
Abstract
Long interspersed nucleotide element 1 (LINE-1 or L1) is a family of non-LTR retrotransposons that can replicate and reintegrate into the host genome. L1s have considerably influenced mammalian genome evolution by retrotransposing during germ cell development or early embryogenesis, leading to massive genome expansion. For many years, L1 retrotransposons were viewed as a selfish DNA parasite that had no contribution in somatic cells. Historically, L1s were thought to only retrotranspose during gametogenesis and in neoplastic processes, but recent studies have shown that L1s are extremely active in the mouse, rat, and human neuronal progenitor cells (NPCs). These de novo L1 insertions can impact neuronal transcriptional expression, creating unique transcriptomes of individual neurons, possibly contributing to the uniqueness of the individual cognition and mental disorders in humans.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Neural stem cells (NSC) reside in discrete neurogenic regions of the adult brain . NSC can remain multipotent and continue to replicate in the neurogenic niche. Upon stimulus, NSC can differentiate into glial progenitors, which will mature into astrocytes or oligodendrocytes, or into neuronal progenitor cell s (NPCs). When a committed to the neuronal lineage, a specific gene expression profile is activated. Interestingly, L1 retrotransposon is one of the transcripts upregulated upon neuronal commitment [1]. And since the L1 element is an autonomous retrotransposon s [2], does it actually retrotranspose in NPCs?
Previous studies have indicated that L1 retrotransposition can occur in germ cells or in early embryogenesis, before the germ line becomes a distinct lineage [3, 4], whereas a cultured cell retrotransposition assay has revealed that human and mouse L1 elements can retrotranspose in a variety of transformed or immortalized cultured cell lines [5–7]. Now we know L1 is capable of high levels retrotransposition in NPCs, generating a neuronal genetic mosaicism in differentiated networks [8].
Initial studies on this field had take advantage of an engineered active L1 retrotransposition cassette, a marker that only expresses if the element inserts back into the genome [5, 9]. For example, if the marker in the L1 cassette is eGFP, then a cell expressing eGFP indicates the engineered L1 successfully retrotransposed. Utilizing the L1-eGFP cassette, the ability of NPCs to support L1 retrotransposition was first reported in rat hippocampal NPCs in vitro [1]. Since then, the L1-eGFP cassette has proven L1 can also retrotranspose in human NPCs in vitro and mouse NPCs in vivo [1, 10–13]. Determining the integration sites of de novo L1 sequences in NPCs would yield insights into the potential effects of neuronal retrotransposition . Early sequencing efforts in neurons discovered L1 could integrate into introns of active genes [1, 10]. L1 integration events in rat NPCs have been shown to alter expression of nearby genes by promoter enhancement and epigenetic silencing [1].
Here, we show how to optimize the protocol to test L1 retrotransposition in neural progenitor cells using a tagged L1-eGFP element in a plasmid. Subsequent investigations may include cellular phenotypic assays and genomic analyses for de novo L1 insertions.
2 Materials (See Note 1 )
-
1.
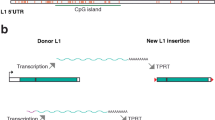
A fresh maxi-prep of the L1-eGFP (Fig. 1) plasmid in a concentration of 1 μg/μL diluted in water. We generally use a Endo-Free plasmid Maxi kit to purify this DNA. See Note 2 for a detailed description of this construct.
Fig. 1 
L1-eGFP retrotransposition assay. (a) The retrotransposition-competent human L1 (L1RP) contains a 5′ untranslated region (UTR) that harbors an internal promoter, two open reading frames (ORF1 and ORF2; not drawn to scale), and a 3′ UTR that ends in a poly (A) tail. ORF2 contains an endonuclease (EN) and reverse transcriptase (RT) domain as well as a cysteine-rich 3′ end (C). The eGFP retrotransposition indicator cassette consists of a backward copy of the eGFP gene whose expression is controlled by the human cytomegalovirus major immediate early promoter (pCMV) and the herpes simplex virus thymidine kinase polyadenylation sequence (pA). This arrangement ensures that eGFP expression will only become activated upon L1 retrotransposition. The black arrows indicate PCR primers flanking the intron present in the eGFP gene. The 343-bp PCR product, diagnostic for the loss of the intron, is indicative of a retrotransposition event. Sequencing of the 343-bp PCR product should validate the precise splicing of the intron
-
2.
A retrotransposition -defective L1 construct (JM111-eGFP) that contains two missense mutations in ORF1 [5, 14, 15] can be used as a negative control (see Note 3 ).
-
3.
A positive using the same plasmid backbone (pCEP4, Invitrogen) containing the eGFP driven by the CMV promoter .
-
4.
A fresh culture of NPCs prepared and cultured as described for rodent adult NPCs [16] or iPSC-derived NPCs [11, 17]. See Note 4 .
-
5.
Dulbecco’s modified Eagle medium (DMEM/F12).
-
6.
