
Abstract
The nonribosomal peptide synthetases are modular enzymes that catalyze synthesis of important peptide products from a variety of standard and non-proteinogenic amino acid substrates. Within a single module are multiple catalytic domains that are responsible for incorporation of a single residue. After the amino acid is activated and covalently attached to an integrated carrier protein domain, the substrates and intermediates are delivered to neighboring catalytic domains for peptide bond formation or, in some modules, chemical modification. In the final module, the peptide is delivered to a terminal thioesterase domain that catalyzes release of the peptide product. This multi-domain modular architecture raises questions about the structural features that enable this assembly line synthesis in an efficient manner. The structures of the core component domains have been determined and demonstrate insights into the catalytic activity. More recently, multi-domain structures have been determined and are providing clues to the features of these enzyme systems that govern the functional interaction between multiple domains. This chapter describes the structures of NRPS proteins and the strategies that are being used to assist structural studies of these dynamic proteins, including careful consideration of domain boundaries for generation of truncated proteins and the use of mechanism-based inhibitors that trap interactions between the catalytic and carrier protein domains.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Structural biology
- Nonribosomal peptide synthetase
- Enzymology
- Modular enzymes
- Peptides
- Siderophores
- Metabolic pathways
1 Introduction
Nonribosomal peptide synthetases deliver amino acid and peptide intermediates, covalently bound to the pantetheine cofactor of a peptidyl carrier protein, to different catalytic domains where the nascent peptide chain is elongated, modified, and ultimately released [1]. The primary catalytic domains are the adenylation domain that activates and loads the pantetheine thiol group and a condensation domain that catalyzes peptide bond formation. During the latter reaction, the condensation domain catalyzes the transfer of the aminoacyl or peptidyl group from an upstream carrier protein domain to the primary amine of an amino acid that has been previously loaded onto the downstream carrier domain. The final core catalytic domain, present only in the terminal module of an NRPS , is a thioesterase domain that catalyzes either hydrolysis or, more commonly, cyclization of the peptide to catalyze release from the final carrier protein domain.
This modular architecture poses important questions for the mechanisms that allow the synthesis to occur in an efficient manner [2]. Namely, for proper peptide synthesis, it is necessary that each carrier protein domain visits the respective catalytic domains in an organized manner. One could easily see how the delivery of a carrier protein domain to the incorrect adenylation domain or the delivery of an amino acid to a downstream condensation domain rather than the upstream would lead to incorrect peptide products. The NRPSs have therefore attracted the attention of many structural biologists who have determined structures of individual catalytic domains and multi-domain components [2, 3]. The cumulative structural understanding is beginning to provide clues to the strategies that NRPS enzymes use to coordinate peptide synthesis. This chapter first presents the structures of NRPS domains and multi-domain complexes (Table 1). The fundamental structural mechanism of NRPS enzymes, requiring that the carrier proteins migrate between neighboring catalytic sites, mandates a degree of conformational flexibility in the NRPS systems. We therefore describe the strategies that have been used to obtain meaningful multi-domain structures that provide insights into the function of these assembly line enzymes.
2 PCP Domain
Similar to the carrier domains of fatty acid synthesis, the NRPS enzymes use a peptidyl carrier protein (PCP) domain that is used to shuttle the substrates and peptide intermediates between different catalytic domains [4]. The PCP domains are the smallest NRPS domains, usually only 70–90 amino acids in length. The PCP domains contain a conserved serine residue that serves as the site for covalent modification with a phosphopantetheine cofactor that is derived from coenzyme A (Fig. 1). This posttranslational modification converts the apo-carrier protein to a holo-state and is catalyzed by a specific phosphopantetheinyl transferase (PPTase) that is often co-expressed with the NRPS cluster [5]. The thiol of the phosphopantetheine group binds covalently to the amino acid and peptide substrates through a thioester linkage with the carboxyl group of the amino acid.

Chemical structure of the phosphopantetheine cofactor attached to a conserved serine residue of the peptidyl carrier protein
Like other acyl carrier proteins, the NRPS PCP domains are composed of four α-helices (Fig. 2a). Helices 1, 2, and 4 are longer, and mostly parallel, while the third helix is shorter and runs approximately perpendicular to the axes of the other three. The serine residue that is the site of addition of the phosphopantetheine group is located at the start of helix α2. This helix is preceded by a long loop that is diverse in sequence and structure between the different NRPS PCP domains.

Structures of core NRPS domains. (a) The structure of the Type I I PCP domain BlmI (PDB 4I4D). The four helices are shown along with Ser44, the site of phosphopantetheinylation. (b) The active site of PheA (PDB 1AMU), the adenylation domain from the gramicidin synthetase NRPS. The ligand molecules AMP and phenylalanine are shown in ball-and-stick representation. Protein side chains are labeled including the residues that form the phenylalanine binding pocket and residues that interact with the nucleotide. (c). The condensation domain of the CDA synthetase (PDB 4JN3) is shown in ribbon representation. The two subdomains are shown, along with active site residues His156, His157, and Asp161, which is partially obscured by His157. (d) The active site of the thioesterase domain from EntF is shown (PDB 3TEJ). The pantetheine, covalently bound to Ser1006, is directed from the PCP domain to the active site, which is composed of the catalytic triad Asp1165, His1271, and Ser1138. The different NRPS domains are shown in specific colors, which are maintained throughout the chapter. PCP domains are shown in blue. Adenylation domain s are shown in pink for the N-terminal sub-domain and maroon for the C-terminal sub-domain. Condensation domain is shown in light green and the thioesterase domain is shown in yellow
The NMR structure of the PCP domain from the third module of the TycC NRPS protein of tyrocidin synthesis [6] demonstrated that the NRPS PCP domains share the prototypic fold of the carrier proteins from related modular polyketide and fatty acid synthesis [7]. A recent crystal structure of a free-standing PCP domain [8] confirms the overall fold in an independent carrier domain. Additional studies have demonstrated that the core helical structure in the apo and holo states [9].
As described below, the structures of PCP domains in complexes with catalytic domains demonstrate the regions of the carrier domains that interact with partner proteins. Not surprisingly, given the presence of the phosphopantetheine cofactor at the start of helix α2, this helix and the loop that joins helix α1 to α2 appear to be the primary determinants for interactions with the catalytic domains. Shotgun mutagenesis of the carrier protein of the EntB protein from enterobactin biosynthesis in E. coli followed by screening to test function in vivo identified regions of the PCP that are involved in interactions with catalytic domains. In addition to the loop and helix α2 mentioned above, these studies also identified residues from the short orthogonal helix α3 that also formed part of the hydrophobic patch that governed interactions with the downstream condensation domain [10, 11].
3 Adenylation Domain
NRPS adenylation domains play a key role in peptide natural product biosynthesis. In the assembly line-like choreography, the adenylation domain is the first domain the substrate encounters before it is added to the nascent peptide natural product. The adenylation domains catalyze a two-step reaction that activates the amino acyl substrate as an adenylate, followed by transfer of the amino acid to the thiol of the pantetheine cofactor of the carrier protein domain (Fig. 3).

Reaction catalyzed by the NRPS adenylation domain
Adenylation domain s belong to a larger adenylate-forming enzyme superfamily containing Acyl-CoA synthetases, NRPS adenylation domains, and beetle luciferase [12]. These enzymes are structural homologs and utilize a similar reaction mechanism that comprises two half reactions. Structural and kinetic results obtained from acyl-CoA synthetases [13–15] and luciferase enzymes [16, 17] have aided in the understanding of the adenylate-forming enzyme family. We focus here specifically on the adenylation domains of NRPS.
