
Abstract
Bioinformatic sequence analysis allows the functional characterization of newly sequenced proteins. Nonribosomal peptide synthetases (NRPSs) are multi-modular enzymes involved in the biosynthesis of natural products. The current omics era has enabled the exponential growth of the sequenced NRPS, and it is important to characterize the final product of these synthetases. Here, how to achieve the prediction of substrates which bind to adenylation domains in NRPS with NRPSsp (www.nrpssp.com) bioinformatic tool is described.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Nonribosomal peptide synthetases (NRPSs) are multi-modular enzymes which biosynthesize many important peptide compounds. These natural products are relevant for the industrial microbiology area and new bioinformatics protocols are being developed for their characterization. The minimal modules which form a NRPS contain the following domains: A-activation (performs the amino acid selection and adenylation), T-thiolation or acyl carrier domain (for the thioesterification), and C-condensation domain (peptide bond formation) [1]. It has been shown that the A-domain recruits a particular type of substrate and the other domains allow the elongation of the nascent peptide. Thus, the primary composition of the final polypeptide is determined by the sequential order of the A-domains along the NRPS.
Until a few years ago, the characterization and identification of NRPS metabolites were a complex process which implies the isolation, purification, and identification of bioactive molecules in order to decipher their chemical structure. Nowadays, the advances of next-generation sequencing technologies and biological databases have enabled the development of new computational biology techniques for their characterization. In particular, structural biology, sequence analysis, and machine learning have been applied to the characterization of NRPS substrates. The first main contribution was performed using the 3D structure of the adenylation domain of gramicidin synthetase A (GrsA) [2] linked to the phenylalanine amino acid. It enabled the identification of key residues in the active site pocket of the A-domain. These key residues were proposed as a specificity-conferring code for the prediction of NRPSs-binding substrates [3] and enabled the development of computational methods which predict the binding constitutive substrates and the biochemical structure of the final product.
These methods perform the analysis in two steps: (1) The constituent domains of NRPS modules are identified and the sequence of adenylation domains is extracted. Hidden Markov models (HMM) have been applied for this purpose. They allow the construction of profiles from a multiple alignment of sequences and the novo identification of these domains in an input sequence. Databases such as Pfam or InterPro have precomputed the characteristic profiles of NRPSs. These profiles are available as hidden Markov models which can be used with HMMER for the domain identification of NRPSs (PF00501 correspond with the adenylation domain). (2) Key residues or whole sequence of adenylation domains is analyzed with machine learning or classification methods, and a prediction of binding substrates is provided. For example, support vector machines have been used in the Web server NRPSpredictor2 [4]. Recent studies have proposed the use of HMM for the prediction as an alternative method to SVM [5, 6].
In this book chapter, a methodology which uses HMM for the characterization of NRPSs substrates is described. It explains how the adenylation domains are extracted and analyzed from a FASTA sequence by means of a Web server and stand-alone commands.
2 Materials
2.1 Web Site Execution
-
1.
Prepare your sequence(s) in a FASTA format file. FASTA format is a text-based format for representing peptide sequences, in which amino acids are represented using single-letter codes. The format also allows the specification of sequence names and comments which precede the sequences. The FASTA format may be used to represent either single sequences or many sequences in a single file. A set of single sequences constitute a multisequence file. The first line of each sequence in a FASTA file starts with a “>” (greater-than) symbol. Following the initial line (used for a unique description of the sequence), the protein sequence is provided in standard one-letter code. NRPSsp allows multisequence files and the analysis is performed for all the sequences in the file.
2.2 Stand-Alone Execution
-
1.
Download the HMM of the AMP-binding domain (ID = PF00501, http://pfam.sanger.ac.uk/family/PF00501/hmm).
-
2.
Download the HMMs of NRPSsp which enable the classification (http://www.nrpssp.com/downloads/nrpshmms).
-
3.
Install HMMER 3 software (http://hmmer.org/) in your local machine. Follow the instructions on its Web site.
-
4.
Prepare your input sequence(s) in a FASTA format file.
3 Methods
3.1 Web Site Execution
-
1.
Paste your sequence(s) or upload your FASTA file with the Web form of NRPSsp and click on the “Run Analysis” button (Fig. 1, vignette 1).
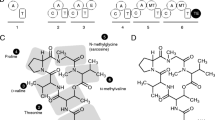
Fig. 1 
NRPSsp analysis example: (1) Sequence of proteins was pasted in the text area and the “Run Analysis” button was clicked on to launch the predictor. (2) Results calculated by the application were shown. Each table corresponds to a sequence analyzed. CVS-formatted results and HMM alignments can be downloaded from this page
-
2.
