
Abstract
Connexins are the structural proteins of gap junctions and their functioning as tumor suppressors is well known. Epigenetic modifications, such as methylation of connexin genes, play important roles in regulating gene expression. Over the past decade, several methods have been applied to characterize DNA methylation-specific loci of connexin genes. This chapter describes analysis of selective connexin32 and connexin43 gene DNA methylation in human gastric tissues using methylation-specific PCR, bisulfite-specific PCR sequencing as well as MassArray techniques.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Connexin
- Gap junction
- Methylation-specific PCR
- Bisulfite-specific PCR sequencing
- MassArray
- DNA methylation
- Normal gastric mucosa
- Non-atrophic gastritis
- Chronic atrophic gastritis
- Intestinal metaplasia
- Dysplasia
- Gastric carcinoma
1 Introduction
Gap junctions are cellular channels necessary to coordinate cell function by allowing adjacent cells to directly share ions and molecules less than 1 kDa [1]. The connexin (Cx) family is a multigene group of gap junction proteins. Gap junctional intercellular communication (GJIC) directly potentiates cells to cooperate electrically or metabolically. In both physiological and pathological conditions, connexin functions are not always linked to their roles in GJIC. For instance, the cytoplasmic carboxy tails of Cx32, also known as gap junction protein β1 or GJB1, and Cx43, also called gap junction protein α1 or GJA1, interact with the PDZ domain of zona occludens protein 1, linking them to the cytoskeleton [2, 3]. Interaction of the Cx43 cytoplasmic carboxy tail with β-catenin may influence Wnt signaling and can reside in the nucleus, resulting in inhibition of cell growth via interfering with key steps of cell cycle regulation [4–6]. Reduced connexin expression and loss of GJIC release initiated cells from growth control exerted by surrounding normal cells and thus allow their clonal expansion, which can be considered as early events in cancer development. Moreover, Cx26, Cx32, and Cx43 have been reported to act as tumor suppressors in human renal cell carcinoma (RCC) and breast cancer. Downregulation of connexin expression in precancerous lesions or cancer tissues may occur at the transcriptional level, of which a prominent mechanism is cancer-specific hypermethylation of their gene promoters [7].
Connexins display high turnover rates with half-lives of 1.5–5 h [8]. In the last few years, it has become clear that epigenetic processes are essentially involved in connexin gene transcription. The most extensively studied epigenetic mechanism is DNA methylation, which occurs almost exclusively at cytosine residues in CpG dinucleotides [9]. Higher frequencies of CpG dinucleotides are found in so-called CpG islands that comprise 1–2 % of the genome [10]. CpG islands are typically present in gene promoter regions and are generally unmethylated. It has been well documented that methylation of CpG islands in connexin gene promoter regions is associated with the transcriptional silencing [11, 12]. DNA hypermethylation contributes to oncogenesis by point mutation and inactivation of tumor suppressor genes. Methylated connexin genes are known to underlie various diseases and accumulation of methylated CpG dinucleotides in connexin gene promoters is frequently observed in neoplastic cells [3, 13, 14]. Not surprisingly, the DNA demethylating agent 5-aza-2′-deoxycytidine suppresses growth of RCC tumors in a xenograft model and breast cancer cell lines by restoring Cx32 and Cx26 expression [5] as well as GJIC [15], indicating the reversed tumorigenicity by re-introduction of functional copies into cells lines using demethylation therapy. Based on the predictable and therapeutic role of DNA methylation in human disease, attention has been paid to this epigenetic modification in gene promoter regions as a gatekeeper of connexin expression [16]. In the present chapter, three methods are described for analyzing the methylation status of connexin CpG islands in normal gastric mucosa as well as tissues in pathological conditions, including non-atrophic gastritis , chronic atrophic gastritis , intestinal metaplasia , dysplasia , and gastric carcinoma .
2 Materials
2.1 Genomic DNA Extraction from Tissues
-
1.
Wizard® Genomic DNA purification kit (Promega, USA).
-
2.
15 mL centrifuge tubes.
-
3.
Small homogenizer.
-
4.
Liquid nitrogen for animal tissue grinding (eventually replacing the small homogenizer).
-
5.
Mortar and pestle for animal tissue grinding (eventually replacing the small homogenizer).
-
6.
Isopropanol at room temperature.
-
7.
70 % ethanol at room temperature.
-
8.
Water bath at 65 °C.
-
9.
