
Abstract
Optogenetic tools to control gene expression have many advantages over the classical chemically inducible systems, overcoming intrinsic limitations of chemical inducers such as solubility, diffusion, and cell toxicity. They offer an unmatched spatiotemporal resolution and permit quantitative and noninvasive control of the gene expression. Here we describe a protocol of a synthetic light-inducible system for the targeted control of gene expression in plants based on the plant photoreceptor phytochrome B and one of its interacting factors (PIF6). The synthetic toggle switch system is in the ON state when plant protoplasts are illuminated with red light (660 nm) and can be returned to the OFF state by subsequent illumination with far-red light (760 nm). In this protocol, the implementation of a red light-inducible expression system in plants using Light-Emitting Diode (LED) illumination boxes is described, including the isolation and transient transformation of plant protoplasts from Arabidopsis thaliana and Nicotiana tabacum.
The original version of this chapter was revised. The erratum to this chapter is available at: DOI 10.1007/978-1-4939-3512-3_28
An erratum to this chapter can be found at http://dx.doi.org/10.1007/978-1-4939-3512-3_28
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Plant synthetic biology
- Plant optogenetics
- Red light-inducible gene expression system
- Plant leaf protoplasts
- Arabidopsis thaliana
- Nicotiana tabacum
1 Introduction
Inducible gene expression systems in plants are essential to study cellular processes, to control target gene expression with minimal or no interference to developmental or growth processes, and for efficient large-scale biopharmaceutical production. Spatial control of gene expression in plants has traditionally been achieved by the use of tissue-specific promoters. This leads to highly specific spatial gene expression, however, once such an expression cassette has been implemented, the promoters can no longer be exogenously controlled [1]. Likewise, classical chemically inducible systems offer temporal control over gene expression in plants (such as ethanol- or dexamethasone-inducible systems [2]) but do not fulfill key requirements of inducible gene expression systems. This is due to the intrinsic limitations of chemical inducers like solubility, diffusion, inability to revert induction without washing steps, inducer-removal in sample processing and pleiotropic effects, limiting their application in vitro and in vivo and their use in long-term treatments [1, 3]. Light as a stimulus overcomes these limitations, offering advantages such as reversibility, fast reactivity, and minimal cell toxicity , therefore allowing a precise control of gene expression in a quantitative and noninvasive manner, with both high spatial and temporal resolution.
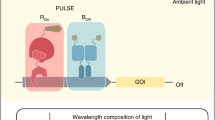
Several light-responsive gene expression systems have been developed for gene control with UVB, blue, or red light and adapted for use in mammalian cell culture and in vivo in animals (reviewed in [4, 5]). However, the application of these optogenetic tools in plants has not yet taken root, mainly due to the fact that light is essential for plant growth and development, therefore having pleiotropic effects. Thus far, only a red/far-red light-inducible system has been applied to plants [6], in principle due to its ability to revert between ON and OFF states with two different wavelengths . In this sense, this toggle switch system is unique and differs from the rest of the optogenetic tools based on photoreceptors which can be activated by light of one wavelength but can only revert to the basal, inactive state nonphotochemically, with shut off kinetics depending on their photobiological properties (dark reversion). The red/far-red light-inducible system is based on the photoreceptor Phytochrome B (PhyB) and phytochrome-interacting factor 6 (PIF6) from Arabidopsis thaliana. This system is a split transcription factor in which the components interact in a light-dependent manner. It is based on three constructs: (1) PIF6 (amino acids 1–100) fused to the mphR(A) (macrolide repressor DNA-binding protein E) and a nuclear localization sequence (NLS) ; (2) PhyB (amino acids 1–650) fused to the Herpes simplex VP16 transactivation domain and an NLS; and (3) multiple repetitions of an etr motif (cognate binding site of the E protein), placed upstream of a CMV minimal promoter followed by a reporter gene, e.g. firefly luciferase (Fig. 1a, Table 1). Upon exposure to red light, PhyB changes its conformation by photoisomerization of the covalently bound chromophore , phytochromobilin (PФB). The activated form of PhyB (Pfr) binds to PIF6 and the VP16 domain is then recruited to the etr motif in close proximity to the minimal promoter, activating transcription of the reporter gene. The PhyB-PIF6 association is readily reversed upon exposure to far-red light, when PhyB changes its conformation to the inactive form (Pr) resulting in the termination of reporter gene expression (Fig. 1b).

