
Abstract
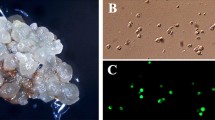
Grapevine embryogenic cultures are ideal target tissues for inserting desired traits of interest and improving existing cultivars via precision breeding (PB). PB is a new approach that, like conventional breeding, utilizes only DNA fragments obtained from sexually compatible grapevine plants. Embryogenic culture induction occurs by placing leaves or stamens and pistils on induction medium with a dark/light photoperiod cycle for 12–16 weeks. Resulting cultures produce sectors of embryogenic and non-embryogenic callus, which can be identified on the basis of callus morphology and color. Somatic embryo development occurs following transfer of embryogenic callus to development medium and cultures can be maintained for extended periods of time by transfer of the proliferating proembryonic masses to fresh medium at 4–6-week intervals. To demonstrate plant recovery via PB, somatic embryos at the mid-cotyledonary stage are cocultivated with Agrobacterium containing the desired gene of interest along with a, non-PB, enhanced green fluorescent protein/neomycin phosphotransferase II (egfp/nptII) fusion gene. Modified cultures are grown on proliferation and development medium to produce uniformly modified somatic embryos via secondary embryogenesis. Modified embryos identified on the basis of green fluorescence and kanamycin resistance are transferred to germination medium for plant development. The resulting plants are considered to prototype examples of the PB approach, since they contain egfp/nptII, a non-grapevine-derived fusion gene. Uniform green fluorescent protein (GFP) fluorescence can be observed in all tissues of regenerated plants.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Agrobacterium
- Culture medium
- Embryogenic cultures
- Growth regulators
- Plant tissue culture
- Precision breeding
- Vitis
1 Introduction
Grapevine is highly prized and grown worldwide for consumption as fresh fruit and processed products, including jam, jelly, juice, raisin, and, particularly, wine. Grape and its products contain a number of flavonoid and non-flavonoid phenols that act as antioxidants and impart health-beneficial properties. Resveratrol and proanthocyanidins present at high levels in wine possess anti-inflammatory activities and are responsible for cardioprotection [1]. A number of grapevine cultivars have been cultivated for centuries and are greatly valued for their specific fruit/enological characteristics. Only 35 elite, mostly ancient, grapevine cultivars account for 66 % of acreage worldwide, as consumers continually seek out the wines produced from them [2]. However, the elite cultivars, having been selected in antiquity with no directed genetic improvement possible since, are susceptible to a number of fungal, bacterial, and viral diseases; they require substantial chemical control in traditional production areas and cannot be grown at all in regions with extreme climatic conditions. Improving abiotic and biotic stress tolerance of these elite cultivars by conventional breeding is impossible because their key sensory attributes are invariably lost. For example, a prized strain of “Pinot Noir” cannot be improved by conventional breeding to create a disease resistant “Pinot Noir.” This is because grapevine, like many woody-perennial crops, are out-crossers, exhibiting self-incompatibility and inbreeding depression. New conventionally bred varieties, despite being resistant and even producing acceptable wine, never correspond to their elite counterparts, since existing enological characteristics are disrupted, thereby meeting consumer recalcitrance [3–5]. Inserting desired traits via precision breeding technology is a viable alternative for improving elite grapevine cultivars without altering their highly prized enological characteristics [6, 7].
Recent advances in grapevine genome sequencing have fostered discovery of desirable traits to instill into elite cultivars, while still maintaining their unique varietal characteristics. Grapevine improvement via precision breeding (PB) involves using DNA sequences found solely in the grapevine genome and it is a logical refinement of conventional breeding, only recently made possible by advances in cell culture, gene insertion, and computational technology [7–9]. Grapevine embryogenic cultures have long been the targets of choice for inserting genes encoding desired traits, since single cells on their surface can be prompted to develop into complete plants. Hence gene insertion into such totipotent cells results in plants that stably express the desired trait [10, 11]. An embryogenic response in a grapevine cultivar involves a complex interaction of the genotype with explant, culture medium and culture conditions [4, 12]. This necessitates protocol optimization for each grapevine cultivar grown in specific regions of the world. We have studied the embryogenic response of leaf and floral explants at various developmental stages, various media compositions, and culture conditions for a large number of cultivars over the last three decades [13, 14]. Embryogenic cultures are maintained on development medium for extended periods of time by careful selection and transfer of proliferating embryonic masses. Following somatic embryo development and germination , regenerated plants are hardened in a growth room and transferred to a greenhouse. We also continue to optimize our previous protocols for gene insertion by improving culture development, cocultivation procedures, reducing culture necrosis, and increasing plant recovery. Genetically modified plants have been recovered from a wide array of Vitis species, cultivars, and interspecific hybrids [15–19].