NRPS adenylation domains consist of approximately 500 residues. The bulk of the enzyme, residues 1–400, makes up the N-terminal subdomain while the final 100 residues form the C-terminal subdomain that sits atop the N-terminal subdomain. Several consensus sequences were identified in adenylation domains and designated A1 through A10 [1, 18]. These regions impart both structural and substrate stabilizing roles. The two-step reaction (Fig. 3) is carried out in a Bi Uni Uni Bi ping-pong mechanism. First Mg-ATP and the carboxylic acid bind to form an acyl-adenylate. After PPi from the ATP leaves the active site, a reorganization of the active site occurs where the C-terminal subdomain rotates changing the active site for the second half reaction. This domain alternation strategy transitions the adenylation domain between the two half reaction conformations, adenylate-forming and thioester-forming [12].
The first two structures of NRPS adenylation domains were PheA (Fig. 2b), a phenylalanine activating adenylation domain dissected from the multi-domain gramicidin synthetase 1, and the free-standing 2,3-dihydroxybenzoic acid (DHB) specific DhbE [19, 20]. Both of these structures are in the adenylate-forming conformation with Phe and AMP in the active site of PheA and no substrate, a DHB-adenylate, and DHB and AMP in the active site of the three DhbE structures. While the bulk of the active site is located in the N-terminal subdomain, a Lys found on the A10 loop of the C-terminal subdomain is required for acyl-adenylate formation [21, 22]. In both PheA and DhbE the Lys is poised in the active site to interact with both the carboxylic acid and the phosphate of the AMP (Fig. 2b). Important N-terminal regions to note are: the phosphate-loop (A3) that orients the β and γ phosphates of ATP and is often unresolved when ATP is not in the active site demonstrating its flexibility, the aromatic residue of the A4 motif (Phe234 in PheA and His207 is DhbE) which interacts with the carboxylic acid, and the aspartic acid of A7 motif that interacts with the ATP ribose hydroxyls. Once the high-energy acyl-adenylate is formed and PPi leaves the active site, domain alternation occurs to prepare the active site for the thioester-forming reaction.
The structures of a related acyl-CoA synthetase [13] provided the first view of a distinct catalytic conformation of a member of this adenylate-forming family. Compared with the previous structures of PheA and DhbE, the C-terminal subdomain of bacterial acetyl-CoA synthetase (Acs) was rotated by ~140° to a new position that created a binding pocket for the CoA nucleotide and a tunnel through which the pantetheine thiol approaches the adenylate of the active site. The hypothesis that all members of this family adopt both catalytic conformations, an adenylate-forming conformation as seen in PheA and a thioester-forming conformation seen in Acs, has now been thoroughly tested. In particular, extensive structural and biochemical analyses with Acs [13, 22] and the related protein 4-chlorobenzoyl-CoA ligase [14, 15, 23, 24] have demonstrated the specific involvement of residues on opposite faces of the C-terminal domain in catalyzing the respective partial reactions.
The 140° domain alternation occurs around a conserved hinge residue, an Asp or a Lys, located in the A8 motif. The importance of the hinge residue and its ability to change rotamers was demonstrated when the hinge in 4-chlorobenzoyl-CoA ligase was mutated to a Pro which essentially trapped it in the adenylate forming conformation [15, 22]. Domain alternation changes the active site without moving the substrate. Notably, the A10 catalytic Lys is removed from the active site by ~25 Å. Also the A8 loop interacts with the N-terminal subdomain where PPi exited and hydrogen bonds with the aromatic residue of the A4 motif rotating it away from the adenylate. This makes room for the pantetheine thiol to attack the carboxylic carbon thus displacing AMP and loading the pantetheine arm of the PCP with the amino acid substrate.
Another model protein that is closely related to NRPS adenylation domains offers more evidence for the role of the rotation of the C-terminal sub-domain. The DltA protein from B. subtilis is involved in cell wall biosynthesis, where it activates a molecule of l-Ala and loads into onto the partner carrier protein DltC. Thus, while not strictly an NRPS adenylation domain, the protein is highly homologous and serves as a useful model for understanding NRPS adenylation domains. The structures of the DltA have been solved with AMP [25] and Mg-ATP [26] in the active site and illustrate the distinct adenylate- and thioester-forming conformations.
In addition to DhbE, many additional bacterial siderophores derived from NRPS systems contain a salicylate or 2,3-dihydroxybenzoate moiety that is involved in iron binding [27]. Often, the aryl acid is activated by an independent adenylation domain. The structures of the E. coli homolog EntE [28, 29], as part of an adenylation-PCP complex, and A. baumannii BasE [30, 31] have also been solved. While DhbE adopts the adenylate-forming conformation, EntE adopts the thioester-forming conformation (Fig. 4). BasE, like several other adenylation domains, shows no electron density for the C-terminal sub-domain, suggesting it is adopting multiple conformations in the crystal lattice.

Domain alternation of NRPS adenylation domains. The structures of two free-standing adenylation domains are shown from two bacterial siderophore synthesis, (a) DhbE from the bacillibactin NRPS of B. subtilis and (b) EntE from the enterobactin NRPS of E. coli . The DhbE structure (PDB 1MDB) is in the adenylate-forming conformation, with the A10 motif of the C-terminal subdomain directed towards the active site. The EntE structure (PDB 4IZ6) adopts the thioester-forming conformation with the A8 motif near the active site. The carrier protein and the pantetheine cofactor of structure 4IZ6 are not shown for clarity
Many NRPS clusters contain a small ~70 residue protein that plays a role in activation of the adenylation activity [32]. The first characterized protein was encoded by the mbtH gene of M. tuberculosis and these proteins are therefore known as MbtH-Like Proteins (MLPs). Biochemical evidence has demonstrated that some adenylation domains require the MLPs for acyl-adenylate formation [33–35]. To date three MLP structures are available: the founding member MbtH, PA2412, and SlgN1 [36–38]. MLPs are thin arrowhead-shaped proteins with three central antiparallel β-sheets followed by two α-helices [37]. Defined in the MLP consensus sequence [32] and presented on one side of the MLP are two Trp residues that stack against each other. These Trp residues were shown to be required for activation of the adenylation domain by MLP [39]. A series of conserved proline residues have also been tested and appear to not be essential for activation of the adenylation domain [40]. A clear understanding of the mechanisms by which MLPs activate the adenylation domains is currently unknown.
4 Condensation Domain
Condensation domain s, usually located at the N-terminus of a module, catalyze amide bond formation between two substrates. The condensation domains transfer the amino acid or peptide from an upstream carrier protein domain to the amino moiety of the substrate that has been previously loaded onto a downstream carrier protein domain (Fig. 5).

Reaction catalyzed by the NRPS condensation domain
These 450 residue domains belong to the chloramphenicol acetyltransferase (CAT) superfamily. Similar to CAT, condensation domains contain a conserved HHxxxDG motif [1]. In CAT, the second His of this motif acts a general base that extracts a proton from chloramphenicol promoting nucleophilic attack and thus acyl transfer [41]. This His is also essential for condensation domain activity [42]; however, its exact role may depend on the substrates [43, 44]. Currently there are four crystal structures of condensation domains. They are: the standalone condensation domain VibH [43], the final condensation domain and its donor PCP dissected from the multi modular TycC [44], the condensation domain in the terminal module SrfA-C solved as a complete module [45], and finally the first condensation domain dissected from Calcium-Dependent Antibiotic synthetase (CDA-C1) [46].
Most commonly, a condensation domain will bind to an upstream donor and a downstream acceptor PCP. In some cases, especially with condensation starter domains, a substrate not bound to a PCP may be used. For example the standalone condensation domain VibH uses norspermidine as the acceptor which accepts DHB from the pantetheine arm of VibB [43]. Despite the current condensation domain structures lacking native ligands in the active site, much can be inferred from the current structures (Fig. 2c). While CAT forms a cyclic trimer [41], the monomeric condensation domains form a pseudo-dimer composed of two subdomains that contain CAT-like folds. The two subdomains adopt a V-shaped structure with a central cleft; at the base of this cleft, the two subdomains are linked by an α-helical linker. Also just above the linker between the two subdomains is the active site with the second His in the HHxxxDG motif located on a portion of the C-terminal subdomain that crosses over to the N-terminal subdomain. A second loop that spans the cleft between the two subdomains has been referred to as a lid or a latch. Despite the name, there is no evidence that this latch opens and closes [46]. Between these two crossovers, a tunnel is formed. As the PCPs bind to the condensation domain, their pantetheine arms must reach through this tunnel to the HHxxxDG active site in order for peptide bond formation to occur.
