
Abstract
The medical use of cannabinoids has been proposed for the control of epilepsy. At present, several studies have focused on investigating how cannabinoids can regulate the expression of epileptic seizures as well as the epileptogenesis process. Some of them suggest that cannabinoids may represent a therapeutic approach for different types of epilepsy. However, experimental evidence indicates that the effects of cannabinoids depend on several experimental and pathological conditions. In this chapter, we provide an overview of these preclinical and clinical research.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
- Endocannabinoid system
- Anandamide (AEA)
- 2-Arachidonoyl glycerol (2-AG)
- Δ9-Tetrahydrocannabinol (Δ9-THC)
- Cannabidiol (CBD)
- CB1/CB2 receptors
- Seizures
- Epilepsy
1 Introduction
During centuries cannabis plants have been used for both medicinal and recreational uses as described in Chinese, Indian, and Arab pharmacopeias [1]. Presently, the medical use of Cannabis extracts has been approved in some European countries [2].
Cannabis plants present a mixture of chemical constituents, the C21 terpenophenolic compounds also called phytocannabinoids. Detailed chemical analysis has allowed the identification of about 70 molecular species of these phytocannabinoids [3] whose amounts depend on each plant and environmental conditions [4, 5]. The most important phytocannabinoids are the psychoactive Δ9-tetrahydrocannabinol (Δ9-THC) and the non-psychoactive cannabidiol (CBD). CBD was isolated in 1940, and its structure was elucidated in 1963 [6, 7]. The Δ9-THC was isolated by Yechiel Gaoni and Raphael Mechoulam [8] and was shown to account for the psychotropic effects of cannabis preparations in rhesus monkeys [9]. During the late 1980s, it was found that Δ9-THC exerts its effects through the activation of two G-protein-coupled receptors: cannabinoid type 1 (CB1) and cannabinoid type 2 (CB2) receptors [10, 11]. Thereafter, the endocannabinoids (eCBs) anandamide and 2-arachidonoylglycerol were identified as the endogenous ligands of CB1 and CB2 receptors [12–14].
A growing body of evidence supports that the eCB system is involved in several functions of the brain and that Cannabis and phytocannabinoids represent pharmacological strategies to induce neuroprotection and control of disorders such as epilepsy, migraine, and pain [15]. In the case of epilepsy, experimental evidence indicates a key role for eCB system in the modulation of neuronal excitability. The present review focuses on providing a better understanding of how and when pharmacological interventions with cannabinoids or phytocannabinoids may control epilepsy.
2 The Endocannabinoid System
The eCB system has a crucial role in different brain functions including cerebral development, cognition, learning, memory, motor behavior, appetite regulation, temperature regulation, and pain [16]. Experimental evidence indicates that the role of eCB in the regulation of physiological responses depends on the gender [17]. The eCB system consists of cannabinoid receptors , their endogenous lipid ligands (eCBs), and the enzymatic machinery for their biosynthesis, cellular uptake, release, and degradation [18].
2.1 Endocannabinoids
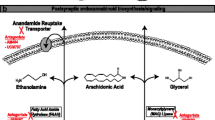
The first eCBs identified in the central nervous system (CNS) [19, 20] were the hydrophobic ligands N-arachidonoyl ethanolamide (anandamide, AEA) [12] and 2-arachidonoyl glycerol (2-AG) [13, 14]. The synthesis of these eCBs depends on specific enzymes using membrane phospholipids as precursors. The N-acylphosphatidylethanolamine-hydrolyzing phospholipase D (NAPE-PLD) is the enzyme responsible for the synthesis of AEA and other N-acylethanolamines [21], whereas different diacylglycerol lipases (DAGLs) are involved in the synthesis of 2-AG [22]. The effects mediated by eCBs are limited by their fast catabolism. The enzyme fatty acid amide hydrolase (FAAH) catabolizes AEA [23]. Monoacylglycerol lipase (MAGL) and serine hydrolase α/β-hydrolase domain 6 (ABHD) induce degradation of 2-AG in the brain [24–26]. Carrier-mediated transport systems are involved in clearing eCBs from the extracellular space [27–29], and their subsequent enzymatic degradation can proceed through either hydrolysis or oxidation [24, 30, 31].
Unlike other neuromodulators and traditional vesicular neurotransmitters, eCBs are believed to be synthesized “on demand” by changes in neural activity [32]. The synthesis of eCBs in postsynaptic neurons can be triggered by the increase in intracellular Ca2+ concentration subsequent to depolarization and activation of voltage-gated Ca2+ channels [33–37] and the activation of certain Gαq/11 protein-coupled receptors [38–40]. Other studies suggest that intracellular storage organelles might accumulate pre-synthesized eCBs [41, 42].
2.2 Endocannabinoid Receptors
CB1 and CB2 receptors belong to the large superfamily of heptahelical G-protein-coupled receptors (GPCR) and couple to Gi/Go proteins. The CB2 receptor is predominately expressed in the immune system [43] and has very limited expression in the CNS. By contrast, the CB1 receptors are highly expressed at presynaptic levels in the brain, and its activation is implicated in inhibition of the synaptic neurotransmission [44–47]. Concerning this notion, it is known that the activation of presynaptic CB1 receptors reduces the release of neurotransmitters like glutamate and γ-aminobutyric acid (GABA) [48] as a consequence of the inhibition of Ca2+ channels and activation of K+ channels [49–54], a situation that may modify the neuronal excitability [55].
