
Abstract
We introduce two GALAXY web servers called GalaxySite and GalaxyPepDock that predict protein complex structures with small organic compounds and peptides, respectively. GalaxySite predicts ligands that may bind the input protein and generates complex structures of the protein with the predicted ligands from the protein structure given as input or predicted from the input sequence. GalaxyPepDock takes a protein structure and a peptide sequence as input and predicts structures for the protein–peptide complex. Both GalaxySite and GalaxyPepDock rely on available experimentally resolved structures of protein–ligand complexes evolutionarily related to the target. With the continuously increasing size of the protein structure database, the probability of finding related proteins in the database is increasing. The servers further relax the complex structures to refine the structural aspects that are missing in the available structures or that are not compatible with the given protein by optimizing physicochemical interactions. GalaxyPepDock allows conformational change of the protein receptor induced by peptide binding. The atomistic interactions with ligands predicted by the GALAXY servers may offer important clues for designing new molecules or proteins with desired binding properties.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
Proteins are involved in numerous biological processes such as enzymatic activities and signal transductions [1–3]. The biological functions of proteins result from their molecular interactions with other molecules such as metal ions, small organic compounds, lipids, peptides, nucleic acids, or other proteins. Typically, proteins interact with other molecules by binding them at specific sites. Therefore, identification of the binding sites on the three-dimensional protein surfaces can be an important step for inferring protein functions [4, 5] and for designing novel molecules that control protein functions [6, 7] or designing new proteins with desired interaction properties [8, 9]. Various methods have been developed to predict ligand binding sites of proteins from protein sequences or structures. Those methods are based on geometry, energy, evolutionary information, or combinations of them [10]. Methods utilizing available experimentally resolved structures of homologous protein–ligand complexes were proven to be successful in predicting binding sites in the community-wide blind prediction experiments [11–13]. Those methods predict binding sites by transferring the available binding information for homologs, assuming that binding sites are conserved among homologs. However, methods based on evolutionary information alone may not be sufficient to predict interactions at the binding sites in atomic detail, and physicochemical interactions may have to be considered in addition.
In this chapter, we introduce two methods that predict binding sites of small organic compounds and peptides that are available on the GALAXY web server called GalaxyWEB [14]. These methods effectively search the protein structure database to find available experimental structures of related proteins complexed with ligands, build three-dimensional protein–ligand complex structures from the available information, and further refine the complex structure to go beyond the available information by optimizing physicochemical energy. The GalaxySite server predicts binding sites of small organic compounds from input protein structure or sequence [15]. Binding ligands are first predicted and the predicted ligands are then docked to the given protein structure or a predicted protein structure if sequence is given. The predicted complex structures are optimized by protein–ligand docking simulations which take into account the binding information derived from related proteins and additional physicochemical energy that do not rely on evolutionary information. GalaxySite was ranked among top methods in the recent critical assessment techniques for protein structure prediction ( CASP) experiments when evaluated in terms of predicted binding site residues [16, 17]. GalaxyPepDock predicts protein–peptide complex structures from input protein structure and peptide sequence [18]. It also combines information on interactions found in homologous complexes in the protein structure database and additional physicochemical energy to optimize the protein–peptide complex structures. The protein structure is allowed to change flexibly according to its interaction with the peptide ligand during optimization.
The method proved its usefulness in the recent critical assessment of prediction of interactions (CAPRI) experiments ([19], http://www.ebi.ac.uk/msd-srv/capri/round28/round28.html). Both GalaxySite ligand binding site prediction server and the GalaxyPepDock peptide binding site prediction server rely on similarity to the protein–ligand complexes of known structures and provide detailed protein–ligand atomic interactions by sophisticated energy optimization.
2 Materials
-
1.
A personal computer or device and a web browser are required to access the GalaxyWEB server through the Internet. A JavaScript enabled web browser is highly recommended to see the results on the web browser: The server compatibility was tested on Google Chrome, Firefox, Safari, and Internet Explorer.
-
2.
The following input materials are required to use GalaxySite and GalaxyPepDock on GalaxyWEB.
