
Abstract
Virologic surveillance is essential to the success of the World Health Organization initiative to eradicate poliomyelitis. Molecular methods have been used to detect polioviruses in tissue culture isolates derived from stool samples obtained through surveillance for acute flaccid paralysis. This chapter describes the use of realtime PCR assays to identify and serotype polioviruses. In particular, a degenerate, inosine-containing, panpoliovirus (panPV) PCR primer set is used to distinguish polioviruses from NPEVs. The high degree of nucleotide sequence diversity among polioviruses presents a challenge to the systematic design of nucleic acid-based reagents. To accommodate the wide variability and rapid evolution of poliovirus genomes, degenerate codon positions on the template were matched to mixed-base or deoxyinosine residues on both the primers and the TaqMan™ probes. Additional assays distinguish between Sabin vaccine strains and non-Sabin strains. This chapter also describes the use of generic poliovirus specific primers, along with degenerate and inosine-containing primers, for routine VP1 sequencing of poliovirus isolates. These primers, along with nondegenerate serotype-specific Sabin primers, can also be used to sequence individual polioviruses in mixtures.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
1.1 Early Poliovirus Diagnostics
Virologic surveillance is essential to the success of the World Health Organization initiative to eradicate poliomyelitis [1, 2]. Expanded surveillance for cases of acute flaccid paralysis has been complemented by the development of improved methods for poliovirus identification. Two independent approaches were employed for routine identification of polioviruses: (1) antigenic characterizations using cross-adsorbed antisera [3] or (2) molecular characterizations using genotype-specific probes ([4, 5] Fig. 1) or PCR primers [6, 7]. Serologic methods can generally differentiate vaccine-related isolates from wild polioviruses [3] but have a limited capacity to differentiate among wild poliovirus genotypes. One of the earliest molecular approaches to poliovirus diagnostics was the use of oligonucleotide RNA fingerprinting [8]. RNA fingerprinting was once widely used for identifying RNA virus isolates by allowing the recognition of viral genetic relationships using a two-dimensional pictorial form of the ribonuclease T1 digested RNA. As diagnostic technologies improved, additional molecular methods were developed which include the use of either probe hybridization or RT-PCR in order to directly identify Sabin and/or some wild genotype viruses. Both of these methods give identifications that are in excellent agreement with results obtained by the more detailed analyses of genomic sequencing [5–7, 9]. In addition, restriction fragment length polymorphism assays of PCR-amplification products were also developed [10, 11].

Dot blot hybridization of poliovirus RNA
Wild polioviruses could be identified indirectly by their non-reactivity with the Sabin-strain-specific molecular reagents. Direct identification of wild polioviruses was only possible with wild genotype-specific probes [4] and PCR primer sets [7]. At that time, the current catalog of wild-genotype-specific molecular reagents did not cover all of the many different poliovirus genotypes still in circulation worldwide [9]. Consequently, identification of wild polioviruses through the exclusive use of Sabin-strain-specific molecular reagents is dependent upon the accurate typing of virus isolates. The standard methods for poliovirus typing with neutralizing antibodies were comparatively time-consuming and laborious [12]. Poliovirus neutralizing antibodies are type specific [12], and no well-characterized poliovirus-specific group antigen has been described [12–14]. Molecular reagents, in the form of nucleic acid probes [4, 5] and PCR primers [7, 15, 16] targeted to highly conserved nucleotide intervals within the 59 untranslated region, have been developed for the reliable detection of nearly all members of the enterovirus group (including polioviruses). Other reagents were described which permitted the detection of some, but not all, polioviruses by PCR [17, 18]. An alternate method to screen out nonpolio enteroviruses (NPEVs) is to isolate virus in recombinant murine cells expressing the human gene for the poliovirus receptor [19]. This approach also permitted the detection of underlying polioviruses in virus mixtures, and avoided the laborious virus neutralization and recultivation steps of standard methods [12]. To overcome these diagnostic limitations, we developed a degenerate, inosine-containing, panpoliovirus (panPV) PCR primer set that could be used to distinguish polioviruses from NPEVs.
2 Materials and Equipment
2.1 Realtime PCR Kit Components
The kit is supplied in one box containing six vials of primers and probes in Buffer A (Serotype 1, Serotype 2, Serotype 3, Pan-Poliovirus, Pan-Enterovirus, Sabin Multiplex), two vials of Buffer B (to which DTT and enzymes should be added prior to the first use) and one vial of DTT. The box also contains the appropriate positive controls for each primer set, a tube of water and one copy of this package insert. Additional required reagent and enzymes, not supplied with the kit, are Protector RNase inhibitor, Transcriptor Reverse Transcriptase, and Taq DNA polymerase from Roche Applied Science. The listed products were used in the development and evaluation of this kit and do not constitute a specific product endorsement. Enzyme availability from manufacturers may vary with each laboratory. Therefore, it is the responsibility of each laboratory to find appropriate substitutes when necessary.
2.2 10× RT-PCR Buffer
RT-PCR buffer is made at a 10× concentration so that when diluted to 1× in a normal PCR reaction, the final concentration of the buffer in reaction mixtures is 67 mM Tris (pH 8.0), 17 mM NH4SO4, 6 μM EDTA, 2 mM MgCl2, and 1 mM dithiothreitol (DTT). Below is the protocol for making 10 ml of 10× RT-PCR buffer. To a 15 ml conical tube, add the following:
-
0.2 ml MgCl2 (1 M).
-
6.7 ml Tris, pH 8.8 (1 M).
-
1.7 ml NH4SO4 (1 M).
-
1.2 μl 0.5 M EDTA
-
1.4 ml PCR grade H2O (nuclease-free).
Mix well and aliquot 1.0 ml of the buffer into 1.5 ml microcentrifuge tubes. Freeze the aliquots at −20 °C. Add 5.0 μl of 1 M dithiothreitol (DTT) to each 1.0 ml aliquot before using for the first time. The buffer + DTT will be stable for at least 1 month after the addition of DTT (re-freeze the buffer after each use).
2.3 Oligonucleotides
Synthetic oligodeoxynucleotides were prepared, purified, and analyzed as described previously (Yang et al. [6]). Primer polarities are indicated by A (antisense or antigenome polarity) or S (sense or genome polarity). See Table 2 for a list of poliovirus Reverse Transcriptase (RT) PCR and sequence primers
-
PCR thermocycler.
-
Certified RNase- and DNase-free 1.5 ml and 200 μl microcentrifuge tubes.
-
Sterile nuclease-free H2O.
-
Ethanol (96–100 %).
-
QIAamp Viral RNA Mini Kit (Qiagen, Inc; Cat. # 52906); 250 extractions (smaller kit available).
-
Sharps container.
-
4 mm disposable biopsy punch (VWR 21909-140).
-
Cutting Mat (VWR WB100020).
-
Bleach (for 10 % solution).
-
Optional: RNase AWAY individual wipes (VWR 89025-862).
-
Tissue wipes (VWR 82003-824).
-
70 °C heat block.
-
Optional: suitable vacuum manifold dedicated to RNA extraction ONLY.
-
Inoculating loop (1 μl) (VWR 90001-096) or
-
Inoculating needle (VWR 90001-104) or
-
Disposable tweezers (Enviro Safety Products 77233).
-
TE-1 buffer: 10 mM TRIS–HCl, 0.1 mM EDTA pH 7.6.
-
Glycogen, Nuclease Free (Ambion, AM9510).
-
1 M Dithiothreitol (DTT).
-
RNA process buffer (200 μl/sample): 191.6 μl TE-1 buffer, 0.4 μl 1 M DTT, 8.0 μl 5 mg/ml nuclease-free glycogen.
-
10× PCR Buffer.
-
dNTP mix (concentration of 10 mM each dNTP, Roche #11814362001).
-
RNase inhibitor (40 U/μl, Roche #3335402001).
-
Reverse transcriptase (20 U/μl, Roche #3531287001).
-
Taq polymerase (5 U/μl, Roche #11596594001).
-
Gel electrophoresis System (gel tray, sample combs, buffer chamber, power supply leads, and power supply).
-
Agarose gel powder (GeneMate Gene Pure LE Agarose, BioExpress #E-3120-500).
-
Electrophoresis buffer (Tris-borate-EDTA = TBE).
-
6× load buffer with bromophenol blue.
-
GelStar, GelRed, or any fluorescent dye that intercalates with DNA.
-
Molecular weight gel marker (such as 100 bp ladder, Roche #11721933001).
-
Ethanol (96–100 %).