Activation of CB1 receptors promotes its interaction with Go proteins, resulting in guanosine diphosphate/guanosine triphosphate exchange and subsequent dissociation of α and βγ subunits with a consequent reduction of adenylate cyclase and cyclic adenosine monophosphate production [56]; inhibition N-, P/Q-, and L-type voltage-gated Ca2+ channels [20, 46, 57, 58]; stimulation of A type K+ channels [44, 59, 60], activation of G-protein-coupled inwardly rectifying K+ channels [61, 62]; and inhibition of the vesicular release machinery [63].
While the CB1 receptor is responsible for the vast majority of the currently known effects of cannabinoids and eCBs in the CNS, it is worth noting that additional cannabinoid receptors may exist. The cannabinoid-sensitive receptor G-protein-coupled receptor 55 (GPR55), identified as a novel cannabinoid receptor that couples to Gα13 protein [64], is activated by some phytocannabinoids such as Δ9-THC. In the brain, GPR55 is present in the caudate, putamen, hippocampus, thalamus, pons, cerebellum, frontal cortex, and thalamus [64]. In human embryonic kidney cells, the activation of GPR55 triggers the release of intracellular Ca2+ from endoplasmic reticulum stores via a pathway dependent on Ras homolog gene family member A (RhoA), phospholipase C, and inositol 1,4.5-trisphosphate receptor [65]. The increases of intracellular Ca2+ levels that result from the activation of GPR55 by L-α-lysophosphatidylinositol (LPI, an endogenous agonist) augment the probability of vesicular release of glutamate at excitatory hippocampal synapses [66, 67]. These results support a relevant role of GPR55 in cerebral excitability.
3 Phytocannabinoids
CBD and Δ9-THC represent the most important phytocannabinoids contained in the Cannabis plants [3]. Δ9-THC is a partial agonist of CB1 receptors that induces most of the behavioral, cognitive, and psychotropic effects of Cannabis. The mechanisms by which Δ9-THC induces these effects also involve the activation and desensitization of the transient receptor potential (TRP) channels of ankyrin type 1 (TRPA1) and vanilloids type 1 (TRPV1) and type 2 (TRPV2) [68–70].
CBD is considered a “multitarget” drug because of its interaction with many other non-eCB signaling systems. It acts as an agonist of TRPV1, TRPV2, and TRPA1 [68, 70–72], 5-hydroxytryptamine1α receptors [73], and glycine receptors [74]. CBD acts as an antagonist of TRP melastatin type-8 channels [69], T-type voltage-gated Ca2+ channels [75], and GPR55 receptors [76]. Also, it exerts dynamic control over intracellular Ca2+ stores [77, 78] and inhibits the uptake and enzymatic degradation of AEA via FAAH [79].
CBD may potentiate some effects induced by Δ9-THC such as analgesia, antiemesis, and anti-inflammation, but it also reduces Δ9-THC-induced psychoactive effects (impaired working memory, sedation, tachycardia, and paranoia) [80–82]. Cannabis products with a high content of CBD induce greater tolerability and lower incidence of psychosis when compared with those with high content of Δ9-THC [83].
4 Effects of Cannabinoids on Seizure Activity and Epilepsy
Several studies indicate that eCBs and cannabinoids play an important role in epilepsy. Here, we summarize evidence from preclinical and clinical studies focused on clarifying this situation.
Concerning experimental models of acute seizure activity, it is described that the i.c.v. administration of arachidonyl-2-chloroethylamide (ACEA, a CB1 receptor agonist) decreases the frequency of penicillin-induced epileptiform activity in rats, an effect blocked by AM-251 (a CB1 receptor antagonist) [84]. Compounds like Δ9-THC, WIN55,212-2, CBD, and AEA and their analog O-1812 induce anticonvulsant effects in the maximal electroshock seizure model [85, 86]. In in vitro models, the activation of CB1 receptors with agonists (methanandamide, 2-AG, AEA, or WIN 55,212-2) reduces the epileptiform activity induced by low or omission of Mg2+ and high K+ [87–89]. The cannabinoid agonist HU210 reduces the epileptiform synchronization in hippocampus induced by kainic acid administration, an effect avoided with the pretreatment with rimonabant, a CB1 receptor antagonist [90]. This group of evidence reveals that cannabinoids may modify both focal and generalized seizures blocking neuronal hypersynchronization associated with epileptic activity.
Studies reveal the participation of cannabinoids in the expression of seizure activity and the epileptogenesis process. Δ9-THC and the cannabinoid agonist WIN55,212 abolish spontaneous epileptic seizures subsequent to pilocarpine-induced SE. Conversely, the administration of the CB1 receptor antagonist SR141716A increases both seizure duration and frequency [91]. The administration of WIN 55,212-2 during 15 days after pilocarpine-induced SE reduces the severity, duration, and frequency of spontaneous recurrent seizures, an effect associated with the preservation of GABAergic neurons, as well as absence of changes in the oxidative stress and expression of NMDA receptor subunits [92]. In the kindling model, the activation of CB1 receptors has been proposed to delay the acquisition of generalized seizures, whereas the inhibition of the enzymatic degradation of AEA did not affect the epileptogenesis process but reduces the neurogenesis associated to it [93]. All these findings support the idea that activation of CB1 receptors can suppress recurrent excitation during epileptogenesis.
