
Abstract
The efficacy of a given drug resides mainly on its ability to specifically target disease mechanisms.
Natural products represent the leading source of bioactive molecules with a broad range of activities.
It is becoming increasingly clear that natural compounds exert their chemopreventive or antitumoral activities targeting simultaneously diverse cellular pathways. Here we describe the use of antibody array to assess the effects of natural compounds on the expression of multiple proteins and of their posttranslational modifications in cellular systems. This might turn to be a very flexible application for cancer chemoprevention studies.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
In the last decade, there was an increase in the investigations of several chemical or natural compounds according for their anticancer activities [1]. In particular, the natural compounds represent a challenge for scientists who often face the problem of finding out the molecular mechanisms through which they can exert their anticancer effects. This is because natural compounds are constituted by many heterogeneous molecules [2, 3]. Usually, a natural extract impacts on several pathways simultaneously and the final biological readout of treating people at high risk of cancer occurrence could be the reprogramming of different molecular perturbations. Recently, we showed that Cynara Scolymus leaf extracts impact on mesothelioma anticancer activities by impinging on different oncogenic signalling pathways [4]. This was evidenced using the Antibody array technology.
Based on these evidences, a high-throughput platform such as the Antibody Microarrays (AM) represents an efficient, accurate, and sensitive technique for screening the protein expression profiles to investigate the molecular mechanisms of a natural extracts [5–10].
Different kinds of protein arrays are available. They differ on the number and the type of antibodies that are spotted on the surface of the slide. The advanced ones permit to analyze simultaneously more than one hundred protein and phospho-protein antibodies in order to discriminate which pathways are modulated by the chemoprevention agent tested. In addition, this kind of assays allows to save resources and to reduce the number of variables that can affect the experimental outcome.
AM are mainly based on the enzyme-linked immunosorbent assay (ELISA) . The antibodies are directly immobilized on the AM slide surface in a specific order. Each antibody is often spotted in replicate and, positive and negative control are loaded on the AM slide in order to discriminate any technical problems such as background problems during data analysis. The detection is often based on an immunofluorescent reaction caused by the binding between the biotinylated protein and the dye-labeled streptavidin substrate. The sandwich assay involves the formation of a three-layered structure that consists in the immobilized antibody, the biotinylated protein and the dye-labeled streptavidin substrate [11].
In general, these assays can be used for analysis of protein extracts from cells, blood samples, and fresh, frozen, and/or FFPE tissues.
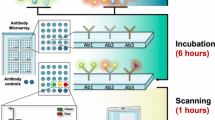
The protocol described below is a more detailed personal modification of the antibody array user’s guide of Full Moon BioSystems, Inc. (754 N Pastoria Ave, Sunnyvale, CA 94085) (Fig. 1).

Workflow
2 Materials
Before starting the experiment prepare all fresh solutions.
Each reagent needs a different temperature of warming before use (see Subheading 3).
The major part of the reagents is provided by the kit (Phospho Explorer Antibody Array, Cat. N. PEX100, Full Moon, BioSystems).
2.1 Protein Extraction
-
1.
One to five million of cells.
-
2.
1× PBS (pH 7.4) (Gibco).
-
3.
Thoma cell counting chamber.
-
4.
Extraction buffer (Full Moon BioSystems).
-
5.
Lysis beads (Full Moon BioSystems).
-
6.
Vortexer.
-
7.
Centrifuge.
2.2 Lysate Purification and Quantification
-
1.
Spin columns (Full Moon BioSystems).
-
2.
Labeling buffer (Full Moon BioSystems).
-
3.
Vortexer.
-
4.
Centrifuge.
-
5.
UV adsorption spectrophotometer (NanoDrop 1000 spectrophotometer, Thermo Scientific).
2.3 Protein Labeling
-
1.
Biotin Reagent (Full Moon BioSystems).
-
2.
N,N-Dimethylformamide (DMF) (Full Moon BioSystems).
-
3.