This chapter describes specific methods for the induction, maintenance, and genetic modification of grapevine embryogenic cultures to insert desired traits of interest in order to produce precision-bred versions of elite cultivars (Fig. 1). Cultures are initiated from leaves and/or floral explants on a wide array of culture media. Rapid proliferation of embryogenic cultures is obtained by growing them in liquid medium [20] and long term maintenance is achieved by culture on specialized X6 medium [12]. Somatic embryo s at the mid-cotyledonary stage of development are cocultivated with disarmed (non-disease causing) Agrobacterium harboring the desired genes of interest. For this demonstration, modified cultures are identified on the basis of non-precision-bred green fluorescence and kanamycin resistance. Plants obtained following germination of embryos are hardened in a growth room and transferred to a greenhouse. The genetically modified status of regenerated plants is confirmed by the uniform expression of the green fluorescence protein gene in various plant tissues.

Somatic embryo genic culture and genetic modification system for grapevine (reproduced from Ref. 26 with permission from Nature Publishing Group)
2 Materials
2.1 Supplies and Equipment
-
1.
Autoclave.
-
2.
Bead sterilizer.
-
3.
Stereomicroscope.
-
4.
Fiber-optic illuminator.
-
5.
Forceps.
-
6.
Scalpels.
-
7.
Clorox® bleach or equivalent.
-
8.
Sterile Whatman 3MM filter paper.
-
9.
Tween 20.
-
10.
Sterile distilled water.
-
11.
100 × 15 mm plastic Petri dishes.
-
12.
GA7 Magenta culture vessels.
-
13.
Laminar airflow sterile culture hood.
-
14.
Growth chamber.
-
15.
pH meter.
-
16.
Micropipettes and micropipette tips.
-
17.
Spray bottles.
-
18.
125 mL Erlenmeyer flasks.
-
19.
960 μM sieves.
-
20.
Rotary shaker.
-
21.
Leica MZFLIII stereomicroscope or equivalent equipped for epi-fluorescence with an HBO 100 W Mercury lamp illuminator and a green fluorescent protein (GFP ) filter set composed of an excitation filter (470/40 nm), a dichromatic beam splitter (485 nm), and a barrier filter (525/50 nm) (Leica Microscopy System Ltd., Heerbrugg, Switzerland).
2.2 Explant Sources
-
1.
One-year-old, dormant grapevine cuttings (see Note 1 ).
-
2.
Established micropropagation cultures.
2.3 Culture Medium Composition
-
1.
Embryogenic culture induction from leaf explants (NB2 medium): Nitsch and Nitsch [21] macro-, micronutrients and vitamins, 0.1 g/L myoinositol , 20 g/L sucrose , 1.0 μM benzyl amino purine (BAP), 5.0 μM 2,4-dichlorophenoxyacetic acid (2,4-D), 7.0 g/L Tissue culture (TC) grade Agar (Phytotechnology labs), pH 6.0 (see Note 2 ).
-
2.
Embryogenic culture induction from stamen and pistil explants (MSI medium): Murashige and Skoog [22], macro-, micro-nutrients and vitamins, 0.1 g/L myo-inositol , 20 g/L sucrose , 4.5 μM BAP, 5.0 μM 2,4-D, 7 g/L TC agar, pH 6.0 (see Note 3 ) [13].
-
3.
The following media may variously be used for embryogenic culture induction from anther and pistil explants:
-
(a)
PIV medium: Nitsch and Nitsch macro- and micronutrients, B5 vitamins, 60 g/L sucrose , 8.9 μM BAP, 4.5 μM 2,4-D, 3.0 g/L Phytagel , pH 5.7 [23].
-
(b)
X1 medium: Modified MS macro-, micronutrients and vitamins, which lack glycine and consisting of modified MS nitrate (X nitrate) consisting of 3.033 g/L KNO3 and 0.364 g/L NH4Cl, 0.1 g/L myoinositol , 20 g/L sucrose , 5.0 μM BAP, 2.5 μM 2,4-D and 2.5 μM beta-naphthoxyacetic acid (NOA ), 7 g/L TC agar, pH 5.8 [13].