Studies indicate that the activation of CB1 receptors can augment or reduce the seizure termination and duration, a situation that depends on the neuronal subpopulation [94]. CB1 receptors are also expressed in astrocytes [95, 96], and their activation is involved in the maintenance of epileptiform discharge [97].
eCBs may interact with other neurotransmitters and neuromodulators. Using the pentylenetetrazol-induced clonic seizure model, it was found that opioids are able to modulate the anticonvulsant effects of cannabinoids [98, 99]. In glutamatergic neurons, activation of CB1 receptors reduces the excitatory neurotransmission and the susceptibility to seizure activity [94, 100]. In experimental models of temporal lobe epilepsy (TLE), the activation of CB1 receptors with agonists (WIN 55,212-2, AEA, or 2-AG) decreases the epileptiform activity, the EPSCs evoked by glutamate, and the excitatory events evoked after antidromic electrical stimulation of mossy fibers in hilus [101].
Several experiments have focused on determining the role of eCB system on seizure activity by enhancing the availability of eCBs. Inhibition of AEA hydrolysis with URB-597, a FAAH inhibitor, results in anticonvulsive effects in the PTZ-induced seizures [102]. The inhibition of the 2-AG hydrolysis using WWL123 (an antagonist of ABHD6) reduces spontaneous seizures in R6/2 mice (a genetic model of juvenile Huntington’s disease seizures) and PTZ-induced tonic-clonic convulsions [103]. Also, the increased levels of 2-AG that result of inhibition of degrading enzyme MAGL have been associated with a delay in the development of the kindling process [104]. The reduced metabolism of eCBs induced by the combination of AM404 (inhibitor of endocannabinoid reuptake) and URB597 (inhibitor of FAAH) results in decreased kainic acid-induced SE in guinea pigs [105]. These studies indicate that the blockage of specific enzymes can represent a new strategy to augment the anticonvulsant effects of eCBs.
In WAG/Rij rats, a genetic animal model of absence seizures, the administration of AEA or WIN55,212-2 (CB1 receptor agonists) reduces the seizure activity, while rimonabant (a CB1 receptor antagonist) increases it [106]. These results suggest that attenuated eCB function may contribute to the generation and maintenance of absence seizures.
5 eCBs and CB1 Receptors in Experimental Models of Seizure Activity and Epilepsy
Several studies indicate that seizure activity and epilepsy modify the eCB system and CB1 receptors. Concerning this issue, it is known that pilocarpine-induced SE increases 2-AG and CB1 receptor expression in hippocampus [91]. Acute seizures induced by kainic acid produce a rapid augmentation of AEA synthesis in the hippocampus and activation of CB1 receptors [107]. Studies indicate that seizure-induced changes in eCBs are age specific. Kainic acid-induced seizures in young rats augment the tissue content of AEA and their biosynthetic enzyme (NAPE-PLD) in the hippocampus, while adult rats present elevated tissue content of 2-AG and its biosynthetic enzyme DAGL [108]. Kindling-induced seizures augment CB1 receptor density in the pyramidal cell layer of the hippocampus [94]. Similar findings have been reported for different mouse models of epilepsy [109, 110]. In contrast, other studies indicate a low expression of CB1 receptors in certain neuronal subpopulation [111, 112]. These contradictory results can be explained by the different epilepsy models used and the period of evaluation after induction of seizures.
Upregulation of CB1 receptors in hippocampus is detected in mice with TLE subsequent to pilocarpine-induced SE [101]. Using the same experimental model of TLE in rats, it was found that spontaneous recurrent seizures are associated with a redistribution of CB1 receptors and changes in expression, binding, and G-protein activation in hippocampus [113]. This situation might depend on the time course of the SE-induced epileptogenesis process [111]. This group of evidence leads to suggest that the redistribution of CB1 receptors is associated with the cerebral plasticity involved in the epileptogenesis process.
6 eCBs and CB1 Receptors in Patients with Epilepsy
In dogs with idiopathic epilepsy, high concentrations of AEA were found in the cerebrospinal fluid, a situation that correlates with the severity of seizures and duration of the disease [114]. This study suggests an important activation of the cannabinoid systems as result of seizure activity. However, the low levels of AEA detected in the cerebrospinal fluid of drug-naïve patients with TLE do not support this hypothesis [115].
Positron emission tomography (PET) imaging revealed increased availability of CB1 receptors in the ipsilateral temporal lobe of patients with TLE, a situation that was more evident in those subjects evaluated within short term after the last seizure and presenting higher number of seizures. These patients also show a decreased availability of CB1 receptors in the ipsilateral superior insular cortex, a condition that may restrict the seizure propagation [116]. However, it is important to consider that in vivo studies using PET imaging cannot avoid the presence of endogenous ligands and the enhanced availability of CB1 receptors can be associated with an increase in their number or affinity, or it is a consequence of low extracellular levels of eCBs.