Design of the red light-controlled gene expression system in plants. (a) Configuration of the vectors. (b) Mode of function. Upon exposure to red light (660 nm), PhyB changes its conformation to its active form (Pfr) that allows the binding to PIF6 and therefore recruitment of the transactivator to the minimal promoter, firefly luciferase is expressed as a consequence. Under far-red (760 nm) light illumination, PhyB is converted to its inactive form (Pr), PhyB-PIF6 disassociates, thus ceasing the transcription of the reporter gene
Here we describe a protocol for a light-inducible expression system that is activated by red light and deactivated by far-red light to control gene expression in leaf protoplasts of Nicotiana tabacum and Arabidopsis thaliana . The control of gene expression with high resolution in time and space overcomes intrinsic limitations of existing systems and facilitates novel applications including the precise interrogation of complex biological signaling processes in a quantitative and noninvasive manner.
2 Materials
Prepare all solutions using double distilled water and p.a. purity grade chemicals. Use plant cell culture tested reagents for plant growth and protoplast isolation media. Prepare and store all reagents at 4 °C unless indicated otherwise.
2.1 Plant Growth
-
1.
SCN (Seedling Culture Nicotiana) (modified from [7]): 0.32 % (w/v) Gamborg B5 basal salt powder with vitamins (bioWORLD, GeneLinx International, Inc., USA), 4 mM MgSO4·7H2O, 58.4 mM sucrose a nd 0.15 % (w/v) gelrite. Mix and adjust to pH 5.8 and autoclave. After autoclaving, add 0.1 % (v/v) of Gamborg B5 Vit Mix (bioWORLD) and pour 50 ml of the medium into each Magenta Plant Culture Box (see Note 1 ).
-
2.
SCA (Seedling Culture Arabidopsis) (modified from [8]): 0.32 % (w/v) Gamborg B5 basal salt powder with vitamins (bioWORLD), 4 mM MgSO4·7H2O, 43.8 mM sucrose and 0.8 % (w/v) phytoagar in H2O. Mix and adjust to pH 5.8. Autoclave and add 0.1 % (v/v) Gamborg B5 Vit Mix (bioWORLD) then pour 50 ml of the medium into each Magenta Plant Culture Box; or alternatively add 1:2000 ampicillin and pour 50 ml of the medium into each 12 cm square plate (see Note 1 ).
-
3.
Seed sterilization solution for A. thaliana (modified from [9]): 5 % (w/v) calcium hypochlorite, 0.02 % (v/v) Triton X-100 in 80 % (v/v) EtOH. Combine the chemicals in a bottle and mix for few hours at room temperature. A precipitate will form. Place the bottle to 4 °C for storage. Allow the precipitate to settle and do not agitate the bottle before use.
-
4.
Seed sterilization solution for tobacco: 5 % active chlorine from NaOCl solution (12 % active chlorine stock solution), 0.5 % (v/v) Tween 20 in autoclaved H2O. Sterilize with a 0.22 μm filter. Prepare fresh prior to each use.
-
5.
Parafilm.
-
6.
Syringe and 22 μm filter.
-
7.
Ampicillin stock (100 mg/ml).
2.2 Protoplast Isolation and PEG Mediated Protoplast Transformation
-
1.
MMC (MES, Mannitol, Calcium) [8]: 10 mM MES, 40 mM CaCl2·H2O, add mannitol until obtaining an osmolarity of 550 mOsm (ca. 85 g/l). Adjust to pH 5.8 and filter sterilize.
-
2.
F-PIN (Fast Protoplast Incubation Nicotiana) (modified from [7]): 10 mM MES, 0.32 % (w/v) Gamborg B5 basal salt powder with vitamins (bioWORLD), 0.38 M sucrose. Adjust to pH 5.8 and filter sterilize.
-
3.
Enzyme solution stock 5 % (10× concentrated): cellulase Onozuka R10 and macerozyme R10 (SERVA Electrophoresis GmbH, Germany) in F-PIN or MMC. Weigh 10 g of cellulase and 10 g of macerozyme and dissolve in F-PIN solution or MMC (preheated to 37 °C) to a total volume of 200 ml H2O (see Note 2 ). Sterile filter the solution with a bottle-top filter into a sterile bottle and make aliquots of 2 ml. Store at −20 °C, avoid thaw–refreeze cycles.
-
4.
