
Abstract
The activity of enzymes involved in active transport of matter across lipid bilayers can conveniently be assayed by measuring their consumption of energy, such as ATP hydrolysis, while it is more challenging to directly measure their transport activities as the transported substrate is not converted into a product and only moves a few nanometers in space. Here, we describe two methods for the measurement of active proton pumping across lipid bilayers and the concomitant formation of a membrane potential, applying the dyes 9-amino-6-chloro-2-methoxyacridine (ACMA) and oxonol VI. The methods are exemplified by assaying transport of the Arabidopsis thaliana plasma membrane H+-ATPase (proton pump), which after heterologous expression in Saccharomyces cerevisiae and subsequent purification has been reconstituted in proteoliposomes.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Cells and organelles depend on electrochemical gradients across their membranes. Membrane -embedded biological pumps are key players in maintaining these gradients through primary, active transport of ions. Among these pumps, plasma membrane H+-ATPases form a subfamily of P-type ATPase s that use the energy derived from hydrolysis of ATP to pump protons out of cells. In protists, fungi, and plants, they are essential for establishing and maintaining the crucial transmembrane electrochemical gradients of protons which provides the driving force for the uptake of nutrients and other cell constituents through an array of secondary transport systems [1–4].
The activity of the plasma membrane H+-ATPases can be conveniently studied when the enzyme resides in the membrane of a closed vesicle. The closed vesicle may be the native membrane of the protein (see, e.g., [5]) or a liposome prepared from a known phospholipid composition. The latter offers a well-defined experimental system where no unknown or unwanted proteins are present. In both cases, the membrane itself must be proton impermeable on the time scale of the experiment. Proton fluxes in this system are typically followed indirectly by use of ΔpH-sensitive dyes that change absorbance or fluorescence properties in response to the formation of pH gradients across the membrane. Acridine orange is a classical ΔpH-sensitive dye, and a number of derivatives of this probe are used for measurement of ΔpH. The spectral properties of acridine orange are relatively insensitive to medium pH at physiological values [6]. However, acridine orange is a monomer in dilute solutions and a dimer at high probe concentrations, and since the monomer and the dimer have different absorbance as well as fluorescence spectra, dimer formation can be followed by assaying the decrease in monomer absorbance or fluorescence [6]. In vesicles with a higher H+ concentration than the surrounding medium, monomeric acridine orange is protonated and trapped, and dimerizes as the concentration inside increases, leading to quenching of the fluorescence [6, 7]. As the degree of dimerization of acridine orange depends strongly on temperature and the anion composition of the vesicle lumen, great care should be taken when using this probe to compare proton pumping activities as a function of temperature and anions in the medium [6].
Here we describe the use of 9-amino-6-chloro-2-methoxyacridine (ACMA ), a derivative of the probe acridine orange. This probe is yellow in solution and can cross bilayers in its unprotonated form. Because of its derived nature ACMA can exist in four states, namely as an anion, as a neutral molecule, as a monocation, and as a dication [8]. The monocation is protonated at the acridinic nitrogen with a pK a ranging between 8.3 [9] and 8.6 [8] implying that at neutral pH the monocation is already the predominant form. The dication is further protonated at the amino group with a pK a ranging between 4.7 [9] and 5.7 [8]. The mechanism by which ACMA reports ΔpH gradients is not clear, as at least the monocation does not dimerize in aqueous solutions even as high as 200 μM [8]. It has been suggested that decrease of fluorescence of ACMA is the result of adsorption of the probe on the membrane, which results in stacking and aggregation at this interface [10]. The related ΔpH probe 9-aminoacridine has been suggested to form a similar dimer-exited state at the membrane [11]. Because of the structural similarities with acridine orange, such stacking would presumably also be sensitive to temperature and anions present. Alternatively, as the mono- and dications have very different fluorescence spectra with the monocation having a fluorescence peak at 480 nm and the dication exhibiting almost no fluorescence at this wavelength [8], it is possible that what is assayed when luminal vesicular pH decreases is simply disappearance of the monocation.
