
Abstract
In order to characterize genetically encoded tools under the most relevant conditions, the constructs need to be expressed in the cell type in which they will be used. This is a major hurdle in developing optogenetic tools for neuronal cells, due to the difficulty of gene transfer to these cells. Several protocols have been developed for transfecting neurons, focusing on improved transfection efficiency. However, obtaining healthy cells is as important. We monitored transfected cell health by measuring electrophysiological parameters, and used them as a guideline to optimize transfection. Here we describe an optimized transfection protocol that achieves reasonably high efficiency (10–20 %) with no discernable impact on cell health, as characterized by electrophysiology.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
1 Introduction
When developing genetically encoded tools, it is critical to assess the level of expression, proper folding, and trafficking of the protein construct in the cell type that they will be used. Systematic metagenomic screens that characterized microbial opsin homologues from a wide range of archaea and alga in mammalian neurons found that successful expression and trafficking of these proteins are key to high-performance tools [1, 2]. This was also true for tool optimization; screens based on primary neurons [3] resulted in dramatic further improvement of genetically encoded calcium indicators which had been extensively optimized using bacteria-based screens [4]. Performing screens in primary neurons is especially challenging, due to the difficulty of gene transfer into neuronal cells. To tackle this challenge, several transfection methods, based on electrical, chemical, or viral gene transfer approaches have been developed [5, 6]. Many of these studies focused on improved transfection efficiency, but reducing toxicity to obtain healthy cells is also critical. Objective criteria on neuronal cell health may be obtained from electrophysiological characterizations, by measuring properties such as resting potential, membrane resistance, and threshold for generating action potentials. We have found that DNA transfection mediated by calcium-phosphate precipitation results in healthy neurons as characterized by electrophysiological properties, and have successfully used it to identify highly efficient channelrhodopsins [2]. The transfected cells using this approach yielded hippocampal mouse neurons that routinely formed giga-ohm seals in whole-cell patch clamp, have membrane capacitance around 50–60 pF, membrane resistance higher than 250 MΩ, and resting potential at −65 mV [2], which are typical values for untransfected control neurons. The method allows transfection of plasmid DNA regardless of its size, and is labor- and cost-effective compared to viral vectors . The transfection efficiency ranges between 10 and 20 %. In addition, we identify specific parameters to tune for improved efficiency.
The transfection method is based on a previous report [7], but has been optimized with fully defined components, instead of relying on commercial kits. As indicated in the previous study [7], the formation of uniform and small calcium phosphate precipitates, and their removal after transfection were found to be critical. We optimized the amount of DNA, calcium chloride concentration, and phosphate concentration for forming uniform small precipitates. We also identified a slightly lower pH wash buffer (pH 6.9) that effectively dissolves all calcium phosphate precipitates after transfection. The protocol has been optimized using primary mouse hippocampal neurons, and has been tested in cortical neurons.
2 Materials
2.1 Neuronal Culture
-
1.
24-well plate.
-
2.
Hemocytometer.
-
3.
Round coverglass (25 mm in diameter, 0.15 mm in thickness), autoclaved.
-
4.
Matrigel: Make 250 μL aliquots. Store at −20 °C. Thaw one vial and dilute in 12 mL Dulbecco’s modified eagle medium (DMEM) (without phenol red). Diluted solution can be stored at 4 °C.
-
5.
Culture Medium (500 mL): 450 mL minimum essential media (MEM) (without phenol red); 5 g/L Glucose (add 2.5 g), 0.1 g/L Transferrin (Bovine holotransferrin, add 50 mg), 2.38 g/L HEPES (add 1.19 g), 2 mM l-Glutamine, 0.025 g/L Insulin (Bovine pancreas ), 50 mL Heat Inactivated Fetal Bovine Serum, 10 mL B27 Supplement. Adjust pH to 7.3 using 10 M NaOH, sterilize with 0.2 μm filter, make 40 mL aliquots, store at −20 °C (see Note 1 ).
-
6.
AraC solution (500 mL): 500 mL MEM, 5 g/L Glucose, 50 mg Transferrin, 1.19 g HEPES, 0.5 mM l-Glutamine, 10 mL B27 Supplement, 4 μM Cytosine β-d-arabinofuranoside hydrochloride (AraC), 25 mL Heat Inactivated Fetal Bovine Serum. Adjust pH to 7.3–7.4, sterilize with 0.2 μm filter, make 40 mL aliquots, store at −20 °C.
