ algorithm [50]. Unfortunately, no software is available so far for performing centrality analysis specifically for viral targets or HFs. However, existing tools for network analysis, e.g., the Cytoscape plugin cytoHubba (http://hub.iis.sinica.edu.tw/cytoHubba/), can be adapted to this task by first calculating centrality values for all proteins in the cellular network and then mapping them to the viral targets and HFs (Fig. 1c).
Although these trends are mostly confirmed with each new large-scale screen, the conclusions that can be drawn from these observations are limited and correlation is often mistaken for causation. Likely targeting of hubs and bottlenecks is not an end in itself but rather a consequence of the targeting of central pathways and biological processes that contain many highly interactive proteins due to their importance for the host. That viruses tend to target such important processes is certainly not surprising. Although it is tempting to speculate that the particular selection of hubs and bottlenecks allows targeting of these pathways and processes more efficiently with fewer interactions, scarcity of current data does not really allow confident conclusions in this respect. Nevertheless, knowledge of this trend—whatever its reason—is relevant for subsequent analyses performed on virus–host interactions as it serves to avoid some pitfalls. For instance, several groups have noted that viral interaction partners and HFs tend to be densely interconnected [3, 4, 6, 46]. This observation would not be remarkable if the density of the subnetwork were compared to any random subnetwork with the same number of proteins, as high-degree proteins tend to have a larger number of interactions between them by default. Instead, subnetwork density has to be compared against the random background of networks with the same number of interactions per protein. These random networks can be obtained by repeatedly switching end-points of two random edges (Fig. 1d). p-Values are obtained by repeating the random permutation several times (>100) and calculating the fraction of subnetworks among the viral targets or HFs in the random networks that have at least the same number of interactions as the true subnetwork. Similar strategies have to be applied whenever a pursued analysis approach might be biased by the increased degree and betweenness centrality of viral targets and HFs.
4 Evaluation of Targeted Pathways and Biological Processes
In order to better describe the mechanisms of virus infection and proliferation, it is crucial to understand which pathways and biological processes are specifically targeted by the virus. This is complicated by the following problems. First, the definition of pathways or processes is often ambiguous and may differ largely between experts or annotation resources. Second, many genes are involved in several processes or pathways and, thus, it may not always be possible to ascertain which of their functions is relevant for virus infection. Finally, for many proteins only some or even none of their functions may be known and, consequently, many pathways or processes have not been described at all or only incompletely. Essentially, there are two general approaches pursued for uncovering the involved pathways and processes in the context of virus infection. The first one focuses on identifying enriched pathways or processes based on existing functional annotations from public databases and statistical methods. The second one—which will be discussed in the next section—aims to identify novel functional modules based on protein interaction networks.
Several resources on protein function annotation are currently available. The most commonly used resource is the Gene Ontology (GO) which provides three hierarchically structured vocabularies (called ontologies) to describe biological processes, molecular functions, and cellular components, respectively [51] (Fig. 2a). Pathway annotation can be obtained from the KEGG [52] and BioCarta (http://www.biocarta.com) databases which also provide interactions between proteins/genes. In principle, any type of annotation can be used for enrichment analysis, including also protein domains from Pfam [53], keywords from Uniprot [54] or disease annotations from OMIM (http://www.omim.org). The main distinction between annotation resources is whether they are hierarchically structured or not. In the first case, proteins will generally be annotated with the most specific term in the hierarchy but any superterm higher up in the hierarchy also applies to the protein. To address this problem, annotations are extended during enrichment analysis such that for any specific annotation term all superterms are also presumed to be annotated to the respective protein (Fig. 2b). Generally, this results in a large degree of redundancy in the results as superterms of significantly enriched terms often also tend to be enriched.

