
Abstract
Phosphoproteomics relies on methods for efficient purification and sequencing of phosphopeptides from highly complex biological systems, especially when using low amounts of starting material. Current methods for phosphopeptide enrichment, e.g., Immobilized Metal ion Affinity Chromatography and titanium dioxide chromatography provide varying degrees of selectivity and specificity for phosphopeptide enrichment. The number of multi-phosphorylated peptides identified in most published studies is rather low. Here we describe a protocol for a strategy that separates mono-phosphorylated peptides from multiply phosphorylated peptides using Sequential elution from Immobilized Metal ion Affinity Chromatography. The method relies on the initial enrichment and separation of mono- and multi-phosphorylated peptides using Immobilized Metal ion Affinity Chromatography and a subsequent enrichment of the mono-phosphorylated peptides using titanium dioxide chromatography. The two separate phosphopeptide fractions are then subsequently analyzed by mass spectrometric methods optimized for mono-phosphorylated and multi-phosphorylated peptides, respectively, resulting in improved identification of especially multi-phosphorylated peptides from a minimum amount of starting material.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others


Key words
- Phosphopeptide enrich ment
- Multi-phosphorylated peptides
- Immobilized metal affinity chromatography
- Sequential elution
- Titanium dioxide chromatography
- Mass spectrometry
1 Introduction
Several techniques exist for phosphopeptide enrichment prior to mass spectrometric analysis. Today the most commonly used methods are Immobilized Metal Affinity Chromatography (IMAC ) [1–3] and titanium dioxide (TiO2 ) chromatography [4–7] (see Chapters 8 and 9). Recent studies comparing three different phosphopeptide enrichment methods including phosphoramidate chemistry (PAC) [8], IMAC and TiO2 chromatography showed that each method isolated distinct, partially overlapping segments of a phosphoproteome, whereas none of the tested methods was able to provide a whole phosphoproteome [9]. This is in itself not surprising as the three different methods apply completely different chemistries for phosphopeptide capture, numerous protocols for IMAC and TiO2 exist and the purification efficiency can be very variable for both IMAC and TiO2 depending on the person who is performing the analysis.
One of the challenges in large-scale phosphoproteomics is the analysis of multi-phosphorylated peptides. Multi-phosphorylated peptides are in general suppressed in the ionization process in the mass spectrometric (MS) analysis in the presence of mono- or non-phosphorylated peptides and therefore the chance to detect them by tandem MS (MS/MS ) analysis is limited. In addition, most mass spectrometers are only able to perform a limited number of MS/MS in a given time period resulting in the negligence of the less abundant multi-phosphorylated peptides. Furthermore, in collision induced dissociation (CID ) the major fragmentation pathway is the loss of phosphoric acid usually resulting in poor peptide backbone fragmentation. Consequently, little sequence information and lower identification rates are obtained. This is especially evident for multi-phosphorylated peptides which lose more phosphoric acid molecules. Several other kind of fragmentation methods exist which can increase the identification of multi-phosphorylated peptides. Optimized phosphorylation-directed multistage tandem MS (pdMS3) [10, 11], multistage activation (MSA) [12], higher energy collision dissociation (HCD ) [13] or Electron capture/transfer dissociation (ECD/ETD ) [14, 15] could provide better identification for multi-phosphorylated peptides. However, in order to set up the special experimental parameters optimal for analysis of multi-phosphorylated peptides, such as normalized collision energy, fragmentation time and number of ions used for fragmentation, the multi-phosphorylated peptides have to be separated from the mono-phosphorylated peptides prior to LC -MS/MS analysis.
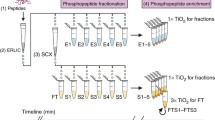
Previously, we developed a method for separation of mono-phosphorylated peptides from multiply phosphorylated peptides where we are using Sequential elution from IMAC (SIMAC ) [11]. In this strategy the peptide mixture is incubated with IMAC beads, which have a stronger selectivity for multi-phosphorylated peptides than for mono-phosphorylated peptides [16]. After incubation, the sample is split in three “elution” fractions (see Fig. 1); an IMAC flow-through fraction, an acidic (1 % TFA ) fraction and a basic (pH 11.3) fraction. The IMAC flow-through and acidic fractions which contain predominantly mono-phosphorylated and a significant number of non-phosphorylated peptides are further submitted to TiO2 chromatography to achieve pure phosphorylated fractions prior to tandem MS analysis. Alternatively, the two fractions can be pooled prior to TiO2 enrichment. The basic fraction is analyzed directly by MS/MS analysis without further TiO2 purification, as this sample in general is relative free of non-phosphorylated peptides.