-
(c)
X2 medium: Modified MS macro-, micronutrients and vitamins, which lacks glycine and MS nitrate being replaced with X nitrate consisting of 3.033 g/L KNO3 and 0.364 g/L NH4Cl, 0.1 g/L myoinositol , 20 g/L sucrose , 5.0 μM BAP, 15.0 μM 2,4-D and 2.5 μM NOA , 7 g/L TC agar, pH 5.8 [13].
-
(d)
NI medium: Nitsch and Nitsch macro-, micronutrients and vitamins, 0.1 g/L myoinositol , 20 g/L sucrose , 5.0 μM BAP, 2.5 μM 2,4-D and 2.5 μM NOA , 7 g/L TC agar, pH 5.8 [13].
-
(e)
NII medium: Nitsch and Nitsch macro-, micronutrients and vitamins, 0.1 g/L myoinositol , 20 g/L sucrose , 5.0 μM BAP, 15.0 μM 2,4-D and 2.5 μM NOA , 7 g/L TC agar, pH 5.8 [13].
-
(a)
-
4.
Embryogenic culture maintenance in liquid medium (B5/MS medium ): B5 macro-nutrients, MS micronutrients and vitamins, 0.4 g/L glutamine , 60 g/L sucrose , 4.5 μM 2,4-D, pH 5.8 [19].
-
5.
Embryo development and maintenance medium (X6 medium): Modified MS macro-, micronutrients and vitamins, which lacks glycine and MS nitrate being replaced with X nitrate consisting of 3.033 g/L KNO3 and 0.364 g/L NH4Cl, 60.0 g/L sucrose , 1.0 g/L myoinositol , 7.0 g/L TC agar , 0.5 g/L activated charcoal , pH 5.8 (see Note 4 ).
-
6.
Agrobacterium solid culture medium (YEP medium): 10 g/L yeast extract, 10 g/L peptone, 5.0 g/L NaCl, 20 g/L agar , pH 7.0.
-
7.
Agrobacterium liquid culture medium (MG/L medium): 5.0 g/L mannitol , 1.0 g/L l-Glutamate, 5.0 g/L tryptone , 2.5 g/L yeast extract, 5.0 g/L NaCl, 150.0 mg/L KH2PO4, 100.0 mg/L MgSO4·7H2O, 2.5 mL/L Fe–EDTA, pH 7.0 (see Note 5 ).
-
8.
Agrobacterium liquid transfer medium (X2 medium): X6 medium modified to contain 20.0 g/L sucrose without TC agar and activated charcoal , pH 5.8.
-
9.
Liquid cocultivation medium (DM medium): DKW basal salts [24], 0.3 g/L KNO3, 1.0 g/L myo-inositol , 2.0 mg/L each of thiamine –HCl and glycine , 1.0 mg/L nicotinic acid , 30 g/L sucrose , 5.0 μM BAP, 2.5 μM each NOA and 2,4-D, pH 5.7.
-
10.
Callus induction medium (DM medium): DKW basal salts, 0.3 g/L KNO3, 1.0 g/L myo-inositol , 2.0 mg/L each of thiamine –HCl and glycine , 1.0 mg/L nicotinic acid , 30 g/L sucrose , 5.0 μM BAP, 2.5 μM each NOA and 2,4-D, 7.0 g/L TC agar , 200 mg/L each of carbenicillin and cefotaxime , and 100 mg/L kanamycin, pH 5.7.
-
11.
Embryo germination medium (MS1B): MS macro-, micronutrients and vitamins, 0.1 g/L myoinositol , 30.0 g/L sucrose , 1.0 μM BAP, 7.0 g/L TC agar , pH 5.8.
2.4 Antibiotic Stock Solutions
-
1.
Rifampicin : Filter-sterilized stock solutions containing rifampicin at 20 mg/mL (see Note 6 ).
-
2.
Kanamycin sulfate: Filter-sterilized stock solutions containing kanamycin sulfate at 100 mg/mL.
-
3.
Carbenicillin and cefotaxime : Filter-sterilized stock solutions containing either carbenicillin or cefotaxime at 200 mg/mL.
2.5 Agrobacterium Culture
-
1.
Binary vector containing the gene of interest and an egfp/nptII fusion gene (reporter marker fusion) under the control of a constitutive promoter.
-
2.
Agrobacterium stock (containing the binary vector) stored in glycerol at −70 °C.
3 Methods
Carry out all surface sterilization, explant isolation, and transfer procedures using established aseptic techniques in a laminar airflow hood. Clorox®. Wrap all dishes with Parafilm®.
3.1 Embryogenic Culture Induction from Leaf Explants
-
1.