Water bath at 37 °C.
2.2 Sodium Bisulfite Conversion of Unmethylated Cytosines in DNA
EpiTect Bisulfite kit (Qiagen, USA).
2.3 PCR Reagents
-
1.
10× PCR buffer (Qiagen, USA).
-
2.
10 mM dNTPs (Amersham Pharmacia Biotech Products Inc., USA).
-
3.
10 mM forward primer.
-
4.
10 mM reverse primer.
-
5.
HotStarTaq DNA polymerase (Qiagen, USA).
-
6.
Bisulfite-treated genomic DNA.
-
7.
DNAse-free and RNAse-free water.
2.4 Methylation-Specific PCR
GelDoc XR system (Bio-Rad, USA).
2.5 Bisulfite-Specific PCR Sequencing
-
1.
BigDye® XTerminator™ purification kit (Applied Biosystems, USA): XTerminator solution and SAM™ solution.
-
2.
BigDye® Terminator v1.1 cycle sequencing kit (Applied Biosystems, USA): M-13 tailed sequencing primers.
-
3.
QIAprep® Miniprep kits (Qiagen, USA).
-
4.
QIAquick gel extraction kit (Qiagen, USA).
2.6 MassArray Assay
-
1.
MassARRAY ® system (Sequenom Inc., USA).
-
2.
MassCLEAVE kit (Sequenom Inc., USA).
3 Methods
As such, three methods, namely methylation-specific PCR (MSP) , bisulfite-specific PCR (BSP) sequencing, and MassArray analysis, were selected to detect Cx32 and Cx43 gene promoter methylation (Table 1). These three approaches are based on the same principle of genomic DNA extraction and bisulfite-modified conversion according to the steps as described below (Fig. 1).

Overview of DNA methylation detection techniques. (MALDI-TOF MS, matrix-assisted laser desorption/ionization time-of-flight mass)
3.1 Genomic DNA Extraction from Human Gastric Tissues
-
1.
Grind tissue in liquid nitrogen using a pre-chilled mortar and pestle.
-
2.
Allow the liquid nitrogen to evaporate, add 10–20 mg fresh tissue to 600 μL of chilled nuclei lysis solution in a 1.5 mL microcentrifuge tube, and homogenize for 10 s (see Note 1 ).
-
3.
Add 3 μL of RNase solution to the cell or animal tissue nuclei lysate and mix. Incubate for 15–30 min at 37 °C and cool to room temperature.
-
4.
Add 200 μL of protein precipitation solution, mix vigorously at high speed for 20 s, and chill the sample on ice for 5 min.
-
5.
Centrifuge at 13,000–16,000 × g for 4 min. The precipitated protein will form a tight white pellet.
-
6.
Carefully transfer the supernatant containing DNA, leaving the protein pellet behind, to a new tube and add 600 μL isopropanol (see Note 2 ).
-
7.
Mix gently by inversion until the white thread-like strands of DNA form a visible mass and centrifuge at 13,000–16,000 × g for 1 min.
-
8.
Carefully decant supernatant and add 600 μL 70 % ethanol. Gently invert the tube several times to wash the DNA and centrifuge at 13,000–16,000 × g for 1 min.
-
9.
Carefully vacuum-aspirate the ethanol and air-dry the pellet for 15 min (see Note 3 ).
-
10.
Redissolve the DNA in 100 μL of DNA rehydration solution overnight at 4 °C or for 1 h at 65 °C. Mix the solution periodically by gently tapping the tube.
-
11.
Store the DNA at 2–8 °C.
3.2 Sodium Bisulfite Conversion of Unmethylated Cytosine
Bisulfite reaction-based assays rely on the chemical conversion of cytosine to uracil. Sodium bisulfite rapidly deaminates position 5 methylated cytosine (5mC) residues to uracil compared to the slower deamination of 5mC to thymine [15, 16]. Bisulfite-based techniques allow for the analysis of this non-CpG methylation, while other techniques cannot, making this one of the major advantages of using bisulfite reaction-based methods.
3.2.1 Bisulfite DNA Conversion
-
1.
Thaw DNA and dissolve the required number of aliquots of bisulfite mix by adding 800 μL RNase-free water to each aliquot. Mix until completely dissolved, which may take up to 5 min.
-
2.
Prepare the bisulfite reactions in 200 μL PCR tubes up to a total volume of 140 μL:
-
(a)
1 ng to 2 μg DNA solution.