The SIMAC strategy used for the enrichment and separation of mono- from multi-phosphorylated peptides. The peptide sample is mixed with the IMAC beads and incubated for 30 min in a Thermomixer at room temperature. After incubation, the beads are packed into a GELoader tip forming an IMAC micro-column. The IMAC flow-through is collected and further enriched using TiO2 chromatography. The mono-phosphorylated peptides are eluted from the IMAC micro-column using acidic elution conditions (1 % TFA , pH 1.0) and for complex samples this eluate is also further enriched using TiO2 chromatography or combined with the IMAC-FT prior to TiO2 enrichment. The multi-phosphorylated peptides are subsequently eluted from the IMAC micro-column using basic elution conditions (ammonia water, pH 11.3). The Figure is taken from [25]
SIMAC greatly improves the number of phosphorylation sites identified even from very low amounts of starting material and offers a way to identify and characterize multi-phosphorylated peptides at large-scale levels [11] (see also Chapter 11).
2 Materials
2.1 Model Proteins
-
1.
Transferrin (human) was a gift from ACE Biosciences A/S. Serum albumin (bovine), beta-lactoglobulin (bovine), carbonic anhydrase (bovine), beta-casein (bovine), alpha-casein (bovine), ovalbumin (chicken), ribonuclease B (bovine pancreas), alcohol dehydrogenase (Baker yeast), myoglobin (whale skeletal muscle), lysozyme (chicken), and alpha-amylase (bacillus species) were from Sigma (St. Louis. MO, USA).
2.2 Reduction, Alkylation, and Digestion of Proteins
-
1.
Triethylammonium bicarbonate .
-
2.
Dithiothreitol (DTT ).
-
3.
Iodoacetamide .
-
4.
Modified trypsin.
-
5.
Acetone .
2.3 Reduction, Alkylation, and Digestion of HeLa Proteins
-
1.
Lysis Buffer: 6 M urea, 2 M thiourea, 1× PhosSTOP phosphatase inhibitors.
-
2.
Dithiothreitol (DTT ).
-
3.
Iodoacetamide .
-
4.
Endoproteinase Lys-C.
-
5.
Triethylammonium bicarbonate .
-
6.
Modified trypsin.
-
7.
PhosStop.
2.4 Immobilized Metal ion Affinity Chromatography (IMAC )
-
1.
Iron-coated PHOS-select™ metal chelate beads (Sigma®), stored at −20 °C.
-
2.
IMAC Loading Buffer: 0.1 % trifluoroacetic acid (TFA ), Protein Sequencer Grade, 50 % acetonitrile, HPLC Grade.
-
3.
GELoader tips (Eppendorf (20 μL) or Bio-Rad (200 μL)).
-
4.
Low-binding microcentrifuge tubes 1.7 mL.
-
5.
1–5 mL disposable syringes fitted to GeLoader tip or p200 tips by using a pipette tip cut in both ends.
-
6.
IMAC Elution Buffer 1: 1 % TFA , 20 % acetonitrile.
-
7.
IMAC Elution Buffer 2: 1 % ammonia water (40 μL ammonia solution (25 %), 980 μL UHQ water (pH ~ 11)), make fresh as required.
-
8.
Formic acid.
- 9.
2.5 Titanium Dioxide (TiO2 ) Chromatography
-
1.
Titanium dioxide (TiO2 ) beads (Titansphere, 5 μm, GL sciences Inc.).
-
2.
Low-binding microcentrifuge tubes 1.7 mL.
-
3.
3 M Empore C8 disk (3 M, Bioanalytical Technologies, St. Paul, MN, USA).
-
4.
Acetonitrile , HPLC Grade.
