
Abstract
Heart failure (HF) is a common clinical endpoint to several underlying causes including aging, hypertension, stress, and cardiomyopathy. It is characterized by a significant decline in the cardiac output. Cardiomyocytes are terminally differentiated cells and therefore, apoptotic death due to beta adrenergic (β-AR) signaling contributes to high attrition rate of these cells. Past treatments of HF offer some survival benefit to patients (e.g., the beta blockers), but at the expense of blocking the compensatory beta-adrenergic signaling in surviving cells. One prerequisite for developing new therapeutics is to be able to grow cardiomyocytes ex vivo, and test their apoptotic response to drugs. Here we describe methods for isolation and culturing of neonatal and adult calcium tolerant cardiomyocytes. Similarly, cardiofibroblasts can also be isolated using the same protocol and subsequently, immortalized with SV40 T-Antigen for ex vivo studies.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Various risk factors such as aging, hypertension, valvular disease, myocardial infarction, cardiomyopathy, myocarditis, among other causes, predispose patients to heart failure (HF) [1]. The disease is characterized by a gradual decline in cardiac output, especially in response to oscillations in metabolic demands produced by other organs of the body. In an acute phase, these effects can be reversed by the existing compensatory mechanisms such as neuro-hormonal system and physiological hypertrophy [2]. However, chronic activity of these compensatory mechanisms can revert the heart homeostatic phenotype to that of a pathogenic state, which in turn contributes to worsened prognosis in heart failure. Overall perturbations in the functionality of the heart are associated with significantly high odds of morbidity and mortality, with damage to the myocardium (contractile portion of the heart) a major determinant of disease severity and progression.
At the cellular and molecular level, apoptotic damage to the myocardium, being a tissue composed of a syncytium of contractile cardiomyocytes, is responsible for end stage HF. Apoptotic loss of terminally differentiated cardiomyocytes cannot be replenished and therefore leads to the loss of contractile function of the organ [3, 4]. Indeed, there are various lines of evidence corroborating the contribution of the cardio-damaging effects of the myocardium, as well as successive chronic cardiomyocyte apoptosis leading to progression of HF [5–7]. Current treatments are aimed at reversing these physiological perturbations to that of a normal homeostatic state [8]. Almost all the existing HF treatments block receptor activity for (Beta-blockers or β-blockers). This has been part of the standard care of HF for several decades, however, median 5-year survival has not improved beyond 50 %. Additionally, the undesirable side effects of this class of drugs warrant investigation into novel therapeutics to target and treat HF.
A thorough understanding of the molecular mechanisms underlying disease progression is paramount for the development of new therapeutics. Recently, Lee and colleagues [9] have deciphered the molecular mechanisms leading to the apoptosis of cardiomyocytes. This group provided unequivocal evidence implicating the role of pro-apoptotic BH3 only protein BIM-mediated apoptosis in several HF disease models. The phenotype of the model was associated with progression of HF, while BIM ablation offered protection from the disease. A clear understanding of the transcriptional regulation of BIM during beta-Adrenergic Receptor (β-AR) activation provides an opportunity for developing novel therapeutics through high throughput screening strategies. Validation of drug hits through these screens requires reliable and amenable tools especially ex vivo cell culture methods.
To study cardiac function in healthy and disease conditions, viable animal models such as the mouse and rat have been extensively used for the past 50 years [10]. The availability of such models has provided valuable information regarding the pathophysiology of heart failure , which has led to therapeutic development. Despite strength of evidence derived from whole organism studies, it is technically challenging to derive information on molecular events leading to the disease progression using in vivo models. Besides, the expense, large animal numbers and sophisticated technical requirements associated with animal experiments limit their use in translational science [11]. Therefore, ex vivo experiments in primary and/or cardiomyocyte cell lines can provide reliable and reproducible alternatives for in vivo experimental studies.