MSC (MES, Sucrose, Calcium) [8]: 10 mM MES, 0.4 M sucrose, 20 mM MgCl2·6H2O, add mannitol until obtaining an osmolarity of 550 mOsm (ca. 85 g/l). Adjust to pH 5.8 and filter sterilize.
-
5.
W5 solution (modified from [10]): 2 mM MES, 154 mM NaCl, 125 mM CaCl2·2H2O, 5 mM KCl, 5 mM glucose. Adjust to pH 5.8 and filter sterilize.
-
6.
MMM (MES, Mannitol, Magnesium) [8]: 15 mM MgCl2, 5 mM MES, mannitol to 600 mOsm (ca. 85 g/l). Adjust to pH 5.8 and filter sterilize.
-
7.
PEG solution: Mix 2.5 ml of 0.8 M mannitol, 1 ml of 1 M CaCl2 and 4 g PEG4000 and 3 ml H2O. Made fresh for each experiment. Not filtered, prepare fresh and place the tube at 37 °C for PEG dissolution, then use directly.
-
8.
PCA (Protoplast Culture Arabidopsis) (modified from [8]): 0.32 % (w/v) Gamborg B5 basal salt powder with vitamins (bioWORLD), 2 mM MgSO4·7H2O, 3.4 mM CaCl2·2H2O, 5 mM MES, 0.342 mM l-glutamine, 58.4 mM sucrose, glucose 550 mOsm (ca. 80 g/l), 8.4 μM Ca-panthotenate, 2 % (v/v) biotin from a biotin solution 0.02 % (w/v) in H2O (warm up the biotin solution to dissolve). Adjust to pH 5.8 and filter sterilize, add 0.1 % (v/v) Gamborg B5 Vitamin Mix and 1:2000 ampicillin to the PCA before use.
-
9.
Scalpel.
-
10.
Disposable 100 μm and 40–70 μm pore size sieve (Greiner bio-one international, Germany).
-
11.
Petri dish 94 × 16 mm.
-
12.
Parafilm.
-
13.
200 μl and 1 ml large orifice pipette tips.
-
14.
Round-bottom 15 ml Falcon tubes.
-
15.
Rosenthal cell counting chamber.
-
16.
Nontreated 6-, and 12-, or 24-well plates.
2.3 Illumination Treatment
-
1.
660 and 760 nm light-emitting diode (LED) illumination boxes.
In brief, the LED illumination boxes are custom-made boxes of PVC that exclude external light and at the same time allow gas exchange. The light boxes contain panels of LEDs (Roithner Lasertechnik GmbH, Austria) of one or several wavelengths . In addition, the irradiation intensity and illumination schemes can be set by using a programmable control unit (for full description see [11] and [12]). As an example, such a box is shown in Fig. 2. The light box is composed of three parts: a base for placing the cell culture plate, the walls, and the lid where the LEDs of specific emission wavelengths are built-in. In this protocol, boxes equipped with either red (660 nm) or far-red (760 nm) LEDs were used.

LED illumination box. (a) Illumination box for one cell culture plate. (b) Opened illumination box. The LEDs are located in the lid of the box. (c) Three components of the light box
2.4 Luminescence Reporter Assay
-
1.
Costar® 96-well flat-bottom white plate.
-
2.
Firefly luciferase substrate: 20 mM tricine, 2.67 mM MgSO4·7H2O, 0.1 mM EDTA·2H2O, 33.3 mM DTT, 0.52 mM ATP , 0.27 mM acetyl-CoA, 0.47 mM d-luciferin (Biosynth AG), 5 mM NaOH, 264 μM MgCO3·5H2O, in H2O. Prepare a beaker with a magnetic stirrer and add the components in the order as above, then add the luciferin and H2O and mix the solution, proceed with the addition of the last two components (NaOH and MgCO3·5H2O). Adjust to pH 8, aliquot the substrate in precooled black Falcon tubes and freeze them at −80 °C (see Note 3 ).
3 Methods
3.1 Seed Sterilization and Plant Material
3.1.1 Arabidopsis thaliana (Wild Type, Columbia-0)
-
1.
Seed sterilization should be done in 1.5 ml tubes in a sterile working hood. For large-scale seed sterilization, fill tubes to a maximum of approximately 250 μl volume. Avoid sterilizing a larger volume in a single tube, as results (efficiency) may vary.
-
2.
Rinse seeds multiple times with 80 % (v/v) ethanol until all large dirt and other plant particles are removed.
-
3.