-
QIAquick PCR Purification Kit (Qiagen, Inc; Cat. # 28106; 250 extractions (smaller kit available)).
-
QIAquick Gel Extraction Kit (Qiagen, Inc; Cat. # 28706; 250 extractions).
-
3 M sodium acetate, pH 5.0.
-
ABI Automated DNA sequencer (models 3130, 3130XL).
-
BigDye® Terminator™ v1.1 or v3.1 Cycle Sequencing kit.
-
MicroAmp Optical 96-well Reaction Plates (Applied Biosystems #N801-0560).
-
Digital Vortex-Genie 2 or any vortex with shock absorbing feet, microplate adapter for automated capillary sequencer plates and elastic bands.Swinging bucket centrifuge (with microplate adapters).
-
Multichannel pipettor (20–200 μl).
-
Troughs (for holding BigDye® Terminator™ reaction mix).
-
MicroAmp clear adhesive film (Applied Biosystems #4306311) or PCR cap strips of 8 count (ABgene #AB-0602).
-
Adhesive film applicator (Applied Biosystems #4333183).
-
Wide bore pipette tips (for pipetting BigDye® Terminator™ solution).
-
BigDye® Terminator™ purification kit (Applied Biosystems #4376487, processes ~2000 10 μl reactions, smaller sizes available).
-
Swinging Bucket Centrifuge with microplate carriers.
-
Centri-Sep 8-well prehydrated strips (Princeton Separations, Inc #CS-912, box of 12)
OR
-
Centri-Sep 96-well prehydrated plates (Princeton Separations, Inc #CS-963, 25 plates).
-
Multichannel pipettor (2–20 μl).
-
Collection plate (deep-well reservoir for effluent from prehydrated columns/plates).
-
PCR tube/plate rack.
-
Hi-Di formamide (Applied Biosystems, #4311320C).
-
POP6 or POP7 (depending on the model of sequencer; see Applied Biosystems sequence products for order numbers and details).
-
10× Running Buffer (Applied Biosystems part number depends on quantity ordered).
3 Methods
3.1 Use of Deoxyinosine
The key to developing an assay specific for any group of viruses is to identify a structural feature unique to that group. The high degree of nucleotide sequence diversity among polioviruses presents a challenge to the systematic design of nucleic acid-based reagents. Genomic sequences that encode intervals of strong amino acid conservation can still be highly degenerate. To accommodate this wide variability, degenerate codon positions on the template were matched by mixed-base or deoxyinosine residues on the primers [20]. In the case of polioviruses, virion surface determinants are unsuitable targets because they are type specific [12, 21]. Internal capsid antigens and nonstructural protein antigens also appear to be unsuitable because they tend to be shared among enteroviruses [13, 14]. However, polioviruses bind to a cell receptor that is distinct from those used by other enteroviruses [19]. The canyon structure on the poliovirion surface that is postulated to bind the cell receptor is primarily formed from conserved intervals of VP1 and VP2. Genetic studies have suggested that VP1 residue Met-132, at the end of the “TYSRFDM” amino acid interval, whose codons are targeted by panPV PCR-2, interacts with the cell receptor [22]. In contrast, the structural role of the highly conserved “NNGHALN” amino acid sequence, whose codons are targeted by panPV PCR-1, is unknown. Many different synonymous codon combinations could potentially occur within the primer-binding sequences (432 for panPV PCR-1 and 512 for panPV PCR-2, assuming usage only of the observed codons). If all degenerate codon combinations are permitted within both target sequences, these panPV PCR primers would have to match 200,000 different sequence combinations. All polioviruses tested with this degenerate panPV primer set have been successfully identified..
3.2 Molecular Serotyping
The approach taken for the development of poliovirus serotype-specific PCR primers followed that taken earlier to develop the panPV group primers ([23]; Fig. 2). We first identified amino acid sequences that were characteristic for each serotype. We then prepared sets of candidate primers for testing against a large collection of wild poliovirus isolates representing all known contemporary genotypes. The primers showing the best specificities and sensitivities were used in routine characterizations of recent wild isolates from many different countries. When a template was inefficiently amplified in our PCR assays, its target sequences were determined, and the design of the primers was further optimized. As with the poliovirus group-specific primers, the target nucleotide sequences were highly degenerate. These new primers fill the gap between group-specific PCR primers that recognize all polioviruses and genotype-specific primers that recognize Sabin vaccine strain-related isolates or particular wild poliovirus genotypes. The typing of polioviruses by PCR offered several advantages over typing by the standard serologic methods [3, 24]. First, the primers are chemically defined reagents having uniform and predictable properties. Second, the primers can be prepared in effectively inexhaustible quantities. Third, the PCR typing assays are rapid, highly specific, and readily standardized. Finally, the exceptional sensitivity of PCR permitted the detection of very low amounts of underlying polioviruses in mixtures of poliovirus or NPEV serotypes.

RT-PCR of noninfectious poliovirus RNAs with polio diagnostic primers
3.3 Real-Time RT-PCR
In order to increase the sensitivity and timeliness of the poliovirus group and serotype assays within the WHO Global Polio Laboratory Network (GPLN) , these assays were adapted to the Real-time TaqMan™ probe platform. To accommodate the wide variability and rapid evolution of poliovirus genomes, degenerate codon positions on the template were matched to mixed-base or deoxyinosine residues on both the primers and the TaqMan™ probes [25]. Designing the degenerate TaqMan™ probes (10–20 pmol probe for each assay) was especially challenging because of the need to use longer sequences to obtain good hybrid stabilities while simultaneously compensating for the high level of degeneracy of sequences between primer binding sites. Although hybrid stabilities can be estimated by physicochemical calculations [26], development of the optimal primer and probe sets was a highly empirical process because variation within the target sequences was not predictable. All of the TaqMan™ probes contain numerous mixed-base and inosine-containing residues to compensate for the high levels of variability in capsid region target sequences within and across poliovirus serotypes. We also developed a novel Real-time run method which takes advantage of the lower anneal temperatures required by the presence of inosine residues which reduce the T m of the oligo binding. These lower anneal/extension temperatures were combined with higher anneal/extension temperatures (which adds to the increased amplicon yields), all within the same amplification run. We have found that this single tube dual-stage amplification run method (15 cycles of 95–44–60 °C, followed immediately with 40 cycles of 95–47–65 °C) increases the sensitivities of both the inosine- and non-inosine-containing assays (Fig. 3). These group and serotype intratypic differentiation (ITD) Real-time assays are routinely used within the GPLN.

Specificities of poliovirus TaqMan rRT-PCR primers and probes, demonstrated by using Sabin strain RNA templates (S1, Sabin 1; S2, Sabin 2; S3, Sabin 3; NTC, no-template control). Individual assays used the following fluorophores: FAM = panEV, panPV, seroPV1, seroPV2, and seroPV3. A multiplex Sabin assay used a combination of primers/probes for the following assays; CY5 = Sab 1, FAM = Sab 2, and ROX = Sab 3. The no-template control was used to manually set the zero baseline fluorescence emissions. The efficiency of each assay was ~90 % or greater, based on tenfold dilutions of control RNAs (10 ng to 1 pg). Cycle threshold values of 30 or more were observed to approach the sensitivity limits of the real-time detection system; therefore, cycle threshold values of < or =30, were considered positive detections of the target template
3.3.1 Real-Time RT-PCR Reactions
-
1.
Fill out PCR worksheet with name, date, primers, samples, and sample order, as well as thermocycler and program identifiers.
-
(a)
Name wells using thermocycler software for samples and controls (positive and reagent).
-
(b)
One positive control: noninfectious control RNA supplied with Polio rRT-PCR kit.
-
(c)
One reagent control: Buffer A + B with no template.
-
(a)
-
2.
Thaw virus isolates and PCR reagents at room temperature.
-
3.
Making Buffer B + enzyme mix: The first time a vial of Buffer B 1 mL is used, add 2.8 μl 1 M DTT, 27.6 μl 40 U/μl RNase inhibitor, 18.0 μl 20 U/μl RT (or 14.4 μl 25 U/μl RT), and 54.8 μl 5 U/μl Taq polymerase (CAUTION, do not use error correcting Taq polymerases like Pfu and Pwo; they will not work with inosine primers) and mix. The enzyme mix should be stable for 6 months at 4 °C. Once the enzymes have been added, mark “+E” on the cap with an indelible marker. For long term storage (>6 months), aliquot and freeze Buffer B + E at −20 °C.
-
4.