Primary cardiomyocyte isolation has been a fundamental procedure vital towards addressing questions in physiology and molecular biology concerning the heart. However, the process has endured technical difficulties, which in turn forced researchers toward the use of cell lines [12, 13]. The use of cell lines has its own drawbacks as there are a limited number of established cardiomyocyte cell lines, including atrial tumor derived HL-1 [14] and embryonic rat ventricular myocyte derived H9C2 [15]. Such lines have been used in studying various cardiac functions such as atrial fibrillation [16], heart metabolism [13] and hypertrophy [12]. Of note, the use of H9C2 cells as an ex vivo model of cardiac studies has some limitations as they are a proliferating cell line, compared to the non-proliferating primary cardiomyocytes [12]. Therefore, their representativeness to an in vivo condition may confound results. There is increasing demand to harness, adapt, and standardize methods for isolation and establishment of primary cardiomyocytes ex vivo for both short and long term experimentation. In doing so, this will provide reliable and reproducible data with a high degree of homogeneity which translates to an in vivo animal environment during HF modeling. Finally, isolating primary cardiomyocytes/fibroblasts from mouse strains with specific gene(s) ablation will greatly help to understand the role of those genes in cellular processes.
In this chapter, we describe the current methods for isolation and establishment of adherent, rod-shaped cardiomyocytes at two developmental stages. Particularly, the methods for isolation of neonatal and adult calcium tolerant cardiomyocytes from mice (a main model for most genetic manipulation studies in the lab) will be emphasized here. The rationale for choice of cell type, i.e., neonatal or adult cardiomyocytes depends mainly on the experiment to be performed. The advantages of harboring neonatal cardiomyocytes include: simpler isolation procedure and culturing methods and they are the system of choice for studying myofibrillogenesis and myofibrillar functions. On the other hand, adult cardiomyocytes are widely accepted as a good model for cardiac cellular physiology and pathophysiology, as well as for pharmaceutical intervention. Genetically modified mice preclude the need for complicated cardiomyocyte infection processes to generate the desired genotype, which are inefficient due to cardiomyocytes’ terminal differentiation. Furthermore, these cells are prone to calcium transients that impair their survival in ex vivo settings; therefore, calcium tolerant cardiomyocytes isolation methods have to be well optimized. The protocol described here is adapted in part from Jacobson and Piper [17] and O’Connell et al. [18] with minor modifications for calcium tolerant adult cardiomyocytes. Specifically, isolation of neonatal cardiomyocytes, as well as cardiofibroblasts for a wide range of ex vivo studies, including cell signaling networks and apoptosis regulation.
2 Materials
2.1 Material for Isolation of Neonatal Mouse Cardiomyocytes
-
1.
Sterile Hanks’ Balanced Salt solution with calcium (Hanks’ medium).
-
2.
Pre-sterilized scalpel for dissection.
-
3.
Sterile trypsin, aliquots stored at 4 °C.
-
4.
Dulbecco’s Modified Eagle’s Medium (complete DMEM ) 1 g/L d-glucose, l-glutamine, 110 mg/L sodium pyruvate; supplemented with 10 % fetal calf serum (FCS).
-
5.
Hanks/Collagenase II : 2.5 g collagenase II in 30 ml Hanks’ medium.
-
6.
Incubators at 37 °C at either 2 or 10 % CO2.
-
7.
100 μM 5-bromo-2′-deoxyuridine (BRDU) (10 mM stocks stored −20 °C).
-
8.
0.1 μM Vitamin C (100 μM stocks stored at −20 °C).
-
9.
96-well flat-bottom plates.
-
10.
50 ml conical tubes.
-
11.
10 ml conical tubes.
-
12.
All pipette types ranging 2–1000 μl and sterile tips.
2.2 Materials for Immortalization of Neonatal Mouse Cardiofibroblasts
-
1.
Lentiviral constructs expressing pSV40 large T antigen, pCMVδ8.2 and pCAG (immortalization, packaging and mouse lentiviral receptor constructs, respectively).
-
2.
5 mg/ml Polybrene (1000×).
-
3.
0.1 % gelatin coated 6-well tissue culture plates (coating of gelatin to be performed by experimenter prior to use of 6-well plates) (see Note 1 ).
-
4.
Fugene 6 Transfection Reagent.
-
5.
3 μg/ml puromycin (3 mg/ml stocks stored at 4 °C).
-
6.
Freeze mix: FCS with 10 % dimethyl sulfoxide (DMSO).
-
7.
Serum free DMEM (SFM).
2.3 Material for Isolation and Culture of Primary Adult Mouse Cardiomyocytes
-
1.
10× perfusion buffer: 70.3 g of NaCl, 11 g of KCl, 0.82 g of KH2PO4, 0.85 g of Na2HPO4 3 g of MgSO4-7H2O, 100 ml of 1 M Na-HEPES per liter. To make fresh single use 1× perfusion buffer, dilute 10× buffer in water and add 0.39 g of NaHCO3, 3.75 g of taurine , 1 g of 2,3-butanedione monoxime (BDM) , 1 g of glucose per liter (see Note 2 ).
