
Abstract
Busulfan is an alkylating agent widely used in the ablation of bone marrow cells before hematopoietic stem cell transplant. Due to large intraindividual and interindividual variations, and narrow therapeutic window, therapeutic drug monitoring of busulfan is warranted. A quick and reliable HPLC-MS/MS method was developed for the assay of plasma busulfan. HPLC involved C18 column, and MS/MS was used in electrospray ionization (ESI) positive mode. Quantitation and identification of busulfan was made using various multiple reactions monitoring (MRMs). Isotopic labeled busulfan-d8 was used as the internal standard. The method is linear from 50 to 2500 ng/mL and has with-in run and between-run imprecision of <10 %.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Busulfan is an anti-leukemic DNA-alkylating agent widely used in combination with cyclophosphamide for myeloablative conditioning regimens prior to hematopoietic stem cell transplantation [1–3]. Busulfan has a narrow therapeutic range with significant toxic side effects at high systemic exposure and risk of incomplete myeloablative and graft rejection at low exposure. Therefore, measurement of busulfan is warranted in busulfan dose adjustment and optimal drug exposure [4].
Various methods including immunoassays, gas chromatography (GC) coupled with electron capture detector or mass spectrometry, liquid chromatography coupled with UV detectors or mass spectrometry or fluorescence detectors have been described [5–16]. Due to better specificity, chromatographic methods are preferred. Since busulfan is not a volatile drug, its measurement by gas chromatography is tedious and time-consuming and requires sample derivatization and extraction [5, 9, 16]. Liquid chromatography mass spectrometry methods often require sample extraction but are preferred as they do not require sample derivatization. Here, we describe a simple protein precipitation no-extraction LC-MS/MS method for the determination of busulfan. The method uses positive ion electrospray ionization (ESI), multiple reactions monitoring (MRM), and D8-busulfan as internal standard.
2 Materials
2.1 Samples
1 mL blood in sodium heparin (no gel). Process the sample and analyze within 4 h of collection or freeze plasma at −70 °C until analysis (see Note 1 ). Children receiving busulfan every 6 h with a 120 min infusion have plasma samples drawn at 120, 135, 150, 180, 240, 300, and 360 min from the start of the infusion. Children receiving busulfan every 24 h with a 180 min infusion have plasma samples drawn at 180, 195, 240, 300, 360, and 480 min from the start of the infusion.
2.2 Reagents
-
1.
7.5 M Ammonium acetate (Sigma Chemicals, St. Louis, MO).
-
2.
0.3 N Zinc Sulfate (Sigma Chemicals, St. Louis, MO).
-
3.
Busulfan Powder (Sigma Chemicals, St. Louis, MO).
-
4.
Busulfan, 1 mg/mL (Cerilliant, Rockwood, CA).
-
5.
Busulfan-d8, 1 mg/mL (Cambridge Isotope Laboratories, Inc.).
-
6.
Mobile phase A (20 mM ammonium acetate/water/0.5 % formic acid): To 1 L of HPLC grade water add 2.7 mL of 7.5 M ammonium acetate and 570 μL of 88 % formic acid. Mix and degas. Stable for 1 month when stored at room temperature.
-
7.
Mobile phase B (20 mM ammonium acetate/methanol/0.5 % formic acid): To 1 L of methanol add 2.7 mL of 7.5 M ammonium acetate and 570 μL of 88 % formic acid. Mix and degas. Stable for 1 month when stored at room temperature.
-
8.
Precipitating reagent: Combine 350 mL methanol, 150 mL 0.3 N Zinc Sulfate Solution, 125 μL of 1 mg/mL busulfan-d8 (primary internal standard).
2.3 Calibrators and Controls
-
1.
Primary internal standard, Busulfan-d8, 1 mg/mL in acetone: Dissolve 10 mg in 10 mL acetone. Stable for 1 year when stored at −70 °C.
-
2.
Primary (1°) standard, Busulfan 1 mg/mL in acetone: Dissolve 100 mg into a 100 mL volumetric flask and q.s. with acetone, stable for 1 year at −70 °C.
-
3.
Secondary (2°) standard, Busulfan 10 μg/mL in negative plasma: Add 250 μL of primary standard into a 25 mL volumetric flask and q.s. with negative plasma to 25 mL. Stable for 1 year at −70 °C.
-
4.
Tertiary (3°) standard, Busulfan 2500 ng/mL in negative plasma: Add 2.5 mL of secondary standard into a 10 mL volumetric flask and q.s. with negative plasma to 10 mL, stable for 1 year at −70 °C (see Note 2 ).
-
5.
Quaternary (4°) standard, Busulfan 1000 ng/mL in negative plasma: Add 1 mL of secondary standard (2°) into a 10 mL volumetric flask and q.s. with negative plasma to 10 mL, stable for 1 year at −70 °C (see Note 3 ).
-
6.