Outline of functional enrichment analysis. (a) The most commonly used annotation resource for functional enrichment analysis is the Gene Ontology (GO), which is structured as a directed acyclic graph. Thus, a term may have more than one parent, e.g., cell death has the parents cellular process and death. Here, only the part of the biological process ontology above the term cell death is shown. (b) As only the most specific GO term is usually annotated directly to a protein, the annotation has to be extended for functional enrichment analysis by any superterm of the annotated terms. (c) Workflow for performing functional enrichment analysis. For this example, we assume 100 proteins identified as viral interactors or HFs and 20,000 proteins in the background population, e.g., all human proteins. p-Values were calculated with Fisher’s exact test and multiple testing correction performed using the Bonferroni method. (d) Exemplary case illustrating the relevance of interactions when evaluating targeted pathways. The pathway consisting of proteins A–F is targeted by the virus at two proteins, either D and C (solid and dashed line) or C and E (solid and dotted line). In the first case, the complete pathway is influenced; in the second case, only the right-hand path from A to E but not the one on the left side. In functional enrichment analysis, however, the pathway is simply represented as a set of proteins and the two situations cannot be distinguished
The most commonly used approach for identifying the relevant functional categories among the large list of annotated functions is based on assessing the statistical difference between the observed frequency of a function among the targets or HFs and the frequency in the background (Fig. 2c). The reason for using statistical testing is that neither the absolute counts nor the ratios of frequencies are informative on their own. For functional categories that are very frequent in the overall protein population, large counts among the targets are expected. In contrast, for a very infrequent category, a few hits among the targets may be sufficient for statistical significance. The most commonly used statistical tests for this purpose are Fisher’s exact test and the hypergeometric test, which are both based on the hypergeometric null distribution and thus equivalent [55]. As these tests are applied individually to each functional category, one additional aspect becomes important, namely multiple testing correction. Essentially, a p-value quantifies the probability that a specific value of the test statistic is expected at random according to the null distribution. Thus, the standard cut-off of 0.05 for significance tests indicates that the probability of seeing this result at random is about 1 in 20 if only one significance test is performed. However, if thousands of tests are performed as in the case of functional enrichment analysis, this means that we can expect a lot of random results with this value. To address this problem multiple testing correction is applied. Here, the most rigorous and straightforward correction method is the Bonferroni method which simply multiplies all p-values by the number of significance tests. As this method is very stringent and discards many truly significant results, several other multiple testing correction methods have been developed. The most commonly used one is the method by Benjamini and Hochberg [56] for control of the false discovery rate (FDR), i.e., the number of results erroneously called significant. Most multiple testing correction methods are available in the statistical programming language R, for instance in the multtest package [57].
A large number of software tools and Web servers have been published so far for functional enrichment analysis (see, e.g., [55] for an overview), in most cases focused on the GO. Among these the DAVID Web server should be noted especially for its ease of use as it allows enrichment analysis for a wide range of annotation resources as well as protein identifier types (e.g., gene symbols, Affymetrix IDs, Entrez Gene IDs). In addition to the classical view of enriched functional categories sorted by associated p-values, a clustering of categories based on the overlap of annotated proteins can be performed, which in light of the inherent redundancy within and between annotation resources provides a better overview of the relevant categories. One important feature generally provided by all tools is that a list of genes can be provided as background population by the user instead of the complete genome. This is important if only a nonrandom subset of the genome was selected for screening, such as the druggable genome. In this case, an enrichment analysis against the genome would generally pick up any functional category already enriched in the background population.
The advantage of functional enrichment analysis is that it is easy to perform using existing tools even without programming skills and provides a first “quick-and-dirty” overview which processes may be involved in virus infection. For instance, in our recently published study on SARS-host interactions [12], GO enrichment analysis provided the first clue that immunophilins might be suitable drug targets for coronavirus treatment. However, there are also several problems associated with enrichment analysis as it is standardly performed. First, it is based on gene lists, requiring a cut-off in case the readout from the experiment is continuous, as, e.g., for RNAi screens. This problem can be addressed by using statistical tests to compare distributions instead of frequencies, such as the Kolmogorov-Smirnov test, and some tools provide this option, e.g., GeneTrail [58]. Second, functional categories are assumed to be independent of each other, which is a very simplifying assumption as categories can overlap in many genes. This does not only result in a large redundancy in the output and affects its interpretability, but may also violate the assumptions behind the statistical tests and multiple correction methods. Despite this problem, more statistically sound approaches as discussed by Goeman and Bühlmann [59] have not gained wide-spread acceptance. Finally, when focusing on functional categories as gene sets only, interactions between genes and proteins are ignored and consistency of the results is not evaluated (Fig. 2d). As a consequence, results from the functional enrichment analysis should always be taken with a grain of salt and not be considered as an important finding by itself but rather be used to derive hypotheses that are followed up and validated by other means.