The evaluation of hippocampal tissue obtained from patients with refractory TLE indicates a reduced expression of cannabinoid receptor-interacting protein-1a (CRIP1a) mRNA and the metabolic enzymes DGAL-α (enzyme involved in the synthesis of 2-AG). There is also a decrease in the mRNA and protein expression of CB1 receptors, mainly at glutamatergic axons, but not in GABAergic boutons, in the dentate gyrus [117]. Considering that CB1 receptors reduce the excitatory neurotransmission in glutamatergic neurons [94, 100], their lower expression at glutamatergic axons can facilitate the excitatory neurotransmission in the epileptic hippocampus. In contrast, CB1 receptors are preserved in dentate gyrus and CA1 region of patients with TLE, suggesting increased expression of these receptors in the GABAergic sprouting axons [118]. These results indicate that the disruption of the inhibitory effects of eCB system on GABAergic transmission in hippocampus of patients with TLE may facilitate the seizure activity.
Concerning TRPV1, studies revealed no significant changes in their expression in hippocampus of animals submitted to repetitive seizures [94]. However, patients with pharmacoresistant temporal lobe epilepsy show increased TRPV1 expression in the hippocampus [119]. Considering that cannabinoids may act as agonists of TRPV1, TRPV2, and TRPA1 [68, 70–72], the activation of these receptors by eCBs may contribute to the modulation of synaptic plasticity in human epileptic hippocampus.
7 The Phytocannabinoids and Epilepsy
There are new well-documented cases reporting remarkably strong beneficial effects of cannabinoids on seizure activity. This situation has triggered an upsurge in exploiting medical marijuana in patients with refractory epilepsy.
CBD is the major constituent of marijuana; it lacks psychoactive side effects and does not act as a CB1 receptor agonist. CBD induces anticonvulsant effects in the seizure activity induced by maximal electroshock test, pentylenetetrazol, pilocarpine-induced temporal lobe seizures, and penicillin [120–123]. However, CBD does not modify the seizure activity induced by cortical administration of cobalt [124]. In kindled rats, CBD reduces the seizure susceptibility and reduces the afterdischarge amplitude, duration, and propagation [125]. Clinical studies also support the anticonvulsant effect of CBD [126]. On the other hand, results obtained from in vitro and in vivo models indicate that cannabidivarin and Δ9-tetrahydrocannabivarin represent the two most important phytocannabinoids with therapeutic potential as anticonvulsant agents [127–131]. At present it is evident that CBD and other phytocannabinoids exert their antiseizure effects at CB1 receptors and other pharmacological targets [128].
In patients with Dravet syndrome, in which epilepsy is usually refractory to standard antiepileptic drugs, medical marijuana with a high CBD/Δ9-THC ratio has been successful to reduce the seizure activity [132]. CBD reduces the seizure frequency in patients with Lennox–Gastaut syndrome, who experience multiple refractory seizures everyday in spite of antiepileptic drugs [133]. Epidiolex (GW Pharmaceuticals), a new phytocannabinoid obtained from Cannabis extracts that contains about 98 % of CBD and 2 % of other cannabinoids, is now approved as a drug to be evaluated in pediatric patients with Dravet and Lennox–Gastaut syndromes [134]. However, proper controlled clinical trials are necessary to establish efficacy and safety of these phytocannabinoids in patients with epilepsy. In addition, future studies have to explore the cellular mechanisms and the signaling pathways involved in the anticonvulsant effects of CBD and other phytocannabinoids.
8 Is the Administration of Cannabinoids a Good Option to Control Epilepsy in Humans ?
It is clear that epilepsy modifies the eCB system (e.g., CB1 receptors). However, as many other neuromodulatory systems, the activation of CB1 receptors can augment or reduce the seizure termination and duration, a situation that depends on the neuronal subpopulation and the experimental model used. Concerning this issue, cannabinoids may induce excitatory effects if CB1 receptors are overexpressed in GABAergic neurons. In contrast, the overexpression of these receptors in glutamatergic neurons can produce inhibitory effects. Therefore, the findings obtained from the evaluation of CB1 receptors in patients with epilepsy using PET or in in vitro conditions have to include a clear identification of the cells in which those changes are produced. In addition, it is relevant to demonstrate that CB1 receptors are functionally active. This situation will help in the clarification of the mechanisms that underlie the anticonvulsant effects of cannabis and cannabinoids in different types of human epilepsy. It will also facilitate the establishment of compounds with therapeutic efficacy to reduce the seizure activity.
Although the results obtained from experimental models are relevant to understand the role of eCBs in epilepsy, they do not reproduce totally the pathological conditions of the human epilepsy. Therefore, the analysis of cerebral tissue obtained from patients with pharmacoresistant epilepsy and submitted to epilepsy surgery is essential to clarify if cannabinoids represent a good therapeutic strategy for epilepsy.