-
2.
Digestion buffer: 1× perfusion buffer supplemented with 2.4 mg/ml Collagenase II .
-
3.
Myocyte stopping buffer: 1× perfusion buffer, supplemented with 10 % FCS, 12.5 μM CaCl2 (see Note 2 ).
-
4.
Myocyte culture medium (1× final concentration): To make 50 ml, add 47 ml of Minimum Essential Medium Eagle with Hanks’ Balanced Salt solution , 0.5 ml Bovine Serum Albumin (1 mg/ml), 0.5 ml Penicillin (100 IU/ml), 1 ml BDM (10 mM), 0.5 ml of ITS cocktail comprising of: 5 μg/ml Insulin, 5 μg/ml Transferrin and 5 ng/ml Selenium/ITS (1 g/L d-glucose, l-Glutamine, 110 mg/L Sodium pyruvate, supplemented with 10 % FCS) (see Note 2 ).
-
5.
5 % isoflurane anesthetic.
-
6.
100 IU/ml heparin in PBS .
-
7.
27 gauge needles.
-
8.
0.5 ml insulin needles.
-
9.
Fine tip forceps.
-
10.
Small clamp.
-
11.
5-0 suture nylon thread.
-
12.
Bright field light dissection microscope (minimum magnification 4×).
-
13.
0.22 μm micrometer filters.
-
14.
100 μm strainer.
-
15.
Sterile 1× phosphate buffered saline (10× PBS stocks).
-
16.
96-well tissue culture plates.
-
17.
The peristaltic pump with associated tubing immersed in 37 °C water bath.
-
18.
Hemocytometer .
-
19.
100 ml Schott bottle.
-
20.
2 mM ATP (200 mM stock).
-
21.
Refrigerated centrifuge 4 °C.
3 Methods
3.1 Method for Isolation of Neonatal Mouse Cardiomyocytes and Cardiofibroblasts
-
1.
Decapitate 0–2 days old neonatal mice under 10× magnification on a dissecting microscope.
-
2.
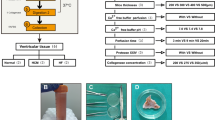
Remove the heart and place it on a 10 cm dish containing 15–20 ml of Hanks’ medium (see Fig. 1 and Note 3 ).
Fig. 1 
Schematic for isolation of neonatal cardiomyocytes and cardiofibroblasts. (a, b) Remove the hearts and place them on a 10 cm dish containing 15–20 ml of Hanks’ medium twice. (c) Slice the ventricles with a scalpel to increase the surface area for enzymatic digestion. (d) Transfer the heart tissues to a digestion bottle (blue cup bottles) containing 30 ml of Hanks’ solution with 2.5 % trypsin and shake overnight at 4 °C. (e) Cellular homogenate treated with equal amounts of DMEM supplemented with 10 % FCS. (f) Cardiofibroblasts, and (g) cardiomyocytes fractions. (h) Cardiomyocytes appearance under ×4 magnification with light microscope
-
3.
Remove surrounding tissues using a scalpel, this includes the atria (see Fig. 1).
-
4.
Transfer the heart onto a 10 cm dish containing 20 ml fresh Hanks’ medium.
-
5.
Make minor cuts on the ventricles to increase the surface area for enzymatic digestion (see Fig. 1).
-
6.
Transfer the heart tissue for digestion into 50 ml conical tubes containing 30 ml of Hanks’ medium with 2.5 % trypsin (see Note 3 ).
-
7.
Gently shake on an orbital shaker at 4 °C (usually performed in the cold room) for overnight tissue digestion.
-
8.
Add equal amounts of DMEM supplemented with 10 % FCS to stop digestion.
-
9.
Incubate in a water bath at 37 °C with agitation for 10 min.
-
10.
Discard the supernatant after incubation, and add 7.5 ml of Hanks’ medium/Collagenase II.
-
11.
Incubate at 37 °C in a water bath with agitation for further 10 min.
-
12.
Harvest the supernatant into a 50 ml conical tube containing 5 ml pre-warmed complete DMEM.
-
13.
Incubate the tube at 37 °C and 10 % CO2.
-
14.