-
(b)
RNase-free water to 20 μL total.
-
(c)
85 μL bisulfite mix.
-
(d)
35 μL DNA protect buffer.
-
(a)
-
3.
Close the PCR tubes and mix the bisulfite reactions thoroughly. Store the tubes at room temperature (see Note 4 ).
-
4.
Set a thermal cycler with the following bisulfite conversion conditions: denaturation 5 min at 99 °C, incubation 25 min at 60 °C, denaturation 5 min at 99 °C, incubation 85 min at 60 °C, denaturation 5 min at 99 °C, incubation 175 min at 60 °C and hold up to overnight at 20 °C.
-
5.
Split each tube into three different tubes and start the PCR program.
3.2.2 Clean-Up of Bisulfite-Converted DNA
-
1.
Briefly centrifuge the PCR tubes containing bisulfite reactions and transfer the complete bisulfite reactions to new 1.5 mL microcentrifuge tubes.
-
2.
Add 560 μL freshly prepared buffer BL containing 10 μg/mL carrier RNA. Mix solution using a vortex and centrifuge briefly (see Note 5 ).
-
3.
Place an EpiTect spin column and collection tube in a suitable rack. Transfer the mixture into EpiTect spin columns.
-
4.
Centrifuge columns at 16,000 × g speed for 1 min. Discard the flow-through and place the spin columns back in the collection tubes.
-
5.
Add 500 μL buffer BW to the spin column and centrifuge at 16,000 × g for 1 min. Discard the flow-through and place spin columns back into the collection tubes.
-
6.
Add 500 μL buffer BD (i.e., desulfonation buffer) to the spin columns and incubate for 15 min at room temperature (see Note 6 ).
-
7.
Centrifuge the columns at 16,000 × g for 1 min. Discard the flow-through and place the columns back into the collection tubes.
-
8.
Add 500 μL buffer BW and centrifuge at 16,000 × g for 1 min. Discard and replace the spin columns. Repeat this step.
-
9.
Place the spin columns into new 2 mL collection tubes and centrifuge at 16,000 × g for 1 min to remove residual liquid.
-
10.
Place the spin columns into clean 1.5 mL microcentrifuge tubes. Add 20 μL buffer EB to the center of the membrane. Elute purified DNA by centrifugation for 1 min at approximately 15,000 × g.
-
11.
Store the DNA at −20 °C (see Note 7 ).
3.3 Primer Design and Methylation Positive Control Preparation
-
1.
The primers for detection of Cx32 and Cx43 gene promoter methylation are designed with MethPrimer for MSP (see Note 8 ) and BSP (see Note 9 ), and EpiDesigner for MassArray analysis (Fig. 2) (Table 2).
Fig. 2 
Locations of primers for polymerase chain reaction for DNA methylation detection with amplicon of Cx32 and Cx43. Reprinted with permission from ref. 19
Table 2 Cx32 and Cx43 primer sequences, amplified fragment size, and annealing temperature. Reprinted with permission from ref. 19 -
2.
Given that the connexin expression is negatively associated with the gene methylation status, the primers for detection of Cx32 and Cx43 expression designed with Primer5 are required as a control for RT-PCR analysis, including the internal reference β-actin.

3.4 Connexin DNA Methylation Analysis
3.4.1 Methylation-Specific PCR
MSP is used to evaluate and quantify the connexin gene methylation status based on bisulfite reactivity. MSP uses two distinct methylation-specific primer sets for the sequence of interest. An unmethylated primer will only amplify sodium bisulfite-converted unmethylated DNA, whereas a methylated primer is specific for sodium bisulfite-treated methylated DNA [16].
-
1.
Following bisulfite modification of tissue DNA, unmethylated and methylated reactions of MSP are carried out in a total volume of 25 μL containing 12.5 μL 2× EpiTect master mix, 2 μL 200 μM dNTP, 0.5 μL 0.4 μM of forward or reverse primer, and 10 μL RNase-free water.
-
2.
Place the PCR tubes in the thermal cycler and start the cycling program using the following conditions: 1 cycle at 95 °C for 10 min, 38 cycles at 94 °C for 15 s, annealing temperature for 30 s, 72 °C for 30 s, and 1 cycle at 72 °C for 10 min.
-
3.
Treat DNA of normal human peripheral blood lymphocytes with SssI methyltransferase and subject to bisulfite modification. This serves as the GC-sites methylation positive control (see Note 10 ). Water can be used as a negative PCR control.