-
(a)
To run GalaxySite for ligand binding site prediction, a sequence in FASTA format or a structure file in standard PDB format for the protein of interest is required. The input target protein sequence/structure file must contain 20 standard amino acids in one/three-letter codes. The input should be a single-chain protein, and the number of amino acids should be greater than 30 and less than 500. The user may judiciously delete irrelevant protein chains or termini before job submission to meet this requirement and/or to save computational cost. An example input sequence (Fig. 1, Label 1) and structure file (Fig. 1, Label 2) can be obtained from the GalaxySite web page.
Fig. 1 
The GalaxySite input page
-
(b)
To run GalaxyPepDock for peptide binding site prediction, a structure file in standard PDB format for the receptor protein of interest and a sequence file in FASTA format for the peptide of interest are required. The number of amino acids of the receptor protein should be less than 900 and that of the peptide less than 30. The input peptide sequence file must contain 20 standard amino acids in one-letter codes. Example input files (Fig. 2, Label 1) can be obtained from the GalaxyPepDock web page.
Fig. 2 
The GalaxyPepDock input page
-
(a)


3 Methods
3.1 Ligand Binding Site Prediction Using GalaxySite
-
1.
Go to GalaxyWEB, http://galaxy.seoklab.org. Click “Site” in the “Services” tab at the top of the page.
-
2.
In the “User Information” section, enter job name (defaults to “None”). The user can provide e-mail address so that the server sends progress reports of the submitted job automatically. Otherwise, the user should bookmark the report page (Fig. 3b) after submitting the job.
Fig. 3 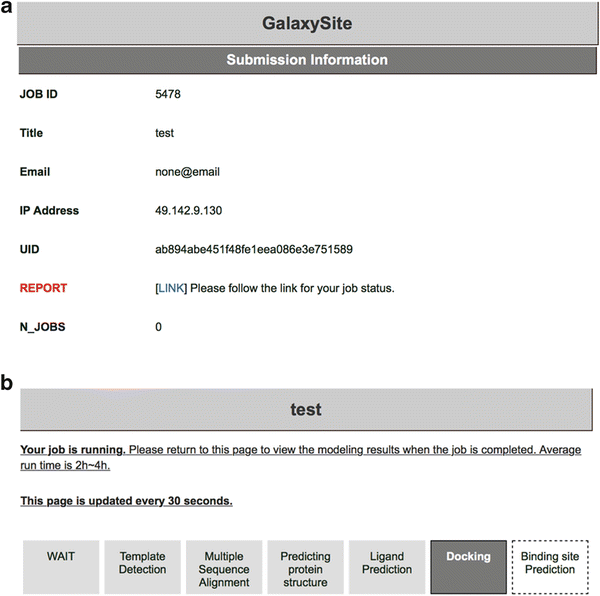
(a) A summary page showing the submission information of a GalaxySite job. (b) An example report page showing the status of the GalaxySite job
-
3.
In the “Query Protein Information” section, provide a FASTA-formatted protein sequence or a standard PDB-formatted protein structure file. If the structure of query protein has been already determined or predicted, the user may simply upload the protein structure file in PDB format (Fig. 1, Label 3). If only the sequence of the query protein is known, the user may provide a FASTA-formatted protein sequence by copying the sequence and pasting it into the text box (Fig. 1, Label 4). When sequence information is provided, the GalaxySite server predicts its protein structure by using a simplified version of GalaxyTBM [20], a template-based protein structure prediction method (see Note 1 ).
-
4.
Press the submit button to queue the job. If any errors occur with the provided input, the user will get a notice about the errors that need to be corrected. If the submission is successful, the user will be directed to the summary page of the submission information which has a link to the report page (Fig. 3a). The number of jobs in the “WAIT” or “RUN” status allowed per user is limited to three.
-
5.
Click “LINK” in the submission information page to access to the report page. The user can track the status of the submitted job in the report page which will be refreshed every 30 s (Fig. 3b). When the job is completed, predicted results will be automatically presented. Average run time of GalaxySite is 2–4 h.
-
6.
Ligands predicted to bind: GalaxySite predicts up to three ligands that are likely to bind to the target protein (see Note 2 ). The predicted ligands are presented in the descending order of the estimated likelihood of binding (Fig. 4). For each ligand, ligand name in a three-letter code (Fig. 4, Label 1) and two-dimensional chemical structure (Fig. 4, Label 2) are shown. Ligand name is hyperlinked to the ligand summary page of RCSB PDB (http://www.rcsb.org) [21] for detailed information on the molecule. PDB IDs for protein–ligand complexes used for the prediction are also provided and hyperlinked to the structure summary page of RCSB PDB (Fig. 4, Label 3).