Labeling buffer (Full Moon BioSystems).
-
4.
Stop Reagent (Full Moon BioSystems).
2.4 Blocking and Coupling
-
1.
Blocking reagent (Full Moon BioSystems).
-
2.
Petri dish 100 × 15 mm.
-
3.
Antibody microarray slides (Full Moon BioSystems).
-
4.
Milli-Q grade water or dd H2O.
-
5.
Coupling reagent (Full Moon BioSystems).
-
6.
Vortexer.
-
7.
Coupling chambers (Full Moon BioSystems).
-
8.
Labeled protein solution.
-
9.
10× Wash buffer (Full Moon BioSystems).
-
10.
Orbital shaker [2].
-
11.
50 ml conical tube.
2.5 Detection
-
1.
Cy3-Streptavidin (1 mg/ml) (#SA-5549, Vector).
-
2.
Detection buffer (Full Moon BioSystems).
-
3.
Petri dish 100 × 15 mm.
-
4.
Milli-Q-grade water or dd H2O.
-
5.
10× Wash buffer (Full Moon BioSystems).
-
6.
Orbital shaker
-
7.
50 ml conical tube.
-
8.
Centrifuge.
2.6 Scanning
-
1.
Agilent Technologies G2505B Micro Array Scanner (Agilent Technologies).
2.7 Data Extraction
-
1.
Gal file (Full Moon BioSystems).
-
2.
Agilent feature extraction software (Agilent technologies).
2.8 Data Analysis
-
1.
Antibody list (Full Moon BioSystems).
-
2.
Array map (Full Moon BioSystems).
-
3.
Matlab software (The MathWorks Inc.).
3 Methods
Please warm all the solution at the right temperature before use. The protocol allows to stop the procedure and stored the sample at −80 °C at the end of the lysate quantification and/or the protein labeling section.
3.1 Protein Extraction (See Note 1 )
-
1.
Wash twice the cell culture dishes with warm 1× PBS (37 °C).
-
2.
Detach the cells by scraping (see Note 2 ) them from the dish in cold 1× PBS (4 °C).
-
3.
Collect cells in a 15 ml conical tubes and centrifuge at 4 °C at 800 rpm for 5 min. Discard supernatant and suspend pellet with cold 1× PBS.
-
4.
Calculate the amount of cells by using a Thoma counting cells chambers. Collect 1–2.5 million or 2.5–5 million of cells and centrifuge at 4 °C at 800 rpm for 5 min. Discard supernatant.
-
5.
Resuspend the pellet in 100 μl (1–2.5 million) or 200 μl (2.5–5 million of cells) of extraction buffer.
-
6.
Add a tube of lysis beads in each sample and mix by vortexing for 1 min.
-
7.
Incubate the mixture on ice for 10 min.
-
8.
Repeat vortexing every 10-min intervals for 60 min. Remember to place sample on ice between vortexing.
-
9.
Centrifuge samples at 17500 × g for 20 min at 4 °C.
-
10.
Transfer the supernatant (protein extract) to a clean tube (see Note 3 ).
3.2 Lysate Purification and Quantification
Warm to room temperature the following reagents before use: spin column and labeling buffer.
-
1.
Remove the top column cap of the spin column and reconstitute the dry gel contained inside by adding 650 μl of labeling buffer.
-
2.
Close the top column cap and vortex vigorously for 5 s (see Note 4 ).
-
3.
Leave the hydrated column at room temperature for 30 min before use (see Note 5 ).
-
4.
After hydration, remove excess fluid by spin the column in its wash tube at 800 × g for 2 min. Remove the top and the bottom tap before centrifuging.
-
5.
Immediately transfer 100 μl of protein extract by dispensing the sample onto the center of the column without touch the gel surface.
-
6.
Place the column in a new collection tube and spin them at 800 × g for 2 min.
-
7.
Discard the spin column and collect the purified protein.
-
8.
Measure the UV adsorption of the protein sample (see Note 6 ). Use labeling buffer as blank.
