
Abstract
With the advent of new generations of high-throughput sequencing technologies, the catalog of human genome variants created by retrotransposon activity is expanding rapidly. However, despite these advances in describing L1 diversity and the fact that L1 must retrotranspose in the germline or prior to germline partitioning to be evolutionarily successful, direct assessment of de novo L1 retrotransposition in the germline or early embryogenesis has not been achieved for endogenous L1 elements. A direct study of de novo L1 retrotransposition into susceptible loci within sperm DNA (Freeman et al., Hum Mutat 32(8):978–988, 2011) suggested that the rate of L1 retrotransposition in the germline is much lower than previously estimated (<1 in 400 individuals versus 1 in 9 individuals (Kazazian, Nat Genet 22(2):130, 1999). Based on these revised estimates of the L1 retrotransposition rate, we modified the ATLAS L1 display technique (Badge et al., Am J Hum Genet 72(4):823–838, 2003) to investigate de novo L1 retrotransposition in human genomes. In this chapter, we describe how we combined a high-coverage ATLAS variant with high-throughput sequencing, achieving 11–25× sequence depth per single amplicon, to study L1 retrotransposition in whole genome amplified (WGA) DNAs.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
In 2003, Brouha et al. suggested that although there are 90 full-length L1s with intact ORFs in the reference human genome, only six “hot-L1s” are responsible for the majority (86 %) of the total L1 retrotransposition activity [4]. Since then, studies of human-specific L1 retrotransposition using different approaches such as comparative bioinformatics analyses, or transposon display techniques such as ATLAS ([2, 3, 4]; reviewed in [5]) have revealed many more active elements segregating in human populations. However, it has proven very difficult to find de novo L1 retrotransposition events, largely due to the low copy number of active L1s, the low frequency of such events ([1]), and the lack of high-resolution and high-coverage molecular genomic techniques. Recently, high-throughput sequencing approaches using, as well as array-based systems have begun enable high-throughput analysis of L1s as genomic structural variants. Currently, there are several approaches to study L1 retrotransposition in the genome. One set of approaches are PCR-based transposon display techniques used to characterize polymorphic human L1s in the human genome [3]. Generally, these techniques rely on the selective amplification of groups of retrotransposon sequences based on diagnostic nucleotide polymorphisms specific for each subfamily (e.g., the trinucleotide sequence ACA at positions 5954–6 of the reference element L1.3 (Accession L19088) discriminates the Ta subfamily from older subfamilies). In these methods, selective PCR is applied to gDNA libraries, and amplicons that show presence/absence variation between individuals are isolated for characterization by sequencing.
One such PCR-based display technique used to study active and polymorphic L1s is ATLAS [3]. Using the ATLAS technique, Badge et al. (2003) identified seven novel full-length L1 insertions, of which three were classified as “hot” (highly active) L1s in cell culture retrotransposition assays [3]. The ATLAS technique has several advantages compared to other transposon amplification techniques as it can analyze L1s at many loci genome-wide, and most importantly it can target the polymorphic, and so likely active, subset of L1 elements. Moreover, this technique is versatile, allowing researcher to adapt it to study different aspects of L1 biology; for example the Transduction Specific variant (TS-ATLAS) allows specific lineages of active elements that share a 3′ transduction, to be studied exhaustively [6]. In addition, as library production only involves the ligation of appropriate linkers, covalent epigenetic modifications, such as cytosine methylation are preserved, making it possible to study these modifications at L1 promoters genome-wide: differential digestion with methylation sensitive restriction enzymes during library preparation reveals methylation status by comparison of the pattern of insertions amplified relative to undigested gDNA libraries (Rahbari et al., unpublished). In the current chapter, we present a high-resolution, high-coverage ATLAS variant, which combined with Roche 454 sequencing platform, enables the analysis of near full-length L1 retrotransposon insertions. In this new approach, genomic coverage is improved to ~80 % of the genome, by using the NIaIII restriction enzyme for library construction. Since higher genome coverage increases the complexity of the amplicon distribution, it becomes unlikely that different insertions can be distinguished by amplicon size alone, necessary to characterize single-molecule L1 retrotransposon insertions. However, we overcome this problem by combining high-coverage ATLAS variant with high-throughput sequencing. This technique not only identifies polymorphic L1s in the genome, but also ables to identify very rare de novo L1 retrotransposition insertions. This technique is able to recover single molecules carrying L1 sequences using small pools of diluted DNA representing 10–20 sperm genome s (unpublished data). A full description of materials and methods for conducting this technique is detailed below. Finally, we conclude this chapter with some notes on high-throughput data analysis.