5 Identification of Novel Functional Modules Involved in Virus Infection
The approaches described in the previous section rely on existing knowledge of pathways and biological processes and predefined functional categories. As this knowledge is likely incomplete, several methods have been developed to identify previously undescribed processes associated with virus infection. In general, these are based on identifying functional modules of virus targets or HFs from the network of cellular interactions using network clustering approaches. The clustering method most commonly used for this purpose is MCODE [60], which aims to detect densely connected subnetworks within large cellular networks (Fig. 3a). Density of a subnetwork is defined as the number of edges in the subnetwork divided by the maximum possible number of interactions among the involved proteins and ranges between 0 and 1. MCODE uses three steps, including protein weighting, determination of dense modules, and optional postprocessing of the detected modules. In the first step of MCODE, proteins are weighted based on the density of interactions among their neighbors in the network. In the second step, modules are extended starting from the highest weighted protein not yet contained in a cluster until the density in the module falls below a density threshold. This threshold is defined as a percentage of the weight of the seed protein. Using this percentage parameter, the number and density of the predicted modules can be adjusted. As MCODE is available as a Cytoscape plugin, it can be easily applied to the network of host interactions among the viral targets and HFs and the results can be immediately inspected visually. It is likely this ease of use that led to its predominance for the analysis of virus–host interactions (e.g., in [7, 28, 33]) and not necessarily a better performance in identifying functional modules. At least for the application of protein complex detection, other network clustering approaches have shown superior performance compared to MCODE [61], e.g., Markov Clustering (MCL) [62], but have yet to be applied for the analysis of virus–host interactomes.

Identification of targeted modules and prediction of virus–host interactions. (a) Clustering of viral targets and HF based on the density of cellular interactions between them. In this example, two clusters are identified in which the proteins are tightly connected. (b) Additional properties, such as co-expression, can be taken into account into the module identification by assigning weights to the interactions. In the example, proteins C, E, G are upregulated among infection and the others are downregulated resulting in stronger weights for some interactions (thick lines) and different identified modules compared to (a). (c) Prediction of novel host factors using the guilt-by-association concept. Associations in this case are the same as in (b) and calculated from protein interactions and co-expression. Proteins C, E, I have previously been identified as HFs. As G and F interact with all of these HFs and are co-expressed, they are predicted as HFs. The other proteins are only weakly associated with an HF and, thus, not predicted as a novel HF. (d) Outline of the approach for predicting direct virus–host interaction. To derive a prediction model, supervised machine learning algorithms (such as support vector machines (SVM)) require a set of known positive (i.e., interacting protein pairs) and negative interactions (i.e., pairs of proteins not interacting) with additional features describing these interactions, e.g., correlation of expression. This set is called the training data. The resulting model is then applied to a set of potential interactions for which the same features have been calculated and for each interaction the model predicts whether this is a true interaction or not
The major challenge in the de novo detection of functional modules involved in virus infection is not the detection of these modules. Depending on the parameters, MCODE or other graph clustering algorithms will always identify some densely connected subnetworks among the viral targets and HFs. Accordingly, the difficulty consists in the assessment of the significance of the results and the biological interpretation of the modules. So far, the problem of module significance has been mostly ignored for this application and all focus has been put on the biological interpretation of the results. This is unfortunate as significance analysis not only serves to distinguish truly relevant results from mere random observations. It can also help to limit the list of identified modules to the most interesting ones for which more in-depth analysis is performed. As the number of identified modules can be large (e.g., 152 in case of the König et al. study on influenza virus HFs [28]), such detailed analysis is often omitted. Instead modules are commonly mapped to known processes and pathways, e.g., from the GO or KEGG, and only considered further if they are significantly enriched in at least one functional category. As a consequence, a large fraction of detected modules are often discarded (e.g., almost 50 % in the König et al. study mentioned above [28]), most notably the so far undescribed and likely novel functional modules. Accordingly, in most studies on virus–host interactions, network clustering so far provided only little incremental insights compared to a simple enrichment analysis. Thus, the advantage lies mostly in the extension of known processes by additional proteins as well as interactions between the proteins.
Apart from network clustering based only on the interactions between viral targets and HFs, additional approaches have been developed to identify modules which are not only connected by many interactions but also similar with regard to other properties, such as phenotype after RNAi knockdown (Fig. 3b). One straightforward way to do this is to assign edge weights to the interaction networks based on the other properties considered. This allows applying state-of-the-art weighted graph clustering approaches such as MCL or even standard distance-based clustering approaches such as average linkage clustering. The latter approach was used by Gonzalez and Zimmer [63] to identify clusters of interacting proteins that also show a similar phenotype in an RNAi screen. In this case, the challenging aspect is the definition of an appropriate weight function/distance metric to quantify different types of similarities between proteins. Given the edge weights, existing implementations of clustering algorithms for instance in R or Matlab can then be easily applied.
