
Abstract
Intercellular communication is essential for the coordination and synchronization of cellular processes. Gap junction channels play an important role to communicate between cells and organs, including the brain, lung, liver, lens, retina, and heart. Gap junctions enable a direct route for ions like calcium and potassium, and low molecular weight compounds, such as inositol 1,4,5-trisphosphate, cyclic adenosine monophosphate, and various kinds of metabolites to pass between cells. Intercellular calcium wave propagation evoked by a local mechanical stimulus is one of the gap junction assays to study intercellular communication. In experimental settings, an intercellular calcium wave can be elicited by applying a mechanical stimulus to a single cell. Here, we describe the use of monolayers of primary bovine corneal endothelial cells as a model to study intercellular communication. Calcium wave propagation was assayed by imaging fluorescent calcium in bovine corneal endothelial cells loaded with a fluorescent calcium dye using a confocal microscope. Spatial changes in intercellular calcium concentration following mechanical stimulation were measured in the mechanical stimulated cell and in the neighboring cells. The active area (i.e., total surface area of responsive cells) of a calcium wave can be measured and used for studying the function and regulation of gap junction channels as well as hemichannels in a variety of cell systems.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Bovine corneal endothelial cells
- Calcium imaging
- Calcium wave propagation
- Connexin 43
- Gap junction assay
- Intercellular communication
- Intracellular calcium
- Mechanical stimulation
1 Introduction
Intercellular communication is essential for tissue homeostasis, control of cell proliferation and synchronization of response to extracellular stresses, thereby coordinating the physiological process between or within a variety of organs and tissues, including the brain, lung, liver , lens, retina, and heart [1]. Connexin proteins have been shown to serve as crucial intercellular communication channels in a variety of cell systems and tissues. In vertebrates, 20 different connexin isoforms are expressed [2]. These connexin isoforms are members of the highly conserved multigenic family of transmembrane proteins, serving as the building blocks for both gap junction and hemichannels . Connexin nomenclature is based on predicted molecular weight of the isoform and is based on sequence similarity and length of the cytoplasmic domain of the connexins, thereby classifying them into α, β, and γ subgroups [3]. As such, six connexins, radially arranged around a central pore, form a connexon . Head-to-head docking of two connexons , also called hemichannels , of adjacent cells results in the formation of a gap junction channel. A plaque of proteinaceous gap junction channels, interconnecting the cytoplasm of adjacent cells forms a gap junction. Gap junctions communicate directly between cells via the diffusion of calcium (Ca2+) or inositol 1,4,5-trisphosphate through gap junctions that couple adjacent cells causing release of Ca2+ from intracellular stores. In contrast, hemichannels can communicate via the release of diffusible extracellular messengers, like adenosine triphosphate that can cause a Ca2+ transient in neighboring cells via Ca2+ influx or via Ca2+ release from intracellular stores (Fig. 1). A more detailed discussion on the mechanisms underlying the initiation and occurrence of intercellular Ca2+ waves and their physiological relevance is provided elsewhere [4].

A schematic model for Ca2+ wave propagation in bovine corneal endothelial cells (BCEC). In normal BCEC , it is hypothesized that mechanical stimulation leads to a moderate rise in cytosolic Ca2+ concentration via inositol 1,4,5-trisphosphate (IP3)-dependent signaling mechanisms, which leads to the opening of Cx43 hemichannels and the flux of adenosine triphosphate (ATP) from the cytosol into the extracellular environment. This allows the propagation of Ca2+ from the “stimulated cell” (SC) to neighboring (NB) cells via activation of purinergic receptors and downstream IP3-induced Ca2+ signaling. This figure and its legend have been taken and adapted from D’hondt C, Iyyathurai J, Himpens B, Leybaert L, Bultynck G. (2014) Cx43-hemichannel function and regulation in physiology and pathophysiology: insights from the bovine corneal endothelial cell system and beyond. Front Physiol. 5:348. doi: 10.3389/fphys.2014.00348. eCollection 2014
A number of techniques is used to study gap junctional communication including microinjection [5], scrape loading [6], fluorescence recovery after photobleaching [7], preloading assay [8], local activation of a molecular fluorescent probe [9], electroporation [10], and dual whole-cell patch clamp [11] and mechanical stimulation [12]. Each gap junction assay has its advantages and limitations [13].