While acridine orange and derivatives measure formation of a ΔpH, oxonol V and VI can be used to measure differences in charge balance across a liposomal membrane [12–15]; the latter is not limited to proton transport, but is highly applicable to other ions as well [12]. The oxonol dyes are blue in solution, negatively charged (pKa ~4.2, [12]), and show a large increase in fluorescence upon insertion into a hydrophobic environment. Generation of an inside positive potential causes the dye to preferentially accumulate inside the liposome, and leads to an increased interaction with the liposomal membrane. Oxonol VI does not interact equally well with all membranes, and particularly high phosphatidylethanolamine concentrations can interfere with the probe [16]. The assay presented here is intended for obtaining relative levels of the gradient achieved in relation to a reference standard (e.g., for comparing variants of the H+-ATPase); if a quantification of the actual membrane potential is desired, the system can be calibrated by the method of Apell and Bersch [12]. Reduction of the temperature to 10 °C can help to resolve the steady state fluorescence level.
The assays described here are based on proteoliposomes prepared from protein heterologously expressed in S. cerevisiae, and purified by affinity chromatography [17]. Measurement of H+ transport is also directly applicable to sealed vesicles from plant plasma membrane preparations [18], as well as measurement of both H+-transport and membrane potential in proteoliposomes prepared from crude membranes from S. cerevisiae [19].
2 Materials
2.1 Instrumentation
-
1.
A fluorometer with excitation and emission filters in the 350–700 nm range. It is preferable to have a temperature control unit and a magnetic stirrer mounted.
-
2.
Fluorometer cuvettes, preferably suitable for magnetic stirring. This protocol is scaled for a 3 mL cuvette.
2.2 Stock Solutions
-
1.
ACMA solution: Prepare 10 mM ACMA in DMSO. Weigh 2.59 mg of ACMA and dissolve in 1 mL DMSO. Dilute to 1 mM in DMSO and store in 100 μL aliquots at −20 °C in the dark.
-
2.
Oxonol VI solution: Prepare 2.5 mM solution in 50 % ethanol. Weigh 0.79 mg of oxonol VI and dissolve in 1 mL 50 % ethanol. Dilute to 50 μM in 50 % ethanol and store at −20 °C in the dark.
-
3.
Valinomycin solution: Prepare 1.25 mM valinomycin in 96 % ethanol. Weigh 1.39 mg of valinomycin and dissolve in 1 mL 96 % ethanol. Dilute to 125 μM in 96 % ethanol and store in aliquots of 100 μL at −20 °C.
-
4.
Vanadate solution: Prepare 0.1 M Sodium orthovanadate in ultrapure water. Weigh 183.9 mg of Na3VO4 and dissolve in 10 mL ultrapure water. Adjust to pH 7 using HCl.
-
5.
ATP -KOH, 0.5 M: Dissolve 5.51 g Na2ATP · H2O in 10 mL ultrapure water. Adjust pH to 7.0 with KOH, and dilute to 20 mL. Store in aliquots of 1 mL at −20 °C.
-
6.
Carbonyl cyanide m-chlorophenylhydrazone (CCCP) solution: Prepare 5 mM in 70 % ethanol. Weigh 1.023 mg of CCCP and dissolve in 1 mL 70 % ethanol. Store in aliquots of 100 μL at −20 °C.
-
7.
0.5 M MOPS-KOH, pH 7.0: Weigh 52.32 g MOPS and dissolve in 300 mL ultrapure water. Adjust pH to 7.0 using KOH, and add ultrapure water to a final volume of 0.5 L.
-
8.
0.5 M K2SO4: Weigh 4.36 g K2SO4 and dissolve in 50 mL ultrapure water.
-
9.
0.5 M MgSO4: Weigh 616.25 mg MgSO4 · 7H2O and dissolve in 5 mL ultrapure water.
2.3 Assay Buffers
-
1.
Proton pumping buffer: Typically 20 mL is prepared, containing 20 mM MOPS-KOH pH 7.0, 50 mM K2SO4, 1 μM ACMA , 1 mM ATP , and 60 nM Valinomycin. Store at 4 °C (see Note 1 ).
-
2.
Membrane potential buffer: Typically 20 mL is prepared, containing 10 mM MOPS-KOH pH 7.0, 50 mM K2SO4, 50 nM oxonol VI, 1 mM ATP . Store at 4 °C (see Note 2 ).
2.4 Biological Materials
-
1.