Sterilize the seeds with 1 ml of the A. thaliana sterilization solution under agitation for 10 min.
-
4.
Remove the solution and replace with 1 ml of 80 % (v/v) EtOH. Incubate 5 min under agitation.
-
5.
Repeat step 4 but incubating for 2 min.
-
6.
Replace the solution with 1 ml absolute ethanol (≥99.5 %) and incubate for 1 min under agitation.
-
7.
Remove all ethanol and let the seeds dry completely under the sterile hood.
-
8.
Add autoclaved water and plate in a line on autoclaved filter paper strips (200–300 seeds/strip) placed on 12 cm square plates containing SC A medium and seal with parafilm. Multiple strips may be placed in one plate. Alternatively, place 1–16 seeds, evenly dispersed, in a Magenta Box containing 50 ml SCA medium.
-
9.
Place the plates in a growth chamber with a 16 h light regime at 22 °C. Two- to three-week old plantlets from 12 cm square plates can be used for protoplast isolation. Three- to four-week old plants grown in Magenta boxes can be used for protoplast isolation.
3.1.2 Nicotiana tabacum cv Petit Havana
-
1.
Incubate the desired number of seeds with 1 ml of seed sterilization solution for tobacco for 5 min at room temperature under agitation. Large-scale seed sterilization for N. tabacum has not been tested, due to the small amount of seeds necessary when growing plants in Magenta boxes.
-
2.
Remove the solution (centrifuge if necessary to sediment the seeds) and rinse the seeds 3–4 times with 1 ml of H2O in the same manner.
-
3.
Place one or two seeds in the middle of a Magenta Box containing 50 ml SCN medium. When more than one seed germinates, the seedlings must be separated to different boxes (around day 4–6 after germination) in order to have only one plant per box for optimal growth.
-
4.
Place the Magenta boxes in a growth chamber with a 16 h light regime at 22–25 °C (plants will grow faster at higher temperatures). Leaves from 2- to 3-week old plants can be used for protoplast isolation.
3.2 Protoplast Isolation and Polyethylene Glycol-Mediated Transformation
A. thaliana and N. tabacum protoplast isolation per flotation and polyethylene glycol-mediated transformation were performed as described before ([8] and [13], respectively) with a few alterations. All pipetting is done with wide orifice tips to avoid damaging the protoplasts. Preferentially use medium acceleration and lowest deceleration settings for the centrifugation steps (140 s acceleration and 300 s deceleration according to DIN58970).
-
1.
Cut the tobacco leaves in 1 mm strips with the abaxial surface facing up starting from the middle lamella with a sterile scalpel (see Note 4 ). Finely slice the plant leaves of A. thaliana with the scalpel in 2 ml of MMC (see Note 5 ).
-
2.
Transfer the cut leaf material into a new Petri dish containing 9 ml F-PIN (tobacco) or 7 ml of MMC (A. thaliana).
-
3.
Proceed with the enzymatic digestion of cut plant material by adding 1 ml of 10× enzyme stock solution (the final concentration of each enzyme should be 0.5 %).
-
4.
Seal the dish with parafilm and cover it with aluminum foil. Incubate overnight (12–16 h) in the dark at 22 °C.
-
5.
Carefully homogenize the digested leaf material by pipetting the leaf-enzyme mixture up and down to release the protoplasts from the plant material.
-
6.
Pass through a disposable 100 μm (tobacco) or 40–70 μm (A. thaliana) pore size sieve.
-
7.
Transfer the filtered protoplast solution to 15 ml round bottom Falcon tubes. One tube should be used for each plate of digested leaf material. The remaining steps should be completed in these tubes.
-
8.
For A. thaliana, centrifuge the filtered protoplast solution in round bottom Falcon tubes at 100 × g for 10–20 min to sediment the protoplasts. Remove supernatant and resuspend in 10 ml of MSC. For tobacco protoplasts, centrifugation is not necessary, as the flotation of protoplasts can be done directly in the F-PIN solution.
-
9.
Very carefully overlay 10 ml of protoplast solution with 2 ml of MMM (see Note 6 ).
-
10.
For A. thaliana protoplasts, centrifuge for 10 min at 80 × g for accumulation of the protoplasts at the interphase of MSC and MMM. For tobacco protoplasts, instead of centrifugation, incubate the tubes at room temperature for 20–30 min, in which time the protoplasts will float to the interphase of F-PIN and MMM (see Note 7 ).