References
Abel EL (1980) Marihuana: the first twelve thousand years. Plenum, New York
Kmietowicz Z (2010) Cannabis based drug is licensed for spasticity in patients with MS. BMJ 340:c3363
Elsohly MA, Slade D (2005) Chemical constituents of marijuana: the complex mixture of natural cannabinoids. Life Sci 78:539–548
Mechoulam R, Hanus L (2000) A historical overview of chemical research on cannabinoids. Chem Phys Lipids 108:1–13
Pertwee RG (2008) The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol 153:199–215
Adams R, Pease DC, Clark JH (1940) Isolation of cannabinol, cannabidiol, and quebrachitol from red oil of Minnesota wild hemp. J Am Chem Soc 62:2194–2196
Michoulam R, Shvo Y, Hashish I (1963) The structure of cannabidiol. Tetrahedron 19:2073–2078
Gaoni Y, Mechoulam R (1964) Isolation, structure and partial synthesis of an active constituent of hashish. J Am Chem Soc 86:1646–1647
Mechoulam R, Shani A, Edery H et al (1970) Chemical basis of hashish activity. Science 169:611–612
Devane WA, Dysarz FA, Johnson MR et al (1988) Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol 34:605–613
Matsuda LA, Lolait SJ, Brownstein MJ et al (1990) Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 346:561–564
Devane WA, Hanus L, Breuer A et al (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258:1946–1949
Mechoulam R, Ben-Shabat S, Hanus L et al (1995) Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50:83–90
Sugiura T, Kondo S, Sukagawa A et al (1995) 2-Arachidonoylglycerol: a possible endogenous cannabinoid receptor ligand in brain. Biochem Biophys Commun 215:89–97
Benbadis SR, Sanchez-Ramos J, Bozorg A et al (2014) Medical marijuana in neurology. Expert Rev Neurother 14(12):1453–1465
Mechoulam R, Parker LA (2013) The endocannabinoid system and the brain. Annu Rev Psychol 64:21–47
Tabatadze N, Huang G, May RM et al (2015) Sex differences in molecular signaling at inhibitory synapses in the hippocampus. J Neurosci 35(32):11252–11265
Pertwee RG, Howlett AC, Abood ME et al (2010) International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62:588–631
Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29:717–727
Wilson RI, Kunos G, Nicoll RA (2001) Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31:453–462
Okamoto Y, Morishita J, Tsuboi K et al (2004) Molecular characterization of a phospholipase D generating anandamide and its congeners. J Biol Chem 279:5298–5305
Bisogno T, Howell F, Williams G et al (2003) Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163:463–468
McKinney MK, Cravatt BF (2005) Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 74:411–432
Dinh TP, Carpenter D, Leslie FM et al (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A 99:10819–10824
Blankman JL, Simon GM, Cravatt BF (2007) A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol 14:1347–1356
Marrs WR, Blankman JL, Horne EA et al (2010) The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat Neurosci 13:951–957
Di Marzo V, Fontana A, Cadas H et al (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372:686–691
Hillard CJ, Edgemond WS, Jarrahian A, Campbell WB (1997) Accumulation of N-arachidonoylethanolamine (anandamide) into cerebellar granule cells occurs via facilitated diffusion. J Neurochem 69:631–638
Piomelli D, Beltramo M, Glasnapp S et al (1999) Structural determinants for recognition and translocation by the anandamide transporter. Proc Natl Acad Sci U S A 96:5802–5807
Deutsch DG, Ueda N, Yamamoto S (2002) The fatty acid amide hydrolase (FAAH). Prostaglandins Leukot Essent Fatty Acids 66:201–210
Yates ML, Barker EL (2009) Inactivation and biotransformation of the endogenous cannabinoids anandamide and 2-arachidonoylglycerol. Mol Pharmacol 76:11–17
Di Marzo V, Deutsch DG (1998) Biochemistry of the endogenous ligands of cannabinoid receptors. Neurobiol Dis 5:386–404
Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29(3):729–738
Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature 410:588–592
Wallmichrath I, Szabo B (2002) Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience 113:671–682
Kim J, Alger BE (2004) Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat Neurosci 7:697–698
Szabo B, Urbanski MJ, Bisogno T et al (2006) Depolarization-induced retrograde synaptic inhibition in the mouse cerebellar cortex is mediated by 2-arachidonoylglycerol. J Physiol (Lond) 577:263–280
Maejima T, Hashimoto K, Yoshida T et al (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31:463–475
Galante M, Diana MA (2004) Group I metabotropic glutamate receptors inhibit GABA release at interneuron–Purkinje cell synapses through endocannabinoid production. J Neurosci 24:4865–4874
Straiker A, Mackie K (2007) Metabotropic suppression of excitation in murine autaptic hippocampal neurons. J Physiol (Lond) 578:773–785
Maccarrone M, Dainese E, Oddi S (2010) Intracellular trafficking of anandamide: new concepts for signaling. Trends Biochem Sci 35:601–608