Fig. 4 
An example of the “Ligands predicted to bind” section on the GalaxySite report page
-
7.
Predicted ligand binding residues: For each predicted ligand, information on the predicted ligand binding residues is provided (Fig. 5a). Ligand binding residues are defined from the protein–ligand complex structure obtained by molecular docking in GalaxySite (Fig 4a, Label 1). If the distance of any amino acid residue from any ligand atom is less than the sum of van der Waals radii of the two atoms + 0.5 Å, the residue is considered to bind the ligand. In addition, detailed atomic interactions between ligand and ligand binding residues are analyzed by using LIGPLOT [22] and can be seen through LINK (Fig 4a, Label 2). On the LIGPLOT page (Fig. 5b), the ligand molecule and the protein amino acid residues are depicted in violet and brown, respectively. Hydrogen bonds are shown in green dashed lines with their lengths, and hydrophobic contacts are shown in red spikes. Ideas for designing ligands or ligand binding site residues may be gained from this interaction analysis.
Fig. 5 
(a) An example of the “Predicted ligand binding residue” section on the GalaxySite report page. (b) An example of interaction analysis between ligand and ligand binding residues made by LIGPLOT. (c) An example of the “Predicted binding poses” section on the GalaxySite report page
-
8.
Predicted binding poses: For each predicted ligand, a predicted protein–ligand complex structure can be seen on the page using PV (http://biasmv.github.io/pv/), a JavaScript protein viewer, if the web browser supports JavaScript (Fig. 5c). Users can zoom in and out by scrolling mouse wheel and change the focusing center by double clicking. Different predicted protein–ligand complex structures are shown by clicking the model number in the “View in PV” line (Fig 4c, Label 3). Predicted protein–ligand complex structures can be downloaded in PDB-formatted file for further analyses (Fig 4c, Label 4).
-
9.
Re-submission with other ligands: Other ligands that are likely to bind to the query protein are listed in another table (Fig. 6). Similarly to the top three ligands with the highest estimated likelihood of binding (see step 6), ligand names, two-dimensional chemical structures, and PDB IDs for the corresponding protein–ligand complexes are shown in the table. By clicking the “Submit” button (Fig. 6, Label 1), the user can re-submit a new ligand binding site prediction job with a selected ligand.
Fig. 6 
An example of the “Re-submission with other possible ligands” section on the GalaxySite report page
-
10.
Detailed explanations on the GalaxySite web server are also provided on the GalaxySite help page; click “Help” tab at the top of the page, and then click “ GalaxySite” on the right of the help page. The prediction method used for the GalaxySite program is described in the original paper [15].




3.2 Peptide Binding Site Prediction Using GalaxyPepDock
-
1.
Go to GalaxyWEB, http://galaxy.seoklab.org. Click “ PepDock” in the “Services” tab at the top of the page.
-
2.
In the “User Information” section, enter job name (defaults to “None”). The user can provide e-mail address so that the server sends progress reports of submitted job automatically. Otherwise, the user should bookmark the report page after submitting job.
-
3.
In the “Protein–peptide Docking” section, provide a standard PDB-formatted protein structure file (Fig. 2, Label 2) and a FASTA-formatted peptide sequence file (Fig. 2, Label 3).
-
4.
Press the submit button to queue the job. If the submission is successful, a “Submission Information” page will appear (Fig. 7a).
Fig. 7 
(a) A summary page showing the submission information of a GalaxyPepDock job. (b) An example report page showing the status of the GalaxyPepDock job
-
5.
Click “LINK” of the submission information page to access the report page. The report page will be refreshed every 30 s, updating the status of the submitted job. When the job is completed, the predicted results will be presented. Average run time of GalaxyPepDock is 2–3 h (Fig. 7b).
-
6.
Predicted protein–peptide complex structures: Predicted structures of the query protein–peptide complex can be visualized on the report page using PV (http://biasmv.github.io/pv/), a JavaScript protein viewer, if the web browser supports JavaScript (Fig. 8). Users can zoom in and out by scrolling mouse wheel and change the focusing center by double clicking. Template structures selected from the database of protein–peptide complex structures to be used in the prediction are shown in light colors; protein and peptide structures are in light red and blue, respectively. Different protein–peptide complex model structures can be seen by clicking the model number in the “View in PV” line (Fig. 8, Label 1). Predicted protein–peptide complex structures can also be downloaded in PDB-formatted files for further analyses (Fig. 8, Label 2).