-
5.
TiO2 Loading Buffer: 1 M glycolic acid in 5 % trifluoroacetic acid (TFA ), 80 % acetonitrile.
-
6.
TiO2 Washing Buffer 1: 1 % TFA , 80 % acetonitrile.
-
7.
TiO2 Washing Buffer 2: 0.1 % TFA , 10 % acetonitrile.
-
8.
TiO2 Elution Buffer: 1 % ammonia water (40 μL ammonia solution (25 %) in 960 μL UHQ water).
-
9.
Formic acid.
2.6 Reversed Phase (RP) Micro-columns
-
1.
POROS Oligo R3 reversed phase material (PerSeptive Biosystems, Framingham, MA, USA).
-
2.
GELoader tips (Eppendorf, Hamburg, Germany) or p200 pipette tips depending on the size of the column needed.
-
3.
3 M Empore C18 disk (3 M, Bioanalytical Technologies, St. Paul, MN, USA).
-
4.
1–5 mL disposable syringes fitted to GeLoader tip or p200 tips by using a pipette tip cut in both ends.
-
5.
RP Washing Buffer: 0.1 % TFA .
-
6.
RP Elution Buffer (for LC -ESI MS/MS analysis): 70 % acetonitrile, 0.1 % TFA .
-
7.
2,5-dihydroxybenzoic acid (DHB ) Elution Buffer (for MALDI MS analysis): 20 mg/mL DHB in 50 % acetonitrile, 1 % ortho-phosphoric acid.
2.7 Other Materials
-
1.
Tabletop centrifuge.
-
2.
pH meter.
-
3.
Thermomixer.
-
4.
Shaker.
-
5.
Vacuum centrifuge.
2.8 Analysis by Mass Spectrometry
-
1.
Mass spectrometer capable of performing MS/MS —preferentially a high-resolution/high mass accuracy instrument (Q-TOFs (Waters, ABSciex, Bruker, and Agilent) or Orbitrap based mass spectrometer (Thermo Fisher Scientific)) interfaced to a nanoHPLC (e.g., Dionex 3000 ultimate LC system (Thermo Fisher Scientific)) with a 50–100 μm i.d. RP capillary column setup for highly sensitive online peptide separation can be used. For simpler samples a MALDI MS instrument can be used (e.g., Bruker Ultraflex (Bruker Daltonics, Bremen, Germany)).
-
2.
Software for processing of raw mass spectrometry data files and generation of peak lists for searching against a protein database (e.g., Uniprot ) Analysis software such as Mascot/Mascot Distiller (Matrix Science, London, UK) (data from most vendors and instruments), Proteome Discover er (Thermo Scientific, Bremen, Germany) (data from Thermo instruments), MaxQuant [15] (high resolution data from Thermo Orbitrap instruments and certain Bruker and ABSciex Q-TOFs) and the TransProteomicPipeline [16] (vendor independent).
3 Methods
The principle of the SIMAC method is illustrated in this chapter firstly using a peptide mixture originating from tryptic digestions of 12 standard proteins (Model proteins) (see Notes 3 and 4 ). The protocol is then applied to enrich for phosphorylated peptides from whole cell lysates from 150 μg of proteins from HeLa cells.
The SIMAC purification method is a simple and very straightforward method. It is fast and efficient for enrichment of phosphopeptides from even highly complex samples [17, 18]. The experimental setup of the method is illustrated in Fig. 1.
3.1 Digestion of Model Proteins and the HeLa Cell Lysate
-
1.
Dissolve each protein in 50 mM triethylammonium bicarbonate (TEAB ), pH 7.8, 10 mM DTT and incubate at 37 °C at 1 h. After reduction, add 20 mM iodoacetamide and incubate the samples at room temperature for 1 h in the dark.
-
2.
Digest each protein using trypsin (1–2 % w/w) at 37 °C for 12 h.
-
3.