-
4.
Create an unmethylated DNA positive control by using the methyltransferase inhibitor 5-aza-2-deoxycytidine (see Note 11 ). Water can be used as a negative PCR control.
-
5.
Detect the PCR products by gel electrophoresis using 2 % agarose gel (Fig. 3).
Fig. 3 
Representative results for Cx32 and Cx43 gene promoters at different gastric carcinogenesis stages using methylation-specific polymerase chain reaction. (a) Agarose gel electrophoresis of MSP bands. (b) Quantified MSP bands (CAG chronic atrophic gastritis , DW diluted water as negative control, DYS dysplasia , GC gastric cancer, IM intestinal metaplasia , M methylated, MP methylation positive control, NAG non-atrophic gastritis , NGM normal gastric mucosa , U unmethylated, UP unmethylated positive control). Reprinted with permission from ref. 19
-
6.
Calculate the methylation level according to the formula [M/(M + U) × 100 %] using the gray values of methylation (M) and unmethylation (U) bands.

3.4.2 Bisulfite-Specific PCR Sequencing
BSP amplifies targets regardless of the gene methylation state of the internal sequence and is considered the gold standard for this type of analysis. One of the primers is fluorescently labeled at the 5′ end, so that the resulting amplicons can be identified by electrophoresis and subjected to sequencing [17]. It thus provides an inherently more accurate assessment of the gene methylation state compared to MSP that selects for presupposed fully methylated or fully unmethylated complementary sequences.
-
1.
The BSP PCR reaction mixture of 25 μL contains 2 μL bisulfite-modified DNA template, 12.5 μL TaKaRa Premix Taq HS, 1 μL each 10 μmol/L forward or reverse primers, and 8.5 μL deionized distilled water.
-
2.
The annealing temperature (Ta) is set using a gradient thermal cycler (see Note 12 ): 1 cycle at 95 °C for 5 min, 35 cycles at 94 °C for 1 min, the target Ta for 2.5 min, 72 °C for 1 min, and 1 cycle at 72 °C for 5 min.
-
3.
Load a fraction of the PCR amplification onto a 1.5 % agarose gel with mass ladder, allow electrophoretic migration, and view the products between 200 and 500 base pairs using standard methods. If the amplified product is of the correct molecular weight and free of nonspecific by-products, proceed to traditional cloning and sequencing as described in step 14. If not, proceed to step 4.
-
4.
Follow the instructions in the QIAquick gel extraction kit to extract and purify the PCR bands:
-
(a)
Excise the DNA fragment from the agarose gel with a clean sharp scalpel.
-
(b)
Weigh the gel slice in a colorless tube and add 3 volumes buffer QG to 1 volume gel.
-
(c)
Incubate at 50 °C until the gel slice is completely dissolved and mix the tube every 2–3 min to help dissolve. Check that the color of the mixture is yellow without dissolved agarose.
-
(d)
Add 1 gel volume of isopropanol to the sample and mix.
-
(e)
Place a QIAquick spin column in a 2 mL collection tube.
-
(f)
Apply the sample to the QIAquick column and centrifuge for 1 min. Discard the flow-through and place the QIAquick column back in the same collection tube to bind DNA.
-
(g)
Add 0.75 mL buffer PE to the QIAquick column and centrifuge for 1 min. Discard the flow-through and centrifuge the QIAquick column for an additional 1 min at 12,000 × g.
-
(h)
Place the QIAquick column into a new 1.5 mL microcentrifuge tube.
-
(i)
Add 50 μL water to the center of the QIAquick membrane and centrifuge the column for 1 min at 16,000 × g to elute DNA.
-
(a)
-
5.
Set up ligation reactions for amplicon cloning by mixing 3 μL of PCR purified product, 1 μL plasmid, 1 μL T4 ligase, and 2.5 μL reaction buffer. Mix the reactions by pipetting. Incubate the reactions for 1 h at room temperature.
-
6.
Transform normal cells as follows:
-
(a)
Carefully transfer 50 μL cell suspension into each prepared tube.
-
(b)
Gently flick the tubes to mix and place them on ice for 20 min.
-
(c)
Heat-shock the cells for 45–50 s in a water bath at exactly 42 °C and do not shake.
-
(d)
Immediately return the tubes to ice for 2 min.
-
(a)
-
7.