2 Materials
2.1 Chemical Reagents and Laboratory Equipment
All chemicals were supplied by local suppliers.
2.2 Restriction Enzymes
New England Biolabs supplied the restriction enzyme, NIaIII. Taq DNA polymerase and T4 DNA ligase can be obtained from other commercial sources.
2.3 Molecular Weight Markers
50 bp, 100 bp, and 1 kb molecular weight markers were supplied by local suppliers.
2.4 Standard Solutions
Southern blot solutions (denaturing and neutralizing), 20× Sodium Chloride Sodium-Citrate (SSC) buffer and 10× Tris-borate/EDTA (TBE) electrophoresis buffer, were made following standard recipes.
Other solutions and buffers are listed below:
-
1.
1× PCR buffer: 45 mM Tris pH 8.8, 11 mM NH4SO4, 4.5 mM MgCl2, 6.7 mM β-mercaptoethanol, 113 μg/ml BSA, and 1 mM dNTPs.
2.5 Oligonucleotides
DNA oligonucleotides were synthesized and HPLC-purified by local suppliers.
3 Methods
All steps of the ATLAS procedure were performed in a Class II laminar flow hood that had been decontaminated by UV exposure for at least 30 min prior to use. All reagents were PCR clean (i.e. opened only in the Class II hood and used only for PCR). For all buffers 18 MΩ water was used, either from a laboratory distillation unit or supplied by Sigma-Aldrich.
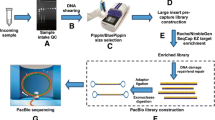
Below we describe the methodology for high-coverage ATLAS, starting with genomic DNA preparation for ATLAS library construction, followed by the generation of amplicons for next-generation sequencing. The schematic diagram (Fig. 1) summarizes all the steps of library preparation, amplification, sequencing, and data analysis.

Summary of the steps involved in modified ATLAS combined with NGS to isolate bona fide de novo LINE-1 insertions from human WGA gDNA
3.1 Genomic DNA Purification
Standard procedures can be applied to purify genomic DNA. If required, and in order to retain molecules evidencing de novo L1 retrotransposition events for further validation by PCR, we recommend performing WGA on the input genomic DNA. The WGA method described by Spits et al. [7] can be used without any additional modifications. Following WGA, samples with a DNA concentration of higher than 3 ng/μl and whose negative controls for WGA are lower than 1 ng/μl should be selected. A Nanodrop or a spectrophotometer can be used for DNA quantification. Next, selected samples are equally divided between three Eppendorf microcentrifuge tubes (preferably DNA LoBind), and an additional round of WGA should be performed on each. By subsequently pooling these three independent WGA reactions allele dropout, resulting from biased amplification in the initial WGA reaction, can be minimized.
3.2 ATLAS Library Construction
200 ng of WGA DNA or genomic DNA is incubated with NIaIII for 3 h at 37 °C in a final reaction volume of 20 μl (enzyme concentration 20U). Controls should be included in the digestion step including: DNA negative (DNA replaced by H2O); digestion enzyme negative (enzyme replaced by 50 % glycerol); and a DNA positive. Following digestion, inactivate the enzyme by heating the reaction at 65 °C for 20 min. Note that after this step the digested DNA can be aliquot into smaller volumes and stored at −80 °C until required (Fig. 1, part 1).