Lyse HeLa cells in 6 M Urea , 2 M ThioUrea containing phosphatase inhibitors (PhosStop). Precipitate proteins using 10 volume excess of ice-cold acetone and incubate over night at −20 °C. Centrifuge the sample at 14,000 × g and wash the pellet twice with ice-cold acetone. Redissolve the pellet in 50 μL 6 M urea, 2 M thiourea, 10 mM DTT containing 1 μg endoproteinase Lys-C and incubate at room temperature for 2 h. After incubation, dilute the sample 10× with 50 mM TEAB , pH 7.8 containing 20 mM iodoacetamide and incubate for 1 h in the dark at room temperature. After incubation, add trypsin (1–2 % w/w) and place the sample at room temperature overnight.
3.2 Batch Mode Sequential Enrichment and Separation with IMAC Beads
Always adjust the amount of IMAC beads to the amount of sample in order to reduce the level of nonspecific binding from non-phosphorylated peptides. For 1 pmol tryptic digest use 7 μL IMAC beads (see Chapter 8). For more complex samples where more material is available, more IMAC beads should be used. This section is describing a protocol for using 150 μg tryptic digest from HeLa cells.
-
1.
Transfer 50 μL IMAC beads to a fresh low-binding microcentrifuge tube 1.7 mL.
-
2.
Wash the IMAC beads twice using 200 μL IMAC Loading Buffer (see Note 5 ).
-
3.
Resuspend the beads in 200 μL IMAC Loading Buffer and add the sample (see Note 6 ).
-
4.
Incubate the sample with IMAC beads in a Thermomixer for 30 min at room temperature.
-
5.
Generate an IMAC micro-column essentially as described in Chapter 10.
-
6.
Squeeze the tip of a 200 μL GELoader tip to prevent the IMAC beads from leaking.
-
7.
After incubation, pack the beads in the constricted end of the GELoader tip by application of air pressure forming an IMAC micro-column [19].
-
8.
It is critical to collect the IMAC flow-through (FT) in a new 1.7 mL low-binding microcentrifuge tube for further enrichment by TiO2 chromatography (see Subheading 3.3).
-
9.
Wash the IMAC column using 70 μL IMAC Loading Buffer.
-
10.
Elute the mono-phosphorylated peptides bound to the IMAC beads using 80 μL of IMAC Elution Buffer 1. Collect the eluate into the IMAC-FT tube. The IMAC-FT and 1 % TFA elution fractions can be analyzed separately.
-
11.
Pool the eluate with the IMAC -FT to obtain the SIMAC-mono fraction and lyophilize it prior to TiO2 enrichment (see Subheading 3.3).
-
12.
Elute the multi-phosphorylated peptides bound to the IMAC micro-column using 80 μL of IMAC Elution Buffer 2 directly into a p200 pipette tip containing a Poros Oligo R3 microcolumn (approximately 1 cm long).
-
13.
Acidify with 100 % formic acid, typically 1 μL per 10 μL eluate (pH should be ~2–3), and 5 μL 100 % TFA , and desalt/concentrate the eluted multi-phosphorylated peptides on the Poros Oligo R3 micro-column (see Subheading 3.4).
-
14.
Elute the peptides from the column using 60 μL RP Elution Buffer into a fresh 1.7 mL low binding microcentrifuge tube.
-
15.
Lyophilize the sample prior to LC -MS/MS .
3.3 TiO2 Batch Mode Purification of the “Mono”-Phosphorylated Peptides
-
1.
Add acetonitrile, TFA , and glycolic acid to the SIMAC -mono peptide fraction to obtain TiO2 Loading Buffer conditions (80 % acetonitrile, 5 % TFA, and 1 M glycolic acid) (see Note 7 ) or dilute the sample at least 10× with the TiO2 Loading Buffer.
-
2.
Add 0.6 mg TiO2 beads per 100 μg peptide solution (see Note 8 ).
-
3.
Place the tubes on a shaker (highest shaking) at room temperature for 5–10 min.
-
4.
After incubation, centrifuge to pellet the beads (table centrifuge <15 s).
-
5.
Transfer the supernatant to another low-binding tube and incubate it with another round of TiO2 beads using half of the amount of TiO2 beads as used in the first incubation. This can be repeated to recover larger amounts of phosphopeptides.
-
6.
Pool the TiO2 beads from the incubations using 100 μL Loading Buffer and transfer the solution to a new low-binding microcentrifuge tube (see Note 9 ).