Add 950 μL SOC medium at room temperature to the tubes containing cells transformed with ligation reactions. Incubate for 1.5 h at 37 °C with shaking at approximately 150 × g.
-
8.
Plate 150 μL of each transformation culture onto LB/ampicillin/IPTG/X-Gal plates. The cell pellets are obtained by centrifugation at 8000 × g for 1 min, resuspended in 150 μL SOC medium. Incubate the plates overnight (i.e., 16–24 h) at 37 °C.
-
9.
Use Plasmid Miniprep to isolate the recombinant plasmid DNA as instructed:
-
(a)
Resuspend pelleted bacterial cells in 250 μL buffer P1 and transfer to a microcentrifuge tube.
-
(b)
Add 250 μL buffer P2 and gently invert the tube 4–6 times to mix.
-
(c)
Add 350 μL buffer N3 and invert the tube immediately but gently 4–6 times.
-
(d)
Centrifuge for 10 min at 13,000 × g in a table-top microcentrifuge.
-
(e)
Transfer supernatants to the QIAprep spin column by decanting.
-
(f)
Centrifuge for 60 s and discard the flow-through.
-
(g)
Wash the Column by adding 0.75 mL buffer PE and centrifuging for 60 s.
-
(h)
Discard the flow-through and centrifuge for an additional 1 min to remove residual wash buffer.
-
(i)
Place the column in a clean 1.5 mL microcentrifuge tube.
-
(j)
Add 50 μL water to the center of QIAprep and spin column to elute DNA.
-
(k)
Let stand for 1 min and centrifuge for 1 min.
-
(a)
-
10.
For sequencing, use the purified recombinant plasmid DNA as templates and perform a cycle sequencing reaction using the BigDye Terminator V1.1 kit. For each reaction, mix 5 μL DNA template, 1.5 μL reaction mix, 3 μL 5× reaction buffer, 1.5 μL 10 μM T7 primer, and 4 μL sterile water. Mix briefly by pipetting and amplify DNA using the following PCR conditions: 1 cycle of denaturing at 1 min and 96 °C, 30 cycles of 10 s at 96 °C, 5 s, 50 s, 4 min at 55 °C and 1 cycle of 4 °C for 10 min.
-
11.
After cycle sequencing, clean up the reaction by centrifuging the reaction plate and pipetting the SAM™ solution into each well (i.e., 20 μL per well for a 96-well plate).
-
12.
Aspirate XTerminator solution and add to each well using a wide-bore pipette tip (i.e., 20 μL per well for a 96-well plate).
-
13.
Seal the plate using a clear adhesive film. Mix for 30 min and centrifuge the reaction plate briefly.
-
14.
Perform sequencing using the DNA analyzer. Fragment analysis is applied to obtain the ratio of amplicons derived from bisulfite-converted methylated and unmethylated connexin DNA (Fig. 4).
Fig. 4 
Screenshots of bisulfite polymerase chain reaction sequencing of Cx43 gene promoter CpG islands. Blue curve indicates the peak of the sulfonated methylCpG site as C. Red curve demonstrates the sulfonated non-methylated CpG site as T. Reprinted with permission from ref. 19

3.4.3 MassArray Analysis
This method is ideal for detection of gene methylation, for discrimination between methylated and non-methylated samples, and for quantifying the methylation levels of DNA. In essence, cleavage products are generated for the reverse transcription reactions for both U (T) and C in separate reactions. Each cleavage product encloses either a CpG site, called a CpG unit, or an aggregate of multiple CpG sites [18]. For both T and C reactions, the resulting cleavage products have the same length and differ only in their nucleotide composition. A distinct signal pair pattern results from the methylated and non-methylated template DNA and is analyzed by the matrix-assisted laser desorption/ionization time-of-flight mass spectrometry technique (MALDI-TOF) (Fig. 5).

Schematic diagram of the MassArray system. The bisulfite treatment converts non-methylated cytosine into uracil, thus generating methylation-dependent sequence changes in the genomic DNA template. PCR, with T7-promoter tagged reverse primers, is used to amplify the template, while preserving the induced sequence changes. These RNA products are processed by T-base-specific cleavage, yielding small RNA fragments
-
1.
Perform PCR amplification of DNA following bisulfite modification using MassArray primers. Each PCR is split into two cleavage (i.e., T and C) reactions. The volume for single reaction is as follows: 0.5 μL 1× Hot Star buffer, 0.04 μL 200 μM dNTP mix, 0.04 μL 0.2 U/μL Hot Star Taq, 1 μL 200 nM forward primer, 1 μL 200 nM reverse primer, and 1 μL DNA template.