Repeat the collagenase II digestion in step 10, four times, and each time harvest the supernatants in individual tubes as described in step 12 above.
-
15.
Spin all four tubes at 151 × g for 5 min at 4 °C.
-
16.
Discard the supernatants and resuspend the cell pellet in 2 ml of complete DMEM per tube. At this stage the solution contains both cardiomyocytes and cardiofibroblasts.
-
17.
Transfer the cell suspension from the four tubes into a 10 cm culture dish and incubate it at 37 °C and 2 % CO2 for 60 min.
-
18.
Transfer the cell suspension containing both cardiofibroblasts and cardiomyocytes into a fresh 10 cm dish. At this point, all the fibroblasts would have adhered to plastic.
-
19.
Add 100 μM BRDU and 0.1 μM vitamin C to the culture and incubate again at 37 °C and 2 % CO2.
-
20.
To the 10 cm dish with adhered cardiofibroblasts from step 17, add fresh 10 ml complete DMEM and incubate at 37 °C and 10 % CO2
-
21.
Maintain these cells in culture for approximately 72 h before proceeding with further experimentation.

3.2 Immortalization of Neonatal Mouse Cardiofibroblasts With SV40 T-Antigen
-
1.
To generate lentiviral particles; seed 1.5 × 106 HEK 293 T cells per 10 cm plate and incubate them for 24 h at 37 °C in 10 % CO2.
-
2.
Transiently co-transfect HEK 293 T cells from step 1 above with pCMVδR.2 (the packaging plasmid), pCAG4 (the mouse lentiviral receptor) and pSV40 T antigen (the immortalization construct) at a ratio of 5: 2: 3 respectively using Fugene 6 (ratio of 1: 3 DNA to Fugene 6 respectively) (see Note 4 ).
-
3.
Harvest the lentiviral supernatant from HEK 293 T cells 48 h post-transfection and store it at −80 °C or use immediately.
-
4.
Seed isolated primary cardiofibroblasts at density of 1 × 105 cells/well in a 0.1 % gelatin coated 6-well plate and incubate at 37 °C and 10 % CO2 for 24 h.
-
5.
Infect adhered cardiofibroblasts from step 4, with 3 ml of filter-sterilized lentiviral supernatant supplemented with 1× polybrene .
-
6.
Spin the cells to be infected cells at 37 °C at 808 × g for 45 min.
-
7.
Incubate the infected cells for 48 h in conditions as described above in 1.
-
8.
To the infected cells from step 7 above, add fresh DMEM containing 3 μg/ml of puromycin to select for stably transfected cells and grow cells to confluency for approximately 1–4 days.
-
9.
To maintain immortalized cells, treat cells with 2 ml of trypsin and expand plate to a gelatin free 10 cm plate along with fresh DMEM and incubate at 37 °C and 10 % CO2.
-
10.
Spin the remaining cell suspension from step 9 at 151 × g at 4 °C and add freezing mix to the pellet and store them at −80 °C for approximately 3 months and long term in liquid nitrogen (keep cells on ice for ~30 min before storage at −80 °C).
3.3 Isolation and Culture of Primary Adult Mouse Cardiomyocytes
3.3.1 Harvesting of the Mouse Heart
-
1.
Anesthetize the mouse with 5 % isoflurane inhalation and confirm anesthesia onset using pinch reflexes.
-
2.
Transfer the anesthetized mouse to a surgery area and fix it on a dissecting board (see Fig. 2).
Fig. 2 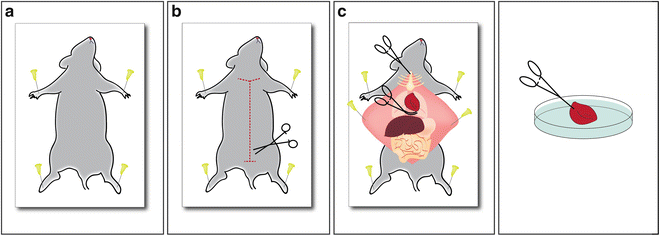
Schematic of surgical procedures of mouse heart harvesting. (a) Pin mouse on board (b) Open the mouse starting at the pubis to the chin. (c) Open the thoracic cavity by cutting through the diaphragm and the costal cartilages at their point of union. (d) Harvested heart on 10 cm petri dish ready for perfusion
-
3.