3.3 ATLAS Linker Preparation and Ligation
Prepare the linkers by mixing an equal volume of each linker primer RRNBOT2: 5′-ACTGGTCTAGAGGGTTAGGTTCCTGCTACATCTCCAGCCTCATG-3′ and RRNDUP1: 5′-AGGCTGGAGATGTAGCAG-3′ at a concentration of 50 μmol. Denature and anneal the mixed linkers by heating to 65 °C for 10 min and cooling to room temperature at the rate of 1 °C every 15 s (Fig. 1, part 2). Note that in the standard ATLAS protocol [3], 100 ng of the digested DNA is ligated to a 40-fold molar excess of the annealed suppression linker. This amount of linker is calculated by assuming the enzyme completely digests the genome into “X” number of fragments with two ligatable ends, and 3 pg of DNA represents one haploid genome equivalent. “X” varies with respect to the enzyme’s cutting frequency (but all calculations are necessarily approximate). For NIaIII, 2.7 μl of the 25 μmol annealed linker was used for each ligation, in a final volume of 20 μl.
Next ligate 100 ng of genomic DNA with the annealed linker overnight at 15 °C, in a final reaction volume of 20 μl. Linker negative (H2O), and enzyme negative (50 % glycerol) and reaction positives should be included as controls for the ligation step. The 20 μl ligation reaction final volume should consist of: 100 ng digested DNA, 2.7 μl annealed linker, 1.34 μl (4 Weiss units) T4 ligase, 2 μl 10× ligase buffer, and 8.96 μl H2O.
Inactivate the ligation by incubating the reaction at 70 °C for 10 min. Remove the excess linkers and short gDNA fragments (<100 bp) using the Qiaquick PCR purification system (Qiagen), according to the manufacturer’s instructions. Elute the purified DNA in PCR clean 5 mM Tris-pH 7.5 to a final volume of 30 μl. Note at this stage the ligated DNA can be aliquot into three volumes of 10 μl and stored at −80 °C (Fig. 1, part 2).
3.4 ATLAS Primary PCR
This stage involves a standard suppression PCR. A 15 μl final reaction volume, consisting of 13.5 μl PCR mix and 1.5 μl of constructed library DNA, is assembled under PCR clean conditions (Fig. 1, part 3). The PCR mix comprises 1× PCR buffer, 0.5 μl of 50 μM RVECPA1 (L1-specific linker primer): RVECPA1 5′ ACTGGTCTAGAGGGTTAGG 3′ and RV5SB2 (L1 5′ UTR internal primer) RV5SA2 5′-ATGGAAATGCAGAAATCACCGT-3′, 0.4 units of Taq polymerase.
Perform PCR using the following cycle conditions: an initial denaturing step at 96 °C for 30 s, followed by 25 cycles of 96 °C for 30 s, 62 °C for 2 min, and then extension at 72 °C for 10 min.
3.5 ATLAS Secondary PCR
The rationale for fusion primer design for the secondary PCR (Subheading 3.5.1) is explained below. The design is based on the assumption that the PCR products are to be sequenced using the Roche 454 platform, incorporating the 454-specific adapters by PCR. However, the sequencing adaptors can be modified based on the preferred sequencing platform. In the following, we have explained the procedure to conduct ATLAS secondary PCR (Subheading 3.5.2) upstream of 454 sequencing using the GS FLX Titanium chemistry.
3.5.1 Fusion Primers Design
For this technique, the amplicon length needs to be given careful consideration. We selected the Roche 454 technology to achieve long read lengths but other technologies such as Illumina and PacBio could be considered. To extract maximum information, the amplicons need to be fully sequenced to enable their accurate mapping to the reference genome, and the determination of their structure. The estimated sequence read length of the 454 Sequencing System with the GS FLX Titanium chemistry used here is about 450 nucleotides, but some of the amplicons generated by the full-length L1-specific suppression PCR are bigger than 450 bp (up to 750 bp). As a result 450 bp bidirectional reads were used to ensure sufficient overlap in the middle of the larger amplicon sequences, such that long amplicons will be reliably covered.
We allowed up to ~50 nucleotides from each end of the amplicon for the fusion primers. For example, for a read to cover an amplicon entirely, it must traverse its key (4 nucleotides) at the proximal end and the template-specific primers (20 nt) and MID (Multiplex IDentifier), sequences (10 nucleotides each) at both ends.