Making reaction solution: For each primer set, mix 19 μl Buffer A (vortex to resuspend probe before use) and 5 μl Buffer B + E; dispense 24 μl reaction solution into each well. For testing large sample numbers, create a master mix of Buffers A + B (i.e., 8 samples × 19 μl Buffer A = 152 μl; 8 × 5 μl Buffer B = 40 μl), and dispense 24 μl of the A + B master mix per reaction well. (We recommend using the first well on your 8-well strip to make the A + B master mix since some commercial eppendorf tubes may bind the probe.)
-
5.
Sample preparation: Take 50 μl virus cell culture and place into a tube and spin it (benchtop microcentrifuge at 2000 × g or full speed (2000 × g) of Tube-Strip PicoFuge) at room temperature for 2 min. (Once the samples have been spun, they can be stored at −20 °C and reused if needed. You need to respin the sample after being stored at −20 °C.)
-
6.
Take 1.0 μl of cell culture supernatant (or 1 μl of Control RNA) for each sample and add into the appropriate reaction strip/plate well. rRT-PCR does NOT require a 95 °C heat step. One microliter of extracted RNA can be used, but it’s not generally required.
-
7.
Place strips in real-time thermocycler and cycle as shown below. If using a thermocycler with a rapid ramp speed, program the ramp from 44 to 60 °C for 45 s (note for the ABI 7500, you can use 25 % ramp speed between the anneal and extension temperatures for all assays). Thermocyclers with regular ramp speeds can use the default ramp time; Stratagene Mx3000P and similar machines do not have adjustable ramp capabilities. An additional intermediate step between the lower and higher temperature in the PCR cycle compensates for the inability to adjust the ramp time between anneal and extension:
-
(a)
RT reaction, 42 °C, 45 min.
-
(b)
Inactivate RT, 95 °C, 3 min.
-
(c)
PCR cycles (all primer sets):
-
Using a Stratagene MX3000P: 95 °C for 24 s, 44 °C for 24 s, 52 °C for 30 s, 60 °C for 24 s for 7 cycles, followed by Stage 2: 95 °C for 24 s, 47 °C for 24 s, 57 °C for 10 s, 65 °C for 24 s for 40 cycles.
-
Using an ABI 7500: Stage 1: 95 °C for 24 s, 44 °C for 30 s, then a 25 % ramp speed to 60 °C for 24 s, for 15 cycles, followed by Stage 2: 95 °C for 24 s, 47 °C for 30 s, then a 25 % ramp speed to 65 °C for 24 s for 40 cycles. The end point fluorescent data is collected at the end of the Stage 2 anneal step.
-
-
(d)
Select the appropriate dye filter to correspond with the assay being used.
-
(a)
-
8.
The dual-stage run method for the VDPV assay is: Stage 1: 95 °C for 24 s, 44 °C for 30 s, 60 °C for 24 s for 5 cycles, followed by Stage 2: 95 °C for 24 s, 50 °C for 30 s, then a 25 % ramp speed to 65 °C for 24 s, for 40 cycles. The end point fluorescent data is collected at the end of the Stage 2 anneal step. VDPVs were identified by VP1 sequencing (Burns et al. [34]).
3.4 VDPV Dual-Stage Assay
In addition to the ITD Real-time assays, specific assays have been developed to screen for the presence of vaccine-derived polioviruses [27]. Vaccine-related viruses excreted by patients with vaccine-associated paralytic poliomyelitis (VAPP) show only limited sequence divergence from the parental OPV strains. However, more highly divergent circulating vaccine-derived polioviruses (cVDPVs), indicative of prolonged replication or circulation, can arise in areas with suboptimal vaccine coverage ([28]). New cVDPV outbreaks continue to emerge as vaccine coverage declines in key high-risk countries and population immunity gaps widen, particularly to type 2. In order to develop VDPV screening assays, the primary amino acid targets for each serotype were identified by sequence analysis as those amino acids which are most likely to change in VDPVs, relative to the normal Sabin virus sequences [29–34]. Once an isolate appears non-Sabin like by screening with these new VDPV assays, they are further analyzed by sequencing the VP1 gene to confirm whether the virus is indeed a VDPV. The reaction set up for the VDPV assays is similar to that described in Subheading 3.3.1, with a modified run method. These Real-time assays also use a single tube dual-stage run method (5 cycles of 95–44–60 °C, followed immediately with 40 cycles of 95–50–65 °C [27]; Fig. 4, Table 1). These VDPV assays have several advantages including (1) sharply reducing the workload to sequence vaccine-related isolates to screen for cVDPVs ; (2) being more sensitive (at least for detecting S2 VDPVs) than the ELISA assay in detecting early genetic changes associated with VDPV emergence [34] and (3) yielding accurate results on serotype mixtures without the need for neutralizing one or more of the serotypes in the mixture, as now required when using the ELISA assay.

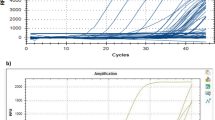
Amplification of reference Sabin strain (S, serotype indicated by number) sequences in the rRT-PCR VDPV assays. Assays used the FAM fluorophore for targeting the VP1 gene. The no-template controls were used to manually set the zero baseline fluorescence emissions. The efficiency of each assay was >90 %, based on tenfold dilutions of control RNA (10 ng to 1 pg). Cycle threshold values of 30 or more were observed to approach the sensitivity limits of the real-time detection system; therefore, cycle threshold values of <30 were considered positive detections of the target template
4 Sequencing
Once poliovirus isolates of program importance have been identified through diagnostic testing, additional vital information is obtained through sequence analysis of the viruses [28]. This information determines genetic relationships to other wild poliovirus isolates or VDPVs to demonstrate chains of transmission, sources of imported viruses causing outbreaks, and estimates of the duration of virus circulation [35]. To achieve this, VP1 sequences are determined for all wild polioviruses and potential VDPVs isolated worldwide. Systematic design of nucleic acid-based poliovirus diagnostic reagents targeting the capsid region (or any coding region) is especially challenging because nucleotide sequence diversity among poliovirus isolates is exceptionally high [20, 23] and nucleotide intervals encoding conserved amino acid sequences are typically highly degenerate. To accommodate the wide variability and rapid evolution of poliovirus genomes [35], degenerate codon positions on the template were matched to mixed-base or deoxyinosine residues on both PCR primers. A particular challenge is to sequence the individual components of isolates containing poliovirus mixtures. The standard method to resolve the mixtures has been by blocking the growth of individual components with type-specific neutralizing antibodies followed by limit-dilution culture or plaque-purification , procedures that are both time-consuming and laborious. Generic poliovirus specific primers, along with degenerate and inosine-containing primers are used for routine VP1 sequencing of poliovirus isolates (Table 2). These primers, along with nondegenerate serotype-specific Sabin primers, can also be used to sequence individual polioviruses in mixtures. By using various combinations of these primers, the VP1 regions of >7000 wild polioviruses, representing all known genotypes were amplified and sequenced, including all combinations of serotype mixtures (including ternary mixtures), whether wild or vaccine-related [36]. The VP1 regions of separate components of homotypic mixtures (e.g, Sabin 1-related poliovirus + type 1 wild poliovirus) can also be sequenced. These mixtures are difficult to resolve using type-specific polyclonal antisera and may also be difficult to resolve using highly specific neutralizing monoclonal antibodies in some situations because of the rapid antigenic reversion of vaccine-related poliovirus isolates [3].
The complete VP1 region can be amplified as a single DNA amplicon and then sequenced using four primers (sense and antisense). An alternative method is to amplify and sequence two overlapping fragments of the VP1 region into a single contig representing a consensus sequence; however, the preferred method is to amplify the complete VP1 region.
4.1 Poliovirus Sequencing
Poliovirus sequencing can be broken down into nine basic steps as follows:
-
RNA extraction from tissue cell culture isolates of poliovirus or from an FTA card spotted with poliovirus isolate.
-
Reverse transcription of the RNA and PCR amplification of the resulting cDNA using the primers Y7R and Q8 (covering the entire VP1 region of the poliovirus genome).
-
Visualizing the DNA amplicon on an agarose gel.
-
Purifying the DNA amplicon to remove unincorporated nucleotides and any nonspecific PCR products (optional step).
-
Performing the sequence PCR protocol for automated capillary DNA sequencing using the primers Y7, PV1A, PV4S, and Q8 (covering the VP1 region in both sense and antisense directions).
-
Removal of unincorporated BigDye terminators from the sequence PCR reactions.
-
Analyzing the reactions on the automated capillary sequencer.