TycC and SrfA-C are both multi-domain structures each with a condensation domain that natively binds two PCPs. The TycC structure has an upstream donor PCP attached via a short linker. While the two domains are interacting, it does not appear that this is the catalytically active state. The Ser on the PCP which the pantetheine is loaded onto is 46.5 Å from the HHxxxDG motif, however the pantetheine is only ~16 Å long. Also several residues on the PCP that have been shown to be required for PCP–C domain interaction [47] are not involved in the interactions found in TycC. On the other hand, SrfA-C, as described below, does appear to form a valid domain interaction despite the pantetheine accepting Ser of the PCP being mutated to an Ala, thus making SrfA-C catalytically dead. This mutated residue is located ~16 Å from the HHxxxDG motif. Also the PCP residues required for PCP–C interactions are interacting with the condensation domain. Specifically, the PCP residues Met1007 and Phe1027 form hydrophobic interactions with Phe24 and Leu28 of the condensation domain [45].
The CDA-C1 structure [46] is the most recent condensation domain structure solved and provides a unique insight into the possible dynamics of NRPS condensation domains. CDA-C1 is in a distinctly more closed conformation than any of the other three condensation domains. In the CDA-C1 structure the N-terminal subdomain is 15°, 22°, and 25° more closed than VibH, TycC and SrfA-C respectively. Both SAXS and biochemical data suggest that this closed conformation seen in CDA-C1 is not due to crystal packing and is biochemically active [46]. Furthermore, normal mode analysis, morphing with energy minimization, and molecular dynamics all confirm that this opening and closing is possible. It is plausible however that condensation domains do not undergo this dynamic motion and are instead locked into a more opened or closed conformation based on the size of the substrate they need to accommodate and thus their location in the NRPS biosynthetic pathway. This, however, does not appear to be the case as SrfA-C is in a more opened conformation than TycC despite TycC being located on the ninth module of the system and SrfA-C being located on the seventh. More work is needed to fully understand the dynamics of condensation domains. Also since all current condensation domain structures lack ligands in the active site it is unclear how terminal condensation domain accommodates such large peptide substrates.
5 Thioesterase Domain
Within the final module of an NRPS pathway, the activity of the final condensation domain catalyzes the transfer of the upstream peptide to the amino acid substrate that is loaded onto the terminal PCP domain. To release the peptide and free the NRPS enzyme for another round of synthesis, the activity of a thioesterase domain is required (Fig. 6).

Reaction catalyzed by the NRPS thioesterase domain
The thioesterase domains are approximately 30 kDa in size and, as a class, can function as either hydrolases (as shown in Fig. 6) or as cyclases, where they can catalyze either lactam or lactone formation with an upstream heteroatom from the peptide chain. The thioesterase domains form an acyl-enzyme intermediate with an active site serine residue that subsequently is released from the enzyme through either hydrolysis with a water nucleophile or cyclization. For enzymes that catalyze lactone or lactam formation, the active site pocket therefore must bind the peptide substrate in an orientation that favors cyclization over hydrolysis by positioning the nucleophilic group to resolve the acyl-enzyme intermediate.
Structures of the genetically truncated thioesterase domains from the SrfA-C subunit [48] of the surfactin NRPS cluster and the FenB protein [49] of the fengycin cluster have been determined. The structure of the thioesterase domain from SrfA-C (Srf-TE) showed the domain belongs to the family of α/β hydrolases composed of a central, mostly parallel β-sheet that is surrounded by α-helices. Srf-TE contains a catalytic serine residue that serves as a nucleophile, attacking the terminal carbonyl of the peptidyl thioester with the pantetheine on the terminal PCP (Fig. 2d).
Three helices form a lid that in Srf-TE adopts two different conformations in the two molecules of the asymmetric unit, referred to as open and closed. The closed state is suggested to be a ground state of the enzyme however the helices in this state also exhibit some degree of disorder. Surfactin , the product of the Srf NRPS system, is an acyl-heptapeptide that contains seven amino acids and an N-terminal β-hydroxy fatty acid. The molecule cyclizes via lactone formation between the C-terminal carboxylate and this β-hydroxyl moiety. The authors soaked an N-acylheptapeptide-N-acetylcysteamine analog lacking the β-hydroxyl into the crystal. Portions of this molecule could be observed in the active site pocket, although the density was not of sufficient quality to enable complete modeling. The density did demonstrate a bent configuration that suggested the contour of the active site directs the cyclization reaction [48].
The thioesterase domain of the fengycin FenB protein (Fen-TE) has also been structurally characterized [49]. Like Srf-TE, Fen-TE was also genetically truncated to enable crystallization of the thioesterase domain. Fengycin is an acyl decapeptide lactone that contains a tyrosine at the fourth position that cyclizes through the phenolic hydroxyl with the C-terminal carboxylate of the peptide. To further characterize the active site, the structure of the enzyme covalently acylated with phenylmethylsulfonyl fluoride, the common inhibitor of hydrolases and proteases containing a serine nucleophile, was determined. The phenyl group from the inhibitor bound in a pocket that likely is used by the last Leu residue of the fengycin peptide. An oxyanion hole that stabilizes the generation and cleavage of the acyl-enzyme intermediate is formed by the backbone amides and is conserved throughout the α/β-hydrolase family. A molecule of fengycin was modeled into the substrate binding pocket and examined with molecular dynamics that showed no dissociation and limited movement of the ligand after an initial equilibration period [49]. This supported the position derived from docking and identified residues that could form interactions with the peptide ligand. In particular, the Tyr hydroxyl from the fourth position of the fengycin peptide was directed through a hydrophobic ridge to a position that allowed attack on the acyl-enzyme intermediate for the cyclization portion of the reaction.
6 Additional Integrated Domains
Additional protein activities are used in NRPS pathways for the complete synthesis of the final product. These proteins often act upon the substrate precursors prior to incorporation into the NRPS assembly line or on the immature peptide in steps that result in the final product maturation [50]. Certain auxiliary proteins do catalyze reactions on the amino acyl or peptidyl intermediates that are bound to a carrier protein. Most of these domains are expressed from isolated genes and most function as independent single domain proteins. However, some proteins are integrated into the NRPS assembly line where they are co-expressed. The most common of these are epimerization and methylation domains, as well as the alternate termination domains that thioester cleavage via an NAD(P)H-dependent reduction.
6.1 Epimerization Domains
Two different types of epimerization domains have been identified in NRPS systems where they catalyze conversion of l-amino acids to d-amino acids. Canonical epimerization domains of ~450 residues are inserted between PCP and condensation domains. While several systems, notably the PchE protein of pyochelin biosynthesis [51] and the HMWP2 protein of yersiniabactin biosynthesis [52], have a shorter, ~350 residue, noncanonical epimerization domain.
Interestingly, the canonical epimerization domains show sequence and structural homology to condensation domains. Only a single structure exists of an NRPS epimerization domain of the epimerization domain from tyrocidin synthetase A [53]. The structure displays the same overall symmetrical fold as the condensation domains, with a large cavity in the center of two subdomains. The epimerization domain contains the same conserved HHxxxDG motif present in the condensation domains. Mutation of the second histidine (His753) and the aspartic acid (Asp757) of this motif, as well as several downstream residues including a glutamate at position 892 and a conserved asparagine and tyrosine pair 975 and 976, were defective for proton wash-out in the epimerization domain of the first module of the gramicidin synthetase, GrsA [54]. In the structure of the TycA epimerization domain, these histidine and the glutamate residues, His743 and Glu882, are positioned on opposite sides of the active site cavity and are potential candidates for a two-base epimerization reaction. The glutamate is part of a conserved EGHGRE motif that is common to epimerization domains, but not the homologous condensation domains [1].