Each pair of the fusion primers consisted of forward and reverse primers. All the forward fusion primers (5′ to 3′ direction) were constructed of the following segments: Roche-LibL-primer A: 5′-CGTATCGCCTCCCTCGCGCCATCAG-3′, a 10 nucleotide MID, and a linker-specific primer, RVECPA2 5′-CCTGCTACATCTCCAGCC-3′. All the reverse fusion primers were constructed from the following segments: Roche-LibL-primer B: 5′-CTATGCGCCTTGCCAGCCCGCTCAG-3′, a 10 nucleotide MID and an L1-specific primer RV5SB2 5′-CTTCTGCGTCGCTCACGCT-3′. The schematic diagram in Fig. 2 indicates the primers orientation. The use of 10 bp MIDs is optional. Shorter MIDs can enable unambiguous de-covolution of multiplexed experiments, but 10 bp MIDs enable higher levels of multiplexing, with less chance of read miss-assignment.

Schematic diagram of the arrangement of suppression PCR L1-specific, linker-specific, and sequencing fusion primers relative to an example full-length L1 insertion site
3.5.2 ATLAS Secondary PCR
Perform the secondary PCR in a 50 μl final reaction volume consisting of 45 μl PCR mix and 5 μl of the primary PCR reaction (Fig. 1, part 4). The PCR mixture comprises 1× PCR buffer, 0.125 μM fusion primer A (containing a linker-specific primer) and 0.125 μM fusion primer B (containing an L1 internal primer), and 0.4 units of Taq polymerase. Use the following thermo-cycling conditions: an initial denaturing step at 96 °C for 30 s; followed by 25 cycles of 96 °C for 30 s, 75 °C for 2 min; followed by an extension step at 72 °C for 10 min. Figure 3 shows an example of ATLAS secondary PCR products, fractionated by agarose gel electrophoresis.

An example of ATLAS secondary PCR products (RVECPA2 + RV5SB2) (2 μl) from 12 individuals (NIaIII libraries) separated on a 1.5 % agarose gel (100 bp molecular weight markers, NEB). The secondary products range from 200 to 750 bp in length, including the 454 fusion primers
3.6 Pooling the Barcoded Amplicons
Prior to sequencing, an equimolar concentration of the secondary PCR products from each library should be pooled together. Note that in order to obtain an equal coverage from all the barcoded samples, it is important to quantify all the secondary PCR products accurately prior to the pooling. Methods for accurately quantifying the range of amplicons are picogreen analysis [8] as well as using an Agilent Bioanalyzer (Invitrogen) (Fig. 1, part 5).
3.7 Size Selection of the Pooled Samples
The ATLAS secondary products contain a range of PCR products with variable lengths (200–750 bp). To minimize length biasing during the emulsion phase PCR (emPCR), we recommend dividing pooled libraries into two size-fractionated batches, with different ranges of amplicon length (Fig. 1, part 6). Each batch can be sequenced separately on physically separate regions of a picotiter plate (Roche 454) or different lanes for Hiseq. One batch (pool number two) contains smaller amplicons ranging from 200 to 350 bp and the other batch contains the longer length amplicons ranging from 300 to 750 bp. The lower and upper range products were made by fractionating the pooled secondary PCR products on an agarose gel and extracting the lower range sizes >350 bp and the upper range products <300 bp. A 50 bp size range overlap was allowed between the upper and lower ranges to avoid losing products at the size fraction junction (300–350 bp).
To achieve fractionation, load two equal volumes (100 μl) of the pooled samples on a 2.5 % agarose gel and run at 120 V for 2 h. Transfer the gel onto a Dark Reader visible light transilluminator (Clare Chemical Research) and cut gel blocks to divide the pooled products into two different size ranges: 200–350 bp and 300–750 bp. Extract DNA from the gel blocks using the Qiaquick gel extraction system (Qiagen) according to the manufacturer’s instructions. Elute the purified DNA in 30 μl of PCR clean 5 mM Tris-pH 7.5. The purified amplicons can be aliquot for sequencing and remaining aliquots stored at −80 °C. To verify that the pooled samples are size-selected correctly, run 1 μl of each batch on an Agilent Bioanalyser (high sensitivity DNA kit, Invitrogen). An example of size-selected libraries is shown in Fig. 4.