Inject it with 0.5 ml heparin (100 IU/ml PBS) intraperitoneally, and allow the heparin to circulate for 2–3 min.
-
4.
Wipe the chest with 70 % ethanol while checking pinch reflexes (see Note 3 ).
-
5.
Make a small incision at the level of the pubis for entrance of the scissors and proceed with a median longitudinal cut superiorly to the chin and separate the skin carefully from the underlying musculature (see Fig. 2b).
-
6.
Open the thoracic cavity by cutting through the diaphragm and the costal cartilages at their point of union on both sides until reaching the articulation of the sternum with the ribs. Raise the inferior part of the sternum with the clamp to expose the heart (see Fig. 2c).
-
7.
Once the heart is exposed, lift it gently using fine curved head forceps. Identify the pulmonary vessels and the aorta, cut the transverse aorta between the carotid arteries and immediately place the heart in a 10 cm dish containing 10 ml of perfusion buffer at room temperature (see Fig. 2d).
-
8.
Remove the extraneous tissues (thymus and lungs) and transfer the heart into a second 10 cm dish with perfusion buffer at room temperature.

3.3.2 Heart Cannulation, Perfusion and Enzymatic Digestion
-
1.
Place the 10 cm dish with the heart under a dissecting microscope (see Note 3 ).
-
2.
Slide the aorta onto a 27 gauge needle cannula with the help of fine tip forceps, insert the tip of the cannula just above the aortic valve as fast as possible (less than 60 s) (see Fig. 3).
Fig. 3 
Schematic for cannulation and perfusion setup. The heart is cannulated with a syringe a 27 gauge needle through the aorta and the well perfused becomes swollen and turns slightly pale, and flaccid
-
3.
Attach a small clamp to the end of the aorta on the cannula to keep the heart in place and tighten the junction with 5-0 suture nylon thread tied to the cannula (see Fig. 3).
-
4.
Set up the perfusion apparatus assembling the perfusion system with two pre-warmed buffer reservoirs at 37 °C in a water bath; one with the perfusion buffer and the other with the digestion buffer in 50 ml conical tubes (see Fig. 3 and Note 2 ).
-
5.
Keep all the tubings at 37 °C in water bath before pumping perfusion fluids through the peristaltic pump (see Fig. 3 and Note 5 ).
-
6.
Connect the outlet of the pump to a cannula open end directly above the aorta.
-
7.
Turn on the peristaltic pump and perfuse the heart with 1× perfusion buffer for 4 min at the rate of 4 ml/min to flush blood away from the vasculature, and to remove extracellular calcium so as to stop contractions (see Fig. 4, and Note 2 ).
Fig. 4 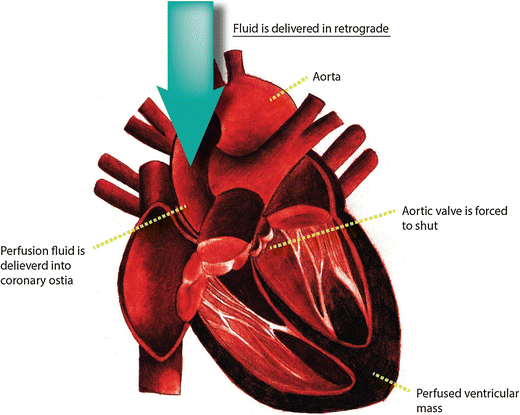
Schematic for heart perfusion. Retrograde introduction of perfusion and digestion buffers to shut the aortic valve and to allow complete ventricular perfusion
-
8.
Switch the perfusion buffer with myocyte digestion buffer and perfuse the heart for another for 3 min at the same flow rate as in step 7 above (see Notes 2 , 5 and 6 ).
-
9.
Add 15 μl of 100 mM CaCl2 to the myocyte digestion buffer reservoir and continue with the perfusion for another 8 min at 4 ml/min.
-
10.
Perfuse the heart until it appeared swollen with slight color change from red to pale (see Fig. 3).
-
11.
Stop digestion if the heart feels spongy upon pinching.
-
12.
Critical step: Turn off the peristaltic pump during the fluid swap to avoid bubble
-
13.
Cut the perfused heart just below the atria to release it from the cannula.
-
14.
Place the ventricles in a 10 cm sterile dish containing 5 ml CaCl2 and topped up with myocyte digestion buffer.
-
15.