-
9.
Store the sample at −80 °C or proceed to the next step.
3.3 Protein Labeling
Warm to room temperature the following reagents before use: DMF, biotin reagent, labelling buffer, stop reagent.
-
1.
Prepare the DMF/biotin solution to final volume of 10 μg/μl by adding 100 μl of warm DMF to 1 mg of biotin.
-
2.
Aliquot 100 μg of protein lysate in a new tube (collect 10–25 μl of lysate basing on the OD measured) (see Note 7 ).
-
3.
Bring the protein lysate to 75 μl by adding labeling buffer.
-
4.
Add 3 μl of DMF/biotin solution to the protein lysate and incubate it for 2 h at room temperature by mixing.
-
5.
At the end of the 2 h stop the reaction by adding 35 μl of stop reagent. Incubate for 30 min at room temperature with mixing.
-
6.
Store the sample at −80 °C or proceed to the next step.
3.4 Blocking and Coupling
Warm to 25–30 °C in a water bath the following reagents before use: blocking reagent, coupling reagent, and 10× wash buffer. Warm to room temperature the antibody microarray slides (see Note 8 ).
-
1.
Prepare the blocking solution by adding 60 ml of warm blocking reagent to 1.8 g of dry milk.
-
2.
Add 30 ml of blocking solution in a 100 × 15 mm Petri dish and submerge the slide in this solution. Submerge only one slide for Petri dish.
-
3.
Incubate at room temperature for 45 min on an orbital shaker at 50 rpm.
-
4.
After that, place the slide in a 50 ml conical tube.
-
5.
Add 45 Milli-Q grade water or dd H2O. Avoid to add water directly on the upper side of the slide. Shake gently for 10–20 s, discard the water and refill the tube with other 45 ml of water.
-
6.
Repeat step 5 ten times (see Note 9 ).
-
7.
Place the slide in a free well of the coupling chamber. Before to place, shake off the excess of fluid from the slide.
-
8.
During step 6, prepare the coupling solution by adding 12 ml of warm coupling reagent to 0,36 g of dry milk.
-
9.
Add in a 15 ml conical tube, 6 ml of coupling solution and one tube of labeled samples previously stored at −80 °C (step 6 of Subheading 3.3). Prepare a 15 ml of conical tube for each labeled sample.
-
10.
Slowly pour the solution of point 8 into a free well. Submerge the slide of the coupling chamber containing the slide (see Note 10 ). Avoid to add the solution directly on the surface of the slide.
-
11.
Incubate at room temperature for 2 h on an orbital shaker at 30 rpm.
-
12.
During step 10, prepare the 1× wash solution by adding 100 m of warm 10× wash buffer to 900 ml of dd H2O.
-
13.
Transfer the slide in a new Petri dish previously filled with 30 ml of 1× wash solution.
-
14.
Incubate at room temperature for 10 min on an orbital shaker at 60 rpm.
-
15.
Discard 1× wash solution from the Petri dish and fill it with other 30 ml of 1× wash solution.
-
16.
Repeat steps 14 and 15 for other two times.
-
17.
After that, place the slide in a 50 ml conical tube and add 45 Milli-Q-grade water or dd H2O. Avoid to add water directly on the upper side of the slide. Shake gently for 10–20 s, discard the water, and refill the tube with other 45 ml of water.
-
18.
Repeat step 17 ten times (see Note 9 ).
3.5 Detection (see Note 11 )
Warm to room temperature the following reagents before use: detection buffer, 1× wash solution.
-
1.
Prepare Cy3-streptavidin solution by adding 30 μl of Cy3-streptavidin (1 mg/ml) to 60 ml of warm detection buffer.
-
2.
Transfer the slide in a new Petri dish and slowly add 30 ml of Cy3-streptavidin solution.
-
3.
Incubate at room temperature for 20 min on an orbital shaker at 50 rpm.
-
4.