The noncanonical epimerization domains are present in the pyochelin producing NRPS , protein PchE of P. aeruginosa and Burkholderia pseudomallei. It shares the interesting property with N-methylation domains of being inserted within the C-terminal sub-domain of the NRPS adenylation domain. There is no structural information regarding these epimerization domains, nor is there any information how the adenylation domain accommodates these insertions.
6.2 N-Methylation Domains
N-methylation of the peptides of NRPS products is seen primarily in fungal NRPS systems. Presumably, this confers proteolytic stability to the methylated peptide bonds. Like the noncanonical epimerization domains, the N-methylation domains are inserted into the C-terminal subdomain of the NRPS adenylation domains. The methylation domain is ~420 residues in length and shows limited homology with other S-adenosylmethionine dependent transferase enzymes. The C-terminal subdomain of the NRPS adenylation domain contains a central three-stranded β-sheet that contains two long anti-parallel strands that each range in length from 7 to 11 residues. Following these two strands is a loop that leads to an amphipathic helix that packs against the central β-sheet. It is into this large loop that the N-methylation domains are inserted. In the structure of the complex between the adenylation and the PCP [28, 29, 55], this loop from the adenylation C-terminal subdomain is directed towards the PCP, suggesting that perhaps the amino acid substrate could be directed to the neighboring methyl transferase domain without a major structural rearrangement of the catalytic domains.
There are no reported structures of NRPS methyl transferase domains. A comparison with the Protein Data Bank finds only short stretches of homology (80–100 residues) to related SAM-dependent methyltransferases, for example the NodS protein of Bradyrhizobium japonicum [56]. Additionally, the structure of a SAM dependent N-methyltransferase from the glycopeptide NRPS cluster of Amycolatopsis orientalis has been determined [57] however this protein is an independent tailoring enzyme with limited homology to the integrated methyl transferase domains of NRPSs.
6.3 Reductase Domains
Certain NRPS clusters terminate not with a thioesterase domain but rather with a NAD(P)H-dependent reductase domain that cleaves the bound peptide to release a C-terminal alcohol or aldehyde rather than a carboxylate. The ~280 residue reductase domains show homology to nucleotide cofactor binding short-chain dehydrogenase/reductase (SDR) enzymes. The structure of a reductase domain from a functionally uncharacterized NRPS protein from Mycobacterium tuberculosis (designated RNRP) was determined to confirm the expected Rossmann fold common to SDR enzymes [58]. A small helical C-terminal domain was identified as well as a short helix-turn-helix insertion in the standard Rossmann fold. Although the structure of a liganded reductase domain was not obtained, examination of cofactor binding by small-angle X-ray scattering identified a small change in the radius of gyration of the domain, suggesting a conformational transition from an open to a closed state. A second structure of the aldehyde-producing reductase domain of AusA, catalyzing therefore only a 2 e− reduction rather than the sequential 4 e− reduction of RNRP, has also been structurally characterized showing a similar overall structure [59]. A di-domain construct, composed of the carrier protein and reductase domain was also crystallized; however, no electron density for the carrier protein domain was observed in this structure preventing insights into the nature of the interaction between the catalytic and carrier domains.
7 Multi-domain NRPS Crystal Structures
Due to the plasticity of NRPSs, crystal structures of multi-domain constructs are difficult to obtain. Similarly, the large size of these proteins presents challenges for NMR. Despite these difficulties there are currently five multi-domain NRPS structures that are in or appear to be in a catalytically relevant state. They are: the four-domain module of SrfA-C [45], the PCP-thioesterase domains of EntF [60, 61], the chimeric adenylation-Carrier Protein EntE-B [28, 29], the native adenylation-PCP protein PA1221 [55], and the MLP-adenylation domain SlgN1 [38]. As noted earlier, the structure of the excised PCP-condensation domain from TycC has been determined [44]; however, the two domains do not appear to interact in a functional manner. These crystal structures help to shed light on how the domains of NRPSs interact to carry out natural product biosynthesis.
7.1 SrfA-C: A Complete NRPS Module
SrfA-C is the largest NRPS crystal structure to date and offers a complete view of an NRPS module. Composed of a domain architecture of condensation-adenylation-PCP-thioesterase, the 144 kDa SrfA-C is the terminal module from the surfactin biosynthetic cluster of Bacillus subtilis [45]. In order to crystallize SrfA-C the Ser of the PCP that is pantetheinylated was mutated to an Ala to produce a homogenous protein sample. Nonetheless, the PCP is interacting with the condensation domain in a catalytically relevant way (Fig. 7a). The condensation and N-terminal subdomain of the adenylation domain share a large interdomain interface and are believed to form a stable platform, upon which the PCP is thought to migrate between different catalytic domains. The C-terminal subdomain of the adenylation domain is neither in the adenylate or thioester-forming conformations. Instead it has adopted an intermediate conformation that is closest to the adenylate forming conformation. Because of this intermediate conformation the catalytic Lys on the A10 motif is positioned slightly outside of the active site, which may prevent the adenylation domain from activating another substrate before the PCP is ready to accept it. The thioesterase domain is positioned near the condensation domain and, interestingly, the pantetheine channel on the thioesterase is facing directly into the core of the SrfA-C protein. A simple rotation of the PCP is therefore insufficient to allow a functional interaction between the PCP and thioesterase domains. This is evident from aligning SrfA-C with the EntF PCP-thioesterase structure [60, 61]. It therefore seems that rotation of the thioesterase domain relative to the core of the SrfA-C module is necessary to adopt a catalytic conformation.

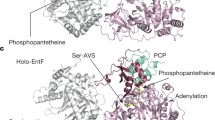
Crystal Structures of multi-domain NRPS proteins. (a) The structure of the complete termination module of SrfA-C (PDB 2VSQ) shows the PCP interactions with the downstream side of the condensation domain. (b) The adenylation-PCP structure of PA1221 (PDB 4DG9) illustrates a functional interface between the PCP and the thioester-forming conformation of the adenylation domain. (c) The structure of the SlgN1 protein (PDB 4GN5) shows the interaction between the MbtH-like domain (forest green) interacting with the N-terminal subdomain of the adenylation domain. The conserved tryptophan residues are highlighted in the MLP. (d) The PCP-thioesterase domain of EntF (PDB 3TEJ) is shown illustrating the binding of the pantetheine into the active site of the thioesterase domain
7.2 EntE-B and PA1221: Adenylation-Carrier Protein Complexes
The structures of the adenylation-carrier protein complexes of the chimeric EntE-B and the native PA1221 (Fig. 7b) were solved in the thioester-forming conformation with the use of aryl/acyl-adenosine vinylsulfonamides [28, 29, 55]. In both structures, the carrier protein is interacting with the adenylation domain and the pantetheine is positioned in the adenylation domain tunnel forming a covalent bond with the adenosine inhibitors. The chimeric EntE-B, which was genetically combined using a linker of similar sequence length and composition to multi-domain NRPSs, formed a domain swapped dimer where the carrier protein from molecule 1 was interacting with the adenylation domain of molecule 2 and vice versa. A comparison to SrfA-C suggests that while the EntE-B linker was of similar sequence length as SrfA-C, the EntE-B linker formed an α-helix which rendered the linker too short for an intramolecular interaction.