Here, we describe the method of studying intercellular communication by investigating Ca2+ wave propagation elicited by mechanical stimulation of a single cell. This technique provides a tool to quantify the spread of the Ca2+ wave over time in cell line models and primary cell systems and to compare different cell treatments quantitatively. The Ca2+ wave propagation is assayed by intracellular Ca2+ imaging. This is done by loading the bovine corneal endothelial cells (BCEC) with the Ca2+-sensitive dye Fluo-4 AM to monitor cytoplasmic Ca2+ concentration. The fluorescence intensity of Fluo4 is a quantitative readout for cytoplasmic Ca2+ concentration. Fluo4 is excited at 488 nm and its emission is recorded at 530 nm. A neutral density filter is used to minimize photobleaching.
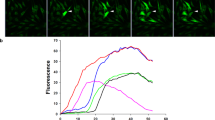
Mechanical stimulation of a single cell consists of an acute short-lasting deformation of the cell by briefly touching less than 1 % of the cell membrane with a glass micropipette (i.e., tip diameter < 1 μm) coupled to a piezoelectric crystal nanopositioner, mounted on a micro-manipulator. In BCEC , mechanical stimulation results in a rapid initial Ca2+ rise that originates at the point of stimulation and spreads throughout the mechanically stimulated cell, slowly diminishing to the baseline level. Subsequently, the intracellular Ca2+ wave propagates to the surrounding neighboring cells as an intercellular Ca2+ wave, upon reaching the cell boundaries. The mechanotransduction-induced Ca2+ increase in the mechanically stimulated cell has not yet been fully elucidated, but could be attributed to Ca2+ influx and/or to Ca2+ release in response to local inositol 1,4,5-trisphosphate production in the mechanical stimulated cells (Fig. 1).
In BCEC , intercellular Ca2+ wave propagation is mainly driven by connexin 43 (Cx43)-based hemichannels mediating the release of adenosine triphosphate in the extracellular environment and only a minor part is driven by gap junctional coupling [14, 15]. Using a combination of genetic tools, like small interfering RNA against Cx43, peptide tools that inhibit Cx43 gap junctions and/or hemichannels and adenosine triphosphate-degrading enzymes, it was shown that the active area (i.e., the maximal total surface area of responsive cells) was heavily reduced in cells treated with (1) small interfering RNA against Cx43, (2) TAT-L2, a cell-permeable peptide inhibiting Cx43 hemichannels , while keeping Cx43 gap junctions in an open state or (3) apyrase, an adenosine triphosphate-degrading enzyme (Fig. 2).

A graph depicting the characteristics of intercellular communication in BCEC , based on mechanical stimulation-induced Ca2+-wave propagation data (active area) [14–16]. Data were further normalized to their respective controls set at 100 %. The graph is intended to indicate the relevance of adenosine triphosphate (ATP) release (blue bars), hemichannels (red bar) and Cx43-based hemichannels (green bars). In general, the data indicate that in BCEC mechanical stimulation-induced Ca2+ wave propagation is almost completely driven by release of ATP into the extracellular environment (i.e., about 90 % inhibition by ATP-degrading enzymes) and that Cx43 hemichannels are a major release pathway for this ATP (i.e., about 60 % inhibition upon Cx43 knockdown or inhibition), although other connexin and/or pannexin isoforms likely contribute to ATP release. Since this graph is intended to provide a general view, readers should access the original research paper for obtaining information about the original mean data and their respective standard error of the mean values. This figure and its legend have been taken from D’hondt C, Iyyathurai J, Himpens B, Leybaert L, Bultynck G. (2014) Cx43-hemichannel function and regulation in physiology and pathophysiology: insights from the bovine corneal endothelial cell system and beyond. Front Physiol. 5:348. doi: 10.3389/fphys.2014.00348. eCollection 2014
Here, we describe in detail the experimental protocol for the measurement of mechanical stimulation -induced Ca2+ wave propagation , as performed in BCEC . Besides mechanical stimulation , the properties of intercellular Ca2+ waves can also be studied in a quantitative manner through their initiation in a controlled manner upon a local photo-release of caged inositol 1,4,5-trisphosphate, which is described elsewhere [17].