Min R, Di Marzo V, Mansvelder HD (2010) DAG lipase involvement in depolarization induced suppression of inhibition: does endocannabinoid biosynthesis always meet the demand? Neuroscientist 16:608–613
Berdyshev EV (2000) Cannabinoid receptors and the regulation of immune response. Chem Phys Lipids 108:169–190
Hampson RE, Evans GJ, Mu J (1995) Role of cyclic AMP dependent protein kinase in cannabinoid receptor modulation of potassium “A-current” in cultured rat hippocampal neurons. Life Sci 56:2081–2088
Mackie K, Lai Y, Westenbroek R et al (1995) Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in Att20 cells transfected with rat-brain cannabinoid receptor. J Neurosci 15:6552–6561
Twitchell W, Brown S, Mackie K (1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 78:43–50
Schweitzer P (2000) Cannabinoids decrease the K(+) M-current in hippocampal CA1 neurons. J Neurosci 20:51–58
Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296(5568):678–682
Huang CC, Lo SW, Hsu KS (2001) Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol 532:731–748
Robbe D, Alonso G, Duchamp F et al (2001) Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21:109–116
Azad SC, Eder M, Marsicano G et al (2003) Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learn Mem 10:116–128
Brown SP, Safo PK, Regehr WG (2004) Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci 24:5623–5631
Lovinger DM (2008) Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol 184:435–477
Li Y, Krogh KA, Thayer SA (2012) Epileptic stimulus increases Homer 1a expression to modulate endocannabinoid signaling in cultured hippocampal neurons. Neuropharmacology 63:1140–1149
Katona I, Freund TF (2012) Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci 35:529–558
Glass M, Felder CC (1997) Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17:5327–5333
Mackie K, Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci U S A 89:3825–3829
Szabo GG, Lenkey N, Holderith N et al (2014) Presynaptic calcium channel inhibition underlies CB(1) cannabinoid receptor-mediated suppression of GABA release. J Neurosci 34:7958–7963
Deadwyler SA, Hampson RE, Mu J et al (1995) Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP dependent process. J Pharmacol Exp Ther 273:734–743
Mu J, Zhuang SY, Hampson RE et al (2000) Protein kinase-dependent phosphorylation and cannabinoid receptor modulation of potassium A current (IA) in cultured rat hippocampal neurons. Pflugers Arch 439:541–546
Henry DJ, Chavkin C (1995) Activation of inwardly rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci Lett 186:91–94
McAllister SD, Griffin G, Satin LS et al (1999) Cannabinoid receptors can activate and inhibit G protein-coupled inwardly rectifying potassium channels in a xenopus oocyte expression system. J Pharmacol Exp Ther 291:618–626
Photowala H, Blackmer T, Schwartz E et al (2006) G protein beta gamma-subunits activated by serotonin mediate presynaptic inhibition by regulating vesicle fusion properties. Proc Natl Acad Sci U S A 103:4281–4286
Ryberg E, Larsson N, Sjögren S et al (2007) The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol 152:1092–1101
Lauckner JE, Jensen JB, Chen HY et al (2008) GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A 105:2699–2704
Oka S, Nakajima K, Yamashita A et al (2007) Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem Biophys Res Commun 362:928–934
Sylantyev S, Jensen TP, Ross RA et al (2013) Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci U S A 110:5193–5198
Qin N, Neeper MP, Liu Y et al (2008) TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci 28:6231–6238
De Petrocellis L, Vellani V, Schiano-Moriello A et al (2008) Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J Pharmacol Exp Ther 325:1007–1015
De Petrocellis L, Ligresti A, Moriello AS et al (2011) Effects of cannabinoids and cannabinoid enriched Cannabis extracts on TRP channel s and endocannabinoid metabolic enzymes. Br J Pharmacol 163:1479–1494
Thomas BF, Gilliam AF, Burch DF et al (1998) Comparative receptor binding analyses of cannabinoid agonists and antagonists. J Pharmacol Exp Ther 285:285–292
Bisogno T, Hanus L, De Petrocellis L et al (2001) Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol 134:845–852
Russo EB, Burnett A, Hall B et al (2005) Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem Res 30:1037–1043
Ahrens J, Demir R, Leuwer M et al (2009) The nonpsychotropic cannabinoid cannabidiol modulates and directly activates alpha-1 and alpha-1-Beta glycine receptor function. Pharmacology 83:217–222
Ross HR, Napier I, Connor M (2008) Inhibition of recombinant human T-type calcium channels by Delta9-tetrahydrocannabinol and cannabidiol. J Biol Chem 283:16124–16134
Ross RA (2009) The enigmatic pharmacology of GPR55. Trends Pharmacol Sci 30:156–163
Drysdale AJ, Ryan D, Pertwee RG et al (2006) Cannabidiol-induced intracellular Ca2+ elevations in hippocampal cells. Neuropharmacology 50:621–631
Ryan D, Drysdale AJ, Lafourcade C et al (2009) Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J Neurosci 29:2053–2063