-
Viewing and editing the raw sequence data.
-
Construction of contigs and consensus poliovirus VP1 sequences.
A flow chart outlining these basic steps can be seen in Fig. 5.

Sequence workflow diagram
4.1.1 RNA Extraction from Cell Culture Isolates
-
1.
Freeze-thaw the tissue cultures twice to release intracellular virus. Collect the supernatant after the second freeze-thaw and centrifuge at 13,000 × g for 1 min to pellet cell debris. Remove the supernatant to a sterile, RNase-free microcentrifuge tube.
-
2.
Follow the QIAamp Viral RNA Mini Spin Protocol or the QIAamp Viral RNA Mini Vacuum Protocol.
4.1.2 RNA Extraction from FTA Cards Spotted with Poliovirus Isolates
-
1.
Prepare RNA processing buffer for the FTA cards to be extracted; 200 μl will be needed per card processed. Aliquot into 1.7 ml nuclease-free microcentrifuge tubes and place on ice.
-
2.
Clean the cutting mat with 10 % bleach followed by high quality DI water and 70 % ethanol OR wipe the mat with an RNAse AWAY wipe. Dry the mat with a clean tissue wipe.
-
3.
Take the FTA card with air dried poliovirus isolate and place the area to be punched on the cutting mat.
-
4.
Using a 4 mm disposable biopsy punch, cut out 6 punches per card and place the punches into a microfuge tube with 200 μl of RNA processing buffer. All six punches can be made sequentially and then removed from the biopsy punch with any of the following: a sterile disposable inoculating loop (with the end removed), a sterile disposable inoculating needle, or sterile disposable forceps.
-
5.
Dispose of the biopsy punch in a sharps container. Use a new biopsy punch for each FTA card processed, as well as new inoculating loop/needle or forceps.
-
6.
If more than one FTA card is processed, always place each card on a different spot on the cutting mat to avoid any chance of cross contamination between specimens. Repeat steps 4 and 5.
-
7.
Vortex the tubes containing the punches in RNA processing buffer. Incubate the tubes at 70 °C for 5 min, vortexing at the halfway point of the incubation period. Open the tubes during the first 2 min of the incubation period to release any pressure inside the tube.
-
8.
After incubation is complete, spin the tubes for 1 min at full speed in a microcentrifuge.
-
9.
Remove the supernatant after centrifugation and place into an appropriate number of labeled microfuge tubes containing 620 μl of AVL buffer (QIAmp Viral RNA mini kit) plus the appropriate amount of carrier RNA according to the QIAmp protocol.
-
10.
Mix the tubes containing the AVL buffer and supernatant by pulse vortexing for 15 s. Briefly spin the tubes in a microcentrifuge to remove any liquid from the cap.
-
11.
Add 620 μl of absolute ethanol to each tube with FTA supernatant plus AVL buffer. Mix by pulse vortexing for 15 s and briefly spin the tubes in a microcentrifuge as in step 10.
-
12.
At this point, follow the QIAamp Viral RNA Mini Spin Protocol or the QIAamp Viral RNA Mini Vacuum Protocol at the stage of adding the supernatant, AVL buffer, ethanol mixture to the QIAmp columns.
-
13.
Store the extracted RNA at −70 °C (preferred) or at −20 °C (~1–2 year storage).
4.1.3 Degenerate RT-PCR for Sequence Amplicons
-
1.
Log into a laboratory notebook a record of the RNA samples to be used in the PCR run. If RNA was extracted the same day of the PCR amplification, keep the tubes containing the RNA on ice throughout the procedure. If frozen RNA is used, thaw the samples on ice and keep them on ice for the remainder of the procedure. DO NOT THAW AT ROOM TEMPERATURE.
-
2.
Remove reagents from the freezer and thaw on ice or place into a tube rack (dedicated for PCR reactions) at 4 °C until the reagents are thawed; then place the reagents on ice.
-
3.
Take out the required number of PCR tubes needed for the PCR run (the number of RNA samples to be processed plus a positive and a negative RNA control sample). Label the tubes appropriately for cross reference to the lab notebook log of the RNA samples to be processed. Place the tubes on ice.
-
4.
Prepare a master mix for the PCR reactions to give a final volume of 47 μl/PCR tube. Below is an example for making a master mix for eight reactions plus two controls.
Reagent
Amt/PCR tube
×10 Rxns
PCR water (DNase–RNase free)
37 μl
370 μl
10× PCR buffer
5 μl
50 μl
dNTPs (10 mM each)
2 μl
20 μl
RNase inhibitor (40 U/μl)
0.25 μl
2.5 μl
Reverse transcriptase (20 U/μl)
0.25 μl
2.5 μl
Y7R primer (40 pmol/μl)
1 μla
10 μl
Q8 primer (10 pmol/μl)
1 μlb
10 μl
Taq polymerase (5 U/μl)
0.5 μl
5 μl
Total
47 μl
470 μl
aConcentrated stock of 40 pmol/ul contains 10 pmol/μl of each primer species because of degeneracy.
bConcentrated stock of 10 pmol/μl contains 5 pmol/μl of each primer species because of degeneracy.
-
5.
Combine the master mix by gently pipetting up and down, avoiding formation of air bubbles; centrifuge the tube briefly. Aliquot 47 μl of master mix into each PCR reaction tube kept on ice. Keeping the reaction tubes on ice, aliquot 3 μl of the sample RNA into each tube and mix by gently pipetting the reaction up and down several times. When you have finished aliquoting all the RNA samples to be tested, store the poliovirus RNA at −20 °C.
-
6.
Place the PCR reactions into the thermocycler and cycle as shown below:
-
(a)
RT reaction, 42 °C, 45 min.
-
(b)
Inactivate RT, 94 °C, 3 min.
-
(c)
PCR cycle (all primers): 94 °C for 30 s , 42 °C for 30 s, ramp at 0.4 °C/s to 60 °C, 60 °C for 2 min for 35 cycles.
-
(d)
Immediately follow the cycle program with a final extension time of 5 min at 60 °C.
-
(a)
-
7.
At the completion of the PCR run, reaction products may be immediately examined by agarose gel or may be frozen at −20 °C for later evaluation.
4.1.4 Visualizing DNA Amplicons
-
1.
Assemble the gel tray into a casting assembly and place a sample comb with the desired number of sample wells into the gel tray.
-
2.
Prepare a 1 % agarose gel by weighing out an appropriate amount of agarose powder (e.g., 1.0 g for 100 ml gel) and mix with 1× TBE buffer in a glass flask. Heat the gel mixture in a microwave oven until melted (the agarose is completely dissolved), cool to approximately 55 °C, and add fluorescent dye directly to the gel according to the dye manufacturer guidelines.
-
3.
Allow the gel to cool slightly, pour into the gel tray and allow it to solidify at room temperature or at 4 °C.
-
4.
Place the solidified gel into the electrophoretic buffer chamber. Pour enough 1× TBE buffer into the chamber to cover the gel; remove the sample comb at this point and flush out the sample wells with 1× TBE buffer.
-
5.
Take out enough PCR tubes to account for the number of reactions you want to run on the gel. Aliquot 1 μl of 6× load dye into each tube and then add 5 μl of each PCR reaction. Place the remainder of the PCR reaction either on ice or at 4 °C.
-
6.
Load the PCR reaction plus load dye (total 6 μl) into the wells made by the sample comb. In the first and last lanes of the gel, load the molecular weight marker (with load dye) to help determine the size of the DNA amplicon of the PCR reaction.
-
7.
Close the gel chamber, attach the leads to a power supply and apply current (~70 V for 100 ml gel). DNA will migrate towards the positive electrode, which is colored red. Continue to run the gel at constant voltage/current.
-
8.
The distance the DNA has migrated through the gel can be judged by visually monitoring the migration of the bromophenol blue dye in the load buffer (bromophenol blue migrates at about the same rate of double stranded DNA fragments of ~300 bp). Also note that interference from the blue dye may be noticed.
-
9.
After sufficient migration has occurred, turn off the power supply and remove the gel tray (wear disposable gloves while handling the gel tray), Place the gel tray on a UV transilluminator with a plexiglass cover and check for PCR amplicons of ~1.1 kb size (WARNING: wear proper PPE (personal protection equipment) including protective eye gear and a UV blocking face shield to minimize exposure to UV light). Photograph the gel to document the results.
-
10.