Initiate in vitro micropropagation cultures from field- or greenhouse-grown grapevine shoot tips (see Note 7 ).
-
2.
Excise unopened leaves, 1.5–5.0 mm in size, from in vitro-grown micropropagation cultures and transfer them to Petri dishes containing NB2 medium (see Note 8 ).
-
3.
Incubate cultures in darkness at 26 °C for 5–7 weeks for the induction of embryogenic callus.
-
4.
After 5–7 weeks, transfer cultures to light (65 μM m−2 s−1 and 16 h photoperiod) at 26 °C for 5 weeks. Screen callus cultures for growth and possible contamination at weekly intervals.
-
5.
Explants will produce callus cultures, which can be distinguished into cream-colored embryogenic callus and dark brown non-embryogenic callus.
-
6.
Carefully transfer the cream colored embryogenic callus to X6 medium for proliferation of proembryonic masses (PEM ) and development of somatic embryo s (SE).
3.2 Embryogenic Culture Induction from Stamen and Pistil Explants
-
1.
Obtain grapevine inflorescences from field-grown grapevines or one year old dormant cuttings.
-
2.
Surface-sterilize dormant cuttings in 25 % Clorox® solution with constant agitation for 5 min, followed by two washes with sterile distilled water.
-
3.
Make fresh cuts at the top and base of the cuttings and transfer 30 cm long cuttings to 500 mL conical flasks containing 250 mL sterile distilled water.
-
4.
Transfer flasks under light (65 μM m−2 s−1 and 16-h photoperiod) at 26 °C for 3–5 weeks for inflorescence growth and development.
-
5.
Determine development stages of stamens and pistils using a stereomicroscope to identify the optimum stage for the specific cultivar (see Note 9 ) (Fig. 2).
Fig. 2 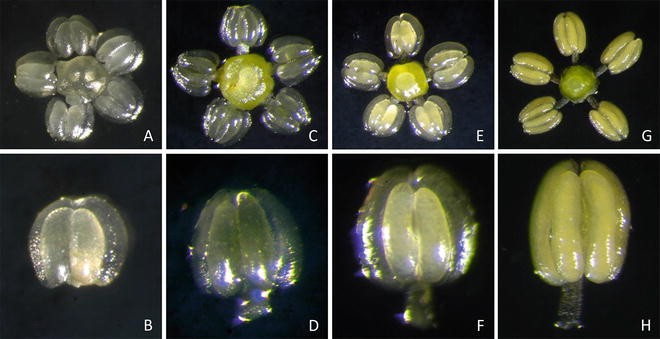
Stamen and pistil explant developmental stages in Vitis. Four stages, I (a, b), II (c, d), III (e, f), and IV (g, h) can be identified on the basis of inflorescence size, stamen color and size, and microspore development stage. Stage I flower clusters are about 2.5–3.0 cm long, individual flower buds 0.5–0.7 mm in diameter, anthers 0.1–0.2 mm in length, white in color and clear in appearance. Stage II flower clusters are about 6–8 cm long with individual flower buds being approximately 1.5 mm in diameter. Anthers are 0.8–1.0 mm long, yellowish in color, and appear translucent with clear walls. Stage III flower clusters are 9–10 cm long and individual flower buds 1.5–2.0 mm in diameter. Anthers are 1.0 mm in length, yellowish in color, and cloudy in appearance with clear walls. The locule appears cloudy and yellowish in color. Microspore walls are thicker and well developed. Stage IV flower clusters are greater than 10 cm in length and individual flower diameter similar to Stage III. Anthers are 1.0 mm long and yellowish in color with completely opaque walls. The locule appears yellow in color and opaque. Microspore walls are thicker and pores in the cell wall are evident (reproduced from Ref. 13 with permission from American Society of Horticultural Sciences)
-
6.
Surface-sterilize inflorescences by rinsing briefly in 70 % ethanol followed by washing them in 25 % Clorox® solution containing a small drop Triton X-100 for 5 min with a periodic manual high degree of agitation. Following washing with Clorox® solution, treat explants with three 5-min washes in sterile distilled water.
-
7.
Using a stereomicroscope, carefully excise intact stamens by separating them from the calyptra and pistil. Place stamens from five inflorescences as a clump in the center of the Petri dish containing induction medium and corresponding pistils with the filament stubs at the perimeter. Seal Petri dishes and place in the dark at 26 °C for 5 weeks (see Note 10 ).
-
8.