2 Materials
2.1 BCEC Medium Preparation
BCEC growing medium: Dulbecco’s modified Eagle’s medium (high glucose, no glutamine and no pyruvate) (Thermo Fisher Scientific, Belgium) supplemented with 6.6 % GlutaMAX™ (Thermo Fisher Scientific, Belgium), 10 % fetal bovine serum (Sigma, Belgium), 1 % antibiotic–antimycotic (Thermo Fisher Scientific, Belgium), and 1 % Fungizone® antimycotic (Thermo Fisher Scientific, Belgium).
2.2 BCEC Isolation
-
1.
Cell culture dish, 100 × 20 mm (Sigma, Belgium).
-
2.
Earle’s balanced salt solution (EBSS) (Thermo Fisher Scientific, Belgium).
-
3.
Fire-polished hook-shaped glass Pasteur pipette.
2.3 Cell Culture
-
1.
Versene solution (Thermo Fisher Scientific, Belgium).
-
2.
Trypsin–ethylenediaminetetraacetic acid, 0.05 % (Thermo Fisher Scientific, Belgium).
-
3.
Laminar air flow cabinet.
-
4.
Hemocytometer.
-
5.
CO2 incubator (37 °C and 5 % CO2).
-
6.
Culture flasks, 25 and 75 cm2 (Sigma, Belgium).
-
7.
Chambered slides, 4.2 cm2 (Sigma, Belgium).
2.4 Measurement of Intercellular Calcium Waves Using Mechanical Stimulation
-
1.
Dulbecco’s phosphate-buffered saline (DPBS) buffer with Ca2+ and Mg2+ (Thermo Fisher Scientific, Belgium).
-
2.
DPBS buffer without Ca2+ and Mg2+ (Thermo Fisher Scientific, Belgium).
-
3.
Fluo-4 AM (Thermo Fisher Scientific, Belgium).
-
4.
LSM510 confocal microscope (Zeiss, Germany).
-
5.
Glass capillaries for nanoliter 2010, 3.5 in. long (World Precision Instruments, UK).
-
6.
Piezoelectric crystal nanopositioner (Piezo Flexure NanoPositioner P-280, operated through E463 amplifier/controller, PI Polytech, Karlsruhe, Germany).
-
7.
Zeitz DMZ-puller (Zeitz Instruments, Germany).
3 Methods
3.1 Procedure of Cell Isolation
-
1.
Isolate the fresh eyes from cow (see Note 1 ).
-
2.
Place the eye on a cell culture dish (i.e., 100 × 20 mm) in a laminar air flow and sterilize by spraying with 70 % ethanol.
-
3.
Remove the 70 % ethanol from the surface of the cornea by rinsing with EBSS–1 % iodine solution.
-
4.
Carefully dissect the cornea from the eye (see Note 2 ) and place it in a new cell culture dish (i.e., 100 × 20 mm), which contains EBSS, with the epithelial cell layer facing upward.
-
5.
Remove any remaining iris tissue still attached to the cornea.
-
6.
Transfer the cornea to another cell culture dish with the endothelial cell layer upward and rinse twice with EBSS.
-
7.
Transfer the cornea to an hourglass and cover with growth medium.
-
8.
Remove the growth medium with a suction pipette, add 300 μL of a trypsin solution to the endothelial layer of the cornea and immediately remove the trypsin solution.
-
9.
Add 300 μL of fresh trypsin solution to the endothelial layer of the cornea and incubate for 30 min at 37 °C with 5 % CO2.
-
10.
Gently scrape the endothelial cells away from the cornea (see Note 3 ) and add to culture flasks (i.e., 25 cm2) containing 4 mL of growth medium (see Note 4 ).
3.2 Cell Culture
-
1.
The next day, add 5 mL of growth medium to the culture flasks (see Note 5 ).
-
2.
Refresh the growth medium every second day until confluency is reached (see Note 6 ).
-
3.
Remove the culture medium and wash the cells twice with 5 mL Versene solution.