2.2 Transfection
-
1.
Transfection buffer (500 mL): 500 mL MEM (without phenol red). pH to 7.15, sterilize with 0.2 μm filter, store at 4 °C (see Note 2 ).
-
2.
Calcium chloride solution (50 mL): 50 mL double-deionized H2O, 2 M CaCl2 (add 14.7 g). Sterilize with 0.2 μm filter, make 1 mL aliquots, store at −20 °C.
-
3.
Sodium phosphate solution (10 mL): Start with about 8 mL double-deionized H2O, 1.5 M Na2HPO4·7H2O (add 4.02 g). Add double-deionized H2O to final volume of 10 mL. Sterilize with 0.2 μm filter, make 1 mL aliquots, store at −20 °C (see Note 3 ).
-
4.
2× HBS solution (50 mL): 50 mL double-deionized H2O, 50 mM HEPES (add 0.65 g), 280 mM NaCl (add 0.8 g), 1.5 mM sodium phosphate (add 50 μL of sodium phosphate solution). pH to 7.0, sterilize with 0.2 μm filter, make 1 mL aliquots, store at −20 °C.
-
5.
Sterile water: Filter sterilize double-deionized H2O, make 1 mL aliquots, store at 4 °C.
-
6.
Wash buffer (500 mL): 500 mL MEM (without phenol red). pH to 6.9, sterilize with 0.2 μm filter, store at 4 °C.
3 Methods
3.1 Neuronal Culture
-
1.
Round coverglasses are placed into each well of a 24-well plate. 75 μL of diluted matrigel is added at the center of coverglass to form a droplet. Transfer the plate carefully to a 37 °C 5 % CO2 incubator without disturbing the droplets, and incubate for 2 h or longer.
-
2.
Remove the matrigel by aspiration, and dry for 30 min with lid open in a biosafety cabinet.
-
3.
Procedure for preparing dissociated hippocampus follows previously published protocols [8, 9], but uses the culture media described here. 75 μL of dissociated mouse hippocampus (at a density of 50,000 total cells per well) in culture medium are added at the center of coverglass and incubated for 3 h in a 37 °C 5 % CO2 incubator.
-
4.
After incubation, the cells are inspected under a microscope to check for adhesion, and 1 mL of prewarmed culture media is added to each well. Cells are placed in a 37 °C 5 % CO2 incubator.
-
5.
After 1–2 days the glial cells cover 50–70 % of the coverglass, and axonal projections of neurons are visible. At this stage, 1 mL AraC solution is added to each well to inhibit glial proliferation. The cells can be transfected starting 1–2 days after this step.
3.2 Transfection
-
1.
For transfection, total of 1.25 μg of DNA is required for each well (see Note 4 ).
-
2.
Prewarm sterile water, calcium chloride, and 2× HBS to room temperature.
-
3.
Remove culture media from wells using a serological pipette, and collect in a 50 mL tube. The collected culture media is placed in a 37 °C bath, and will be added back after the transfection is completed (see Note 5 ).
-
4.
Add 0.5 mL of transfection buffer to each well, and place the plate in a 37 °C 5 % CO2 incubator.
-
5.
For each well, prepare the following transfection mix according to Table 1:
Table 1 Preparation of transfection mixture -
6.
For each transfection mix, prepare equal volume of 2× HBS in a 1.5 mL tube. Take the cells out from the incubator.
-
7.
Transfer transfection mix into the 1.5 mL tube containing equal volume of 2× HBS and mix by pipetting 10 times to start calcium phosphate precipitation. Incubate the mixture for 30 s without mixing (see Note 6 ).
-
8.
Add 50 μL of the transfection mix plus HBS drop-wise to each well (see Note 7 ).
-
9.
Repeat steps 7 and 8 until all transfection mix are added. Transfer the plate into a 37 °C 5 % CO2 incubator, and incubate for 20–30 min (see Note 8 ).
-
10.
After incubation, view cells under the microscope to check calcium phosphate precipitate size (Fig. 1).
Fig. 1 
Bright-field microscope images of small and even calcium phosphate precipitates. Since the precipitates are hard to visualize in neuronal cultures , HeLa cells are shown to give a sense of precipitate size. (a) Untransfected HeLa cells. (b) HeLa cells being incubated with calcium phosphate precipitates. Small black dots covering the cells are the precipitates. Scale bars indicate 20 μm
-
11.