-
7.
Vortex the solution for 10 s and then centrifuge in a table centrifuge to pellet the beads. Remove the supernatant.
-
8.
Wash the beads with 70–100 μL (see Note 10 ) Washing Buffer 1, mix for 10 s and then centrifuge to pellet the beads.
-
9.
Wash the beads with 70–100 μL Washing Buffer 2, mix for 10 s and then centrifuge to pellet the beads. This step is important to remove peptides that bind to TiO2 in a HILIC mode (see Note 11 ).
-
10.
Dry the beads for 5–10 min in the vacuum centrifuge or on the table.
-
11.
Elute the phosphopeptides with 100–200 μL Elution Buffer—mix well and leave the solution on a shaker for 10 min to allow an efficient elution.
-
12.
Centrifuge the solution for 1 min and pass the supernatant over a small stage tip filter [20] (C8 stage tip) into a new low-binding tube to recover the liquid without any TiO2 beads.
-
13.
Wash the beads with 30 μL Elution Buffer and pool the wash (eluate) with the eluate from the previous step.
-
14.
Elute potential bound peptides from the C8 filter with 5 μL 30 % acetonitrile and pool with the eluate from steps 12 to 13.
-
15.
Lyophilize the eluted peptides or acidify the eluate with 1 μL formic acid per 10 μL eluate for direct cleanup of the phosphopeptides using RP material prior to downstream analyses as described for the multi-phosphorylated peptides above (see Subheading 3.4) (e.g., HILIC fractionation [17]).
3.4 Poros Oligo R3 Reversed Phase (RP) Micro-column Desalting/Concentration of the Sample
Use GELoader tip micro-columns of ~6–10 mm or p200 pipette tips micro-columns (1–2 cm) depending on the amount of material to be purified. Here, it is illustrated for the p200 pipette tip (150 μg peptides from HeLa cell lysate).
-
1.
Suspend Poros Oligo R3 reversed phase (RP) material in 200 μL 100 % acetonitrile.
-
2.
Prepare a p200 pipette tip micro-column by stamping out a small plug of C18 material from a 3 M Empore™ C18 extraction disk and place it in the constricted end of the tip.
-
3.
Pack Poros Oligo R3 RP beads on top of the p200 stage tip until the size of the column is 1–2 cm.
-
4.
Load the acidified phosphopeptide sample slowly onto the RP micro-column (~1 drop/s).
-
5.
Wash the RP micro-column using 60 μL RP Washing Buffer.
-
6.
Elute the phosphopeptides from the RP micro-column using 40–60 μL RP Elution Buffer, followed by lyophilization of the phosphopeptides. (N.B. For MALDI MS analysis the peptides can be eluted off the GeLoader tip RP micro-column directly onto the MALDI target using 1 μL DHB solution. After crystallization the sample is ready for MALDI MS analysis).
-
7.
Redissolve the lyophilized phosphopeptides in 0.5 μL 100 % formic acid and dilute immediately to 10 μL with UHQ water. The sample is then ready for LC -ESI-MSn analysis.
3.5 μHPLC Tandem Mass Spectrometry (LC -MS/MS ) Analysis
For LC -MS/MS analysis of purified phosphopeptides a standard strategy as described below can be used. A typical nanoLC setup would include a 0.075 mm × 200 mm analytical column packed with 3 μm RP resin interfaced with a high resolution/mass accuracy mass spectrometer as described in our original paper [17]. The number of phosphopeptides identified in the analysis can be increased by maximizing the resolution of the nanoLC separation via longer columns (e.g., 50 cm) and smaller chromatographic particle sizes (e.g., 1.9 μm). Alternatively, a two column system can be utilized using a 0.1 mm × 20 mm pre-column packed with RP resin (3–5 μm) combined with an analytical column as described above. A two column system is described below.
-
1.
The phosphopeptides are redissolved in 0.1 % TFA and loaded onto a pre-column as described above using a μHPLC system (e.g., Dionex or EASY-LC ) at a loading speed of 5 μL/min.
-
2.