Transfer the slide in a new Petri dish previously filled with 30 ml of 1× wash solution.
-
5.
Incubate at room temperature for 10 min on an orbital shaker at 60 rpm.
-
6.
Discard 1× wash solution from the Petri dish and fill it with other 30 ml of 1× wash solution.
-
7.
Repeat steps 5 and 6 for other two times.
-
8.
After that, place the slide in a 50 ml conical tube and add 45 Milli-Q-grade water or dd H2O. Avoid to add water directly on the upper side of the slide. Shake gently for 10–20 s, discard the water, and refill the tube with other 45 ml of water.
-
9.
Repeat step 8 for ten times (see Note 9 ).
-
10.
Shake off excess water from the slide and place the slide in a new 50 ml conical tube.
-
11.
Dry the slide by centrifuging the 50 ml conical tube filled with the slide at 100 × g for 2 min at room temperature (see Note 12 ).
3.6 Scanning (see Note 13 )
-
1.
Put the slide in a slide holder for the Agilent microarray scanner with the barcode facing up in the slide holder.
-
2.
Place assembled slide holder into an empty slot of the carousel.
-
3.
Use an Agilent HD_GX_1 color protocol, with 5 μm scanning resolution, and green color channel, to scan the slide.
3.7 Data Extraction
-
1.
Open the scanner slide image (.tiff) with the Agilent Feature Extraction software (see Note 14 ).
-
2.
Flip the scanner slide image from left to lower right (landscape/portrait).
-
3.
Convert the gal file (see Note 15 ) to a grid.csv file compatible with the Agilent software.
-
4.
Open the grid (grid.csv) and adjust it on the scanner slide image thus to center each spot of the slide to each point of the grid.
-
5.
Create a new project, change project properties, and run Feature Extraction for a non-Agilent image (see Note 16 ).
-
6.
Proceed with data extraction.
3.8 Data Analysis
For data analysis and features selection we use Matlab software (The MathWorks Inc.).
-
1.
A quality control of the signals is performed on negative and positive controls and checking intensity values of two different housekeeping protein such as GAPDH and ACTIN.
-
2.
A quantile normalization between arrays and Z score transformation is performed to express the background corrected spot intensity values as unit of a standard deviation from the normalized mean of zero.
-
3.
Features is selected basing on Z ratios calculated by taking the difference between the averages of the observed protein Z scores and dividing by the standard deviation of all the differences for that particular comparison [12]. A Z ratio higher than 1.5 is inferred as significant.
-
4.
Unsupervised Hierarchical Clustering is used to investigate clusters of samples (Fig. 2).
Fig. 2 
Unsupervised hierarchical clustering example. Heat maps depicting phospho-protein levels of a control and three different treatments

4 Notes
-
1.
Usually the kit provides only two antibody array assay slides; for this reason it is important to know if the treatment, that we want to test, has worked before performing the experiment. It is advisable to use an aliquot of the cells to test a positive read out.
-
2.
Do not use trypsin to detach the cells: it could impair cell protein activity. Use only the lysis buffer provided by the kit to lysate the samples. Other buffers could impair the ELISA reaction.
-
3.
The supernatant should appear clear. If it appears cloudy, centrifuge again at 17500 × g for 20 min at 4 °C. If it is still cloudy, store tubes at −70 °C for 15 min. Remove tubes from the freezer and immediately centrifuge at 14000 rpm for 20 min at 4 °C.
-
4.
Spin column cap is provided of two caps one on top and one at bottom. After adding labelling buffer, remove air bubbles by sharply tapping the bottom of the column.
-
5.
Reconstitute the spin column during the centrifuge of step 9 of the protein extraction thus to allow an immediately purification of the protein samples.
-
6.
Use proteins A280 program for the NanoDrop 1000 spectrophotometer (Thermo Scientific). The absorbance should be greater than four OD and the UV absorption spectrum should reveal two peaks one at 200–230 nm and one at 240–280 nm (Fig. 3). If the OD is lower than four, the protein must be concentrated at 4 °C in a vacuum centrifuge. Moreover, if the protein lysate is not clear the peaks will not appear well separated. In this case it should necessary repeat procedure described in Note 3 .