-
11.
Collect the protoplasts at the interphase and transfer into a new Falcon tube with 7 ml of W5 solution. For each floatation tube to be used, prepare two W5-filled collection Falcon tubes. Multiple rounds of protoplast collection can be done (if necessary overlay again with MMM) until no further protoplasts float to the interphase or enough protoplasts are obtained.
-
12.
Centrifuge the collected protoplasts for 10 min at 100 × g to pellet and resuspend in a defined volume of W5 for counting (see Note 8 ).
-
13.
Determine the cell density using a Rosenthal cell counting chamber.
-
14.
Sediment the protoplasts by centrifuging for 5 min at 80 × g. Discard supernatant and adjust with MMM solution to a density of 5 × 105 cells/ml for tobacco and 5 × 106 cells/ml for A. thaliana.
-
15.
-
(a)
For the transformation of tobacco protoplasts, prepare 50 μg of DNA in H2O (see Note 9 ) in a round bottom Falcon tube and add 1 ml of the protoplasts in MMM. Carefully mix by pipetting and incubate for 5 min.
-
(b)
For A. thaliana protoplasts, prepare 15–30 μg of DNA in H2O (see Note 9 ) adjusted to a maximum volume of 20 μl (volume adjustment with MMM). Transfer the 20 μl DNA solution to the rim of a well of a 6-well culture plate (slightly tilt the plate for easier pipetting in the following steps). Dispense 100 μl of the protoplast solution to each well with DNA and mix by gentle pipetting. Incubate for 5 min.
-
(a)
-
16.
-
(a)
For tobacco protoplast transformation, add 1 ml PEG4000 solution to the protoplasts in a drop-wise manner with a tip-in-tip method while slowly rotating the Falcon tube (see Note 10 ). After 8 min (see Note 11 ), consecutively add 1, 2, 3, and 4 ml of W5 per minute to the tube as a stepwise dilution of the transformation, and gently tilt the tube after each step for mixing.
-
(b)
For A. thaliana protoplast transformation, gently shake the 6-well plate from side to side to distribute the protoplasts and DNA along the rim before directly adding 120 μl of PEG4000 solution drop-wise, tip-in-tip. Do not mix after the addition of PEG. Incubate for 8 min (see Notes 11 and 12 ) and quickly add 120 μl of MMM and, directly afterwards, at least 1.2 ml of PCA. Gently mix by tilting the plate after the addition of PCA (final volume should be at least 1.6 ml).
-
(a)
-
17.
Only for tobacco, sediment the cells at 5 min at 80 × g, discard the supernatant and resuspended in at least 1.6 ml PCA.
-
18.
After transformation, if only one condition is to be tested, leave the A. thaliana protoplast suspension in a well of a 6-well plate. In the case of tobacco protoplasts transfer the 1.6 ml from the tube into a well of a 6-well plate.
If more than one condition is to be tested, split the protoplasts in different plates according to the number of light conditions to be assayed. The volume pipetted to each well in the new plates will depend on the number of replicates per condition. Considering that 25,000 protoplasts (see below) will be used for each measurement (80 μl protoplast suspension), it follows that for 6 replicates 150,000 protoplasts are needed, amounting to 480 μl protoplast suspension. Scale down to 12- or 24-well plate to avoid high evaporation rates. Seal the plate(s) with parafilm.
3.3 Illumination Treatment and Reporter Assay
-
1.
After transformation of the protoplasts, illuminate the plates with the appropriate wavelength (i.e. 660, 760 nm) and intensity of light with LED arrays, or incubate in the dark prior to reporter quantification. The spectra of the LEDs and the radiation intensity can be determined with a spectroradiometer (e.g. AvaSpec-ULS2048-USB2 FC/PC and FC-UVIR200-2-ME-1FCPC, Avantes, Netherlands).