-
4.
Add 1.5 mL trypsin solution and place it in the incubator for 3–4 min to detach the cells.
-
5.
Thereafter add 12 mL of growth medium to inhibit the trypsin action and pipette the medium three times in and out to disperse the cells, subsequently count the cells using a hemocytometer.
-
6.
Seed the cells in chambered slides with an area of 4.2 cm2 with a cell count of 165,000 cells (i.e., cell density is 39,286 per cm2) (see Note 7 ).
-
7.
Incubate the cells in an incubator at 37 °C with 5 % CO2. Refresh the medium every 2 days until 95 % confluency is reached (see Note 8 ).
3.3 Measurement of Intercellular Calcium Waves Using Mechanical Stimulation
-
1.
Wash the chambered slide two times with DPBS buffer (see Note 9 ).
-
2.
Load the cells with the 500 μL of 10 μM Ca2+-sensitive dye Fluo-4 AM solution (see Note 10 ) and incubate for 30 min at 37 °C.
-
3.
Wash the dye three times with DPBS and add 500 μL of DPBS buffer (see Note 11 ).
-
4.
The Ca2+ wave propagation is assayed by measure spatial changes in intracellular Ca2+ concentration following mechanical stimulation using the LSM510 confocal microscope.
-
5.
Use an oil immersion 40× objective and set the confocal microscope using Argon laser by excite at 488 nm (i.e., use beam splitter HFT 488) and collect the fluorescence emission at 530 nm (i.e., using a long pass emission filter LP 505), set the pinhole at minimum.
-
6.
Search the confluent cells and position the glass micropipette at 45° in respect to the chambered slide (see Note 12 ).
-
7.
Operate the nanopositioner with voltage between 0.2 and 1.5 V during the mechanical stimulation .
-
8.
Collect and store images.
-
9.
Draw a polygonal region of interest to define the total surface area of responsive cells (i.e., active area, AA) using the software of the confocal microscope.
4 Notes
-
1.
Isolate BCEC from fresh eyes of maximal 18 months old cows, obtained from the slaughterhouse, in order to isolate primary culture of BCEC . Eyes are enucleated at the slaughterhouse within 5 min postmortem and preserved in EBSS-1 % iodine solution on ice for transportation to the laboratory, where cell isolation takes place.
-
2.
Use a sterile sharp razor blade to make a deep enough cut through the sclera. Do not press much on eyeball while cutting to avoid ooze out of fluids. Then use sterile forceps to peel the cornea away from the underlying tissue.
-
3.
Use a fire-polished hook-shaped glass Pasteur pipette to scrape the endothelial cells.
-
4.
Repeat the procedure once more.
-
5.
When the BCEC are still not attached to the surface of the flask, put the flask for another 2 days in the incubator.
-
6.
Once attached to the surface of the flask, change the cell culture medium. On average, the cells are confluent in 10 days (i.e., 7–12 days).
-
7.
Transfer the remaining cells to a 75 cm2 culture flasks at a density of 6250 per cm2 and refresh the culture medium every 2 days with total volume of 20 mL. When confluency is reached, trypsinize the cells and redistribute the cells into chambered slides. This procedure can be repeated twice and cell cultures up to passage 2 can be used for experiments.
-
8.
On average, confluency is reached within 3–4 days.
-
9.
Use DPBS containing Ca2+ and Mg2+.
-
10.
In order to open the gap junction channels, prepare 10 μM Fluo-4 AM in DPBS without Ca2+ and Mg2+.
-
11.
Now, use DPBS containing Ca2+ and Mg2+ to close the gap junction channels.
-
12.
Prepare a glass pipette (i.e., tip diameter < 1 μm) using microelectrode puller and couple it to a piezoelectric crystal nanopositioner, which is operated through an amplifier, mounted on a micro-manipulator.