6 Prediction of Virus–Host Interactions
The small overlaps between screens of viral targets or HFs for the same species indicate that a large number of interactions are missed in each screen and, thus, a substantial number of interactions still remain to be detected. Accordingly, several methods have been developed to identify novel virus–host interactions or HFs. Just as for the large-scale screening methods, two objectives can be distinguished here: (1) the identification of proteins either interacting functionally with the virus (similar to RNAi) or (2) the identification of binary physical interactions between a viral and a host protein (similar to Y2H). Most approaches for the first application can be roughly subsumed by the term “guilt-by-association” (Fig. 3c). Accordingly, proteins are predicted as HFs if they are closely associated either functionally or physically with other HFs. What distinguishes the individual methods is the definition of the associations and the prediction of the HFs based on these associations. Usually, associations and confidence scores for these associations are calculated by integrating several types of evidence, such as co-expression, and domain co-occurrence, for instance using Bayesian methods [64]. Alternatively, functional associations including confidence scores for each type of evidence are also readily available from the STRING database, which integrates evidence from genomic context, high-throughput experiments, co-expression, and literature mining [65].
Using the association scores and information on known HFs, the likelihood of a protein to be an HF can be scored. The most straightforward way to do this involves a summing up of the association scores of this protein to known HFs, either with or without normalization to the total sum of association scores of this protein [64, 66]. A number of more sophisticated methods are described in a recent article by Murali et al. [66], including their novel SinkSource algorithm. As all of these methods only provide likelihood scores for a protein being an HF, a cut-off has to be applied to obtain the final predictions and the quality of the predictions depends strongly on the choice of the cut-off. Thus, to compare different methods, evaluation procedures should be used that are independent of a particular choice of cut-off, such as receiver operating characteristic (ROC) curves or precision-recall curves. In both cases, proteins are sorted by their confidence scores calculated based on all other proteins and all possible cut-offs are evaluated. For each cut-off, true positive rate ( = fraction of HFs correctly predicted = recall) and false positive rate ( = fraction of non-HFs wrongly predicted as HF), in case of ROC curves, or recall and precision ( = fraction of predictions that are HFs), in case of recall-precision curves, are calculated and plotted against each other. If the curve for one method is always above the curve for another method, the first method is clearly superior. If no such clear trends are observed, the area under the curve (AUC) can be calculated which provides one single measure of performance. For ROC curves, the AUC quantifies the probability that a true HF is ranked before a random non-HF.
For the prediction of physical binding between a virus and a host protein, in principle the same methods can be used that have been developed for the prediction of intraspecies interactions. Generally, these approaches exploit similarities of a protein pair to known interacting protein pairs either from the same or a different species. These similarities may be quantified in terms of sequence or structural similarities between the proteins (e.g., [67, 68]) or other evidence as used for scoring associations for the prediction of HFs (e.g., [69, 70]). In the latter case, so-called supervised machine learning approaches are generally applied to learn a classification model that identifies true interactions based on certain features of the interaction. To learn the model, both known true interactions are required (positive examples) as well as protein pairs that do not interact (negative examples). Here, the challenging aspect is the selection and calculation of the interaction features and the collection of positive and negative examples (training data). Given this training data, any out-of-the-box supervised learning algorithm can be used, for instance support vector machines (SVM) or any other algorithm included in the WEKA software library [71].
The limitation of these approaches for the prediction of direct virus–host protein interactions consists in the scarcity of training data. For most viruses, the number of known interactions to the host is very small even when including closely related species. Accordingly, sequence and structure similarity to known interacting pairs is in most cases not large enough to confidently transfer interactions. Furthermore, other types of experimental evidence commonly used to infer interactions, such as gene expression studies, are also generally not available. Despite these difficulties efforts have been undertaken with some success to predict virus–host interactions mostly based on sequence homology and other sequence features but also protein centrality measures and GO annotations [66, 72, 73]. In all of these cases, however, predictions were focused on HIV-1–human interactions for which the largest amount of data is available. It remains to be seen how successful these approaches can be for less well-studied virus–host interactomes.