Both the EntE-B and PA1221 structures reveal the various interactions between the adenylation and carrier protein domains which includes both hydrophobic interactions and hydrogen bonding [28]. Biochemical studies also help to confirm that while the EntE-B forms a domain swapped dimer due most likely to the linker, the interaction between the ArCP and the A domain seem to be the same interaction that would occur in the intra-domain interaction. In particular, the insights derived from the EntE-B crystal structure were used to guide mutations in the EntE homolog from A. baumannii to improve the ability to recognize the heterologous EntB carrier protein as a partner [29].
7.3 SlgN1: An MLP-Adenylation Domain Structure
As noted earlier, some adenylation domains require MLPs for acyl adenylate formation [32–35]. The crystal structure of SlgN1, containing a MLP fused to the N-terminus of an adenylation domain, showed for the first time were MLPs bind to adenylation domains [38]. The MLP binds the N-terminal subdomain of the adenylation domain distal from the active site (Fig. 7c). The closest active site motif appears to be A7 aspartic acid which binds the ribose hydroxyls of the ATP. A key interaction is an Ala residue presented on the surface of SlgN1 that is inserted between two stacked Trp on the MLP. For MLP dependent adenylation domain this Ala (Ala 433 in SlgN1) is highly conserved while for nondependent MLP adenylation domains this residue varies. Unfortunately the C-terminal subdomain of the adenylation domain of SlgN1 was removed to facilitate crystallization. Therefore it is unclear if the MLP is bound to the adenylation domain for just acyl adenylate formation or also for the subsequent thioester formation.
7.4 EntF: PCP-Thioesterase Domain Structures
Finally, the structures of the PCP-TE domains of EntF were solved by both X-ray diffraction and NMR [60, 61]. While the NMR structure was that of an apo PCP and shows a dynamic interaction, the crystal structure used an α-chloroacetyl amide coenzyme A analog which was loaded onto the PCP domain using the phosphopantetheinyl transferase Sfp. This made it possible to structurally analyze the thioesterase pantetheine channel along with the domain interactions (Fig. 7d). The pantetheine is inserted into the thioesterase channel which complements the pantetheine with a hydrophobic pocket around the di-methyls of the pantetheine, hydrogen bonding with the two amide carbonyls, and also van der Waals interactions [61]. At the end of the channel the loaded pantetheine encounters the thioesterase catalytic triad. The PCP and thioesterase form extended hydrophobic interactions which includes the thioesterase lid region which interacts with the PCP.
7.5 Modeling of Dynamics of the SrfA-C Module
While only a few structures of multi-domain NRPSs exist, the various conformations available assist in decoding the NRPS choreography for natural product biosynthesis. In order for substrate loading onto the pantetheinylated PCP the adenylation domain undergoes domain alternation after activating the substrate. Now in the thioester-forming conformation the PCP can interact with the adenylation domain and the pantetheine can enter the adenylation domain tunnel and attack the substrate (Fig. 8a). With the substrate loaded the PCP can now migrate over to the condensation domain where it awaits the delivery of the upstream peptide or amino acid substrate (Fig. 8b). Based on the SrfA-C structure it appears that A domain needs to adopt or come close to adopting the adenylate-forming conformation. It seems plausible that the movement of the C-terminal subdomain of the adenylation domain facilitates the movement of the PCP between the adenylation and condensation domains. The C-terminal subdomain and the PCP do not move as a rigid body; rather there appears to be two components to the movement to allow the PCP to adopt the proper position bound to the adenylation domain [55]. Finally the PCP must interact with the thioesterase domain. Since the thioesterase active site channel is directed toward the core of the condensation domain, the PCP and thioesterase domains must move relative to the condensation and adenylation domains, as also relative to each other, to allow a functional interaction to form.

Model for the delivery of the PCP to the adenylation domain active site. The SrfA-C protein is shown in (a). A model conformation that adopts the thioester-forming conformation where the PCP is bound to the adenylation domain and (b) the crystallographic structure where the PCP interacts with the condensation domain. The N-terminal subdomain of the adenylation domain, as well as the condensation and thioesterase domains, are all shown in surface representation, while the PCP and the C-terminal subdomain of the adenylation domain are shown as ribbons. The structure in panel (a) is derived by modeling the SrfA-C (PDB 2VSQ) adenylation and PCP domains onto the thioester-forming conformation observed with EntE-B (PDB 4IZ6) or PA1221 (PDB 4DG9). The serine residue that is the site of phosphopantetheinylation is highlighted with the yellow sphere
8 Strategies to Crystallize NRPS Proteins
Structurally characterizing NRPS proteins through either NMR or X-ray crystallography is challenging given that a complete module is over 1000 residues in length and multi-domain proteins containing multiple modules are not uncommon. The large size limits the use of NMR and the size and flexibility make crystallization anything but routine. Despite these challenges, the previous sections have described numerous successful examples of structural characterization of the NRPS proteins. We present here a summary of the strategies that have been used to overcome the difficulties inherent to these large modular proteins.
8.1 Structural Studies of Individual Domains
Examination of single domains can provide insights into the action of multi-domain proteins. Initial attempts at structural characterization of NRPSs therefore focused on individual domains; only more recently have multi-domain structures been achieved. These early studies took advantage of both the rare type II nature of some NRPS clusters, where individual catalytic domains were expressed as a single domain protein, as well as the use of molecular tools to produce genetically truncated proteins.
Specific examples of the study of type II NRPS domains include the VibH condensation domain [43] and the DltA [25] and BasE [30] adenylation domains. Insights into the carrier domain architecture resulted from the type II PCP domain that has recently been determined [8], as well as the full length structure of EntB, a two-domain protein that contains a catalytic isochorismatase domain fused to a carrier protein domain [62]. Comparison of these structures with truncated and multi-domain proteins has failed to demonstrate any features that differ between the type I and type II protein domains.
Type I I NRPS proteins are relatively rare, however, and most studies of single NRPS domains have used genetically truncated protein constructs. The production of isolated domains presents additional challenges as it is necessary to identify accurately the domain termini. A truncation that is too short can result in exposure of hydrophobic residues that normally reside within the protein core. Alternately, defining a domain boundary that is too large may result in the inclusion of linker sequences or portions of neighboring domains that are poorly ordered. Often, these extraneous regions can hinder protein folding or solubility and preclude functional or structural analysis. Likely for this reason, many of the genetically truncated NRPS domains that have been studied lie at the N- or the C-terminus of the native multi-domain protein. Indeed, of the many NRPS proteins that have been structurally characterized (Table 1), only three truncated proteins are truly “internal,” where a choice needed to be made about the precise locations of both the N- and C-terminal truncations. Two structural studies on the TycC protein from tyrocidin synthesis, describing the PCP domain from the third module [6, 9] and the PCP-condensation di-domain crystal structure [44], were internal domain constructs. Additionally, an adenylation domain from SidN [63], a protein that is involved in the production of a fungal siderophore [64], was also excised from the middle of a larger NRPS protein. In this last example, the authors note that successful structure determination of SidN required optimization of the N- and C-termini and that changes of as few as 14 residues could impact protein solubility.
The choice of the boundaries between domains is therefore a critical decision to be made in expression of a genetically truncated NRPS domain. The availability of existing structures provides useful clues to guide the design of a protein construct. Identification of the boundary between the adenylation and PCP domains is aided by the presence of a conserved A10 catalytic motif near the C-terminus of the adenylation domain. This nearly identical motif is defined as PXXXXGK, where the X can represent any amino acid [1, 12]. Following this motif is a loop that leads to the start of helix 1 for the downstream PCP domain. In both the structures of SrfA-C and PA1221, the natural NRPS protein structures showing the adenylation-PCP linker, the 9 residues following the catalytic lysine pack against the C-terminal domain. In the structure of the PheA genetically truncated adenylation domain, the 11 residues following the A10 lysine interact with the body of the C-terminal subdomain. Therefore, at least ten residues following the A10 lysine should be considered to be part of the adenylation domain and should not be disrupted through a genetic truncation.