The phosphopeptides are eluted directly onto the analytical column (e.g., 0.075 mm × 200 mm) using a gradient (60–120 min) from 0 to 35 % B-Buffer (e.g., A-Buffer: 0.1 % formic acid; B-Buffer: 90 % acetonitrile, 0.1 % TFA ) at an elution speed of 2–300 nL/min.
-
3.
The phosphopeptides are eluted directly into a tandem mass spectrometer and analyzed by Data Dependent Analysis.
LC -ESI-MS/MS analysis of multi-phosphorylated peptides is improved by redissolving the phosphopeptides by sonication in an EDTA containing buffer prior to LC-ESI-MS/MS analysis [21].
An example of the results obtained by the SIMAC method using a relatively low complexity sample consisting of tryptic peptides derived from 12 standard proteins is shown in Fig. 2. The Figure shows the MALDI MS results obtained on a Bruker Ultraflex from a direct analysis of 1 pmol of the tryptic digest (Fig. 2a), the MALDI MS peptide mass map from the purification of the IMAC flow-through from 1 pmol peptide mixture using TiO2 chromatography (Fig. 2b), the MALDI MS peptide mass map of the mono-phosphorylated peptides eluted from the IMAC material using 1 % TFA (Fig. 2c) and the MALDI MS peptide mass map obtained from the basic elution from the IMAC material (Fig. 2d). The phosphopeptides are illustrated by #P (see Note 12 ).

Results obtained from 1 pmol peptide mixture using the SIMAC strategy. (a) MALDI MS peptide mass map of the direct analysis of the tryptic peptides. (b) MALDI MS peptide mass map of peptides identified from the IMAC flow-through after further enrichment using TiO2 chromatography. (c) MALDI MS peptide mass map of peptides eluted from the IMAC micro-column using 1 % TFA . (d) MALDI MS peptide mass map of peptides eluted from the IMAC microcolumn using ammonia water (pH 11.30). The number of phosphate groups on the individual phosphopeptides is indicated by “#P”. Asterisk indicates the metastable loss of phosphoric acid
An example of the results obtained using the present SIMAC protocol for enrichment of phosphopeptides from a total of 150 μg peptides derived by tryptic digestion from a HeLa cell lysate is shown in Fig. 3. The enriched phosphopeptides were separated on a Dionex 3000 ultimate LC system using a homemade RP capillary column (25 cm) directly into a Q-Exactive Plus ESI-MS/MS instrument. The peptides were separated using a 90 min gradient from 0 to 25 % B Buffer (90 % acetonitrile in 0.1 % formic acid). The MS instrument was set to isolate and fragment 12 parent ions per MS cycle (MS and MS/MS resolution was set to 70,000 and 35,000 at 200 m/z, respectively; MS and MS/MS AGC target was 1E6 and 5E4, respectively; normalized collision energy was 30; isolation window was 1.5 Da). Here a total of 3370 unique phosphopeptides were identified from the 150 μg of starting material, using the Proteome Discover er 1.4.1.14 (SwissProt_2014_04 (20340 entries)) with an enrichment percentage of about 88 % phosphopeptides using TiO2 only (see Fig. 3a). When SIMAC was applied to the same sample a total of 5337 unique phosphopeptides (enrichment percentage 89 %) could be identified, whereof 3804 and 2499 were identified in the SIMAC mono and multi fractions, respectively. Of these, only 966 unique phosphopeptides were shared between the two fractions (see Fig. 3b) indicating a good separation. When looking at the number of phosphate groups on the unique phosphopeptides identified in each fraction a clear enrichment of multi-phosphorylated peptides could be seen when using the SIMAC procedure, as the SIMAC multi fraction contained 56.5 % phosphopeptides (see Fig. 3d) with 2 or more phosphate groups compared to only 14 % in the mono fraction (Fig. 3c) and 22 % in the TiO2 enrichment (see Chapter 9). In total the SIMAC procedure resulted in the identification of 31 % multi-phosphorylated peptides.