As an example, Fig. 3a shows time-course and dose-response curves for the red light-inducible gene expression system. Protoplasts were isolated from A. thaliana plantlets and 10 μg of each plasmid (pMZ827, pMZ828 and pROF100) were transformed into the protoplasts. Several transformations were made in parallel (22 transformations) and after transformation all the protoplasts were pooled. Aliquots of 3.5 ml of the protoplasts suspension were transferred into one well of seven different 6-well plates (one plate for each illumination condition). The luminescence determination was made for each condition at different points in time (0, 6, 12, 18, and 24 h). As a dark control, 1 ml of protoplast suspension was transferred into one well of four different 24-well plates. In this way, a single plate per time point was used and accidental exposure of the plate to ambient light avoided. The results of the kinetics and expression levels of the red light-inducible system in A. thaliana protoplast depicted in Fig. 3a indicate between 1 and 4 μE/m2/s as optimal illumination conditions for maximum expression rates. The highest expression levels are achieved at 24 h but a better dynamic range (399 and 395 × fold induction) is obtained at 18 h of gene expression for 2–4 μE/m2/s red-light intensities (Fig. 3b). It is, however, recommended to adjust the protocol to the application of interest.

Time- and dose-response curves of the red light inducible gene expression system in protoplasts of A. thaliana. Protoplasts from A. thaliana were transformed for red light–inducible firefly luciferase expression (pMZ827, pMZ828, and pROF100). After transformation, 3.5 ml aliquots of protoplast suspensions containing approximately 1.09 × 106 protoplasts, were illuminated either at different intensities of 660 nm (0.5, 1, 2, 4, 8, and 16 μE/m2/s), at 760 nm (17 μE/m2/s) light, or were kept in the dark as a control. (a) Samples were taken at the indicated points (0, 6, 12, 18, and 24 h after transformation) and firefly luciferase expression was determined. Fig. 3 (continued) The graph shows the reporter luminescence values at different time points and different illumination conditions. (b) Reporter luminescence values after 18 h expression at the indicated light intensities. Data are means ± SEM (n = 6 technical replicates)
-
2.
To determine reporter expression, first gently mix the protoplast suspension with the pipette and transfer 80 μl (25,000 protoplasts) into a Costar® 96-well flat-bottom white plate, including 4–6 replicates for each condition (see Note 13 ).
-
3.
Add 20 μl of firefly luciferase substrate and monitor the luminescence in a plate reader [14]. 10 s of shaking plate for homogeneous substrate availability and directly luminescence measurement for 20 min kinetics (interval of 2 min) are advisable.
4 Notes
-
1.
Prepare the plates or Magenta Plant Culture boxes directly after autoclaving because the gelrite and phytoagar will not dissolve upon reheating.
-
2.
Both enzyme extracts are not to be inhaled and are poorly soluble. For these reasons: solve under a fume hood by adding 10 ml of prewarmed (37 °C) MMC/F-PIN to each bottle directly, shake, and pour into beaker and rinse bottles repeatedly. Fill beaker to 200 ml. The solution will not be clear, should, however, be a clear brown after filtration.
-
3.
For certain solutions, a stock solution can be prepared in advance; however, tricine, DTT, ATP , and acetyl-CoA should be prepared fresh. From the addition of DTT on, all steps should be performed under a fume hood. Moreover, luciferin is sensitive to light, oxygen, and high temperature so that from its addition on, the preparation should be performed in darkness and as quickly as possible. Due to the high price of acetyl-CoA, it is also preferable to purchase this substrate in small amounts (50 mg for the preparation of 200 ml of firefly luciferase substrate) and use the entire content in a single preparation of substrate to avoid freeze–thaw cycles and waste.
-
4.
Choose healthy leaves not showing nutritional deficiency, chlorosis, or mechanical damage.
-
5.
A. thaliana plant material grown in plates should be carefully cut from the plate with a scalpel in a way that avoids including roots and seeds, and should then be cut finely into small pieces. When cutting the plant material from Magenta Plant Culture Boxes, take only the leaves and either cut them in strips as described for tobacco or slice them finely. Sterile featherweight forceps are helpful in holding A. thaliana leaves from Magenta boxes to be cut in strips without inflicting damage to them.
-
6.
Gentle inversion of the tube before adding the MMM solution slowly helps for a clear separation of phases. For addition of MMM use a tip-in-tip technique i.e. placing a smaller tip into the tip of a bigger tip for a slow solution dispense.
-
7.
Collecting the first band of protoplasts at the interphase after 10–15 min increases the speed of protoplast floatation.
-
8.
Protoplasts will not be successfully pelleted if the collection tube contains less W5 than MMM.
-
9.
DNA amounts mentioned in the protocol are total amounts of DNA. When more than one plasmid is used, the amounts of each plasmid mus t be adjusted proportionally, keeping the total DNA amount constant. Purify the plasmid DNA using midiprep or maxiprep kits and check the quality of the plasmid DNA by agarose gel electrophoresis (e.g. RNA content).