-
2.
Seal the plates and cycle as follows: 94 °C for 15 min, 45 cycles at 94 °C for 20 s, melting temperature of primer for 30 s, 72 °C for 1 min and 72 °C for 3 min.
-
3.
Add 2 μL of shrimp alkaline phosphatase to each 5 μL PCR reaction to dephosphorylate unincorporated dNTPs from the PCR. Incubate the plates for 20 min at 37 °C and then incubate at 85 °C for 5 min.
-
4.
Prepare transcription/RNase A cocktail for each cleavage reaction (i.e., T and C). The 5 μL total condition per plate is composed of 3.15 μL RNase-free double distilled water. 0.89 μL 0.64× T7 Polymerase buffer, 0.24 μL T/C cleavage mix, 0.22 μL 3.14 mM DTT, 0.44 μL 22 U T7 RNA and DNA Polymerase, and 0.06 μL 0.09 mg/mL RNase A.
-
5.
Add 5 μL transcription/RNase A cocktail and 2 μL of shrimp alkaline phosphatase PCR sample into a new uncycled microtiter plate. Centrifuge the plates for 1 min and incubate the plates at 37 °C for 3 h.
-
6.
Add 6 mg of clean resin to each well. Rotate for 10 min and spin down for 5 min at 3200 × g.
-
7.
Add EpiTYPER reaction product and acquire spectra from the two cleavage reactions. The molecular weight of each fragment is determined by MALDI-TOF and the EpiTYPER software generates a report that contains quantitative information for each analyzed fragment (Fig. 6).
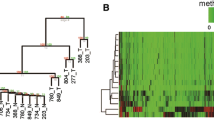
Fig. 6 
Representative results for analysis of DNA methylation of Cx32 and Cx43 gene promoters at different gastric carcinogenesis stages by MassArray analysis. The validated length for Cx32 gene is 484 CpG sites containing 18 CpG sites (15 detected). The validated length for Cx43 gene is 447 bp with a total of 12 CpG sites (11 detected). This panel provides graphical representations of the CpG sites within the selected amplicon. The color code refers to the degree of DNA methylation shown in the methylation panel in order to provide quick and reliable comparison between samples and CpG sites. Reprinted with permission from ref. 19

4 Notes
-
1.
Alternatively, thawed tissue is introduced in the chilled nuclei lysis solution and homogenized for 10 s using a small homogenizer.
-
2.
Some supernatant may remain near the pellets in the original tube containing the protein pellet. Leave this residual liquid in the tube to avoid contaminating the DNA solution with the precipitated protein.
-
3.
The DNA pellet is very loose at this time. Using either a sequencing pipette tip or a drawn pipette is recommended to avoid aspirating the pellet into the pipette.
-
4.
DNA protect buffer should turn from green to blue after addition to DNA bisulfite mix indicating sufficient mixing and accurate pH.
-
5.
Carrier RNA is not necessary when using more than 100 ng DNA.
-
6.
If there are precipitates in buffer BD, avoid transferring them to the spin column. Moreover, it is important to close the lid of the column before incubation.
-
7.
To increase the yield of DNA in the eluate, transfer the spin column to a new 1.5 mL microcentrifuge tube, add an additional 20 μL buffer EB to the center of the membrane, and centrifuge for 1 min at 16,000 × g. Combine both eluates.
-
8.
The methylated primer set assumes the CpGs are fully methylated; thus the primer will have all four bases in the sequence. The unmethylated primer set anneals to genomic DNA that is not methylated in the same primer binding site, and therefore will have T instead of C in the primer sequence. Design guidelines generally have a C or T near the 3′ end of the two primer sets, respectively, where any mismatches will be discriminated against by the polymerase.
-
9.
Design primers to the bisulfite-converted DNA sequence such that each primer is 25–30 bases long, has a melting temperature of approximately 60 °C, and does not hybridize to any CpG-cytosines.
-
10.
It is important to test the primer sets with a control genomic DNA with a known methylation status along with genomic DNA with an unknown methylation status. The properly designed methylated primer set will only amplify the control methylated genomic DNA and not unmethylated genomic DNA, and the unmethylated primer set will only be positive for unmethylated genomic DNA.