N2 supplement (Invitrogen).
-
7.
l-glutamine in solution.
-
8.
Basic fibroblast growth factor (FGF-2). Prepare it following manufacturer’s instructions.
-
9.
Poly-l-ornithine and laminin coated tissue culture grade dishes.
-
10.
A transfection system. Because of the large size of the L1 indicator plasmids and the sensitivity of the NPC, we have successfully used the Nucleofector technology (Lonza). Make sure you have the right kit for your cells (rodent or human NPC). See Note 5 .
-
11.
Puromycin .
-
(a)
Warm (37 °C) the required volume of N2-medium, add fresh FGF-2 just before you start the experiment (see Note 4 ).
-
(b)
Working solution of puromycin or other specific antibiotic to select the L1 reporter in the target cells.
-
(a)

3 Methods (See Note 6 )
-
1.
Prepare plasmid aliquots (1–5 μL) in no more than 10 μL volume in sterile 1 mL microtubes.
-
2.
Prepare 100 μL of Nucleofector solution in sterile 1 mL microtubes by mixing the cell type specific Nucleofector solution with the supplement reagent provided by the kit. The final solution can be store at 4 °C for a month.
-
3.
Harvest the target cell types using appropriated methods, and gently centrifuge (1000 × g) to final volume of 100 μL. See Note 7 .
-
4.
Mix the cells with the Nucleofector solution to a final volume of 200 μL.
-
5.
Add the right amount of plasmid DNA (4 μg) to the mixture. Gently pipette up and down to homogenize the mixture.
-
6.
Immediately, transfer the solution to the electroporation cuvette, avoiding bubbles. Select the cell-type specific program and proceed with the electroporation. See Note 8 .
-
7.
Remove the cuvette from the machine; slowly add 500 μL of pre-warmed media with FGF-2 to the cuvette.
-
8.
Remove the total volume from the cuvette and immediately plate the electroporated cells into pre-warm culture dishes with media. Make sure they are well distributed along the tissue culture dish. Optional: you can add extra add 500 μL of pre-warmed media with FGF-2 to the cuvette to recovery remaining cells.
-
9.
Wait 3–8 h for cells to attach to the bottom of the plate. Time will depend on cell type and viability. Next, change the entire media from the culture dishes. See Note 9 .
-
10.
Cells harboring the L1 expression constructs can be selected by the addition of puromycin (1 μg/mL) 48 h after electroporation to the culture medium. Puromycin -resistant cells can be screened for EGFP expression by flow cytometry.
-
11.
After 3–7 days, eGFP expression can be detected in NPCs (see Note 10 ). After 7 days, transfected puromycin -resistant cells can be analyzed with a Becton Dickenson FACStar Plus containing a blue argon laser (488 nm) and fluorescein filter sets (530/30 bandpass). A total of 10,000 events were analyzed based on forward scatter versus side scatter profiles. Dead cells were excluded by propidium iodide gating. Live cells were analyzed for fluorescence intensity.
-
12.
PCR can confirm the presence of the retrotransposed (i.e., spliced) eGFP gene in NPC-positive cells; sequencing of the PCR products will confirm the precise splicing of the intron (Fig. 1). The eGFP PCR primers were previously described [15]. See Note 11 .
4 Notes
-
1.
Prepare all cell media and washing solutions using ultrapure water (prepared by purifying deionized water to attain a sensitivity of 18 MΩ cm at 25 °C) and analytical grade reagents. Prepare and store all reagents at room temperature (unless indicated otherwise). We do not add sodium azide to the reagents. Diligently follow all waste disposal regulations when disposing waste materials.
-
2.
The indicator cassette consists of the enhanced green fluorescence protein gene (eGFP) in the opposite orientation of the L1 transcript, a heterologous promoter (pCMV), and a polyadenylation signal (pA) in the 3′ UTR region of the element (Fig. 1). The eGFP gene is interrupted by an intron (IVS2 of the γ-globin gene) in the same transcriptional orientation as the L1 transcript. This arrangement ensures that eGFP-positive cells will arise only when a transcript initiated from the promoter driving L1 expression is spliced, reverse-transcribed, and integrated into chromosomal DNA, thereby allowing expression of the retrotransposed eGFP gene from the pCMV promoter [5, 15].
-
3.
Make sure you sequence or use restriction enzymes to verify the identity of your plasmids maxi-prep. The L1-eGFP or other L1 indicator plasmid is available upon request from pioneers in the L1 retrotransposon field, Drs. Haig Kazazian, Joef Boeke, and John Moran. Here, we used the eGFP to exemplify the retrotransposition assay in neural progenitor cells, but other genes as L1 indicators should work just fine.
-
4.
Briefly, NPCs were maintained on poly-l-ornithine and laminin coated flasks in N2-medium. Make sure you wash your coated plates at least three times with PBS before use. Coating left over materials may affect cell survival and proliferation.