Results obtained from the enrichment of phosphorylated peptides from acetone precipitated proteins from HeLa cells using TiO2 chromatography or SIMAC . (a) Overview of the number of unique phosphopeptides identified in the TiO2 and SIMAC experiments. (b) Venn diagram showing the overlap between the SIMAC mono and multi fractions. (c) Percentage distribution of the number of phosphate groups on the phosphopeptides identified in the SIMAC mono fraction. (d) Percentage distribution of the number of phosphate groups on the phosphopeptides identified in the SIMAC multi fraction
4 Notes
-
1.
It is important to obtain the highest purity of all chemicals used.
-
2.
All solutions should be prepared in UHQ water.
-
3.
Always start by testing the method using a model peptide mixture. It is important to freshly prepare the peptide mixture as peptides bind to the surface of the plastic tubes in which they are stored. In addition, avoid transferring the peptide sample to different tubes to minimize adsorptive losses of the sample.
-
4.
The peptide mixture used for the experiment illustrated in this chapter contained peptides originating from tryptic digestions of 1 pmol of each of the 12 proteins. Experiments have shown that the presented method is sensitive down to the low femtomole level [11].
-
5.
The PhosSelect IMAC beads are very fragile so high speed mixing should be avoided in any steps.
-
6.
The sample should be diluted in IMAC Loading Buffer or for larger volume add 100 % TFA and 100 % acetonitrile to make the sample up to the IMAC Loading Buffer. The total volume should not exceed 300 μL.
-
7.
If you have 100 μL peptide sample, you can add 50 μL water, 50 μL 100 % TFA , 800 μL acetonitrile, and 76 mg glycolic acid to make the sample up to the proper TiO2 Loading Buffer.
-
8.
The optimal amount of TiO2 beads to add to the sample in order to reduce non-specific binding and optimize phosphopeptide yield is 0.6 mg TiO2 per 100 μg of peptide starting material (see [17] for further information). This will of course change depending on the source of biological material used as TiO2 selectively enriches other biomolecules (reviewed in [22]) such as sialylated glycopeptides [23] and acidic lipids [24] commonly found in membrane fractions.
-
9.
The transfer to a new tube is performed due to the fact that peptides stick to plastic and can be eluted from the plastic surface in the last elution step resulting in contamination with non-modified peptides.
-
10.
For larger scale analysis, where more TiO2 beads are used, larger volumes of the buffers should be used.
-
11.
TiO2 is an efficient HILIC material and hydrophilic peptides can bind to the material when loaded in high organic solvent. The inclusion of 5 % TFA and 1 M glycolic acid should prevent most hydrophilic non-modified peptides from binding, however, some can still be found in the eluates from TiO2. Therefore in order to eliminate any binding from non-modified hydrophilic peptides this last Washing Buffer is important. For membrane preparations the last washing supernatant will contain neutral glycopeptides which can then be analyzed further.
-
12.
The results obtained using this protocol will differ according to the mass spectrometer used for the analysis of the phosphopeptides, not only between MALDI MS and ESI MS but also within different MALDI MS instruments, depending on laser optics, laser frequency, instrumental Configuration, sensitivity, etc.