There should be only one band per PCR reaction (band size is ~1.1 kb). If this is true, then the remaining product stored at 4 °C may be column-purified for use in DNA sequencing reactions (see Subheading 4.1.5). If there are nonspecific bands in addition to the correct 1.1 kb fragment, the VP1 fragment can be extracted from the gel (see Subheading 4.1.6).
4.1.5 Direct Purification of PCR DNA Amplicon
-
1.
Remove the correct number of microcentrifuge tubes corresponding to the PCR amplicons to be purified and label the tubes accordingly.
-
2.
Using the QIAquick PCR Purification Kit, add 5 volumes of PB buffer (with indicator dye added) to each microcentrifuge tube for each volume of PCR sample (e.g., a 50 μl PCR reaction would require 250 μl of PB buffer).
-
3.
Add the PCR reaction to the corresponding microcentrifuge tube with PB buffer.
-
4.
Mix by vortexing. Check the color of the mixture; it should be dark yellow. If the color is orange or violet, add 10 μl of 3 M sodium acetate, pH 5.0 (color should now be yellow).
-
5.
Follow the QIAquick PCR purification microcentrifuge protocol or the QIAquick PCR purification vacuum manifold protocol. Elute the DNA from the column using either the elution buffer provided in the kit or with nuclease-free water.
-
6.
The purified DNA is now ready for sequencing. The DNA may be quantified by running a sample adjacent to standards containing known quantities of the same-sized DNA fragment or by spectrophotometric analysis for nucleic acids.
4.1.6 Gel Extraction of PCR DNA Amplicon
-
1.
Make a 1 % agarose gel with an appropriate gel stain as outlined in step 3 of this manual. After the gel has solidified, place the gel into the electrophoresis chamber of the gel box and add 1× TBE buffer to cover the gel; remove the sample comb and flush the wells with 1× TBE buffer.
-
2.
Mix 9 μl of 6× load buffer (see step 3) with the remaining 45 μl of PCR product and load into the sample wells of the gel. REMEMBER: In the first and last lanes of the gel, load the molecular weight marker (with load dye) to help determine the size of the DNA amplicon of the PCR reaction.
-
3.
Close the gel chamber, attach the leads to a power supply and apply current (~70 V for 100 ml gel). Keep the voltage/current constant.
-
4.
The distance the DNA has migrated through the gel can be judged by visually monitoring the migration of the bromophenol blue dye in the load buffer (bromophenol blue migrates at about the same rate of double stranded DNA fragments of ~300 bp).
-
5.
After sufficient migration has occurred (i.e., the bromophenol blue dye has migrated ~ halfway through the gel), turn off the power supply and remove the gel tray. Place the gel tray on a UV transilluminator with a plexiglass cover and check for PCR amplicons of ~1.1 kb size (WARNING: wear proper PPE (personal protection equipment) including protective eye gear and a UV blocking face shield to minimize exposure to UV light). Photograph the gel to document the results.
-
6.
Wearing proper laboratory PPE, including protective eyewear and a protective UV blocking face shield, use a surgical scalpel to excise the 1.1 kb bands from the gel. Place each gel piece into a separate labeled microcentrifuge tube.
-
7.
Weigh the gel slice in the tube. Use the QIAquick Gel Extraction Kit to extract the DNA from the gel slice. Add 3 volumes of QG buffer from the kit to 1 volume of gel (100 mg of gel ~100 μl).
-
8.
Incubate the gel slice + QG buffer at 50 °C for at least 10 min, vortexing every 2 min to aid in gel dissolution. If the gel slice is not completely dissolved after 10 min, the incubation time can be extended for a few minutes. THE GEL SLICE MUST BE COMPLETELY SOLUBILIZED.
-
9.
Check the color of the gel solution—it should be the same color (yellow) as the as the QG buffer. If the color is orange or violet, add 10 μl of 3 M sodium acetate, pH 5.0.
-
10.
Add the sample to a labeled QIAquick spin column and follow the QIAquick PCR purification microcentrifuge protocol or the QIAquick PCR purification vacuum manifold protocol. Please allow the PE wash buffer to remain on the column for a 5 min incubation time prior to removal of the wash.
-
11.
Elute the DNA from the column using either the elution buffer provided in the kit or with nuclease-free water.
-
12.
The purified DNA is now ready for sequencing. The DNA may be quantified by running a sample adjacent to standards containing known quantities of the same-sized DNA fragment or by spectrophotometric analysis for nucleic acids.
4.1.7 Degenerate Sequence PCR Reactions
-
1.
Log into a laboratory notebook a record of the DNA samples to be used in the PCR run. If the DNA amplicon was purified the same day of the sequence PCR amplification, keep the tubes containing the DNA on ice throughout the procedure. If frozen DNA is used, thaw the samples on ice and keep them on ice for the remainder of the procedure. DO NOT THAW AT ROOM TEMPERATURE.
-
2.
Take all reagents for the reaction (Ready Reaction Premix, primers, and BigDye Sequencing dilution buffer) out of the freezer and thaw on ice or place into a PCR reaction dedicated tube rack at 4 °C until the reagents are thawed; then place the reagents on ice.
-
3.
Take out the required number of PCR tubes needed for the PCR run (the number of DNA samples × 4). Label the tubes appropriately for cross reference to the lab notebook log of the DNA sample/primer to be processed. Place the tubes on ice.
-
4.
For each DNA template there will be a total of four reactions, one for each of the following primers: Y7, PanPV1A, PanPV2S, and Q8. Oligonucleotide composition and priming position on the poliovirus genome for each of these primers are listed in Table 2. Use of four primers ensures coverage of the entire VP1 region in both the sense and antisense directions (see Fig. 2). The amount of DNA template used in the PCR sequence reaction will depend on the method used to purify the sequence reaction post thermocycling. The PCR reaction (10.0 μl) is prepared as follows: 2.0 μl of Ready Reaction premix (v1.1 or v3.1), 1.0 μl 5× Big Dye Sequence buffer, 3.2 pmol primer (dilute from stock as needed to give a final added volume of 1.0 μl), 20–40 ng DNA template (sephadex column purification) OR 5–10 ng DNA template (BDXterminator purification), nuclease-free water added to a final volume of 10.0 μl.
-
5.
Mix the PCR reactions by pipetting up and down. If necessary, spin the tubes briefly to remove any bubbles.
-
6.
Place the PCR tubes into the PCR thermal cycler and run the following PCR program (all primers): 94 °C 20 s, 42 °C 15 s, ramp at 0.4 °C/s to 60 °C, 60 °C for 4 min for 25 cycles.
-
7.
At the end of the PCR sequencing program, remove the tubes and proceed to Subheading 4.1.8 (Purifying Sequence Reactions with BigDye® XTerminator™) or Subheading 4.1.9 (Purifying Sequence Reactions with Spin Columns).
4.1.8 Purifying Sequence Reactions with BigDye® XTerminator™
-
1.
Remove the PCR sequence reactions from the PCR thermocycler.
-
2.
Follow the BigDye® XTerminator™ purification protocol as outlined in the premix pipetting section of the process. Use a trough to hold the final premix solution (SAM + XTerminator™) and a multichannel pipettor to aliquot 55 μl to each PCR reaction. When pipetting from the trough, keep the premix agitated by rocking the trough back and forth lengthwise to create a wave motion.
-
3.
After the premix is aliquoted as indicated by the BDXterminator protocol, seal the plate using the adhesive film applicator. An alternative method of sealing the plate is to cap the plate wells with PCR strip caps (rows of 8).
-
4.
Place the sealed plate on the Digital Vortex-Genie 2 and vortex for 30 min at 1800 × g at room temperature.
-
5.
After vortexing is complete, spin the plate in a swinging bucket centrifuge for 2 min at 1000 × g at room temperature.
-
6.
The samples may now be analyzed on the DNA sequencer (Subheading 4.1.10) OR you can store the sealed plate for up to 48 h at room temperature or up to 10 days at 4 °C.
4.1.9 Purifying Sequence Reactions with Spin Columns
-
1.
Place a 96-well sequence plate onto a PCR plate rack.
-
2.
Aliquot 15 μl of HiDi Formamide into the wells of the plate for the number of sequencing reactions that you have.
-
3.
Remove the number of strips that you will need to purify your sequence reactions or take out a Centri-Sep plate.
-
4.
Using a swinging bucket centrifuge, pre-spin the columns or Centri-Sep plates into the collection plate according to the manufacturer’s specifications (2 min at 750 × g for the column strips, 1500 × g for the Centri-Sep plate; please see the centrifugation notes at the end of this section) to remove the interstitial fluid present in the columns/plate.