-
11.
It is recommended that DNA from peripheral blood is used as a control for the unmethylated reaction.
-
12.
Ensure that temperatures 1–5 °C below and 1–5 °C above the Ta are tested in a gradient thermal cycler in order to determine the temperature at which a single specific PCR product is amplified. Using more cycles may be necessary if starting with less than 1 μg of genomic DNA.
References
Decrock E, Vinken M, De Vuyst E et al (2009) Connexin-related signaling in cell death: to live or let die? Cell Death Differ 16:524–536
Balda MS, Matter K (2000) The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J 19:2024–2033
Penes MC, Li X, Nagy JI (2005) Expression of zonula occludens-1 (ZO-1) and the transcription factor ZO-1-associated nucleic acid-binding protein (ZONAB)-MsY3 in glial cells and colocalization at oligodendrocyte and astrocyte gap junctions in mouse brain. Eur J Neurosci 22:404–418
Herrero-Gonzalez S, Gangoso E, Giaume C et al (2010) Connexin43 inhibits the oncogenic activity of c-Src in C6 glioma cells. Oncogene 29:5712–5723
Sirnes S, Lind GE, Bruun J et al (2015) Connexins in colorectal cancer pathogenesis. Int J Cancer 137:1–11
Dang X, Doble BW, Kardami E (2003) The carboxy-tail of connexin-43 localizes to the nucleus and inhibits cell growth. Mol Cell Biochem 242:35–38
Mesnil M, Krutovskikh V, Piccoli C et al (1995) Negative growth control of HeLa cells by connexin genes: connexin species specificity. Cancer Res 55:629–639
Leithe E, Rivedal E (2007) Ubiquitination of gap junction proteins. J Membr Biol 217:43–51
Vinken M, De Rop E, Decrock E et al (2009) Epigenetic regulation of gap junctional intercellular communication: more than a way to keep cells quiet? Biochim Biophys Acta 1795:53–61
Costello JF, Plass C (2001) Methylation matters. J Med Genet 38:285–303
Piechocki MP, Burk RD, Ruch RJ (1999) Regulation of connexin32 and connexin43 gene expression by DNA methylation in rat liver cells. Carcinogenesis 20:401–406
Yano T, Ito F, Kobayashi K et al (2004) Hypermethylation of the CpG island of connexin 32, a candidate tumor suppressor gene in renal cell carcinomas from hemodialysis patients. Cancer Lett 208:137–142
Yi ZC, Wang H, Zhang GY et al (2007) Downregulation of connexin 43 in nasopharyngeal carcinoma cells is related to promoter methylation. Oral Oncol 43:898–904
Tsujiuchi T, Shimizu K, Itsuzaki Y et al (2007) CpG site hypermethylation of E-cadherin and Connexin26 genes in hepatocellular carcinomas induced by a choline-deficient L-Amino Acid-defined diet in rats. Mol Carcinog 46:269–274
Hayatsu H, Wataya Y, Kai K et al (1970) Reaction of sodium bisulfite with uracil, cytosine, and their derivatives. Biochemistry 9:2858–2865
Kai K, Tsuruo T, Hayatsu H (1974) The effect of bisulfite modification on the template activity of DNA for DNA polymerase I. Nucleic Acids Res 1:889–899
Pappas JJ, Toulouse A, Bradley WE (2009) A modified protocol for bisulfite genomic sequencing of difficult samples. Biol Proced Online 11:99–112
Jurinke C, Denissenko MF, Oeth P et al (2005) A single nucleotide polymorphism based approach for the identification and characterization of gene expression modulation using MassARRAY. Mutat Res 573:83–95
Wang Y, Huang LH, Xu CX et al (2014) Connexin 32 and 43 promoter methylation in Helicobacter pylori-associated gastric tumorigenesis. World J Gastroenterol 20:11770–11779
Acknowledgements
The authors are grateful to the Endoscopic Unit of Third Xiangya Hospital of Central South University for the supply of clinical samples. This work was financially supported by the National Natural Science Foundation of China (No. 81172301).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Liu, X., Xu, C. (2016). DNA Methylation Analysis of Human Tissue-Specific Connexin Genes. In: Vinken, M., Johnstone, S. (eds) Gap Junction Protocols. Methods in Molecular Biology, vol 1437. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3664-9_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3664-9_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3662-5
Online ISBN: 978-1-4939-3664-9
eBook Packages: Springer Protocols