Use of the 2100 Agilent Bioanalyser 2100 device (High Sensitivity DNA kit, Invitrogen) to check the size selection of the pooled amplicons at different size ranges. Lane 1: Marker, lane 2: lower range product size (200–350 bp), lane 3: upper range product size (300–750 bp), lane 6: No-size selected pooled libraries (control); product size ranges from 200 to 750 bp
3.8 Sequence Coverage Calculation
Prior to sequencing, the required sequence coverage should be calculated to be sure to generate enough reads to confidently characterize single-molecule events. To calculate this, we mapped the L1Hs Ta-specific oligonucleotides used in the primary suppression PCR to the human reference genome resulting in the identification of ~3000 discrete potential priming sites. Data from exhaustive fosmid sequencing studies [9] enable an estimate of the number of novel (i.e. not previously characterized) L1s per screened genome, as between 4 and 6 insertions. Since this is a small fraction of the ~3000 oligo binding sites shared by the majority of human genomes (determined by in silico mapping), failing to account for these in the coverage estimates will only result in a very small overestimation. By contrast, the proportion of polymorphic L1 Ta elements (in any genome) is about 30 % [10], making ~3000 L1 amplicons per average genome a substantial overestimate (as many insertions will be absent from a given genome). By this logic, our simplifying assumptions can only lead to an underestimate of the coverage required. In the current protocol, the NIaIII restriction enzyme is used to construct the genomic DNA library. Knowing (from in silico digestion Badge and Rouillard, personal communication) that about 80 % of the human genome is within 1 kb of a NIaIII site, the number of accessible L1 loci for this experiment would be 80 % of ~3000, i.e. ~2400 L1 loci, assuming a random distribution of L1 insertion loci and restriction sites. Based on this knowledge and the given sequencing coverage for the experiment, it is possible to calculate the minimum expected number of reads per each de novo L1 retrotransposition . For example, we used ¾ of a picotiter plate and the number of beads per quarter plate should be around ~160,000 (according to the manufacturer’s data). Thus the total number of the reads expected from all three regions is 160,000 × 3 = 480,000 reads. When fourteen libraries are sequenced, the number of expected reads per amplicon would be 480,000/3000 × 14, or ~11 reads per amplicon. Therefore, it has been estimated that for a single molecule present in one library we should detect about 11 reads, but the coverage would be much higher for constitutive L1 loci present in all libraries. This calculation is very useful in adjusting the data analysis filters (Subheading 4).
3.9 Post-sequencing Data Analysis (see Note 1 )
Initially, the bulk reads in a FASTA or FASTQ format should be separated according to their MIDs. Following this, it is recommended to either trim off the sequences introduced by PCR (linkers, fusion primers) from both sides except for the sequence of the L1-specific primer (RV5BS2), which should be retained in the sequence structure.
Following trimming, map the sequences to the human genome reference. While various alignment algorithms can be used, one option is to use the public instance of the Galaxy web service [11]: http://main.g2.bx.psu.edu/ and the included LastZ tool to map the FASTA files [11]. Next, the coordinates of the mapped reads should be compared to the coordinates of the L1 oligo data set. The L1 oligo data set can be generated, by mapping the L1-specific primer (RV5BS2) to the reference genome, also using LastZ.
Reads whose coordinates overlap with the L1 oligo data set correspond to the subset of L1 loci that are already present in the reference genome; to select candidate de novo/novel L1 insertion in the remaining sequences, the coordinates of the candidate novel insertion should be checked against the reported polymorphic L1 insertions in published data (previously characterized, but that are absent from the reference genome [12]). Figure 5 shows an example of an L1 sequence isolated from ATLAS libraries, which is absent from the reference genome. However, it has been previously characterized as a polymorphic insertion. We recommend to check the latest published L1 data sources, as euL1DB [12] and others. Further filters such as minimum and maximum number of reads required from an amplicon can be set, depending on the sequencing experiment design.