-
5.
After centrifugation, place the columns into the formamide-filled wells of the 96-well sequence plate OR place the Centri-Sep plate above the formamide filled 96-well plate.
-
6.
Using the multichanneled pipettor, pipette the 10 μl sequencing reaction into the center of each well of the Centri-Sep strip/plate, making sure not to touch the top of the gel in the well.
-
7.
Spin the loaded Centri-Sep column/plate according to manufacturer’s instructions. The purified fluorescently tagged DNA will spin through the column directly into formamide in the 96-well sequence plate.
-
8.
Remove the Centri-Sep columns/ plate from the top of the 96-well sequence plate. The samples (now in the 96-well sequence plate) are ready to be analyzed on the DNA sequencer (Subheading 4.1.10).
4.1.10 Analyze Purified Sequence Reactions on the Automated DNA Sequencer
-
1.
Take the 96-well sequence plate containing the purified sequence reactions and cover it with a plate septa. Place the plate into the plate base /retainer assembly and put it into the automated sequencer according to manufacturer specifications.
-
2.
Prepare 1× Running buffer (dilute 1 part 10× running buffer to 9 parts nuclease-free water (PCR grade)). Add the buffer to the two buffer chambers in the sequencing instrument according to ABI instructions.
-
3.
Prepare a sample sheet according to the user’s guide for the automated DNA sequencer, following a logical naming convention. (Please note that an electronic sample sheet template may be imported.) Make sure that the proper instrument/run protocols are chosen based on the version of BigDye used (v1.1 or v3.1), the length of the capillary in the machine, and the purification method used. (NOTE: if BigDye XTerminator was used to purify the post sequencing PCR reactions, make sure that the proper run modules are available on the sequencer. These run protocols are freely distributed by Applied Biosystems on their website.)
-
4.
Begin the sequence run. After the run is complete, download the sequence data from the data collection software run files and edit the raw sequence data as described in Subheading 4.2—Viewing and Editing Raw Sequence Data.
4.2 Viewing and Editing Raw Sequence Data
4.2.1 Software
You will need a software package capable of analyzing trace files, constructing contigs and editing base pairs. Examples are the Sequencher analysis package distributed by Gene Codes Corporation Ann Arbor, Michigan; Lasergene distributed by DNASTAR Inc. Madison, Wisconsin; and Bionumerics (Applied Maths, Inc., Austin, Texas). All subsequent sequence chromatograms and contig representation presented in this section are generated through Sequencher Ver. 4.8
4.2.2 Sequence Quality and Trimming
-
1.
The trace data files are imported into the Sequencher software, and the chromatograms of each file are examined for quality. Peaks present in the chromatogram should have a good signal to noise ratio (distinct with minimal background) and base-calling should be clean and concise.
-
2.
Trimming—Frequently, the beginning (close to the primer read) of the sequence data will have ambiguous base calls (N’s) and/or will have a great deal of background noise. Generally, the first 15–25 base pairs of the sequence displayed in the chromatogram will need to be trimmed. The tail end (3′ end) of the sequence will also need to be trimmed; read lengths of the sequence will vary based on the following criteria: (1) quality of the sequence read and (2) the primer used for the sequence. As a general rule, more than three ambiguous bases and/or a deterioration of sequence quality (i.e., loss of peak resolution and base calling accuracy) signals the need to trim the end portion of the sequence read.
4.2.3 Troubleshooting Sequence Quality Issues
Noisy background and/or trailing peak width early in the sequence read could be due to a number of factors including
-
1.
Errors generated during sequence run—Check the error log and rerun the sample.
-
2.
Polymer bubbles—Check the sequencer to make sure no bubbles are present in the line from the polymer supply or in the capillary array.
-
3.
High number of sequence runs on the capillary—ABI sequence capillaries are guaranteed for 100 sequence runs only. Capillaries should be changed between 100 and 150 runs. Please note that the capillaries can be run more than 150 runs, but this is not recommended because sequence quality may suffer.
-
4.
Impure DNA amplicon—check the starting DNA material on a 1 % agarose gel to ensure that only the 1.1 kb Y7/Q8 amplicon is present. If other bands can be seen, extract the 1.1 kb band from the gel and gel purify the fragment using a kit such as the QiaQuik gel purification system from Qiagen. Re-sequence the purified DNA.
-
5.
Mixtures of serotypes/genotypes present in the DNA amplicon—Check the original PCR result from the diagnostic PCR to ensure that no mixture is present such as a wild–sabin mix or a wild mixture of different serotypes (e.g., a PV1/PV3 wild poliovirus mixture).
See Fig. 6 for examples of unacceptable sequence quality.

Examples of unacceptable sequence quality. Panel a has high background (a low signal to noise ratio) in a portion of the chromatogram, and Panel b shows trailing peaks in the Y7 primer sequence
4.3 Construction of Contigs and Consensus of Poliovirus VP1 Sequences
4.3.1 Contig Assembly
-
1.
After sequence quality is ascertained, the sequences corresponding to the Y7, PV1A, PV4S, and Q8 primers for each DNA amplicon analyzed can be assembled into a single contig. Minimum contig assembly parameters should be at least 85 % base pair match with a 20 base pair overlap between primer sequences (see Fig. 7).
Fig. 7 
Sequence contig formed using Sequencher software (GeneCodes) demonstrating good overlap with both sense primers (Y7 and PV4S shown in green) and antisense primers (PV1A and Q8 shown in red) in the VP1 gene region of poliovirus. Primers extend into the VP3 and 2A gene regions to ensure complete coverage of VP1
-
2.
Good sequence overlap between the sense and the antisense primers over the VP1 region is critical to ascertain agreement between the sequence strands; each primer provides sequence from a different region of VP1 and strengthens the validity of the base pair calls. THERE MUST BE COMPLETE COVERAGE OF THE VP1 REGION WITH BOTH THE SENSE AND ANTISENSE STRANDS OF THE CONTIG. There should be no region of single-stranded sequence or absence of a sense or antisense strand.

4.3.2 Base Pair Agreement and Editing
Contigs should be checked for ambiguous base pair calls and gaps between the assembled sequences. The software package used should have the ability to check and to edit any ambiguous base call(s) in the contig
The chromatograms for bases in the contig should be examined to get a clear consensus for the proper base pair calls in the VP1 region (Fig. 8). From the three aligned chromatograms in Fig. 8, the ambiguous base in the PV1A sequence is a ‘T’ in agreement with the other two sequence primer strands. The consensus base pair call would be ‘T’. Notice that the location of the base in question differs in each of the three primer sequence strands; contig assembly effectively aids in simultaneous editing base calls in all sequence strands for a specific Y7/Q8 DNA amplicon.

Alignment of three primer sequences in a single poliovirus VP1 contig. The ambiguous base call in the PV1A sequence is a ‘T’ comparing the base calls in the same position in the Y7 and Q8 primer sequences
After all ambiguous base pairs have been resolved, the contig should be renamed with a descriptive nomenclature to help identify the specimen sequenced. An example would be a unique identifier such as a Lab Id that is used only once and not rolled over from year to year.
A consensus sequence may be generated from the edited contig. The consensus sequence should be trimmed to the VP1 region (906 base pairs for PV1, 903 base pairs for PV2, and 900 base pairs for PV3) (Fig. 9).

Alignment of a poliovirus serotype 1 VP1 sequence (generated from the consensus sequence of a VP1 contig) with the reference Sabin 1 strain sequence for appropriate trim length (906 bp). The consensus sequence would be trimmed at the arrow points
4.4 Sequencing Poliovirus Mixtures
When sequencing poliovirus specimens, mixtures of different serotypes and genotypes are often encountered and can present challenges in obtaining good sequence data. The degree of difficulty in resolving individual polio serotypes/genotypes depends on the type of mixture present in the specimen. Mixture types can be classified as follows, from the easiest type of mixture to the most difficult:
-
1.
Sabin–Sabin mixtures—The specimen contains combinations of the three Sabin strains (Sabin, Sabin2, Sabin3). These are the easiest mixtures to resolve due to the availability of Sabin specific primers that can be used in the initial RT-PCR reactions to produce Sabin-specific DNA amplicons and/or used in the sequencing PCR reaction.
-
2.
Wild–Wild mixtures—The specimen contains a combination of two different serotypes (e.g., PV1 and PV3) but no Sabin or Sabin derived polioviruses (VDPVs). Serotype specific primers in the initial RT-PCR reaction produce specific DNA amplicons, and the primers can also be used in sequencing PCR reactions to resolve these mixtures.