To isolate cardiomyocytes from the ventricles, place them in a 10 cm dish containing 5 ml CaCl2 topped up with digestion buffer and cut them into small pieces.
-
16.
Transfer the cut tissues together with the enzyme buffer to a sterile digestion bottle (100 ml Schott bottle containing a 1 cm magnetic stir bar) and stir using a magnetic stirrer gently for 15 min, while keeping the bottle immersed in a 37 °C water bath to ensure further tissue digestion (see Note 3 ).
-
17.
Pass the cell suspension through a 100 μm strainer to filter out tissue debris and retain single cell suspension of cardiomyocytes (see Note 7 ).
-
18.
Transfer the suspension from step 17 above to a 10 ml yellow cap tube, and aliquot 10 μl for cell counting in a hemocytometer.
-
19.
Allow the remaining myocytes to sediment by gravity for 1–2 min at room temperature in the 10 ml tube.
-
20.
Spin the cell suspension at 151 × g for 5 min.
-
21.
Discard the supernatant and resuspend the cell pellet in 10 ml myocyte stopping buffer and add 100 μl of 200 mM ATP to the tube, up to the final concentration of 2 mM (see Note 3 ).
-
22.
Up-titrate the calcium concentration from 12.5 μM to 1.2 mM in a 3-step procedure at room temperature as described in step 23.
-
23.
Label three 10 ml tubes and with increasing calcium concentrations as follows: Tube 1: 100 μM calcium, 10 μl of 100 mM CaCl2 in 10 ml myocyte stopping buffer; Tube 2: 400 μM calcium, 40 μl of 100 mM CaCl2 in 10 ml myocyte stopping buffer; Tube 3: 900 μM calcium, 90 μl of 100 mM CaCl2 in 10 ml myocyte stopping buffer.
-
24.
Spin the myocytes at 151 × g for 5 min at 4 °C.
-
25.
Collect the cardiofibroblasts contained in the supernatant and keep it on ice and resuspend the pellet containing cardiomyocytes in each tube gently using a 1.5 mm plastic pipette (see Note 3 ).
-
26.
Allow the myocytes to stand for 2 min and spin as described in step 24.
-
27.
Repeat steps 24–26 for all the three tubes.
-
28.
Resuspend the final cell pellet in 1 ml of myocyte culturing medium and seed the cells on a laminin coated (10 μg/ml in PBS ) plate (see Note 8 ).


4 Notes
-
1.
To make 0.1 % Gelatin, add 100 mg of gelatin powder to 100 mL of 1× PBS. Dissolve the gelatin with gentle heating, and filter the solution with a 0.2 μM filter. To coat plates, add sufficient volume to cover plate surface, and incubate at 37 °C for 1 h or overnight at 4 °C. Aspirate off the gelatin before use.
-
2.
Adjust the pH of 1× perfusion buffer with sterile HCl. Equilibrate the perfusion buffer, the myocyte stopping buffer, the digestion buffer and the culture medium at 37 °C before starting heart perfusion. Filter-sterilize all the reagents with 0.22-μm filter before use.
-
3.
Strict aseptic techniques must be adhered to throughout all processes and all centrifugation fractions must be kept on ice at all times.
-
4.
For transient transfection, make a total DNA mix of 4 μg and 12 μl of Fugene 6 Transfection Reagent in a ratio of 1:3 respectively, in 100 μl of serum free media (SFM). Add Fugene to SFM but not vice versa for efficient transfection.
-
5.
Wash the perfusion set up with 40 ml of 1 M HCl, followed by 50 ml of distilled water and 40 ml of 70 % ethanol, followed by 50 ml of distilled water at a flow rate of 4 ml/min before perfusion.
-
6.
Introduce perfusion and digestion buffers in retrograde to force the aortic valve to shut and to allow the perfusion fluid to enter the coronary artery via the coronary ostia, thus resulting into complete ventricular perfusion (see Fig. 4). Turn off the peristaltic pump during the fluid swap to avoid bubble formation. The myocyte digestion buffer can be collected and reused the process is complete.
-
7.
To enhance cardiomyocytes isolation from the ventricles, mix the digestion mixture by pipetting up and down with a plastic transfer pipette.
-
8.
To make laminin at 10 μg/ml laminin, adjust concentration from 2 mg/ml stock frozen aliquot and make it up with 1× PBS for all the wells to be seeded. For coating plates (see Note 1 ).