Not including the type II PCP domain [8], which has a longer N- and C-termini, the PCP domains are fairly consistent in size. The type I PCP domain structures, whether in isolation or as part of the multi-domain targets, all have 29–32 residues between the start of the helix α1 and the serine that is phosphopantetheinylated. This defines the N-terminal region of the PCPs that should not be disrupted. Similarly, the distance from the site of cofactor modification to the C-terminal end of helix α4 in the type I PCP domains is 37 or 38 residues in all but PA1221, which is slightly longer at 40 residues. This again defines the boundaries of the PCP fairly consistently.
The definition of the start of the condensation domains is more challenging, given that three of the four available structures are derived from protein domains that lie at the N-terminus of the protein. The shortest N-terminus comes from VibH, the self-standing condensation domain of vibriobactin biosynthesis [43]. An α-helix is the first secondary structural element that is shared by all four. While the VibH structure begins two residues before this helix, the remaining three structures share an additional six residues that should likely be maintained in a condensation domain.
To define the C-terminus of the condensation domain, the SrfA-C structure offers the best view of the linker between the condensation and adenylation domains. This linker, composed of 32 residues, interacts closely with residues from both domains. The first 13 residue interact with the condensation domain, while the remaining helix and loop interact with the adenylation domain. The tighter interaction between the linker and both catalytic domains relates to the fact that the condensation and N-terminal subdomain of the adenylation domain are expected to form a stable interface that likely does not change during the NRPS catalytic cycle.
The thioesterase domain, by definition, resides at the C-terminus of the NRPS protein so the C-terminal end will be defined. The thioesterase domains are nearly always preceded by a PCP domain and the boundary of the PCP is defined, as discussed above. This leaves a short loop, as is seen in the PCP-thioesterase structure that is a suitable site for truncation to produce an isolated thioesterase domain.
8.2 Mutations to the Phosphopante-theinylation Site
Crystallization requires a uniform protein sample that is suitable to form the crystal lattice. One source of heterogeneity in NRPS proteins derives from the PCP domain and their mixture of apo- and holo-proteins that bear the phosphopantetheine modification. The preparation of holo-PCP domains uses either co-expression of a phosphopantetheinyl transferase or biochemical incubation with a specific or general PPTase. While these reactions should proceed to completion, there are no good ways to separate the apo- from holo-PCP domains. Therefore, several structural studies, by both NMR and crystallography, have used mutated PCP domains in which the serine residue is replaced with an alanine. This mutation dictates that the apo-protein is present at 100 % and limits heterogeneity. Of course, there are certain limitations to this strategy as the apo-protein is not the biologically active form of the PCP domain. The differences in overall structure of the TycC PCP domain that depend on the apo- and holo-state of the protein [9] should therefore be considered if this apo-PCP domain is used.
Nonetheless, the mutation of the serine to an alanine removes the potential for modification by the PPTase enzymes and prevents the presence of the flexible cofactor that may hinder crystallization. This strategy has been used in the crystallization of the SrfA-C module [45] and also in the investigation of the solution structure of the PCP-thioesterase domain [60].
8.3 Use of Selective Inhibitors to Minimize Conformational Flexibility
The use of specific ligands that bind in the active sites of NRPS domains to reduce the conformational dynamics has proven to be quite useful and additionally provides some of the most detailed views of ligands bound in the active sites of NRPS domains. The most effective of these inhibitors are mechanism-based inhibitors that mimic intermediates in the reaction pathway and therefore provide the added benefit of demonstrating how the catalytic domains recognize their substrates. Additionally, these compounds have been useful to trap the transient interactions between catalytic and carrier protein domains.
The first example of such a compound was the use of a chloroacetyl thioester of the pantetheine cofactor that was used by Bruner and Liu to crystallize a PCP-thioesterase complex (Fig. 9a). An α-chloroacetyl amide derivative of amino-CoA, where the CoA terminal thiol is replaced with an amine was generated [65]. This modified CoA molecule was loaded onto the PCP domain via a PPTase and shown to inhibit the thioesterase reaction. The proper binding of the α-chloroacetyl moiety in the proximity of the nucleophilic serine of the thioesterase domain was proposed to facilitate the interaction between the PCP and thioesterase domains [65]. Subsequently, this inhibitor was indeed used as a tool to crystallize the di-domain PCP-thioesterase of EntF [61]. Although designed to form a covalent interaction between the pantetheine and the catalytic serine of the thioesterase domain by attack on the α-carbon [65], the inhibitor molecule in the active site was not covalently attached to the protein. The authors proposed a mechanism whereby the Ser1138 attacked the carbonyl carbon of the α-chloroacetyl amide pantetheine derivative to form an oxyanion that displaces the chloride ion and forms an epoxide intermediate. This was then hydrolyzed either directly or following reaction with another nucleophilic group on the enzyme.

Mechanism-based inhibitors used to crystallize NRPS multi-domain structures. (a) The α-chloroacetyl-CoA derivative was designed to react with the catalytic serine of thioesterase domain. The catalytic reaction, the inhibitor rationale, and the observed structure are shown. (b) The vinylsulfonamide inhibitor is shown, along with the two-step reaction catalyzed by the adenylation domain. The covalent inhibitor complex has been observed in two crystal structures
A second strategy to use mechanism-based inhibitors has been used successfully to determine the structure of the interaction between adenylation and PCP domains. This strategy expanded upon the use of sulfonamide inhibitors of the adenylation domain in which the phosphate diester moiety of the acyl adenylate intermediate was replaced with sulfamate or sulfamide analogs [66–68]. To further optimize this inhibitor to react covalently with the pantetheine thiol of a partner carrier protein domain, Aldrich and colleagues introduced a linker containing a double bond between the sulfonamide moiety and the salicylic acid (Fig. 9b) on an inhibitor directed towards the aryl-activating type II adenylating enzyme MbtA [69]. The affinity of this vinylsulfonamide inhibitor is reduced by nearly five orders of magnitude with MbtA compared to the parental sulfonamide inhibitor [70], likely due to the loss of the carbonyl carbon and the nitrogen atom of the linker [69]. However, it still displayed apparent inhibition constants of 100–300 μM against the adenylation reaction and was deemed sufficient for biochemical and structural studies. Additionally, in the presence of the adenylating enzyme MbtA, the vinylsulfonamide inhibitor reacted covalently with the pantetheine moiety of the MbtB carrier protein and the adenylation and PCP proteins co-migrated on a native gel.
Suitable vinylsulfonamide analogs of adenylate intermediates have been used to trap crystallographically proteins from two different NRPS systems. It was first used to determine the structure of the adenylation-PCP interface between the EntE adenylation domain of enterobactin biosynthesis along with its partner EntB [28, 29]. Additionally, to facilitate crystallization of these proteins, a chimeric protein construct was designed that fused genetically the coding sequences for the EntE adenylation domain with the coding sequence for the carrier protein domain of the bifunctional EntB [29]. Subsequently, a vinylsulfonamide inhibitor was also used to determine the structure of an uncharacterized two domain adenylation-PCP protein from P. aeruginosa [55]. In the latter case, it was first necessary to identify the amino acid substrate preference to design the inhibitor with the appropriate amino acid side chain bound to the vinylsulfonamide linker.
Both the structures of PA1221 and the EntE-B chimeric protein demonstrate that the use of the mechanism-based inhibitor can serve to stabilize the domain interactions sufficiently to allow them to be observed crystallographically. In the case of PA1221, crystallization of the apo-protein in the absence of the inhibitor resulted in a structure in which no electron density could be observed for the carrier protein domain. Interestingly, the adenylation domain adopted the thioester-forming conformation that is identical to that observed to interact with the PCP in the structure of the holo-protein in the presence of the ligand yet still failed to bind the PCP. Thus the inhibitor was a critical reagent for sufficiently stabilizing the interaction to allow the full length protein to be observed in the crystal structure.