-
3.
Wild–VDPV–Sabin mixtures—The specimen contains a combination of wild and at least one Sabin or Sabin derived strain of different serotypes (e.g., PV1 wild–PV2 VDPV–Sabin3). Combinations of Sabin specific, serotype-specific and genotype-specific primers are used in both RT-PCR reactions (to produce specific DNA amplicons) and in the PCR sequence reactions to resolve these mixtures.
-
4.
Homotypic mixtures—The specimen contains a combination of wild, Sabin or Sabin-derived strains of the same serotype. This is the most difficult mixture to resolve, as serotype-specific primers will target both wild and Sabin/VDPVs in the mix. Strategies for resolution of these mixtures involve production of several DNA amplicons using both serotype- and genotype-specific primers and also sequencing the amplicons with different combinations of these primers (see Table 2).
The genotype- and serotype-specific primers used in resolving poliovirus mixtures are described in Table 2.
The frequency of each type of mixture encountered is dependent on the specimen type. Poliovirus mixtures from AFP specimens are predominantly Sabin–Sabin mixtures. Environmental specimens, which are composite samples from sewage originating from many households (as opposed to one single patient), can have all four mixture types, particularly in polio endemic regions. Strategies for resolving the four mixture types are described in detail in the next sections.
4.4.1 Sabin–Sabin Poliovirus Mixtures
The following primer pairs may be used in the RT-PCR reaction (same PCR conditions as used for the Y7R/Q8 amplification) to produce Sabin specific DNA amplicons for the VP1 region:
-
1.
246S/Q8—Produces Sabin 1 DNA amplicon. Primers amplify the end of VP3 and the beginning of 2A to give ~1.1 kb PCR product.
-
2.
247S/Q8—Produces Sabin 2 DNA amplicon. Primers amplify the end of VP3 and the beginning of 2A to give ~1.1 kb PCR product.
-
3.
248S/Q8—Produces Sabin 3 DNA amplicon. Primers amplify the end of VP3 and the beginning of 2A to give ~1.1 kb PCR product.
The Q8 primer is used as the antisense primer with the Sabin-specific sense primer to produce the full length Sabin VP1 sequences. A combination of Sabin-specific and generic primers can be used to sequence the purified DNA amplicon. Figure 10 gives an overview of the strategies and primers used to resolve Sabin–Sabin mixtures.

Sequencing Sabin–Sabin mixtures
4.4.2 P1 Wild–P3 Wild Poliovirus Mixtures
The best strategy to resolve wild–wild mixtures is to have serotype- and wild genotype-specific primers in the RT-PCR reaction to produce single serotype DNA amplicons. The following primers are used to resolve each serotype component in the mixture:
-
1.
PV1 wild component—generate 2 DNA amplicons.
-
(a)
P1 Sense/Q8—Produces a serotype 1-specific DNA amplicon (~1 kb PCR product).
-
(b)
Y7R/(AFRO P1W 1A or SOAS P1W 1A)—Produces a serotype 1/wild genotype-specific DNA amplicon (~400 bp PCR product). The choice of genotype-specific primer is dependent upon the suspected wild genotype of the sample.
-
(a)
-
2.
PV3 wild component—generate 2 DNA amplicons.
-
(a)
P3 Sense/Q8—Produces a serotype 3-specific DNA amplicon (~1 kb PCR product).
-
(b)
Y7R/(AFRO P3W 7A or SOAS P3W 5A)—Produces a serotype 3/wild genotype-specific DNA amplicon (~300 bp PCR product). The choice of genotype-specific primer is dependent upon the suspected wild genotype in the sample.
-
(a)
The P1 and P3 Sense primers amplify the beginning of the VP1 region; therefore, in order to obtain a full-length VP1 sequence on the 5′ end, a second, smaller amplicon is generated with Y7R (amplifies the end of VP3) and a genotype/serotype-specific primer. The purified DNA amplicons are sequenced according to serotype with a combination of serotype/genotype-specific primers (P1 Sense, P3 Sense, AFRO P1W1A/SOAS P1W 1A, AFRO P3W 7A/SOAS P3W 5A, 2952S, and 2922S) and generic primers. Figure 11 gives an overview of the strategies and primers used to resolve wild–wild mixtures.

Sequencing wild–wild mixtures. Primers are color coded as follows; primers in red are serotype specific, primers in blue are serotype/genotype specific, primers in black are generic
4.4.3 Wild–VDPV–Sabin Poliovirus Mixtures
Wild–Sabin mixtures that also have a VDPV component are rarely encountered in specimens from AFP cases but are more frequent in environmental specimens from polio-endemic areas. The Sabin components of the mixture can be resolved by using Sabin-specific primers, and the wild component is resolved using a combination of serotype-specific and serotype/genotype-specific primers. An example of the strategy to resolve a poliovirus mixture that was determined by rRT-PCR to be PV1 wild, PV2 discordant, and PV3 discordant is given below:
-
1.
PV1 wild component—generate 2 DNA amplicons.
-
(a)
P1 Sense/Q8—Produces a serotype 1 specific DNA amplicon (~1 kb PCR product).
-
(b)
Y7R/(AFRO P1W 1A or SOAS P1W 1A)—Produces a serotype 1/wild genotype-specific DNA amplicon (~400 bp size PCR product). The choice of genotype-specific primer is dependent upon the suspected wild genotype in the sample.
-
(a)
-
2.
PV2 Sabin component—generate a PV2 Sabin-specific amplicon using 247S/Q8.
-
3.
PV3 Sabin component—generate a PV3 Sabin-specific amplicon using 248S/Q8.
Overall, four DNA amplicons will be produced. The P1 DNA amplicons are sequenced with a combination of serotype 1, wild genotype-specific, and generic primers to get a full-length VP1 PV1 wild sequence. The PV2 and PV3 amplicons are sequenced with a combination of Sabin-specific and generic primers to achieve full length Sabin VP1 sequences for each serotype. It may be noted that if Sabin-specific amplicons cannot be achieved using the Q8 primer on the 3′ end, Sabin-specific primers can be used at this position (e.g., 247S/253A produce an ~940 bp amplicon). However, a full length VP1 sequence may not be obtained with complete sequence coverage in the both the sense and antisense directions. Figure 12 gives an overview of the strategies and primers used to resolve wild–VDPV–Sabin mixtures.

Sequencing components of wild and Sabins (discordant in VDPV assay) from a poliovirus specimen. *The Q8 sequence primer listed for the Sabin amplicons will only sequence the amplicons S2amp1 and S3amp1
4.4.4 Homotypic Poliovirus Mixtures
Homotypic poliovirus mixtures refer to specimens that contain combinations of wild, VDP, and Sabin genotypes that share the same serotype (e.g., PV1 wild and PV1 Sabin). These mixtures are by far the most difficult to resolve of all the mixture types. Serotype-specific primers may not be informative because the viruses are of the same serotype. Genotype-specific primers are helpful, but some cross reactivity can occur. Depending on the complexity of the mixture (titer of each component, presence of multiple Sabins of the same serotype) only partial VP1 sequences may be obtained. Virus plaque purifications of the mixtures can be done to separate the viruses, but this process is time consuming and not done routinely. Figure 13 gives on overview of a strategy to resolve a PV1 wild, PV1 Sabin poliovirus mixture. Overall, at least four RT-PCR reactions with combinations of generic, serotype-, and genotype-specific primers need to be done to target the wild and Sabin components of the mixture. The resulting DNA amplicons are sequenced with generic and specific (serotype- and genotype-) primers to produce VP1 sequences for each genotype.

Example of the strategy to resolve a serotype 1 poliovirus homotypic mixture
4.5 Notes
-
1.
Check all kit reagents for expiration dates.
-
2.
All reagents and disposable plastic ware used in the procedure should be certified nuclease-free.
-
3.
Check Buffer AVL solution for precipitate. If necessary, redissolve precipitate by warming at 80 °C for no more than 5 min.
-
4.
Make all solutions according to kit instructions and write the date of first use on each solution bottle.
-
5.
Perform RNA extractions on a positive poliovirus control and negative cell control, preferably from the same cell line as the isolates being tested.
-
6.
This protocol assumes that the user has a thorough knowledge of how to handle RNA and the hazard to RNA by RNases. All reagents and disposable plastic ware used in the procedure should be certified RNase-free.
-
7.
Store the RNA at −70 °C (preferable) or at −20 °C (1–2 year storage limit). Most viral RNAs are stable for many years (≥4 years) at −70 °C.