After 5 weeks of incubation in the dark, transfer Petri dishes to light (65 μM m−2 s−1 and 16-h photoperiod) at 26 °C. Screen developing cultures using a dissecting microscope for the presence of embryogenic callus at weekly intervals for 12–16 weeks (see Note 11 ).
-
9.
Induction of embryogenic callus is observed either from the filament tip, connective tissue or in some cases from pistil explants.
-
10.
Transfer the embryogenic callus to X6 medium for SE development and proliferation.

3.3 Embryogenic Culture Proliferation in Liquid Medium
-
1.
Transfer 1.0 g rapidly growing embryogenic culture to sterile 125 mL Erlenmeyer flasks containing 40 mL autoclaved liquid medium. Cover the flasks with aluminum foil and seal the neck with Parafilm. Transfer the flasks to a rotary shaker and incubate in diffused light (15 μM m−2 s−1 and 16-h photoperiod) at 120 rpm.
-
2.
After 2 weeks, separate differentiated somatic embryo s by filtering cultures through a sterile 960 μM stainless steel and collecting the fine fraction. Transfer the fine fraction to fresh liquid medium and differentiated SE to X6 medium for embryo development .
-
3.
Maintain suspension cultures by transfer to fresh liquid medium at 2–3-week intervals (see Note 12 ).
3.4 Embryogenic Culture Maintenance
-
1.
Transfer embryogenic cultures obtained from induction medium to X6 medium for development and proliferation of SE (Fig. 3a).
Fig. 3 
Embryogenic culture maintenance in Vitis. Actively proliferating proembryonic masses contained within embryogenic culture masses grown on X6 medium (a) are identified, sub-cultured, and manipulated using a stereomicroscope placed in a laminar flow culture hood and illuminated with a fiber optic light source (b, c). Only microscopic proembryonic tissue masses are selected (d) and these are accumulated so as to create five cultures in each Petri dish containing 30 mL (thick) of freshly made X6 medium (e) and the cycle is repeated at 4–6-week intervals. Proembryonic masses are uniformly composed of small, densely cytoplasmic embryogenic cells (f). It is important to keep a uniform subculture time and a stable incubation temperature to avoid precocious germination
-
2.
Maintain embryogenic cultures by precisely separating PEM from differentiated SE using a stereomicroscope as described below (Fig. 3b, c) and selectively transfer only PEM to fresh X6 medium at 4–6-week intervals (see Note 13 ) (Fig 3d, e).

3.5 Agrobacterium Culture Initiation for Plant Genetic Modification
-
1.
Streak Agrobacterium culture stock containing the binary plasmid onto a Petri dish containing solid YEP medium with 20 mg/L rifampicin and 100 mg/L kanamycin. Incubate dishes in the dark at 26 °C for 2–3 days until single bacterial colonies are visible.
-
2.
Transfer a single bacterial colony to a 125 mL conical flask containing 30 mL MG/L medium with 20 mg/L rifampicin and 100 mg/L kanamycin. Seal the flask with Parafilm and incubate on a rotary shaker at 180 rpm and room temperature for 16–20 h.
-
3.
Transfer the overnight culture to a 50 mL centrifuge tube and spin at 4200 × g for 8 min at room temperature. Discard the supernatant and resuspend the pellet in 20 mL liquid X2 medium. Transfer the culture to a 125 mL conical flask and incubate for an additional 3 h under the same conditions as above. Use this culture for cocultivation .
3.6 Gene Insertion into Embryogenic Cells
-
1.
Carefully transfer cotyledonary-stage SE to sterile Petri dishes. Avoid wounding of embryos during transfer to prevent culture browning (see Note 14 ).
-
2.
Add 5.0 mL Agrobacterium culture to the SE and mix thoroughly by swirling. Incubate for 7–10 min and then remove the bacterial solution completely using a micropipette.
-
3.
Transfer SE to a Petri dish containing two layers of filter paper soaked in liquid DM medium. Seal the Petri dish with Parafilm® and cocultivate in darkness at 26 °C for 72 h (see Note 15 ).
-
4.
Following cocultivation for 72 h, observe SE for transient GFP expression using a stereomicroscope equipped for epi-fluorescence.
-
5.
Transfer cocultivated cultures to a 125 mL conical flask containing liquid DM medium with 200 mg/L each of carbenicillin and cefotaxime , and 15 mg/L kanamycin.
-
6.
Transfer the flask to a rotary shaker at 110 rpm and wash SE for 3 days to inhibit remnant bacterial growth.
-
7.