Fig. 8 
An example of the “Predicted protein–peptide complex structures” section on the GalaxyPepDock report page
-
7.
Additional information: Additional information on predicted models and intermediate results generated during the GalaxyPepDock run is provided in a table (Fig. 9a). Structures of protein template and peptide template are given as PDB IDs and can also be downloaded (Fig. 9a, Labels 1 and 2, respectively). Sequences and alignments of the query and the template used for the prediction are provided (Fig. 9a, Label 3) for both protein and peptide (Fig. 9b). Structure similarity between the predicted protein structure and the protein template structure is presented in terms of TM-score [23] and RMSD (Fig. 9a, Label 4). A score called interaction similarity score [18] that was designed to describe the similarity of the amino acids of the query complex aligned to the interacting residues of the template complex is reported for each prediction. This is to give an idea on the degree of the relative differences in similarity to the selected templates among different models (Fig. 9a, Label 5).
Fig. 9 
An example of the “Additional information” section on the GalaxyPepDock report page. (a) A summary table showing the results of the protein–peptide complex structure predictions. (b) An example of structure/sequence alignments between the query protein/peptide and the template protein/peptide. (c) An example of the list of predicted binding residues of protein
-
8.
Predicted binding site residues: Binding site residues of the protein taken from the predicted complex structure (Fig. 9a, Label 7 and 9c) and the estimated prediction accuracy of the binding site (Fig. 9a, Label 6) are provided (see Note 3 ). Those residues with any heavy atom within 5 Å from any peptide heavy atom in the predicted structure are reported as binding residues.
-
9.
GalaxyPepDock help page is also available; click the “Help” tab at the top of the page, and click “GalaxyPepDock” on the right of the help page. More detailed description of the prediction method of GalaxyPepDock can be found in the original paper [18].



4 Notes
-
1.
When a protein sequence is provided as input, GalaxySite predicts its protein structure first by using a simplified version of the GalaxyTBM template-based protein structure prediction program. Protein structure is required because ligand binding sites are predicted by structure-based protein–ligand docking with additional information from available protein–ligand complex structures in the database. For computational efficiency, loop/termini modeling and further refinement step employed in the original GalaxyTBM are skipped during the GalaxySite runs. If the user desires to use a protein structure predicted by the full components of GalaxyTBM, he/she can run the GalaxyTBM program on GalaxyWEB. Select “TBM” in the “Services” tab at the top of the GalaxyWEB page. The same FASTA-formatted protein sequence described in the Materials section is sufficient to run GalaxyTBM.
-
2.
Because GalaxySite predicts ligand binding sites using available protein–ligand complex structures, it cannot predict ligand binding sites if no structures for similar protein–ligand complexes are identified. In such cases, GalaxySite generates the message, “No template for binding site prediction has been found”.
-
3.
The estimated prediction accuracy in GalaxyPepDock means the estimated fraction of correctly predicted binding site residues. This value is obtained by using the linear regression data obtained from the prediction and experimental results on the PeptiDB test set [24]. A low value of estimated prediction accuracy implies that proper templates were not able to be selected, and the current similarity-based method may not provide reliable results for the query. When a very low value of estimated accuracy is returned, the user is recommended to try an ab initio protein–peptide docking method such as PEP-SiteFinder [25] that does not rely on similarity to the known structures.