N2 media is comprised of Dulbecco’s modified Eagle medium (DMEM/F12) (1:1), N2 supplement (Invitrogen), 2 mM l-glutamine and 20 ng/mLFGF-2. Change the N2/FGF-2 media every 2–3 days. Passage the cells when they reach about 90 % confluency.
-
5.
Other reagents such as Lipofectamine (Invitrogen), Fugene 6 (Promega/Roche) or calcium-based transfection can also work and needs to be optimized for the target cell type. Previously optimize your transfection efficiency using a reporter system (such as the pCEP-eGFP) in your target cells. We observed some variability with different cell preparations, species, and passage number.
-
6.
Carry out all procedures at room temperature unless otherwise specified. All tissue culture experiments should be performed inside safety cabinets, following sterile working practices.
-
7.
We avoid using enzymatic methods but if you do, make sure to inhibit the enzyme or wash the cell pellet before proceed. Also, the number of cells to be electroporated needs to be optimized. We find that 50,000 rat NPCs to be ideal in our conditions. Too much cells may inhibit the reaction and too little may decrease survival.
-
8.
Generally, we perform one reaction a time, and have observed toxicity in cells that stay in the cuvette for more than 10 min.
-
9.
The Nucleofector solution is quite toxic. Thus, the earlier you can replace the culture medium, the better.
-
10.
If you experience cell death in the first hours post-transfection, you may try to use 24–48 h conditioned media (50:50) from healthy-growing cell cultures, instead pure fresh media. Some cells benefit from released factors and survive better in this condition.
-
11.
Puromycin -resistant eGFP-negative NPC clones may also harbor a PCR product that corresponded in size to the retrotransposed eGFP gene. This observation may represent a truncation of the 5′ end or the retrotransposed eGFP gene may undergo epigenetic silencing either during or soon after L1 retrotransposition .
References
Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435:903–910
Muotri AR, Marchetto MC, Coufal NG, Gage FH (2007) The necessary junk: new functions for transposable elements. Hum Mol Genet 16(Spec No. 2):R159–R167. doi:10.1093/hmg/ddm196, 16/R2/R159 [pii]
Ostertag EM, DeBerardinis RJ, Goodier JL, Zhang Y, Yang N, Gerton GL, Kazazian HH Jr (2002) A mouse model of human L1 retrotransposition. Nat Genet 32:655–660
Prak ET, Dodson AW, Farkash EA, Kazazian HH Jr (2003) Tracking an embryonic L1 retrotransposition event. Proc Natl Acad Sci U S A 100:1832–1837
Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr (1996) High frequency retrotransposition in cultured mammalian cells. Cell 87:917–927
Morrish TA, Gilbert N, Myers JS, Vincent BJ, Stamato TD, Taccioli GE, Batzer MA, Moran JV (2002) DNA repair mediated by endonuclease-independent LINE-1 retrotransposition. Nat Genet 31:159–165
Han JS, Szak ST, Boeke JD (2004) Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429:268–274
Muotri AR, Gage FH (2006) Generation of neuronal variability and complexity. Nature 441:1087–1093. doi:10.1038/nature04959, nature04959 [pii]
Freeman JD, Goodchild NL, Mager DL (1994) A modified indicator gene for selection of retrotransposition events in mammalian cells. Biotechniques 17:46, 48-49, 52
Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, Gage FH (2009) L1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131. doi:10.1038/nature08248, nature08248 [pii]
Muotri AR, Marchetto MC, Coufal NG, Oefner R, Yeo G, Nakashima K, Gage FH (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446. doi:10.1038/nature09544, nature09544 [pii]
Muotri AR, Zhao C, Marchetto MC, Gage FH (2009) Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus 19:1002–1007. doi:10.1002/hipo.20564
Garcia-Perez JL, Marchetto MC, Muotri AR, Coufal NG, Gage FH, O’Shea KS, Moran JV (2007) LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet 16:1569–1577
Brouha B, Meischl C, Ostertag E, de Boer M, Zhang Y, Neijens H, Roos D, Kazazian HH Jr (2002) Evidence consistent with human L1 retrotransposition in maternal meiosis I. Am J Hum Genet 71:327–336
Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH Jr (2000) Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Res 28:1418–1423
Gage FH, Ray J, Fisher LJ (1995) Isolation, characterization, and use of stem cells from the CNS. Annu Rev Neurosci 18:159–192
Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR (2010) A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143:527–539. doi:10.1016/j.cell.2010.10.016
Acknowledgements
The work was supported by grants from the National Institutes of Health (NIH) R01 MH094753-01, and the NIH Director’s New Innovator Award Program, 1-DP2-OD006495-01.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Muotri, A.R. (2016). L1 Retrotransposition in Neural Progenitor Cells. In: Garcia-Pérez, J. (eds) Transposons and Retrotransposons. Methods in Molecular Biology, vol 1400. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3372-3_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3372-3_11
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3370-9
Online ISBN: 978-1-4939-3372-3
eBook Packages: Springer Protocols