Disclaimer
Part of this protocol has been previously published as D’hondt C, Himpens B, Bultynck G. (2013) Mechanical stimulation -induced calcium wave propagation in cell monolayers : the example of bovine corneal endothelial cells . J Vis Exp. (77):e50443. doi: 10.3791/50443. A permission from the Journal of Visualized Experiments to reuse parts of the article “D’hondt, C., Himpens, B., Bultynck, G. Mechanical Stimulation -induced Calcium Wave Propagation in Cell Monolayers: The Example of Bovine Corneal Endothelial Cells . J. Vis. Exp. (77), e50443, doi:10.3791/50443 (2013)” for inclusion in Methods in Molecular Biology (Springer) has been obtained.
References
Kar R, Batra N, Riquelme MA et al (2012) Biological role of connexin intercellular channels and hemichannels. Arch Biochem Biophys 524:2–15
Scemes E, Spray DC, Meda P (2009) Connexins, pannexins, innexins: novel roles of “hemi-channels”. Pflugers Arch 457:1207–1226
Sohl G, Willecke K (2004) Gap junctions and the connexin protein family. Cardiovasc Res 62:228–232
Leybaert L, Sanderson MJ (2012) Intercellular Ca2+ waves: mechanisms and function. Physiol Rev 92:1359–1392
Meda P (2000) Probing the function of connexin channels in primary tissues. Methods 20:232–244
El-Fouly MH, Trosko JE, Chang CC (1987) Scrape-loading and dye transfer. A rapid and simple technique to study gap junctional intercellular communication. Exp Cell Res 168:422–430
Abbaci M, Barberi-Heyob M, Stines JR et al (2007) Gap junctional intercellular communication capacity by gap-FRAP technique: a comparative study. Biotechnol J 2:50–61
Goldberg GS, Bechberger JF, Naus CC (1995) A pre-loading method of evaluating gap junctional communication by fluorescent dye transfer. Biotechniques 18:490–497
Dakin K, Zhao Y, Li WH (2005) LAMP, a new imaging assay of gap junctional communication unveils that Ca2+ influx inhibits cell coupling. Nat Methods 2:55–62
Geletu M, Guy S, Firth K et al (2014) A functional assay for gap junctional examination; electroporation of adherent cells on indium-tin oxide. J Vis Exp 92: e51710
Wilders R, Jongsma HJ (1992) Limitations of the dual voltage clamp method in assaying conductance and kinetics of gap junction channels. Biophys J 63:942–953
D’hondt C, Himpens B, Bultynck G (2013) Mechanical stimulation-induced calcium wave propagation in cell monolayers: the example of bovine corneal endothelial cells. J Vis Exp 77:e50443
Abbaci M, Barberi-Heyob M, Blondel W et al (2008) Advantages and limitations of commonly used methods to assay the molecular permeability of gap junctional intercellular communication. Biotechniques 45(33–52):56–62
Gomes P, Srinivas SP, Vereecke J et al (2005) ATP-dependent paracrine intercellular communication in cultured bovine corneal endothelial cells. Invest Ophthalmol Vis Sci 46:104–113
Gomes P, Srinivas SP, Van Driessche W et al (2005) ATP release through connexin hemichannels in corneal endothelial cells. Invest Ophthalmol Vis Sci 46:1208–1218
Ponsaerts R, De Vuyst E, Retamal M et al (2010) Intramolecular loop/tail interactions are essential for connexin 43-hemichannel activity. FASEB J 24:4378–4395
Decrock E, De Bock M, Wang N et al (2015) Flash photolysis of caged IP3 to trigger intercellular Ca2+ waves. Cold Spring Harb Protoc 3:289–292
Acknowledgements
The work has been supported by Concerted Actions of the K.U. Leuven (GOA/09/012), Research Foundation - Flanders (F.W.O.; grant G.0298.11 to GB), Interuniversity Attraction Poles Program (Belgian Science Policy; P7/13 to G.B), and a “Krediet aan Navorser” grant of the FWO (15117.14 N to CDH).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Iyyathurai, J., Himpens, B., Bultynck, G., D’hondt, C. (2016). Calcium Wave Propagation Triggered by Local Mechanical Stimulation as a Method for Studying Gap Junctions and Hemichannels. In: Vinken, M., Johnstone, S. (eds) Gap Junction Protocols. Methods in Molecular Biology, vol 1437. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3664-9_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3664-9_15
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3662-5
Online ISBN: 978-1-4939-3664-9
eBook Packages: Springer Protocols