Transfer washed cultures to each 100 × 15 mm Petri dish containing 25 mL solid DM medium with 200 mg/L each of carbenicillin and cefotaxime and 100 mg/L kanamycin.
-
8.
Place Petri dishes in dark at 26 °C for 4 weeks to permit callus development and proliferation.
-
9.
After 4 weeks, transfer callus cultures to 100 × 15 mm Petri dishes containing 30 mL X6 medium with 200 mg/L each of carbenicillin and cefotaxime and 70 mg/L kanamycin for secondary embryo development . Place Petri dishes in dark and screen at weekly intervals for the presence of modified SE lines.
-
10.
Independent SE lines are identified by bright GFP fluorescence and kanamycin resistance (see Note 16 ).
-
11.
Transfer independent genetically modified embryo lines to individual Petri dishes containing X6 medium with 200 mg/L each of carbenicillin and cefotaxime and 70 mg/L kanamycin.
-
12.
Screen cultures for the development of modified embryo development and proliferation.
3.7 Somatic Embryo Germination and Plant Regeneration
-
1.
Transfer cotyledonary-stage SE to MS1B medium and culture under light (65 μM m−2 s−1 and 16 h photoperiod) at 26 °C for embryo germination (see Note 17 ).
-
2.
After 3 weeks, transfer well-developed plants with a robust shoot and root system to plastic pots containing sterile Pro-Mix BX potting mix (Premier Horticulture Inc., Red Hill, PA) and acclimate in a growth room in light (65 μM m−2 s−1 and 16-h photoperiod) at 26 °C.
-
3.
After 4 weeks, transfer well acclimated, vigorously growing plants to a greenhouse.
-
4.
Confirm gene expression in regenerated plants by observing various plant tissues using a stereomicroscope equipped for epi-fluorescence (see Note 18 ).
4 Notes
-
1.
Dormant cuttings are obtained by pruning annual wood from grapevines during the winter season. Alternatively, certified cuttings can be obtained from grapevine germplasm repositories such as the University of California, Davis, or the USDA cold-hardy grapevine repository in Geneva, NY.
-
2.
An embryogenic response from unopened leaf explants on NB2 medium is observed from all seedless cultivars tested, whereas a majority of seeded cultivars will only respond using the stamen/pistil procedure. This factor must be considered prior to embryogenic culture initiation from leaf explants.
-
3.
Production of embryogenic responses from stamen and pistil explants varies widely with Vitis species and cultivar. Hence untested individual cultivars must be evaluated on each induction medium listed above to obtain an embryogenic response. A list of responsive varieties and their optimum induction media can be found in our reference publication [13].
-
4.
The use of TC agar (Phytotechnology Laboratories, LLC, Shawnee Mission, KS, USA, Catalog No. A 175) or an agar brand of equivalent purity, is paramount for successful induction and maintenance of embryogenic cultures. Use of other gelling agents in X6 medium results in a rapid decline in embryogenic potential and eventual culture death. A simple observation to judge agar purity is the relative translucence of poured dishes: the more translucent, the better.
-
5.
To make a stock solution of Fe–EDTA, dissolve 7.44 g of Na2EDTA·2H2O and 1.86 g FeSO4·7H2O in sterile distilled water and make final volume to 1 L. Although a number of bacterial media were used for Agrobacterium culture, MG/L medium provides better cell quality by avoiding overgrowth and assists in maintaining bacterial virulence.
-
6.
Rifampicin is dissolved in methanol or DMSO for making stock solutions. Carbenicillin , cefotaxime and kanamycin sulfate are dissolved in distilled water and then filter sterilized. All antibiotic solutions are stored at −20 °C and thawed just prior to use. Antibiotics are added to culture medium after autoclaving and cooling the medium to 55 °C. Rifampicin is light labile; preparation of stock solutions must be accomplished in very dim light with storage in the dark.
-
7.
Micropropagation cultures are initiated from the earliest sprouting shoots of previously dormant field-grown plants when they reach approximately 10 cm in length. Micro-dissected shoot apical meristems are used as explants. We previously determined that shoot apical meristems taken at this stage and from the field consistently yield the most sterile and vigorous micropropagation cultures. Cultures are initiated on C2D4B medium, with five meristems per dish [25] and are ultimately used for obtaining unopened leaf explants.
-
8.
It is critical to use only unopened leaves of specific size. Use of larger leaf explants will produce solely non-embryogenic cultures with no regeneration ability.
-
9.