9 Conclusions
Despite the multitude of NRPS structures, including the structures of multi-domain proteins over the last 5 years, there remains much to be done to understand completely the structural basis for NRPS biosynthesis. In particular, more structures are needed of full modules of enzymes that will provide more insights into the nature of the interactions of the NRPS domains. The condensation domain lacks structures in the presence of ligands and more crystal or NMR structures will identify the roles of specific active site residues in binding and catalysis. Additionally, a superposition of the functional interaction between the PCP and thioesterase domains of EntF [60, 61] with the SrfA-C structure [45] shows that the PCP domain in the functional interaction is predicted to overlap with the adenylation-condensation core of SrfA-C. This suggests that the thioesterase domain of SrfA-C is not positioned in a “catalytic” orientation in the crystal lattice. Likely, the thioesterase position in SrfA-C is dynamic and adopts a different relative position when it interacts with the PCP in the course of the catalytic cycle. The use of inhibitors such as the α-chloroacetylamide pantetheine derivative [61, 65] with full-length EntF, for example, would allow the determination of the PCP-thioesterase interface in the context of a full NRPS module.
Another exciting target of NRPS structural biochemistry is the examination of the additional integrated domains, N-methyltransferases or epimerization domains, in the context of a full NRPS module. The carrier protein domains of SrfA-C, or the E. coli homolog EntF, must make their way to three consecutive catalytic domains (adenylation, condensation, and thioesterase domains). In modules that harbor additional integrated domains, the carrier protein must find its way to yet another catalytic domain and we can ask what structural features are required for these specialized modules.
Additional NRPS structures may also begin to address the question of what dynamic features, within or between catalytic domains, drive the proper delivery of the PCP along the catalytic assembly line. The large rotation of the two sub-domains within the NRPS adenylation domain offers a potential major rearrangement that could facilitate interactions with the PCP. Additional conformational changes may also compel the PCP to bind additional catalytic domains in a proper orientation.
Finally, now that the structure of the complete SrfA-C module [45] has toppled what was once the most sought-after structure in NRPS enzymology, a new high profile target is a structure of a multi-module protein. The SrfA-C structure suggests that the condensation domain and N-terminal subdomain of the adenylation domain form a stable interface and, relative to this platform, the PCP and possibly the other catalytic domains migrate to transit through the catalytic cycle. In a larger, multimodule NRPS, might this platform from one module form a larger complex with the condensation and adenylation domains of the next module as well. While atomic resolution crystal structures would provide answers to these questions, additional structural tools such as electron microscopy and small angle X-ray scattering may also provide insights into the larger macromolecular organization of the catalytic domains.
References
Marahiel MA, Stachelhaus T, Mootz HD (1997) Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem Rev 97:2651–2674
Marahiel MA, Essen LO (2009) Chapter 13. Nonribosomal peptide synthetases mechanistic and structural aspects of essential domains. Methods Enzymol 458:337–351
Koglin A, Walsh CT (2009) Structural insights into nonribosomal peptide enzymatic assembly lines. Nat Prod Rep 26:987–1000
Mercer AC, Burkart MD (2007) The ubiquitous carrier protein--a window to metabolite biosynthesis. Nat Prod Rep 24:750–773
Beld J, Sonnenschein EC, Vickery CR et al (2014) The phosphopantetheinyl transferases: catalysis of a post-translational modification crucial for life. Nat Prod Rep 31:61–108
Weber T, Baumgartner R, Renner C et al (2000) Solution structure of PCP, a prototype for the peptidyl carrier domains of modular peptide synthetases. Struct Fold Des 8:407–418
Crosby J, Crump MP (2012) The structural role of the carrier protein--active controller or passive carrier. Nat Prod Rep 29:1111–1137
Lohman JR, Ma M, Cuff ME et al (2014) The crystal structure of BlmI as a model for nonribosomal peptide synthetase peptidyl carrier proteins. Proteins 82:1210
Koglin A, Mofid MR, Lohr F et al (2006) Conformational switches modulate protein interactions in peptide antibiotic synthetases. Science 312:273–276
Lai JR, Fischbach MA, Liu DR et al (2006) A protein interaction surface in nonribosomal peptide synthesis mapped by combinatorial mutagenesis and selection. Proc Natl Acad Sci U S A 103:5314–5319
Lai JR, Koglin A, Walsh CT (2006) Carrier protein structure and recognition in polyketide and nonribosomal peptide biosynthesis. Biochemistry 45:14869–14879
Gulick AM (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol 4:811–827
Gulick AM, Starai VJ, Horswill AR et al (2003) The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry 42:2866–2873
Reger AS, Wu R, Dunaway-Mariano D et al (2008) Structural characterization of a 140° domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47:8016–8025
Wu R, Cao J, Lu X et al (2008) Mechanism of 4-chlorobenzoate:coenzyme a ligase catalysis. Biochemistry 47:8026–8039
Branchini BR, Murtiashaw MH, Magyar RA et al (2000) The role of lysine 529, a conserved residue of the acyl-adenylate- forming enzyme superfamily, in firefly luciferase. Biochemistry 39:5433–5440
Branchini BR, Southworth TL, Murtiashaw MH et al (2005) Mutagenesis evidence that the partial reactions of firefly bioluminescence are catalyzed by different conformations of the luciferase C-terminal domain. Biochemistry 44:1385–1393
Mootz HD, Marahiel MA (1999) Design and application of multimodular peptide synthetases. Curr Opin Biotechnol 10:341–348
Conti E, Stachelhaus T, Marahiel MA et al (1997) Structural basis for the activation of phenylalanine in the non- ribosomal biosynthesis of gramicidin S. EMBO J 16:4174–4183
May JJ, Kessler N, Marahiel MA et al (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc Natl Acad Sci U S A 99:12120–12125
Hamoen LW, Eshuis H, Jongbloed J et al (1995) A small gene, designated comS, located within the coding region of the fourth amino acid-activation domain of srfA, is required for competence development in Bacillus subtilis. Mol Microbiol 15:55–63
Reger AS, Carney JM, Gulick AM (2007) Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46:6536–6546
Gulick AM, Lu X, Dunaway-Mariano D (2004) Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry 43:8670–8679
Wu R, Reger AS, Lu X et al (2009) The mechanism of domain alternation in the acyl-adenylate forming ligase superfamily member 4-chlorobenzoate: coenzyme A ligase. Biochemistry 48:4115–4125
Yonus H, Neumann P, Zimmermann S et al (2008) Crystal structure of DltA. Implications for the reaction mechanism of non-ribosomal peptide synthetase adenylation domains. J Biol Chem 283:32484–32491
Du L, He Y, Luo Y (2008) Crystal structure and enantiomer selection by D-alanyl carrier protein ligase DltA from Bacillus cereus. Biochemistry 47:11473–11480
Quadri LE (2000) Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol Microbiol 37:1–12
Sundlov JA, Gulick AM (2013) Structure determination of the functional domain interaction of a chimeric nonribosomal peptide synthetase from a challenging crystal with noncrystallographic translational symmetry. Acta Crystallogr D Biol Crystallogr 69:1482–1492
Sundlov JA, Shi C, Wilson DJ et al (2012) Structural and functional investigation of the intermolecular interaction between NRPS adenylation and carrier protein domains. Chem Biol 19:188–198