ATLAS and high-throughput sequencing can capture known polymorphic L1 insertions. The screen shot shows the result of a BLAT search using 454 traces (numbered black rectangles) that co-locate with the 5′ flanking DNA (black rectangle labeled “5p”) of a known polymorphic L1 element that is absent from hg19. This novel L1 insertion was previously reported
3.10 Characterizing Candidate Novel and De Novo L1-Mediated Retrotransposition Events Using Site-Specific PCR
Having identified a subset of reads which are confirmed to be absent from the human genome reference sequence and all the available L1 databases, these reads can be proposed as candidate novel L1 retrotransposons, and subjected to independent validation.
The presence of non-reference insertions should be verified via site-specific PCR. In this procedure the 5′ end and flanking regions of non-reference L1s can be amplified using an L1-specific primer and a primer specific for the 5′ flanking genomic region. Amplification of the “empty” site, using primers specific for the 5′ and 3′ flanking genomic regions from gDNA of an individual unrelated to the sequenced donor can be used to verify the PCR primers function, as well as the ability to amplify the insertion region. Amplification of the 5′ and 3′ ends of the insertion enables characterization of the insertion as a canonical (full-length, carrying Target Site Duplication s and terminating in a 3′ poly A-tail) insertion. Long-range PCR of the entire insertion and characterization by direct sequencing from WGA gDNA, even from single cells, is feasible in our hands.
4 Notes
-
1.
Post-sequencing data analysis to identify de novo L1 insertion from the ATLAS data can be done in many different ways, using different software platforms.
References
Freeman P, Macfarlane C, Collier P, Jeffreys AJ, Badge RM (2011) L1 hybridization enrichment: a method for directly accessing de novo L1 insertions in the human germline. Hum Mutat 32(8):978–988. doi:10.1002/humu.21533
Kazazian HH Jr (1999) An estimated frequency of endogenous insertional mutations in humans. Nat Genet 22(2):130
Badge RM, Alisch RS, Moran JV (2003) ATLAS: a system to selectively identify human-specific L1 insertions. Am J Hum Genet 72(4):823–838
Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH Jr (2003) Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A 100(9):5280–5285
Beck CR, Garcia-Perez JL, Badge RM, Moran JV (2011) LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet 12:187–215. doi:10.1146/annurev-genom-082509-141802
Macfarlane CM, Collier P, Rahbari R, Beck CR, Wagstaff JF, Igoe S, Moran JV, Badge RM (2013) Transduction-specific ATLAS reveals a cohort of highly active L1 retrotransposons in human populations. Hum Mutat 34(7):974–985. doi:10.1002/humu.22327
Spits C, Le Caignec C, De Rycke M, Van Haute L, Van Steirteghem A, Liebaers I, Sermon K (2006) Whole-genome multiple displacement amplification from single cells. Nat Protoc 1(4):1965–1970. doi:10.1038/nprot.2006.326
Ahn SJ, Costa J, Emanuel JR (1996) PicoGreen quantitation of DNA: effective evaluation of samples pre- or post-PCR. Nucleic Acids Res 24(13):2623–2625
Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV (2010) LINE-1 retrotransposition activity in human genomes. Cell 141(7):1159–1170
Boissinot S, Chevret P, Furano AV (2000) L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol Biol Evol 17(6):915–928
Taylor J, Schenck I, Blankenberg D, Nekrutenko A (2007) Using galaxy to perform large-scale interactive data analyses. Curr Protoc Bioinformatics Chapter 10:Unit 10.15. doi:10.1002/0471250953.bi1005s19
Mir AA, Philippe C, Cristofari G (2015) euL1db: the European database of L1HS retrotransposon insertions in humans. Nucleic Acids Res 43(Database issue):D43–D47. doi:10.1093/nar/gku1043
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Rahbari, R., Badge, R.M. (2016). Combining Amplification Typing of L1 Active Subfamilies (ATLAS) with High-Throughput Sequencing. In: Garcia-Pérez, J. (eds) Transposons and Retrotransposons. Methods in Molecular Biology, vol 1400. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3372-3_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3372-3_6
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3370-9
Online ISBN: 978-1-4939-3372-3
eBook Packages: Springer Protocols