De Petrocellis L, Di Marzo V (2010) Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: focus on G-protein-coupled receptors and transient receptor potential channels. J Neuroimmune Pharmacol 5:103–121
Karniol IG, Carlini EA (1973) Pharmacological interaction between cannabidiol and delta9-tetrahydrocannabinol. Psychopharmacologia 33:53–70
Russo E, Guy GW (2006) A tale of two cannabinoids: the therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med Hypotheses 66:234–246
Englund A, Morrison PD, Nottage J et al (2013) Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J Psychopharmacol 27:19–27
Schubart CD, Sommer IE, van Gastel WA et al (2011) Cannabis with high cannabidiol content is associated with fewer psychotic experiences. Schizophr Res 130:216–221
Kozan R, Ayyildiz M, Agar E (2009) The effects of intracerebroventricular AM-251, a CB1-receptor antagonist, and ACEA, a CB1-receptor agonist, on penicillin-induced epileptiform activity in rats. Epilepsia 50(7):1760–1767
Wallace MJ, Wiley JL, Martin BR et al (2001) Assessment of the role of CB1 receptors in cannabinoid anticonvulsant effects. Eur J Pharmacol 428:51–57
Wallace MJ, Martin BR, DeLorenzo RJ (2002) Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur J Pharmacol 452:295–301
Ameri A, Wilhelm A, Simmet T (1999) Effects of the endogeneous cannabinoid, anandamide, on neuronal activity in rat hippocampal slices. Br J Pharmacol 126(8):1831–1839
Ameri A, Simmet T (2000) Effects of 2-arachidonylglycerol, an endogenous cannabinoid, on neuronal activity in rat hippocampal slices. Naunyn Schmiedebergs Arch Pharmacol 361(3):265–272
Deshpande LS, Blair RE, Ziobro JM et al (2007) Endocannabinoids block status epilepticus in cultured hippocampal neurons. Eur J Pharmacol 558:52–59
Mason R, Cheer JF (2009) Cannabinoid receptor activation reverses kainate-induced synchronized population burst firing in rat hippocampus. Front Int Neurosci 3:1–6
Wallace MJ, Blair RE, Falenski KW et al (2003) The endogenous cannabinoid system regulates seizure frequency and duration in a model of temporal lobe epilepsy. J Pharmacol Exp Ther 307:129–137
Di Maio R, Cannon JR, Greenamyre JT (2015) Post-status epilepticus treatment with the cannabinoid agonist WIN 55,212-2 prevents chronic epileptic hippocampal damage in rats. Neurobiol Dis 73:356–365
Wendt H, Soerensen J, Wotjak CT et al (2011) Targeting the endocannabinoid system in the amygdala kindling model of temporal lobe epilepsy in mice. Epilepsia 52(7):e62–e65
von Rüden EL, Jafari M, Bogdanovic RM et al (2015) Analysis in conditional cannabinoid 1 receptor-knockout mice reveals neuronal subpopulation-specific effects on epileptogenesis in the kindling paradigm. Neurobiol Dis 73:334–347
Navarrete M, Araque A (2008) Endocannabinoids mediate neuron-astrocyte communication. Neuron 57:883–893
Navarrete M, Araque A (2010) Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron 68:113–126
Coiret G, Ster J, Grewe B et al (2012) Neuron to astrocyte communication via cannabinoid receptors is necessary for sustained epileptiform activity in rat hippocampus. PLoS One 7(5):e37320
Shafaroodi H, Samini M, Moezi L et al (2004) The interaction of cannabinoids and opioids on pentylenetetrazole-induced seizure threshold in mice. Neuropharmacology 47:390–400
Bahremand A, Shafaroodi H, Ghasemi M et al (2008) The cannabinoid anticonvulsant effect on pentylenetetrazole-induced seizure is potentiated by ultra-low dose naltrexone in mice. Epilepsy Res 81:44–51
Monory K, Massa F, Egertová M et al (2006) The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 51(4):455–466
Bhaskaran MD, Smith BN (2010) Cannabinoid mediated inhibition of recurrent excitatory circuitry in the dentate gyrus in a mouse model of temporal lobe epilepsy. PLoS One 5:e10683
Vilela LR, Medeiros DC, Rezende GH et al (2013) Effects of cannabinoids and endocannabinoid hydrolysis inhibition on pentylenetetrazole-induced seizure and electroencephalographic activity in rats. Epilepsy Res 104(3):195–202
Naydenov AV, Horne EA, Cheah CS et al (2014) ABHD6 blockade exerts antiepileptic activity in PTZ-induced seizures and in spontaneous seizures in R6/2 mice. Neuron 83(2):361–371
von Rüden EL, Bogdanovic RM, Wotjak CT et al (2015) Inhibition of monoacylglycerol lipase mediates a cannabinoid 1-receptor dependent delay of kindling progression in mice. Neurobiol Dis 77:238–245
Shubina L, Aliev R, Kitchigina V (2015) Attenuation of kainic acid-induced status epilepticus by inhibition of endocannabinoid transport and degradation in guinea pigs. Epilepsy Res 111:33–44
Citraro R, Russo E, Ngomba RT et al (2013) CB1 agonists, locally applied to the cortico-thalamic circuit of rats with genetic absence epilepsy, reduce epileptic manifestations. Epilepsy Res 106(1-2):74–82
Marsicano G, Goodenough S, Monory K et al (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302:84–88