Fig. 3 
UV absorption spectrum example of a clear cell lysate. The two peaks, 230 and 270 nm, are well separated
-
7.
It is possible to use less than 100 μg of protein lysate, till to a minimum of 40 μg with a concentration of 2 μg/μl.
-
8.
Warm the slides at room temperature for 30–45 min; after that, open the package and leave the slides for 10 min at room temperature in order to dry the surface of the slides. Please note that the side of the slide with the barcode labeled is the upper one and must face up.
-
9.
After the last wash, the slide surface should appear uniformly smooth. If it does not happen repeat step 5 of Subheading 3.4.
-
10.
It is important to do step 9 just after step 6 of Subheading 3.4 in order to avoid the surface of the slide from becoming dry.
-
11.
Perform these steps in the dark or cover with aluminum foil all the steps in which it used the Cy3-streptavidin solution.
-
12.
It is very important to shake off the excess of fluid from the slide as quickly as possible in order to avoid spots formation due to water evaporation, which could compromise the quality of scan image.
-
13.
Here we described the procedure for scanning the slide with the Agilent Technologies G2505B Micro Array Scanner; however, it could be used other scanners which are compatible with 3 × 1 in. slides (76 × 25 mm).
-
14.
Use Agilent Feature Extraction software or another one compatible with the scanner that is used.
-
15.
The gal file is provided by the company that produced the microarray protein assay.
-
16.
See the Agilent Feature Extraction software user’s guide.

References
Steward WP, Brown K (2013) Cancer chemoprevention: a rapidly evolving field. Br J Cancer 109:1–7
Newman DJ, Cragg GM (2012) Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod 75(3):311–335
Danishefsky S (2010) On the potential of natural products in the discovery of pharma leads: a case for reassessment. Nat Prod Rep 27:1114–1116
Pulito C, Mori F, Sacconi A Cynara scolymus affects malignant pleural mesothelioma by promoting apoptosis and restraining invasion.Oncotarget 6(20):18134–50
Fan H, Yang L, Fu F et al (2012) Cardioprotective effects of salvianolic acid a on myocardial ischemia-reperfusion injury in vivo and in vitro. Evid Based Complement Alternat Med 2012:508938
Chung A, Chin YE (2009) Antibody array platform to monitor protein tyrosine phosphorylation in mammalian cells (Chapter 18). Methods Mol Biol 527:247–255
Sanchez-Carrayo M (2006) Antibody arrays: technical considerations and clinical applications in cancer. Clin Chem 52(9):1651–1659
Pierobon M, Wulfkuhle J, Liotta L et al (2015) Phosphoproteomic analysis in oncological practice. Oncogene 34(7):805–814
Eke I, Schneider L, Forster C et al (2013) EGFR/JIP-4/JNK2 signaling attenuates cetuximab-mediated radiosensitization of squamous cell carcinoma cells. Cancer Res 73:297–306
Mitchell SJ, Martin-Montalvo A, Mercken EM et al (2014) The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 6:836–843
Lee JR, Magee DM, Gaster RS et al (2013) Emerging protein array technologies for proteomics. Expert Rev Proteomics 10:65–75
Cheadle C, Vawter MP, Freed WJ et al (2003) Analysis of microarray data using Z score transformation. J Mol Diagn 5:73–81
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Pulito, C., Sacconi, A., Korita, E., Maidecchi, A., Strano, S. (2016). Antibody Array as a Tool for Screening of Natural Agents in Cancer Chemoprevention. In: Strano, S. (eds) Cancer Chemoprevention. Methods in Molecular Biology, vol 1379. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3191-0_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3191-0_17
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3190-3
Online ISBN: 978-1-4939-3191-0
eBook Packages: Springer Protocols