Arabidopsis thaliana plasma membrane H+-ATPase isoform 2, purified and reconstituted into preformed asolectin or dioleoylphosphatidylcholine vesicles as described [20]: 12 μg purified protein per 220 μL reconstitution mixture, containing MOPS-KOH buffer (pH 7), 50 mM octylglucoside, and 10.6 mg/mL asolectin (see Note 3 ).
3 Methods
3.1 Measurement of Proton Transport
-
1.
Turn on the fluorometer and set up the parameters as follows: excitation wavelength 412 nm, emission wavelength 480 nm, and measurement duration approximately 10 min with 1 s resolution. Adjust slits as necessary; a bandpass of 2 nm is usually sufficient.
-
2.
Fill the fluorometer cuvette with 1 mL of proton pumping assay buffer. Add 10 μL proteoliposomes from the reconstitution mixture, corresponding to about 0.5 μg protein, and homogenize gently with a pipette.
-
3.
Introduce the cuvette into the fluorometer (remember to include the stirring bar), and start the monitoring of the emission intensity (see Note 4 ).
-
4.
Wait until fluorescence is stable (60–120 s).
-
5.
Add MgSO4 to start the assay (12 μL 0.5 M) (see Note 5 ).
-
6.
Let the experiment run for a couple of minutes (see Note 6 ).
-
7.
Dissipate the membrane potential by addition of 1 μL of 5 mM CCCP when the fluorescence has stabilized.
-
8.
Optional: Repeat the measurements on a sample supplemented with 3 μL of 0.1 M vanadate as the background control (see Note 7 ).
An example of H+ transport measurement by the ACMA assay is shown in Fig. 1.

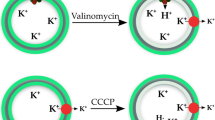
Example of a typical measurement of H+ transport into phospholipid vesicles using the ACMA assay. Proteoliposomes with reconstituted AHA2 were mixed with proton pumping buffer containing the fluorophore ACMA (AC in closed ring), valinomycin (denoted V), and ATP , and the fluorescence intensity at 480 nm was monitored. After a stable fluorescence emission was obtained, proton pumping was initiated by the addition of MgSO4. The ionophore valinomycin ensures that K+ ions are free to diffuse out of the proteoliposomes, thereby eliminating any contribution from the electrical potential against proton pumping. Upon proton accumulation and luminal acidification, ACMA is protonated and forms dimeric or higher order complexes within the proteoliposomes (AC in dashed ring), resulting in a shift in its fluorescence maxima, measured as a drop in fluorescence intensity at 480 nm. After maximum proton influx is achieved, the proton gradient is disrupted by the addition of CCCP (denoted C). This leads to a luminal alkalization followed by deprotonation and dissociation of the ACMA oligomers, resulting in restored fluorescence intensity at 480 nm. Figure is modified from [21]
3.2 Measurement of Membrane Potential
-
1.
Turn on the fluorometer, and set up the parameters as follows: excitation wavelength 610 nm, emission wavelength 640 nm, and measurement duration up to approximately 30 min with 1 s resolution. Adjust slits as necessary; a bandpass of 5 nm is usually sufficient.
-
2.
Fill the fluorometer cuvette with 3 mL membrane potential assay buffer, and 10 μL of proteoliposomes containing approximately 0.5 μg of purified protein.
-
3.
Insert the cuvette into the fluorometer, and allow for the temperature to stabilize to the desired level (usually room temperature or 10 °C), i.e., 5 min (see Note 8 ).
-
4.
Start monitoring the fluorescence emission.
-
5.
Wait for the fluorescence to stabilize (60–120 s).
-
6.
Start the reaction by the addition of MgSO4 (12 μL of 0.5 M) (see Note 5 ).
-
7.
Wait for the fluorescence to stabilize. After an initial stable level, the fluorescence may decrease as the vesicles can become leaky to H+.
-
8.
Dissipate the membrane potential with valinomycin (3 μL of 125 μM).
-
9.
Wait for the fluorescence level to stabilize.
-
10.
The initial rate of transport can be determined from a linear fit of the initial change in fluorescence; this can be correlated to rates derived from measurements with ACMA . The maximal membrane potential achieved can be determined as the difference in fluorescence between the peak fluorescence under pumping, and the level obtained after addition of valinomycin.
An example of the fluorescence response of oxonol VI to energization of proteoliposomes containing incorporated plasma membrane H+-ATPase is shown in Fig. 2.