-
8.
Always wear protective eyewear and a protective UV blocking face shield when observing DNA on a transilluminator to prevent damage to the eyes from UV light.
-
9.
Several commercially available kits can be used for DNA purification and extraction from gels; any of these kits may be used as long as the purified product is suitable to be used in downstream sequence reactions.
-
10.
Thermocycled sequence reactions may be stored dark and frozen at −20 °C for no more than 24 h prior to purification and subsequent analysis on the automated sequencer.
-
11.
Purified sequence reactions may be stored according to the purification method used:
-
(a)
Centri-Sep columns/plates—the purified reactions should be dried in a vacuum centrifuge (no heat or low heat) and stored dark and sealed (adhesive film) at −20 °C for up to 10 days. Do not store at temperatures below −20 °C.
-
(b)
BigDye® Terminator™—reactions cleaned with BigDye XTerminator in plates sealed with adhesive film can be stored as follows: room temperature (20–25 °C) for no more than 48 h, 4 °C up to 10 days, or −20 °C for 1 month.
-
(a)
-
12.
The process described for BigDye XTerminator purification of sequence reactions assumes that the user will be using an automated DNA capillary sequencer (ABI 3130 or 3730 instruments) equipped with BigDye XTerminator run modules to analyze the fluorescently tagged PCR product.
-
13.
The process described for purifying sequence reactions with spin columns mentions Centri-Sep prehydrated 8-well strips (for small numbers of samples) and Centri-Sep prehydrated 96-well plates (for processing large numbers of samples) but any gel purification method specifically formulated for sequence reaction purification could work just as well.
-
14.
If the RCF (relative centrifugal force) or g force of a centrifuge rotor is unknown (i.e., the centrifuge only measures RPM speeds), it can be calculated by the following formula: RCF = r × 11.18 × (rpm ÷ 1000)2 where RCF is relative centrifugal force in “g,” r is the rotor radius in centimeters, and rpm is the rotor speed (revolutions per minute).
-
15.
To assist in trimming the VP1 sequence to the proper length, the contig assembly parameters in the Sequencher software can be adjusted to a 75 % sequence match (Sequencher ver 4.8) and the consensus sequence assembled with the reference strain VP1 sequences for Sabin1 (AY184219), Sabin2 (AY184220), or Sabin3 (AY184221) into a single contig. The consensus sequence may be trimmed according to the sequence length of the reference strain (Fig. 9).
References
Hull H et al (1994) Paralytic poliomyelitis: seasoned strategies, disappearing disease. Lancet 343(8909):1331–1337
Organization WH (2008) Progress towards interrupting wild poliovirus transmission worldwide, January 2007-April 2008. Wkly Epidemiol Rec 83(19):170–177
van der Avoort HG et al (1995) Comparative study of five methods for intratypic differentiation of polioviruses. J Clin Microbiol 33(10):2562–2566
da Silva EE et al (1991) Oligonucleotide probes for the specific detection of the wild poliovirus types 1 and 3 endemic to Brazil. Intervirology 32(3):149–159
De L et al (1995) Identification of vaccine-related polioviruses by hybridization with specific RNA probes. J Clin Microbiol 33(3):562–571
Yang CF et al (1991) Detection and identification of vaccine-related polioviruses by the polymerase chain reaction. Virus Res 20(2):159–179
Yang CF et al (1992) Genotype-specific in vitro amplification of sequences of the wild type 3 polioviruses from Mexico and Guatemala. Virus Res 24(3):277–296
Kew BK et al (1984) Applications of oligonucleotide fingerprinting to the identification of viruses. Methods Virol 8:41–84
Kew OM et al (1995) Molecular epidemiology of polioviruses. Semin Virol 6(6):401–414
Balanant J et al (1991) The natural genome variability of poliovirus analyzed by a restriction fragment length polymorphism assay. Virology 184(2):645–654
Schweiger B et al (1994) Differentiation of vaccine and wild-type polioviruses using polymerase chain reaction and restriction enzyme analysis. Arch Virol 134(1–2):39–50
Melnick J (1990) Poliomyelitis. In: Warren K, Mahmoud A (eds) Tropical and geographical medicines, 2nd edn. McGraw-Hill, New York, pp 558–576
Mertens T, Pika U, Eggers HJ (1983) Cross antigenicity among enteroviruses as revealed by immunoblot technique. Virology 129(2):431–442
Emini E et al (1985) Antigenic conservation and divergence between the viral-specific proteins of poliovirus type 1 and various picornaviruses. Virology 140(1):13–20
Hyypia T, Auvinen P, Maaronen M (1989) Polymerase chain reaction for human picornaviruses. J Gen Virol 70:3261–3268
Olive DM et al (1990) Detection and differentiation of picornaviruses in clinical samples following genomic amplification. J Gen Virol 71(9):2141–2147
Abraham R et al (1993) Shedding of virulent poliovirus revertants during immunization with oral poliovirus vaccine after prior immunization with inactivated polio vaccine. J Infect Dis 168(5):1105–1109
Egger D et al (1995) Reverse transcription multiplex PCR for differentiation between polio- and enteroviruses from clinical and environmental samples. J Clin Microbiol 33(6):1442–1447
Hovi T, Stenvik M (1994) Selective isolation of poliovirus in recombinant murine cell line expressing the human poliovirus receptor gene. J Clin Microbiol 32(5):1366–1368
Kilpatrick DR et al (1996) Group-specific identification of polioviruses by PCR using primers containing mixed-base or deoxyinosine residue at positions of codon degeneracy. J Clin Microbiol 34(12):2990–2996
Minor PD et al (1986) Antigenic and molecular evolution of the vaccine strain of type 3 poliovirus during the period of excretion by a primary vaccinee. J Gen Virol 67(4):693–706
Colson E, Racaniello V (1994) Soluble receptor-resistant poliovirus mutants identify surface and internal capsid residues that control interaction with the cell receptor. EMBO J 13(24):5855–5862
Kilpatrick DR et al (1998) Serotype-specific identification of polioviruses by PCR using primers containing mixed-base or deoxyinosine residues at positions of codon degeneracy. J Clin Microbiol 36(2):352–357
Melnick J (1996) Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In: Fields BN, Knipe DM, Howley PM et al (eds) Fields virology, 3rd edn. Lippincott-Raven, Philadelphia, pp 655–712
Kilpatrick DR et al (2009) Rapid group-, serotype-, and vaccine strain-specific identification of poliovirus isolates by real-time reverse transcription-PCR using degenerate primers and probes containing deoxyinosine residues. J Clin Microbiol 47(6):1939–1941
Rychlik W, Spencer WJ, Rhoads RE (1990) Optimization of the annealing temperature for DNA amplification in vitro. Nucleic Acids Res 18(21):6409–6412
Kilpatrick DR et al (2014) Identification of vaccine-derived polioviruses using dual-stage real-time RT-PCR. J Virol Methods 197:25–28
Kew OM et al (2005) Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu Rev Microbiol 59:587–635
Jegouic S et al (2009) Recombination between polioviruses and co-circulating Coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog 5(5):e1000412
Kew O et al (2002) Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 296(5566):356–359
Martin J et al (2000) Evolution of the Sabin strain of type 3 poliovirus in an immunodeficient patient during the entire 637-day period of virus excretion. J Virol 74(7):3001–3010
Shimizu H et al (2004) Circulation of type 1 vaccine-derived poliovirus in the Philippines in 2001. J Virol 78(24):13512–13521
Yakovenko ML et al (2006) Antigenic evolution of vaccine-derived polioviruses: changes in individual epitopes and relative stability of the overall immunological properties. J Virol 80(6):2641–2653
Burns C et al (2013) Multiple independent emergences of type 2 vaccine-derived polioviruses during a large outbreak in Northern Nigeria. J Virol 87(9):4907–4922
Jorba J et al (2008) Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range. J Virol 82(9):4429–4440
Kilpatrick DR et al (2011) Poliovirus serotype-specific VP1 sequencing primers. J Virol Methods 174(1–2):128–130
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Burns, C.C., Kilpatrick, D.R., Iber, J.C., Chen, Q., Kew, O.M. (2016). Molecular Properties of Poliovirus Isolates: Nucleotide Sequence Analysis, Typing by PCR and Real-Time RT-PCR. In: Martín, J. (eds) Poliovirus. Methods in Molecular Biology, vol 1387. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3292-4_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3292-4_9
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3291-7
Online ISBN: 978-1-4939-3292-4
eBook Packages: Springer Protocols