Negative plasma matrix preparation: Add 850 mg EDTA trisodium salt hydrate to 500 mL pooled expired plasma from blood bank. The plasma first undergoes three cycles of freeze/thaw cycles. Centrifuge the plasma for 5 min at 4600 × g and filter the supernatant. Stable for 1 year at −70 °C (see Note 4 ).
-
7.
Preparation of calibrators: Prepare calibrators in negative plasma as described in Table 1. Stable for 1 year at −70 °C.
Table 1 Preparation of calibrators -
8.
Preparation of controls: Prepare controls in negative plasma as described in Table 2 (see Note 5 ). Stable for 1 year at −70 °C.
Table 2 Preparation of quality controls
2.4 Analytical Equipment and Operating Conditions
-
1.
Liquid chromatography system: Prominence UFLC system (Schimadzu Scientific Instruments) or equivalent.
-
2.
Analytic column: Supelcosil LC-18, 5 cm × 4.6 mm, 5 μm (Sigma-Aldrich).
-
3.
LC parameters: Flow rate, 0.9 mL/min. Column temperature, 55 °C. HPLC gradient is shown in Table 3.
Table 3 HPLC gradient -
4.
Mass spectrometry : 4000 Qtrap (AB Sciex) or equivalent. Use electrospray ionization source (ESI) and positive polarity mode to monitor ion pairs in multiple reactions monitoring (MRM) mode. MRMs are given in Table 4. Mass spectrometry settings are given in Table 5. Optimized mass spectrometry parameters are given in Table 6.
Table 4 MRMs for busulfan and busulfan-D8 Table 5 Mass spectrometry settings Table 6 Mass spectrometry optimization for various ions
3 Methods
3.1 Stepwise Procedure
-
1.
Pipette 100 μL of well-mixed standards, patient plasma and control to a microcentrifuge tube.
-
2.
Add 100 μL 0.9 % NaCl solution and gently vortex to mix.
-
3.
Add 500 μL precipitating/IS reagent, then immediately cap and vortex twice for total 30 s (2× dilutions are performed for each sample at the same time).
-
4.
Centrifuge tubes for 5 min at 12,000 × g.
-
5.
Carefully transfer approximately 100 μL of solution into labeled autosampler vials (see Note 6 ).
-
6.
Inject 20 μL into LC/MS/MS for analysis.
3.2 Data Analysis
-
1.
Data are analyzed using Analyst 4.1 software (AB Sciex).
-
2.
Standard curves are generated based on linear regression of the analyte/IS peak area ratios (y) versus analyte concentration (x) using MRMs provided in Table 4.
-
3.
Typically, coefficient of correlation is >0.99.
-
4.
Runs are accepted if calculated controls fall within two standard deviations of target values.
-
5.
The linearity ranges from 50 to 2500 ng/mL. Any sample exceeding 2500 ng/mL is diluted with negative plasma and re-run.
-
6.
Between and with-in run imprecision are <10 %.
-
7.
Carry-over monitoring is evaluated by injecting negative sample after highest calibrator.
-
8.
Ion suppression is monitored by comparing peak area counts of samples with plasma matrix-free sample and is typically <20 %.
-
9.
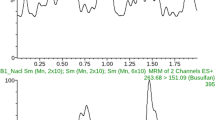
Typical chromatograms for busulfan and busulfan-d8 are given in Fig. 1.
Fig. 1 
HPLC-MS/MS MRM chromatograms for busulfan and busulfan-d8 (1000 ng/mL)

3.3 Pharmacokinetic Modeling
The data are curve fit using a peeling algorithm to generate initial polyexponential parameter estimates with final parameter estimates determined from an iterative, nonlinear weighted least squares regression algorithm with reciprocal (1/y2calc) weighting. Model-dependent pharmacokinetic parameters are calculated from final polyexponential parameter estimates. Alternatively, a model-independent approach can be applied to analyze the data. Area under the plasma concentration versus time curve during the sampling period (AUC0-n) can be calculated using the trapezoidal rule. Extrapolation of the AUC to infinity (AUC0-∞) is calculated by summation of AUC0-n + Cn/λz, where Cn represents the final plasma concentration and λz is the apparent terminal elimination rate constant.
Dose adjustments are driven by clinician defined exposure estimates, typically a desired average steady-state concentration over the entire dosing regimen (Css avg) expressed in ng/mL, or an average AUC over the entire dosing regimen expressed in μmol min. Representative plasma concentration versus time profiles observed with a 6-h and a 24-h dosing interval are illustrated in Fig. 2.

Plasma concentration versus time profiles for 6-h and a 24-h dosing interval
4 Notes
-
1.
Samples that are clotted, hemolysed, or collected in gel tubes are not suitable.
-
2.
Tertiary standard also serves as calibrator 6.
-
3.
Quaternary standard also serves as calibrator 5.
-
4.