References
Li S, Dass C (1999) Iron(III)-immobilized metal ion affinity chromatography and mass spectrometry for the purification and characterization of synthetic phosphopeptides. Anal Biochem 270:9–14
Neville DC, Rozanas CR, Price EM, Gruis DB, Verkman AS, Townsend RR (1997) Evidence for phosphorylation of serine 753 in CFTR using a novel metal-ion affinity resin and matrix-assisted laser desorption mass spectrometry. Protein Sci 6:2436–2445
Posewitz MC, Tempst P (1999) Immobilized gallium(III) affinity chromatography of phosphopeptides. Anal Chem 71:2883–2892
Kuroda I, Shintani Y, Motokawa M, Abe S, Furuno M (2004) Phosphopeptide-selective column-switching RP-HPLC with a titania precolumn. Anal Sci 20:1313–1319
Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ (2005) Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol Cell Proteomics 4:873–886
Pinkse MW, Uitto PM, Hilhorst MJ, Ooms B, Heck AJ (2004) Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2D-NanoLC-ESI-MS/MS and titanium oxide precolumns. Anal Chem 76:3935–3943
Sano A, Nakamura H (2004) Chemo-affinity of titania for the column-switching HPLC analysis of phosphopeptides. Anal Sci 20:565
Zhou HL, Watts JD, Aebersold RA (2001) A systematic approach to the analysis of protein phosphorylation. Nat Biotechnol 19:375–378
Bodenmiller B, Mueller LN, Mueller M, Domon B, Aebersold R (2007) Reproducible isolation of distinct, overlapping segments of the phosphoproteome. Nat Methods 4:231–237
Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP (2004) Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci U S A 101:12130–12135
Thingholm TE, Jensen ON, Robinson PJ, Larsen MR (2008) SIMAC - a phosphoproteomic strategy for the rapid separation of mono-phosphorylated from multiply phosphorylated peptides. Mol Cell Proteom 7(4):661–671
Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ (2004) A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal Chem 76:3590–3598
Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M (2007) Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods 4:709–712
Chalmers MJ, Hakansson K, Johnson R, Smith R, Shen J, Emmett MR, Marshall AG (2004) Protein kinase A phosphorylation characterized by tandem Fourier transform ion cyclotron resonance mass spectrometry. Proteomics 4:970–981
Schroeder MJ, Webb DJ, Shabanowitz J, Horwitz AF, Hunt DF (2005) Methods for the detection of paxillin post-translational modifications and interacting proteins by mass spectrometry. J Proteome Res 4:1832–1841
Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol 20:301–305
Engholm-Keller K, Birck P, Storling J, Pociot F, Mandrup-Poulsen T, Larsen MR (2012) TiSH--a robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. J Proteomics 75:5749–5761
Engholm-Keller K, Hansen TA, Palmisano G, Larsen MR (2011) Multidimensional strategy for sensitive phosphoproteomics incorporating protein prefractionation combined with SIMAC, HILIC, and TiO(2) chromatography applied to proximal EGF signaling. J Proteome Res 10:5383–5397
Gobom J, Nordhoff E, Mirgorodskaya E, Ekman R, Roepstorff P (1999) Sample purification and preparation technique based on nano-scale reversed-phase columns for the sensitive analysis of complex peptide mixtures by matrix-assisted laser desorption/ionization mass spectrometry. J Mass Spectrom 34:105–116
Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75:663–670
Liu S, Zhang C, Campbell JL, Zhang H, Yeung KK, Han VK, Lajoie GA (2005) Formation of phosphopeptide-metal ion complexes in liquid chromatography/electrospray mass spectrometry and their influence on phosphopeptide detection. Rapid Commun Mass Spectrom 19:2747–2756
Engholm-Keller K, Larsen MR (2011) Titanium dioxide as chemo-affinity chromatographic sorbent of biomolecular compounds--applications in acidic modification-specific proteomics. J Proteomics 75:317–328
Jensen SS, Larsen MR (2007) Evaluation of the impact of some experimental procedures on different phosphopeptide enrichment techniques. Rapid Commun Mass Spectrom 21:3635–3645
Calvano CD, Jensen ON, Zambonin CG (2009) Selective extraction of phospholipids from dairy products by micro-solid phase extraction based on titanium dioxide microcolumns followed by MALDI-TOF-MS analysis. Anal Bioanal Chem 394:1453–1461
Thingholm TE, Jensen ON, Larsen MR (2009) Enrichment and separation of mono- and multiply phosphorylated peptides using sequential elution from IMAC prior to mass spectrometric analysis. Methods Mol Biol 527:67–78, xi
Acknowledgements
This work was supported by the Danish Natural Science and Medical Research Councils (grant no. 10-082195 (T.E.T)) and the Lundbeck Foundation (M.R.L—Junior Group Leader Fellowship).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Thingholm, T.E., Larsen, M.R. (2016). Sequential Elution from IMAC (SIMAC): An Efficient Method for Enrichment and Separation of Mono- and Multi-phosphorylated Peptides. In: von Stechow, L. (eds) Phospho-Proteomics. Methods in Molecular Biology, vol 1355. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-3049-4_10
Download citation
DOI: https://doi.org/10.1007/978-1-4939-3049-4_10
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-3048-7
Online ISBN: 978-1-4939-3049-4
eBook Packages: Springer Protocols