7 Conclusions
In summary, a large number of methods have been developed for the computational analysis of virus–host screens focusing either on the role of the viral targets and HFs within the host network or biological processes and pathways targeted by the virus. Mostly, however, these approaches are not readily available as software tools, thus limiting their applicability for biological users. Fortunately, at least in some cases the methods can be replicated using existing implementations for individual steps such that only little programming skills are required.
References
Friedel CC, Haas J (2011) Virus-host interactomes and global models of virus-infected cells. Trends Microbiol 19(10):501–508
Striebinger H, Kögl M, Bailer SM (2013) High-throughput analysis of virus-host interactions by yeast two hybrid assay. In: Bailer SM, Lieber D (eds) Virus-Host Interactions: Methods and Protocols, Methods in Molecular Biology, vol. 1064
Calderwood MA et al (2007) Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci USA 104(18):7606–7611
de Chassey B et al (2008) Hepatitis C virus infection protein network. Mol Syst Biol 4:230
Zhang L et al (2009) Analysis of vaccinia virus-host protein-protein interactions: validations of yeast two-hybrid screenings. J Proteome Res 8(9):4311–4318
Shapira SD et al (2009) A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 139(7):1255–1267
Konig R et al (2008) Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135(1):49–60
Khadka S et al (2011) A physical interaction network of dengue virus and human proteins. Mol Cell Proteomics 10(12):M111.012187
Tafforeau L et al (2011) Generation and comprehensive analysis of an influenza virus polymerase cellular interaction network. J Virol 85(24):13010–13018
Le Breton M et al (2011) Flavivirus NS3 and NS5 proteins interaction network: a high-throughput yeast two-hybrid screen. BMC Microbiol 11:234
Lee S et al (2011) An integrated approach to elucidate the intra-viral and viral-cellular protein interaction networks of a gamma-herpesvirus. PLoS Pathog 7(10):e1002297
Pfefferle S et al (2011) The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 7(10):e1002331
Bourai M et al (2012) Mapping of Chikungunya virus interactions with host proteins identified nsP2 as a highly connected viral component. J Virol 86(6):3121–3134
Gao L et al (2012) A set of host proteins interacting with papaya ringspot virus NIa-Pro protein identified in a yeast two-hybrid system. Acta Virol 56(1):25–30
Simonis N et al (2012) Host-pathogen interactome mapping for HTLV-1 and 2 retroviruses. Retrovirology 9(1):26
Cherry S (2009) What have RNAi screens taught us about viral-host interactions? Curr Opin Microbiol 12(4):446–452
Griffiths SJ (2013) Screening for host proteins with pro- and antiviral activity using high-thoughput RNAi. In: Bailer SM, Lieber D (eds) Virus-Host Interactions: Methods and Protocols, Methods in Molecular Biology, vol. 1064
Cherry S et al (2005) Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev 19(4):445–452
Sessions OM et al (2009) Discovery of insect and human dengue virus host factors. Nature 458(7241):1047–1050
Hao L et al (2008) Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454(7206):890–893
Brass AL et al (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319(5865):921–926
Yeung ML et al (2009) A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem 284(29):19463–19473
Zhou H et al (2008) Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 4(5):495–504
Krishnan MN et al (2008) RNA interference screen for human genes associated with West Nile virus infection. Nature 455(7210):242–245
Tai AW et al (2009) A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5(3):298–307
Ng TI et al (2007) Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology 45(6):1413–1421
Li Q et al (2009) A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci USA 106(38):16410–16415
Konig R et al (2010) Human host factors required for influenza virus replication. Nature 463(7282):813–817
Bortz E et al. (2011) Host- and strain-specific regulation of influenza virus polymerase activity by interacting cellular proteins. mBio 2(4):e00151-11
Karlas A et al (2010) Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 463(7282):818–822
Brass AL et al (2009) The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139(7):1243–1254
Snijder B et al (2012) Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol 8:579
Bushman FD et al (2009) Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog 5(5):e1000437
Fu W et al (2009) Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res 37(Database issue):D417–D422
Navratil V et al (2009) VirHostNet: a knowledge base for the management and the analysis of proteome-wide virus-host interaction networks. Nucleic Acids Res 37(Database issue):D661–D668