Drake EJ, Duckworth BP, Neres J et al (2010) Biochemical and structural characterization of bisubstrate inhibitors of BasE, the self-standing nonribosomal peptide synthetase adenylate-forming enzyme of acinetobactin synthesis. Biochemistry 49:9292–9305
Neres J, Engelhart CA, Drake EJ et al (2013) Non-nucleoside inhibitors of BasE, an adenylating enzyme in the siderophore biosynthetic pathway of the opportunistic pathogen Acinetobacter baumannii. J Med Chem 56:2385–2405
Baltz RH (2011) Function of MbtH homologs in nonribosomal peptide biosynthesis and applications in secondary metabolite discovery. J Ind Microbiol Biotechnol 38:1747–1760
Wolpert M, Gust B, Kammerer B et al (2007) Effects of deletions of mbtH-like genes on clorobiocin biosynthesis in Streptomyces coelicolor. Microbiology 153:1413–1423
Boll B, Taubitz T, Heide L (2011) Role of MbtH-like proteins in the adenylation of tyrosine during aminocoumarin and vancomycin biosynthesis. J Biol Chem 286:36281–36290
Felnagle EA, Barkei JJ, Park H et al (2010) MbtH-like proteins as integral components of bacterial nonribosomal peptide synthetases. Biochemistry 49:8815–8817
Buchko GW, Kim CY, Terwilliger TC et al (2010) Solution structure of Rv2377c-founding member of the MbtH-like protein family. Tuberculosis (Edinb) 90:245–251
Drake EJ, Cao J, Qu J et al (2007) The 1.8 A crystal structure of PA2412, an MbtH-like protein from the pyoverdine cluster of Pseudomonas aeruginosa. J Biol Chem 282:20425–20434
Herbst DA, Boll B, Zocher G et al (2013) Structural basis of the interaction of MbtH-like proteins, putative regulators of nonribosomal peptide biosynthesis, with adenylating enzymes. J Biol Chem 288:1991–2003
Zhang W, Heemstra JR Jr, Walsh CT et al (2010) Activation of the pacidamycin PacL adenylation domain by MbtH-like proteins. Biochemistry 49:9946–9947
Zolova OE, Garneau-Tsodikova S (2012) Importance of the MbtH-like protein TioT for production and activation of the thiocoraline adenylation domain of TioK. Med Chem Commun 3:950–955
Murray IA, Cann PA, Day PJ et al (1995) Steroid recognition by chloramphenicol acetyltransferase: engineering and structural analysis of a high affinity fusidic acid binding site. J Mol Biol 254:993–1005
Stachelhaus T, Mootz HD, Bergendahl V et al (1998) Peptide bond formation in nonribosomal peptide biosynthesis. Catalytic role of the condensation domain. J Biol Chem 273:22773–22781
Keating TA, Marshall CG, Walsh CT et al (2002) The structure of VibH represents nonribosomal peptide synthetase condensation, cyclization and epimerization domains. Nat Struct Biol 9:522–526
Samel SA, Schoenafinger G, Knappe TA et al (2007) Structural and functional insights into a peptide bond-forming bidomain from a nonribosomal peptide synthetase. Structure 15:781–792
Tanovic A, Samel SA, Essen LO et al (2008) Crystal structure of the termination module of a nonribosomal peptide synthetase. Science 321:659–663
Bloudoff K, Rodionov D, Schmeing TM (2013) Crystal structures of the first condensation domain of CDA synthetase suggest conformational changes during the synthetic cycle of nonribosomal peptide synthetases. J Mol Biol 425:3137–3150
Lai JR, Fischbach MA, Liu DR et al (2006) Localized protein interaction surfaces on the EntB carrier protein revealed by combinatorial mutagenesis and selection. J Am Chem Soc 128:11002–11003
Bruner SD, Weber T, Kohli RM et al (2002) Structural basis for the cyclization of the lipopeptide antibiotic surfactin by the thioesterase domain SrfTE. Structure 10:301–310
Samel SA, Wagner B, Marahiel MA et al (2006) The thioesterase domain of the fengycin biosynthesis cluster: a structural base for the macrocyclization of a non-ribosomal lipopeptide. J Mol Biol 359:876–889
Walsh CT, Chen H, Keating TA et al (2001) Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr Opin Chem Biol 5:525–534
Patel HM, Tao J, Walsh CT (2003) Epimerization of an L-cysteinyl to a D-cysteinyl residue during thiazoline ring formation in siderophore chain elongation by pyochelin synthetase from Pseudomonas aeruginosa. Biochemistry 42:10514–10527
Perry RD, Fetherston JD (2011) Yersiniabactin iron uptake: mechanisms and role in Yersinia pestis pathogenesis. Microbes Infect 13:808–817
Samel SA, Czodrowski P, Essen LO (2014) Structure of the epimerization domain of tyrocidine synthetase A. Acta Crystallogr D Biol Crystallogr 70:1442–1452
Stachelhaus T, Walsh CT (2000) Mutational analysis of the epimerization domain in the initiation module PheATE of gramicidin S synthetase. Biochemistry 39:5775–5787
Mitchell CA, Shi C, Aldrich CC et al (2012) Structure of PA1221, a nonribosomal peptide synthetase containing adenylation and peptidyl carrier protein domains. Biochemistry 51: 3252–3263
Cakici O, Sikorski M, Stepkowski T et al (2010) Crystal structures of NodS N-methyltransferase from Bradyrhizobium japonicum in ligand-free form and as SAH complex. J Mol Biol 404:874–889
Shi R, Lamb SS, Zakeri B et al (2009) Structure and function of the glycopeptide N-methyltransferase MtfA, a tool for the biosynthesis of modified glycopeptide antibiotics. Chem Biol 16:401–410
Chhabra A, Haque AS, Pal RK et al (2012) Nonprocessive [2 + 2]e- off-loading reductase domains from mycobacterial nonribosomal peptide synthetases. Proc Natl Acad Sci U S A 109:5681–5686
Wyatt MA, Mok MC, Junop M et al (2012) Heterologous expression and structural characterisation of a pyrazinone natural product assembly line. Chembiochem 13:2408–2415
Frueh DP, Arthanari H, Koglin A et al (2008) Dynamic thiolation-thioesterase structure of a non-ribosomal peptide synthetase. Nature 454:903–906
Liu Y, Zheng T, Bruner SD (2011) Structural basis for phosphopantetheinyl carrier domain interactions in the terminal module of nonribosomal peptide synthetases. Chem Biol 18:1482–1488
Drake EJ, Nicolai DA, Gulick AM (2006) Structure of the EntB multidomain nonribosomal peptide synthetase and functional analysis of its interaction with the EntE adenylation domain. Chem Biol 13:409–419
Lee TV, Johnson LJ, Johnson RD et al (2010) Structure of a eukaryotic nonribosomal peptide synthetase adenylation domain that activates a large hydroxamate amino acid in siderophore biosynthesis. J Biol Chem 285:2415–2427
Johnson LJ, Koulman A, Christensen M et al (2013) An extracellular siderophore is required to maintain the mutualistic interaction of Epichloe festucae with Lolium perenne. PLoS Pathog 9:e1003332
Liu Y, Bruner SD (2007) Rational manipulation of carrier-domain geometry in nonribosomal peptide synthetases. Chembiochem 8:617–621
Ferreras JA, Ryu JS, Di Lello F et al (2005) Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat Chem Biol 1:29–32
Somu RV, Boshoff H, Qiao C et al (2006) Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J Med Chem 49:31–34
Miethke M, Bisseret P, Beckering CL et al (2006) Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J 273:409–419
Qiao C, Wilson DJ, Bennett EM et al (2007) A mechanism-based aryl carrier protein/thiolation domain affinity probe. J Am Chem Soc 129:6350–6351
Vannada J, Bennett EM, Wilson DJ et al (2006) Design, synthesis, and biological evaluation of b-ketosulfonamide adenylation inhibitors as potential antitubercular agents. Org Lett 8:4707–4710
Acknowledgments
The research from our lab that is included in this review has been supported by the National Institutes of Health (GM-068440).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Miller, B.R., Gulick, A.M. (2016). Structural Biology of Nonribosomal Peptide Synthetases. In: Evans, B. (eds) Nonribosomal Peptide and Polyketide Biosynthesis. Methods in Molecular Biology, vol 1401. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3375-4_1
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3375-4_1
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3373-0
Online ISBN: 978-1-4939-3375-4
eBook Packages: Springer Protocols