References
Kovacs A, Papp Z, Nagy L (2014) Causes and pathophysiology of heart failure with preserved ejection fraction. Heart Fail Clin 10(3):389–398
Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O’Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulou VV, Gheorghiade M (2014) Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail 2(2):97–112
Sliwa K, Mayosi BM (2013) Recent advances in the epidemiology, pathogenesis and prognosis of acute heart failure and cardiomyopathy in Africa. Heart 99(18):1317–1322
Shiojima I (2012) Chronic heart failure: progress in diagnosis and treatment. Topics: I. Progress in epidemiology and fundamental research; 2. Molecular mechanisms of chronic heart failure. Nihon Naika Gakkai Zasshi 101(2):314–321
Vatta M, Stetson SJ, Perez-Verdia A, Entman ML, Noon GP, Torre-Amione G, Bowles NE, Towbin JA (2002) Molecular remodelling of dystrophin in patients with end-stage cardiomyopathies and reversal in patients on assistance-device therapy. Lancet 359(9310):936–941
Abbate A, Sinagra G, Bussani R, Hoke NN, Merlo M, Varma A, Toldo S, Salloum FN, Biondi-Zoccai GG, Vetrovec GW, Crea F, Silvestri F, Baldi A (2009) Apoptosis in patients with acute myocarditis. Am J Cardiol 104(7):995–1000
Gurtl B, Kratky D, Guelly C, Zhang L, Gorkiewicz G, Das SK, Tamilarasan KP, Hoefler G (2009) Apoptosis and fibrosis are early features of heart failure in an animal model of metabolic cardiomyopathy. Int J Exp Pathol 90(3):338–346
Reed BN, Sueta CA (2015) A practical guide for the treatment of symptomatic heart failure with reduced ejection fraction (HFrEF). Curr Cardiol Rev 11(1):23–32
Lee YY, Moujalled D, Doerflinger M, Gangoda L, Weston R, Rahimi A, de Alboran I, Herold M, Bouillet P, Xu Q, Gao X, Du XJ, Puthalakath H (2013) CREB-binding protein (CBP) regulates beta-adrenoceptor (beta-AR)-mediated apoptosis. Cell Death Differ 20(7):941–952
Zaragoza C, Gomez-Guerrero C, Martin-Ventura JL, Blanco-Colio L, Lavin B, Mallavia B, Tarin C, Mas S, Ortiz A, Egido J (2011) Animal models of cardiovascular diseases. J Biomed Biotechnol 2011:497841
McGonigle P, Ruggeri B (2014) Animal models of human disease: challenges in enabling translation. Biochem Pharmacol 87(1):162–171
Watkins SJ, Borthwick GM, Arthur HM (2011) The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev Biol Anim 47(2):125–131
Zordoky BN, El-Kadi AO (2007) H9c2 cell line is a valuable in vitro model to study the drug metabolizing enzymes in the heart. J Pharmacol Toxicol Methods 56(3):317–322
Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ Jr (1998) HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 95(6):2979–2984
Kimes BW, Brandt BL (1976) Properties of a clonal muscle cell line from rat heart. Exp Cell Res 98(2):367–381
Rao F, Deng CY, Wu SL, Xiao DZ, Yu XY, Kuang SJ, Lin QX, Shan ZX (2009) Involvement of Src in L-type Ca2+ channel depression induced by macrophage migration inhibitory factor in atrial myocytes. J Mol Cell Cardiol 47(5):586–594
Jacobson SL, Piper HM (1986) Cell cultures of adult cardiomyocytes as models of the myocardium. J Mol Cell Cardiol 18(7):661–678
O’Connell TD, Rodrigo MC, Simpson PC (2007) Isolation and culture of adult mouse cardiac myocytes. Methods Mol Biol 357:271–296
Acknowledgements
This work was supported by the National Health and Medical Research Council Grant No. 1085281 to HP. GWM and CN are supported by La Trobe University Post graduate Research scholarships.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Mbogo, G.W., Nedeva, C., Puthalakath, H. (2016). Isolation of Cardiomyocytes and Cardiofibroblasts for Ex Vivo Analysis. In: Puthalakath, H., Hawkins, C. (eds) Programmed Cell Death. Methods in Molecular Biology, vol 1419. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3581-9_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3581-9_10
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3579-6
Online ISBN: 978-1-4939-3581-9
eBook Packages: Springer Protocols