-
10.
If the PEG has sedimented to the bottom of the tube, mixing by gently tilting the tube will be necessary.
-
11.
The duration of PEG treatment is critical in the transformation; the suggested 8 min treatment leads to high transformation efficiency in our experience.
-
12.
Gently shaking the plate side to side before PEG addition avoids the aggregation of protoplasts.
-
13.
It is recommended to pipette the protoplasts in the following order: 660 nm (highest to low intensities)—760 nm—dark, as the system is rapidly activated by ambient light. Due to the sensitivity of the system, it is also recommended to work in a darkroom with green safelight emitted by LEDs (~520 nm). Green light illumination at moderate intensities does not lead to noticeable activation of PhyB.
References
Corrado G, Karali M (2009) Inducible gene expression systems and plant biotechnology. Biotechnol Adv 27(6):733–743
Junker A, Junker B (2012) Synthetic gene networks in plant systems. In: Weber W, Fussenegger M (eds) Synthetic gene networks, vol 813, Methods in molecular biology. Humana, New York, pp 343–358
Padidam M (2003) Chemically regulated gene expression in plants. Curr Opin Plant Biol 6(2):169–177
Zhang K, Cui B (2015) Optogenetic control of intracellular signaling pathways. Trends Biotechnol 33(2):92–100
Müller K, Naumann S, Weber W, Zurbriggen MD (2015) Optogenetics for gene expression in mammalian cells. Biol Chem 396(2):145–152
Müller K, Siegel D, Rodriguez Jahnke F, Gerrer K, Wend S, Decker EL, Reski R, Weber W, Zurbriggen MD (2014) A red light-controlled synthetic gene expression switch for plant systems. Mol BioSyst 10(7):1679–1688
Dovzhenko A, Bergen U, Koop HU (1998) Thin-alginate-layer technique for protoplast culture of tobacco leaf protoplasts: shoot formation in less than two weeks. Protoplasma 204(1-2):114–118
Dovzhenko A, Dal Bosco C, Meurer J, Koop HU (2003) Efficient regeneration from cotyledon protoplasts in Arabidopsis thaliana. Protoplasma 222(1–2):107–111
Luo Y, Koop H-U (1997) Somatic embryogenesis in cultured immature zygotic embryos and leaf protoplasts of Arabidopsis thaliana ecotypes. Planta 202(3):387–396
Menczel L, Galiba G, Nagy F, Maliga P (1982) Effect of radiation dosage on efficiency of chloroplast transfer by protoplast fusion in nicotiana. Genetics 100(3):487–495
Müller K, Zurbriggen MD, Weber W (2014) Control of gene expression using a red- and far-red light-responsive bi-stable toggle switch. Nat Protoc 9(3):622–632
Müller K, Engesser R, Metzger S, Schulz S, Kämpf MM, Busacker M, Steinberg T, Tomakidi P, Ehrbar M, Nagy F, Timmer J, Zubriggen MD, Weber W (2013) A red/far-red light-responsive bi-stable toggle switch to control gene expression in mammalian cells. Nucleic Acids Res 41(7):e77
Koop H-U, Steinmüller K, Wagner H, Rößler C, Eibl C, Sacher L (1996) Integration of foreign sequences into the tobacco plastome via polyethylene glycol-mediated protoplast transformation. Planta 199(2):193–201
Wend S, Bosco CD, Kämpf MM, Ren F, Palme K, Weber W, Dovzhenko A, Zurbriggen MD (2013) A quantitative ratiometric sensor for time-resolved analysis of auxin dynamics. Sci Rep 3:2052
Acknowledgments
This work was supported in part by the Excellence Initiative of the German Federal and State Governments (EXC294-BIOSS, GSC-4 Spemann Graduate School (SGBM)) and the Alexander von Humbolt Foundation (research Grant no. 1141629). We thank Susanne Knall and Frauke Bartels-Burgahn for experimental assistance. We thank J. Schmidt, D. Schächtele and J. Meßmer (University of Freiburg) for designing and constructing the illumination boxes.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Ochoa-Fernandez, R. et al. (2016). Optogenetics in Plants: Red/Far-Red Light Control of Gene Expression. In: Kianianmomeni, A. (eds) Optogenetics. Methods in Molecular Biology, vol 1408. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3512-3_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3512-3_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3510-9
Online ISBN: 978-1-4939-3512-3
eBook Packages: Springer Protocols