Chatr-aryamontri A et al (2009) VirusMINT: a viral protein interaction database. Nucleic Acids Res 37(Database issue):D669–D673
Kumar R, Nanduri B (2010) HPIDB–a unified resource for host-pathogen interactions. BMC Bioinformatics 11(Suppl 6):S16
Pickett BE et al (2012) ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 40(Database issue):D593–D598
Stark C et al (2011) The BioGRID interaction database: 2011 update. Nucleic Acids Res 39(Database issue):D698–D704
Alfarano C et al (2005) The biomolecular interaction network database and related tools 2005 update. Nucleic Acids Res 33(Database issue):D418–D424
Licata L et al (2012) MINT, the molecular interaction database: 2012 update. Nucleic Acids Res 40(Database issue):D857–D861
Kerrien S et al (2012) The IntAct molecular interaction database in 2012. Nucleic Acids Res 40(Database issue):D841–D846
Salwinski L et al (2004) The database of interacting proteins: 2004 update. Nucleic Acids Res 32(Database issue):D449–D451
Keshava Prasad TS et al (2009) Human protein reference database–2009 update. Nucleic Acids Res 37(Database issue):D767–D772
Dyer MD, Murali TM, Sobral BW (2008) The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog 4(2):e32
Wuchty S, Siwo G, Ferdig MT (2010) Viral organization of human proteins. PLoS One 5(8):e11796
van Dijk D et al (2010) Identifying potential survival strategies of HIV-1 through virus-host protein interaction networks. BMC Syst Biol 4:96
Yu H et al (2004) Genomic analysis of essentiality within protein networks. Trends Genet 20(6):227–231
Jeong H et al (2001) Lethality and centrality in protein networks. Nature 411(6833):41–42
Brandes U (2001) A faster algorithm for betweenness centrality. J Math Sociol 25:163–177
Ashburner M et al (2000) Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet 25(1):25–29
Kanehisa M et al (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38(Database issue):D355–D360
Punta M et al (2012) The Pfam protein families database. Nucleic Acids Res 40(Database issue):D290–D301
UniProt Consortium (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res 40(Database issue):D71–D75
Rivals I et al (2007) Enrichment or depletion of a GO category within a class of genes: which test? Bioinformatics 23(4):401–407
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57(1):289–300
Pollard KS et al (2010) multtest: Resampling-based multiple hypothesis testing. R package version 2.6.0. http://CRAN.R-project.org/package=multtest
Backes C et al (2007) GeneTrail–advanced gene set enrichment analysis. Nucleic Acids Res 35(Web Server issue):W186–W192
Goeman JJ, Buhlmann P (2007) Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics 23(8):980–987
Bader GD, Hogue CW (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4:2
Brohée S, van Helden J (2006) Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7:488
van Dongen S (2000) Graph clustering by flow simulation. University of Utrecht, Utrecht, Netherlands
Gonzalez O, Zimmer R (2011) Contextual analysis of RNAi-based functional screens using interaction networks. Bioinformatics 27(19):2707–2713
Lee I et al (2011) Prioritizing candidate disease genes by network-based boosting of genome-wide association data. Genome Res 21(7):1109–1121
Szklarczyk D et al (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res 39(Database issue):D561–D568
Murali TM et al (2011) Network-based prediction and analysis of HIV dependency factors. PLoS Comput Biol 7(9):e1002164
Ng SK, Zhang Z, Tan SH (2003) Integrative approach for computationally inferring protein domain interactions. Bioinformatics 19(8):923–929
Yu H et al (2004) Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res 14(6):1107–1118
Jansen R et al (2003) A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302(5644):449–453
Qi Y, Bar-Joseph Z, Klein-Seetharaman J (2006) Evaluation of different biological data and computational classification methods for use in protein interaction prediction. Proteins 63(3):490–500
Hall M et al (2009) The WEKA data mining software: an update. SIGKDD Explorations 11(1):10–18
Evans P et al (2009) Prediction of HIV-1 virus-host protein interactions using virus and host sequence motifs. BMC Med Genomics 2:27
Qi Y et al (2010) Semi-supervised multi-task learning for predicting interactions between HIV-1 and human proteins. Bioinformatics 26(18):i645–i652
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Friedel, C.C. (2013). Computational Analysis of Virus–Host Interactomes. In: Bailer, S., Lieber, D. (eds) Virus-Host Interactions. Methods in Molecular Biology, vol 1064. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-62703-601-6_8
Download citation
DOI: https://doi.org/10.1007/978-1-62703-601-6_8
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-62703-600-9
Online ISBN: 978-1-62703-601-6
eBook Packages: Springer Protocols