Once the analysis is performed, the results page is displayed (Fig. 1 vignette 2). It presents one table for each protein sequence and these tables have one row for each adenylation domain that have been identified in the sequences. The following information is provided for each result analysis:
-
Protein identifier: The sequence description which has been provided in the input FASTA file.
-
Adenylation domain start position: The position of the sequence where the analyzed adenylation domain starts.
-
Adenylation domain end position: The position of the sequence where the analyzed adenylation domain ends.
-
Substrate: The predicted substrate which binds with the adenylation domain.
-
Substrate name: The name of the predicted substrate.
-
Score: This is the HMMER bit score. The bit score tells how the input adenylation domain matches the HMM of the predicted substrate. It reflects whether the sequence is a better match of the profile model (positive score) or of the null model of nonhomologous sequences (negative score).
-
Prediction-conditioned fallout: This value is estimated as False Positives/True Positives + False Positives. The calculation is based on the error obtained in the LOO test for a particular score. It represents the estimated error that occurs for the resulting score. The font color reflects the reliability of the result (red = low, yellow = medium, and green = high).
-
Alignments: This links to the resulting HMMER alignments which have been performed to obtain this result.
-

3.2 Stand-Alone Execution
-
1.
Run HMMER in order to locate adenylation domains with the following command:
# Prepare the HMM database.
hmmpress AMP-binding.hmm
# Scan adenylation domains in the input sequences.
hmmscan --domtblout AMP_Domains_Coords.txt AMP-binding.hmm inputProteins.fasta > outputDomainsAlign.txt
-
2.
Extract the adenylation sequence.
# Get a FASTA file with adenylation sequences.
grep -v "^#" AMP_Domains_Coords.txt | awk '{print $4"/"$20"-"$21, $20, $21, $4}' | esl-sfetch -Cf inputProteins.fasta - > inputADomains.fasta
-
3.
Run HAMMER in order to classify the adenylation domain.
# Perform the substrate prediction.
hmmsearch -o hmmAligments.txt --tblout substrateprediction.csv nrpshmms inputADomains.fasta
-
4.
Explore your results.
Results can be explored with spreadsheet software. The file “substrateprediction.csv” contains the results of the prediction done. The result with a bigger score should be selected as the predicted binding substrate. The file “hmmAligments.txt” contains the HMMER alignments.
4 Notes
Some previous considerations should be taken into account after the analysis of sequences. Firstly, the different types of binding substrates which can be predicted are limited to 30, and it is expected that the low score results correspond with substrates that are not considered in the predictor. Moreover, the exploration of sequences and species which are in the training database is recommendable. If input sequences come from species which have high phylogenetic similarity with training datasets, then more reliable results are expected. Finally, it is also recommendable to use other prediction software, such as NRPSpredictor2, in order to compare and evaluate the results obtained.
References
Schwarzer D, Marahiel MA (2001) Multimodular biocatalysts for natural product assembly. Naturwissenschaften 88:93–101
Conti E, Stachelhaus T, Marahiel MA et al (1997) Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J 16:4174–4783
Stachelhaus T, Mootz HD, Marahiel MA (1999) The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol 6:493–505
Röttig M, Medema MH, Blin K et al (2011) NRPSpredictor2 – a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res 39:W362–W367
Khayatt BI, Overmars L, Siezen RJ et al (2013) Classification of the adenylation and acyl-transferase activity of NRPS and PKS systems using ensembles of substrate specific hidden Markov models. PloS One 8:e62136
Prieto C, Garcia-Estrada C, Lorenzana D et al (2012) NRPSsp: non-ribosomal peptide synthase substrate predictor. Bioinformatics 28:426–427
Acknowledgments
This work was supported by Agencia de Inversiones y Servicios de Castilla y León (record CCTT/10/LE/0001) and by Juan de la Cierva programme (JCI-2009-05444) of the Ministry of Science and Innovation (Spain).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Prieto, C. (2016). Characterization of Nonribosomal Peptide Synthetases with NRPSsp. In: Evans, B. (eds) Nonribosomal Peptide and Polyketide Biosynthesis. Methods in Molecular Biology, vol 1401. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3375-4_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3375-4_17
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3373-0
Online ISBN: 978-1-4939-3375-4
eBook Packages: Springer Protocols