Example of a typical measurement of the electrical potential (φ) using the assay based on the oxonol VI dye. Proteoliposomes with reconstituted AHA2 were mixed with membrane potential buffer containing oxonol VI (Ox in dashed ring) and ATP , and the fluorescence intensity at 640 nm was monitored. After a stable fluorescence emission was obtained, proton pumping was initiated by the addition of MgSO4. Upon proton accumulation and buildup of an electrical potential, oxonol VI preferentially accumulates inside the proteoliposomes, leading to an increased interaction with the liposomal membrane (Ox in closed ring), which is monitored by an increase in fluorescence at 640 nm. After maximum electrical potential was achieved it was disrupted by the addition of the ionophore valinomycin (denoted V), which allows K+ to diffuse out of the proteoliposomes. This results in dissociation of oxonol VI from the membrane, and a decrease in fluorescence intensity at 640 nm. This assay was performed at 10 °C. Note that the large spikes at the addition of MgSO4 and valinomycin are due to opening of the fluorometer chambers. Figure is modified from [21]
4 Notes
-
1.
Proton pumping buffer without ACMA , ATP , and valinomycin can be prepared in bulk and stored up to 6 months. Buffer containing all ingredients should be prepared immediately before use and protected from light by wrapping the container of the prepared solution with aluminum foil.
-
2.
Membrane potential buffer without oxonol VI and ATP can be prepared in bulk and stored up to 6 months. Buffer containing all ingredients should be prepared immediately before use and protected from light by wrapping the container of the prepared solution with aluminum foil.
-
3.
We routinely reconstitute the plasma membrane H+-ATPase into asolectin vesicles of mixed phospholipid composition, although we have also successfully reconstituted using vesicles with a defined lipid composition, e.g., dioleoylphosphatidylcholine, palmitoyloleoylphosphatidylcholine, or mixtures of palmitoyloleoylphosphatidylcholine and palmitoyloleoylphosphatidylglycerol.
-
4.
To ensure your setting and the ACMA containing proton pumping buffer are working properly, run an excitation scan from 350 to 450 nm (emission 480 nm) and an emission scan from 450 to 500 nm (excitation 412 nm). For ACMA, the excitation and emission maxima should be at 412 nm and 480 nm, respectively. The fluorescence spectrum for ACMA is shown in Fig. 3.
-
5.
We typically initiate the reaction by the addition of Mg2+ as the true substrate of the plasma membrane H+-ATPase is MgATP. ATP is already present in the assay buffer.
-
6.
The initial rate of H+ flux is used when calculating the relative rate of H+ transport. After 5–10 min the vesicles might become leaky to protons and you will observe a slight increase in fluorescence. The maximal degree of fluorescence quenching is reached when proton influx into vesicles matches efflux and depends on both proton pumping activity and leakiness of the vesicles. Proton fluxes cannot be quantified in terms of numbers of H+ transported due to the non-ideal behavior of the ΔpH probe.
-
7.
Orthovanadate inhibits many NTPases, including P-type ATPase s, and will therefore block all proton pumping that is dependent on the plasma membrane H+-ATPase. Solutions of orthovanadate are prone to polymerization when stored for prolonged periods of time, and these polyvanadate ions are unable to properly inhibit NTPases. To ensure that the orthovanadate solution is properly monomeric before use, the solution should be boiled for 5 min and placed on ice.
-
8.
To ensure your settings and the oxonol VI containing membrane potential buffer are working properly, run an excitation scan from 550 to 630 nm (emission 640 nm) and an emission scan from 620 to 700 nm (excitation 610 nm). The properties of oxonol VI are as follows: Excitation and emission maxima at 610 nm and 640 nm, respectively. It is important to measure the spectra in the presence of liposomes since these both shift and greatly increase the fluorescence yield of oxonol VI. The fluorescence spectrum for oxonol VI is shown in Fig. 3.