Stamen and pistil explants can be divided into 4 developmental stages based on size of inflorescences and individual flowers, anther size and anther color. Stage II and III explants are known to produce an embryogenic cultures in a large percentage of cultivars tested [12].
-
10.
It is critical to carefully excise intact stamens (anther with attached filament) and place all stamens from five flowers in a clump/group to obtain an optimal embryogenic response. No embryogenic response will be obtained with damaged or detached filaments.
-
11.
Embryogenic response from stamen and pistil explants is genotype dependent. In general, a greater number of cultivars produce an embryogenic response from stamen explants [13].
-
12.
A difference in culture proliferation rates and persistence is observed among various cultivars in both solid and liquid medium. This factor must be considered to ensure transfer to fresh medium at the right interval and avoid culture browning.
-
13.
The use of a stereomicroscope in order to select proper tissue for transfer is an absolute requirement to accomplish this procedure (Fig. 3b) and cannot be stressed enough. It is important to selectively transfer rapidly proliferating PEM to fresh X6 medium using a microscope at 4–6-week interval (Fig. 3c, e). Failure to do so will lead to asynchrony of cultures, precocious SE germination , decrease in embryogenic competence and eventual termination of cultures.
-
14.
It is important to use rapidly growing embryogenic cultures for gene insertion. Use of older cultures can result in significantly lower-to-none insertion frequency and poor plant regeneration.
-
15.
Cocultivation of SE on filter paper dramatically improves gene insertion efficiency while preventing bacterial overgrowth and culture necrosis [19].
-
16.
Proliferation of grapevine embryogenic cultures occurs by direct secondary embryogenesis with new embryos arising from the surface cells of existing SE or pre-existing embryogenic calli (Fig. 3f). Thus, surface cells of cotyledonary-stage SE are ideal targets for gene insertion and plant regeneration.
-
17.
Plant recovery from germinated somatic embryo s can be enhanced by trimming enlarged, fleshy cotyledons. This response is species and cultivar dependent and needs to be tested for specific cultivars [16]. A newly published two-step culture procedure dramatically improves plant recovery [26]. This includes culturing embryos on C2D4B medium for a 3-week period followed by transferring the germinated embryos to MSN medium.
-
18.
Uniform GFP expression is observed in plant tissues including leaves, roots, flowers, stamens, and pistils (Fig. 4). Gene insertion efficiency varies widely with Vitis species and cultivars [16]. Among the various species and cultivars tested, “Thompson Seedless” (syn. “Sultania”) will initiate embryogenic cultures from leaves, stamens and pistils at very high efficiencies and produces the highest number of modified embryo and plant lines [16]. Cultures are readily initiated from both leaves and floral organs. Thus, it is an ideal model with which to learn the procedures.
Fig. 4 
GFP expression in a genetically modified grapevine . Uniform expression is observed in somatic embryo s (a), leaves and tendrils (b), roots (c), inflorescences (d), stamens, and pistils (e). Note that the central glowing spot in (e) represents the stigma (reproduced from Refs. 18 and 15 with permission from Springer)

References
Bertelli A, Das D (2009) Grapes, wines, resveratrol and heart health. J Cardiovasc Pharmacol 54:468–476
Anderson K, Aryal N (2014) Database of regional, national and global winegrape bearing areas by variety, 2000 and 2010. Wine Econ. Res. Ctr., Univ. Adelaide, Dec 2013. http://www.adelaide.edu.au/wine-econ/databases/winegrapes/
Gray DJ, Jayasankar S, Li Z (2005) Vitis spp. Grape (Chapter 22). In: Litz RE (ed) Biotechnology of fruit and nut crops. CAB International, Wallingford, Oxfordshire, pp 672–706
Einset J, Pratt C (1975) Grapes. In: Moore JN, Janick J (eds) Advances in fruit breeding. Purdue University Press, Lafayette, IN, pp 130–153
Winkler AJ, Cook JA, Kliewer WM, Lider LA (1974) General viticulture. University of California Press, Berkeley, pp 16–28
Vivier MA, Pretorius IS (2000) Genetic improvement of grapevine: tailoring grape varieties for the third millennium – a review. S Afr J Enol Vitic 21:5–26