Remove the transfection mixture by aspiration, and add 1 mL wash buffer drop-wise to each well. Incubate cells in wash buffer for 10 min in a 37 °C 5 % CO2 incubator. After this incubation calcium phosphate precipitates should be completely dissolved when visualized under a microscope. If the precipitates remain, repeat this wash step.
-
12.
Remove wash buffer by aspiration, and add 1.5 mL of culture media collected from step 3 to cells. Add 0.5 mL of fresh culture media to bring the total volume to about 2 mL.
-
13.
The transfected cells remain healthy in a 37 °C 5 % CO2 incubator for 2 weeks or longer. Typically for high-expressing proteins, expression is detected after 1–2 days. For well-expressing microbial opsins, expression is detected after 1–2 days, and reach optimal levels after a week (Fig. 2).
Fig. 2 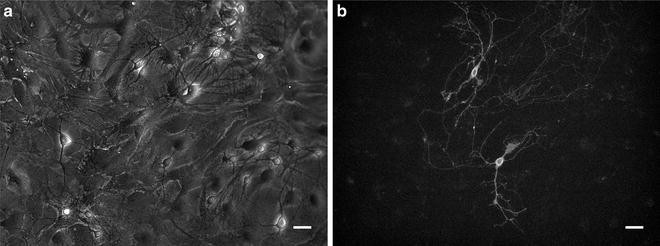
Primary mouse hippocampal neurons transfected with channelrhodopsin-2 (ChR2) fused with EGFP. (a) Bright-field image. (b) ChR2 expression visualized by EGFP fluorescence. Images were taken 1 week after transfection. Scale bars indicate 20 μm


4 Notes
-
1.
The pH of plating medium is critical for neuronal health. Measure pH of an aliquot again after filtration to make sure pH is at 7.3.
-
2.
The pH of the transfection buffer is important for generating even and small precipitates. If the pH is low, no precipitate will form, and if too high, large aggregates tend to form.
-
3.
This sodium phosphate solution is prepared to accurately control the phosphate concentration in the 2× HBS solution. The phosphate concentration is critical for obtaining small and even calcium phosphate precipitation [10].
-
4.
The DNA is typically a plasmid, or can be a mixture of multiple plasmids. DNA in double-deionized water works best and the concentration should be higher than 100 ng/μL.
-
5.
The culture media at this point may contain factors released by cells, and therefore is kept for use after transfection.
-
6.
It seems better not to disturb the mixture during this step for obtaining small uniform precipitates. Incubation over 30 s is not necessary.
-
7.
Drop-wise addition of the transfection mix plus HBS is critical for forming uniform precipitates. When doing this, the pipette tip should be about 1 in. above the liquid surface, so that the mixture is added in droplets.
-
8.
This incubation time can be extended to 1 h to improve transfection efficiency, but it tends to increase toxicity to neurons.
References
Chow BY, Han X, Dobry AS et al (2010) High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463(7277):98–102
Klapoetke NC, Murata Y, Kim SS et al (2014) Independent optical excitation of distinct neural populations. Nat Methods 11(3):338–346
Chen TW, Wardill TJ, Sun Y et al (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499(7458):295–300
Akerboom J, Chen TW, Wardill TJ et al (2012) Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci 32(40):13819–13840
Karra D, Dahm R (2010) Transfection techniques for neuronal cells. J Neurosci 30(18):6171–6177
Washbourne P, McAllister AK (2002) Techniques for gene transfer into neurons. Curr Opin Neurobiol 12(5):566–573
Jiang M, Chen G (2006) High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat Protoc 1(2):695–700
Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1(5):2406–2415
Beaudoin GM 3rd, Lee SH, Singh D et al (2012) Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7(9):1741–1754
Jordan M, Schallhorn A, Wurm FM (1996) Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res 24(4):596–601
Acknowledgements
This work was funded by the University of Connecticut and the Brain and Behavior Research Foundation (NARSAD Young Investigator grant). Shiyao Wang was partially supported by the outstanding scholars program of the UConn graduate school.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Wang, S., Cho, Y.K. (2016). An Optimized Calcium-Phosphate Transfection Method for Characterizing Genetically Encoded Tools in Primary Neurons. In: Kianianmomeni, A. (eds) Optogenetics. Methods in Molecular Biology, vol 1408. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3512-3_16
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3512-3_16
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3510-9
Online ISBN: 978-1-4939-3512-3
eBook Packages: Springer Protocols