References
Meade JC, Li C, Stiles JK et al (2000) The Trypanosoma cruzi genome contains ion motive ATPase genes which closely resemble Leishmania proton pumps. Parasitol Int 49:309–320
Nakamoto RK, Slayman CW (1989) Molecular properties of the fungal plasma-membrane [H+]-ATPase. J Bioenerg Biomembr 21:621–632
Slayman CL, Long WS, Lu CY (1973) The relationship between ATP and an electrogenic pump in the plasma membrane of Neurospora crassa. J Membr Biol 14:305–338
Palmgren MG (2001) Plant plasma membrane H+-ATPases: powerhouses for nutrient uptake. Annu Rev Plant Physiol Plant Mol Biol 52:817–845
Palmgren MG (1990) An H+-ATPase assay: proton pumping and ATPase activity determined simultaneously in the same sample. Plant Physiol 94:882–886
Palmgren MG (1991) Acridine orange as a probe for measuring pH gradients across membranes: Mechanism and limitations. Anal Biochem 192:316–321
Clerc S, Barenholz Y (1998) A quantitative model for using acridine orange as a transmembrane pH gradient probe. Anal Biochem 259:104–111
Marty A, Bourdeaux M, Dell’Amico M et al (1986) 9-amino-2-methoxy-6-chloroacridine monocation fluorescence analysis by phase-modulation fluorometry. Eur Biophys J 13:251–257
Capomacchia AC, Schulman SG (1975) Electronic absorption and fluorescence spectrophotometry of quinacrine. Anal Chim Acta 77:79–85
Casadio R (1991) Measurements of transmembrane pH differences of low extents in bacterial chromatophores. Eur Biophys J 19:189–201
Grzesiek S, Otto H, Dencher NA (1989) delta pH-induced fluorescence quenching of 9-aminoacridine in lipid vesicles is due to excimer formation at the membrane. Biophys J 55:1101–1109
Apell HJ, Bersch B (1987) Oxonol VI as an optical indicator for membrane potentials in lipid vesicles. Biochim Biophys Acta 903:480–494
Kiehl R, Hanstein WG (1984) ATP-dependent spectral response of oxonol VI in an ATP-Pi exchange complex. Biochim Biophys Acta 766:375–385
Cooper CE, Bruce D, Nicholls P (1990) Use of oxonol V as a probe of membrane potential in proteoliposomes containing cytochrome oxidase in the submitochondrial orientation. Biochemistry 29:3859–3865
Holoubek A, Večeř J, Opekarová M et al (2003) Ratiometric fluorescence measurements of membrane potential generated by yeast plasma membrane H + -ATPase reconstituted into vesicles. Biochim Biophys Acta Biomembr 1609:71–79
Gibrat R, Grignon C (2003) Liposomes with multiple fluorophores for measurement of ionic fluxes, selectivity, and membrane potential. Methods Enzymol 372:166–186
Ekberg K, Wielandt AG, Buch-Pedersen MJ et al (2013) A conserved asparagine in a P-type proton pump is required for efficient gating of protons. J Biol Chem 288:9610–9618
Lund A, Fuglsang AT (2012) Purification of plant plasma membranes by two-phase partitioning and measurement of H+ pumping. Methods Mol Biol 913:217–223
Venema K, Palmgren MG (1995) Metabolic modulation of transport coupling ratio in yeast plasma membrane H(+)-ATPase. J Biol Chem 270:19659–19667
Lanfermeijer FC, Venema K, Palmgren MG (1998) Purification of a histidine-tagged plant plasma membrane H+-ATPase expressed in yeast. Protein Expr Purif 12:29–37
Rodriguez A, Benito B, Cagnac O (2012) Using heterologous expression systems to characterize potassium and sodium transport activities. Methods Mol Biol 913:371–386
Acknowledgement
This work was supported by the UNIK research initiative of the Danish Ministry of Science, Technology and Innovation through the “Center for Synthetic Biology” at the University of Copenhagen and the Danish National Research Foundation through the PUMPKIN Center of Excellence (DNRF85).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Additional information
This work is dedicated to the memory of Alex Green Wielandt, who set the basis of this study until his untimely death.
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Wielandt, A.G., Palmgren, M.G., Fuglsang, A.T., Günther-Pomorski, T., Justesen, B.H. (2016). Measuring H+ Pumping and Membrane Potential Formation in Sealed Membrane Vesicle Systems. In: Bublitz, M. (eds) P-Type ATPases. Methods in Molecular Biology, vol 1377. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3179-8_17
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3179-8_17
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3178-1
Online ISBN: 978-1-4939-3179-8
eBook Packages: Springer Protocols