Fezza F, Marrone MC, Avvisati R et al (2014) Distinct modulation of the endocannabinoid system upon kainic acid-induced in vivo seizures and in vitro epileptiform bursting. Mol Cell Neurosci 62:1–9
Karlócai MR, Tóth K, Watanabe M et al (2011) Redistribution of CB1 cannabinoid receptors in the acute and chronic phases of pilocarpine-induced epilepsy. PLoS One 6(11):e27196
Bojnik E, Turunç E, Armağan G et al (2012) Changes in the cannabinoid (CB1) receptor expression level and G-protein activation in kainic acid induced seizures. Epilepsy Res 99(1–2):64–68
Falenski KW, Carter DS, Harrison AJ et al (2009) Temporal characterization of changes in hippocampal cannabinoid CB(1) receptor expression following pilocarpine-induced status epilepticus. Brain Res 1262:64–72
Wyeth MS, Zhang N, Mody I et al (2010) Selective reduction of cholecystokinin-positive basket cell innervation in a model of temporal lobe epilepsy. J Neurosci 30(26):8993–9006
Falenski KW, Blair RE, Sim-Selley LJ et al (2007) Status epilepticus causes a long-lasting redistribution of hippocampal cannabinoid type 1 receptor expression and function in the rat pilocarpine model of acquired epilepsy. Neuroscience 146(3):1232–1244
Gesell FK, Zoerner AA, Brauer C et al (2013) Alterations of endocannabinoids in cerebrospinal fluid of dogs with epileptic seizure disorder. BMC Vet Res 9:262
Romigi A, Bari M, Placidi F et al (2010) Cerebrospinal fluid levels of the endocannabinoid anandamide are reduced in patients with untreated newly diagnosed temporal lobe epilepsy. Epilepsia 51:768–772
Goffin K, Van Paesschen W, Van Laere K (2011) In vivo activation of endocannabinoid system in temporal lobe epilepsy with hippocampal sclerosis. Brain 134(Pt 4):1033–1040
Ludányi A, Eross L, Czirják S et al (2008) Downregulation of the CB1 cannabinoid receptor and related molecular elements of the endocannabinoid system in epileptic human hippocampus. J Neurosci 28:2976–2990
Maglóczky Z, Tóth K, Karlócai R et al (2010) Dynamic changes of CB1-receptor expression in hippocampi of epileptic mice and humans. Epilepsia 51(Suppl 3):115–120
Sun FJ, Guo W, Zheng DH et al (2013) Increased expression of TRPV1 in the cortex and hippocampus from patients with mesial temporal lobe epilepsy. J Mol Neurosci 49(1):182–193
Izquierdo I, Tannhauser M (1973) Letter: the effect of cannabidiol on maximal electroshock seizures in rats. J Pharm Pharmacol 25:916–917
Karler R, Turkanis SA (1978) Cannabis and epilepsy. Adv Biosci 22–23:619–641
Jones NA, Hill AJ, Smith I et al (2010) Cannabidiol displays anti-epileptiform and anti-seizure properties in vitro and in vivo. J Pharmacol Exp Ther 332:569–577
Jones NA, Glyn SE, Akiyama S et al (2012) Cannabidiol exerts anticonvulsant effects in animal models of temporal lobe and partial seizures. Seizure 21:344–352
Colasanti BK, Lindamood C III, Craig CR (1982) Effects of marihuana cannabinoids on seizure activity in cobalt-epileptic rats. Pharmacol Biochem Behav 16:573–578
Turkanis SA, Smiley KA, Borys HK et al (1979) An electrophysiological analysis of the anticonvulsant action of cannabidiol on limbic seizures in conscious rats. Epilepsia 20:351–363
Devinsky O, Cilio MR, Cross H et al (2014) Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 55:791–802
Hill AJ, Weston SE, Jones NA et al (2010) Delta(9)-Tetrahydrocannabivarin suppresses in vitro epileptiform and in vivo seizure activity in adult rats. Epilepsia 51:1522–1532
Hill AJ, Williams CM, Whalley BJ et al (2012) Phytocannabinoids as novel therapeutic agents in CNS disorders. Pharmacol Ther 133:79–97
Hill TD, Cascio MG, Romano B et al (2013) Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br J Pharmacol 170:679–692
Amada N, Yamasaki Y, Williams CM et al (2013) Cannabidivarin (CBDV) suppresses pentylenetetrazole (PTZ)-induced increases in epilepsy-related gene expression. Peer J 1:e214
Bialer M, Johannessen SI, Levy RH et al (2015) Progress report on new antiepileptic drugs: a summary of the Twelfth Eilat Conference (EILAT XII). Epilepsy Res 111:85–141
Maa E, Figi P (2014) The case for medical marijuana in epilepsy. Epilepsia 55:783–786
Hussain SA, Zhou R, Jacobson C et al (2015) Perceived efficacy of cannabidiol-enriched cannabis extracts for treatment of pediatric epilepsy: a potential role for infantile spasms and Lennox-Gastaut syndrome. Epilepsy Behav 47:138–141
Devinsky O, Sullivan J, Friedman D et al (2015) Epidiolex (Cannabidiol) in treatment resistant epilepsy. American Academy of Neurology 67th annual meeting abstract 2015, Washington, DC, USA
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Zavala-Tecuapetla, C., Rocha, L. (2016). Do Cannabinoids Represent a Good Therapeutic Strategy for Epilepsy?. In: Talevi, A., Rocha, L. (eds) Antiepileptic Drug Discovery. Methods in Pharmacology and Toxicology. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6355-3_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6355-3_5
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6353-9
Online ISBN: 978-1-4939-6355-3
eBook Packages: Springer Protocols