References
Kristiansen K (2004) Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors: molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol Ther 103(1):21–80. doi:10.1016/j.pharmthera.2004.05.002
Negri A, Rodriguez-Larrea D, Marco E, Jimenez-Ruiz A, Sanchez-Ruiz JM, Gago F (2010) Protein-protein interactions at an enzyme-substrate interface: characterization of transient reaction intermediates throughout a full catalytic cycle of Escherichia coli thioredoxin reductase. Proteins 78(1):36–51. doi:10.1002/prot.22490
Pawson T, Nash P (2000) Protein-protein interactions define specificity in signal transduction. Genes Dev 14(9):1027–1047
Campbell SJ, Gold ND, Jackson RM, Westhead DR (2003) Ligand binding: functional site location, similarity and docking. Curr Opin Struct Biol 13(3):389–395
Kinoshita K, Nakamura H (2003) Protein informatics towards function identification. Curr Opin Struct Biol 13(3):396–400
Laurie AT, Jackson RM (2006) Methods for the prediction of protein-ligand binding sites for structure-based drug design and virtual ligand screening. Curr Protein Pept Sci 7(5):395–406
Sotriffer C, Klebe G (2002) Identification and mapping of small-molecule binding sites in proteins: computational tools for structure-based drug design. Farmaco 57(3):243–251
Damborsky J, Brezovsky J (2014) Computational tools for designing and engineering enzymes. Curr Opin Chem Biol 19:8–16. doi:10.1016/j.cbpa.2013.12.003
Feldmeier K, Hocker B (2013) Computational protein design of ligand binding and catalysis. Curr Opin Chem Biol 17(6):929–933
Tripathi A, Kellogg GE (2010) A novel and efficient tool for locating and characterizing protein cavities and binding sites. Proteins 78(4):825–842. doi:10.1002/prot.22608
Lopez G, Ezkurdia I, Tress ML (2009) Assessment of ligand binding residue predictions in CASP8. Proteins 77(Suppl 9):138–146. doi:10.1002/prot.22557
Lopez G, Rojas A, Tress M, Valencia A (2007) Assessment of predictions submitted for the CASP7 function prediction category. Proteins 69(Suppl 8):165–174. doi:10.1002/prot.21651
Oh M, Joo K, Lee J (2009) Protein-binding site prediction based on three-dimensional protein modeling. Proteins 77(Suppl 9):152–156. doi:10.1002/prot.22572
Ko J, Park H, Heo L, Seok C (2012) GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res 40(Web Server Issue):W294–W297. doi:10.1093/nar/gks493
Heo L, Shin WH, Lee MS, Seok C (2014) GalaxySite: ligand-binding-site prediction by using molecular docking. Nucleic Acids Res 42(Web Server Issue):W210–W214. doi:10.1093/nar/gku321
Gallo Cassarino T, Bordoli L, Schwede T (2014) Assessment of ligand binding site predictions in CASP10. Proteins 82(Suppl 2):154–163. doi:10.1002/prot.24495
Schmidt T, Haas J, Gallo Cassarino T, Schwede T (2011) Assessment of ligand-binding residue predictions in CASP9. Proteins 79(Suppl 10):126–136. doi:10.1002/prot.23174
Lee H, Heo L, Lee MS, Seok C (2015) GalaxyPepDock: a protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. doi:10.1093/nar/gkv495
Lensink MF, Wodak SJ (2013) Docking, scoring, and affinity prediction in CAPRI. Proteins 81(12):2082–2095. doi:10.1002/prot.24428
Ko J, Park H, Seok C (2012) GalaxyTBM: template-based modeling by building a reliable core and refining unreliable local regions. BMC Bioinformatics 13:198. doi:10.1186/1471-2105-13-198
Bernstein FC, Koetzle TF, Williams GJ, Meyer EF Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M (1977) The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol 112(3):535–542
Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8(2):127–134
Zhang Y, Skolnick J (2005) TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 33(7):2302–2309. doi:10.1093/nar/gki524
London N, Movshovitz-Attias D, Schueler-Furman O (2010) The structural basis of peptide-protein binding strategies. Structure 18(2):188–199. doi:10.1016/j.str.2009.11.012
Saladin A, Rey J, Thevenet P, Zacharias M, Moroy G, Tuffery P (2014) PEP-SiteFinder: a tool for the blind identification of peptide binding sites on protein surfaces. Nucleic Acids Res 42(Web Server issue):W221–226. doi:10.1093/nar/gku404
Acknowledgement
This work was supported by the National Research Foundation of Korea grants funded by the Ministry of Science, ICT & Future Planning (No. 2013R1A2A1A09012229).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Heo, L., Lee, H., Baek, M., Seok, C. (2016). Binding Site Prediction of Proteins with Organic Compounds or Peptides Using GALAXY Web Servers. In: Stoddard, B. (eds) Computational Design of Ligand Binding Proteins. Methods in Molecular Biology, vol 1414. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3569-7_3
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3569-7_3
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3567-3
Online ISBN: 978-1-4939-3569-7
eBook Packages: Springer Protocols