Analyze the negative plasma to assure that it is negative for busulfan and any other unanticipated interference.
-
5.
Controls should be prepared separately and independently from calibrators.
-
6.
Avoid touching the sides of the tube when transferring supernatant.
References
Cavo M, Bandini G, Benni M, Gozzetti A, Ronconi S, Rosti G, Zamagni E, Lemoli RM, Bonini A, Belardinelli A et al (1998) High-dose busulfan and cyclophosphamide are an effective conditioning regimen for allogeneic bone marrow transplantation in chemosensitive multiple myeloma. Bone Marrow Transplant 22:27–32
O’Donnell MR, Long GD, Parker PM, Niland J, Nademanee A, Amylon M, Chao N, Negrin RS, Schmidt GM, Slovak ML et al (1995) Busulfan/cyclophosphamide as conditioning regimen for allogeneic bone marrow transplantation for myelodysplasia. J Clin Oncol 13:2973–2979
Shah AJ, Lenarsky C, Kapoor N, Crooks GM, Kohn DB, Parkman R, Epport K, Wilson K, Weinberg K (2004) Busulfan and cyclophosphamide as a conditioning regimen for pediatric acute lymphoblastic leukemia patients undergoing bone marrow transplantation. J Pediatr Hematol Oncol 26:91–97
Slattery JT, Risler LJ (1998) Therapeutic monitoring of busulfan in hematopoietic stem cell transplantation. Ther Drug Monit 20:543–549
Athanasiadou I, Angelis YS, Lyris E, Archontaki H, Georgakopoulos C, Valsami G (2014) Gas chromatographic-mass spectrometric quantitation of busulfan in human plasma for therapeutic drug monitoring: a new on-line derivatization procedure for the conversion of busulfan to 1,4-diiodobutane. J Pharm Biomed Anal 90:207–214
Courtney JB, Harney R, Li Y, Lundell G, McMillin GA, Agarwal G, Juenke JM, Mathew A, Gonzalez-Espinoza R, Fleisher M et al (2009) Determination of busulfan in human plasma using an ELISA format. Ther Drug Monit 31:489–494
Embree L, Burns RB, Heggie JR, Phillips GL, Reece DE, Spinelli JJ, Hartley DO, Hudon NJ, Goldie JH (1993) Gas-chromatographic analysis of busulfan for therapeutic drug monitoring. Cancer Chemother Pharmacol 32:137–142
French D, Sujishi KK, Long-Boyle JR, Ritchie JC (2014) Development and validation of a liquid chromatography-tandem mass spectrometry assay to quantify plasma busulfan. Ther Drug Monit 36:169–174
Hassan M, Ehrsson H (1983) Gas chromatographic determination of busulfan in plasma with electron-capture detection. J Chromatogr 277:374–380
Juenke JM, Miller KA, McMillin GA, Johnson-Davis KL (2011) An automated method for supporting busulfan therapeutic drug monitoring. Ther Drug Monit 33:315–320
Kellogg MD, Law T, Sakamoto M, Rifai N (2005) Tandem mass spectrometry method for the quantification of serum busulfan. Ther Drug Monit 27:625–629
Lai WK, Pang CP, Law LK, Wong R, Li CK, Yuen PM (1998) Routine analysis of plasma busulfan by gas chromatography-mass fragmentography. Clin Chem 44:2506–2510
Moon SY, Lim MK, Hong S, Jeon Y, Han M, Song SH, Lim KS, Yu KS, Jang IJ, Lee JW et al (2014) Quantification of human plasma-busulfan concentration by liquid chromatography-tandem mass spectrometry. Ann Lab Med 34:7–14
Murdter TE, Coller J, Claviez A, Schonberger F, Hofmann U, Dreger P, Schwab M (2001) Sensitive and rapid quantification of busulfan in small plasma volumes by liquid chromatography-electrospray mass spectrometry. Clin Chem 47:1437–1442
Quernin MH, Duval M, Litalien C, Vilmer E, Aigrain EJ (2001) Quantification of busulfan in plasma by liquid chromatography-ion spray mass spectrometry. Application to pharmacokinetic studies in children. J Chromatogr B Biomed Sci Appl 763:61–69
Vassal G, Re M, Gouyette A (1988) Gas chromatographic-mass spectrometric assay for busulfan in biological fluids using a deuterated internal standard. J Chromatogr 428:357–361
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Deng, S., Kiscoan, M., Frazee, C., Abdel-Rahman, S., Dalal, J., Garg, U. (2016). A Simple Liquid Chromatography Tandem Mass Spectrometry Method for Quantitation of Plasma Busulfan. In: Garg, U. (eds) Clinical Applications of Mass Spectrometry in Drug Analysis. Methods in Molecular Biology, vol 1383. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3252-8_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3252-8_9
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3251-1
Online ISBN: 978-1-4939-3252-8
eBook Packages: Springer Protocols