Gray DJ, Li ZT, Dhekney SA (2014) Precision breeding of grapevine for improved traits. Plant Sci. doi:10.1016/j.plantsci.2014.03.023, Online First
Li ZT, Dhekney SA, Gray DJ (2011) Use of the VvMybA1 gene for non-destructive quantification of promoter activity via color histogram analysis in grapevine (Vitis vinifera) and tobacco. Transgenic Res 20:1087–1097
Li ZT, Kim KH, Jasinski JR, Creech MR, Gray DJ (2012) Large-scale characterization of promoters from grapevine (Vitis spp.) using quantitative anthocyanin and GUS assay. Plant Sci 196:132–142
Kikkert JR, Vidal MJ, Reisch BI (2004) Stable transformation of plant cells by particle bombardment/biolistics. In: Pena L (ed) Transgenic plants: methods and protocols, 286th edn. Humana, Totowa, NJ, pp 61–78
Gray DJ, Dhekney SA, Li ZT, Cordts JM (2012) Genetic engineering of grapevine and progress towards commercial deployment. In: Scorza R, Mou B (eds) Transgenic horticultural crops: challenges and opportunities – essays by experts. CRC, Taylor and Francis, Boca Raton, FL, pp 317–331
Gray DJ (1995) Somatic embryogenesis in grape. In: Jain SM, Gupta PK, Newton RJ (eds) Somatic embryogenesis in woody plants, vol 2. Kluwer Academic, Dordrecht, pp 191–217
Dhekney SA, Li ZT, Compton ME, Gray DJ (2009) Optimizing initiation and maintenance of Vitis embryogenic cultures. HortSci 44:1400–1406
Dhekney SA, Li ZT, Gray DJ (2011) Factors influencing induction and maintenance of Vitis rotundifolia Michx. embryogenic cultures. Plant Cell Tissue Organ Cult 105:175–180
Dhekney SA, Li ZT, Dutt M, Gray DJ (2008) Agrobacterium-mediated transformation of embryogenic cultures and regeneration of transgenic plants in Vitis routundifolia Michx. (muscadine grape). Plant Cell Rep 27:865–872
Dhekney SA, Li ZT, Zimmerman TW, Gray DJ (2009) Factors influencing genetic transformation and plant regeneration of Vitis. Am J Enol Vitic 60:285–292
Dhekney SA, Li ZT, Gray DJ (2011) Grapevines engineered to express cisgenic Vitis vinifera thaumatin-like protein exhibit fungal disease resistance. In Vitro Cell Dev Biol Plant 47:458–466
Li ZT, Dhekney S, Dutt M, Van Aman M, Tattersall J, Kelley KT, Gray DJ (2006) Optimizing Agrobacterium-mediated transformation of grapevine. In Vitro Cell Dev Biol Plant 42:220–227
Li ZT, Dhekney SA, Dutt M, Gray DJ (2008) An improved protocol for Agrobacterium-mediated transformation of grapevine. Plant Cell Tissue Organ Cult 93:311–321
Jayasankar S, Gray DJ, Litz RE (1999) High frequency somatic embryogenesis and plant regeneration from suspension cultures of grapevine. Plant Cell Rep 18:533–537
Nitsch JP, Nitsch C (1969) Haploid plants from pollen grains. Science 163:85–87
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Franks T, He DG, Thomas M (1998) Regeneration of transgenic Vitis vinifera L. Sultana plants: genotypic and phenotypic analysis. Mol Breed 4:321–333
Driver JA, Kuniyuki AH (1984) In vitro propagation of paradox walnut rootstock. HortSci 19:507–509
Gray DJ, Benton CM (1991) In vitro micropropagation and plant establishment of muscadine grape cultivars (Vitis rotundifolia). Plant Cell Tissue Organ Cult 27:7–14
Li ZT, Kim KH, Dhekney SA, Jasinski JR, Creech MR, Gray DJ (2014) An optimized procedure for plant recovery from somatic embryos significantly facilitates the genetic improvement of Vitis. Hort Res 1:1–7
Acknowledgements
S.A. Dhekney holds the E.A. Whitney Endowed Professorship in the UW Department of Plant Sciences. The research that enabled the Precision Breeding approach for grapevine was fostered by long-term support from the Florida Department of Agriculture and Consumer Services Viticulture Trust Fund, the USDA Specialty Crops Research Initiative Grant Program, and the Florida Agricultural Experiment Station, UF/IFAS.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Dhekney, S.A., Li, Z.T., Grant, T.N.L., Gray, D.J. (2016). Somatic Embryogenesis and Genetic Modification of Vitis . In: Germana, M., Lambardi, M. (eds) In Vitro Embryogenesis in Higher Plants. Methods in Molecular Biology, vol 1359. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-3061-6_11
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3061-6_11
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3060-9
Online ISBN: 978-1-4939-3061-6
eBook Packages: Springer Protocols