
Abstract
Duchenne muscular dystrophy (DMD) (the most common form of muscular dystrophy) is caused by a lack of dystrophin protein. Currently, although many therapeutic strategies are under investigation, there is no cure for DMD and unfortunately, patients succumb to respiratory and/or cardiac failure in their second or third decade of life. Preclinical work has focused on the mouse model C57BL/10ScSn-Dmdmdx/J (BL10/mdx ), which does not exhibit a robust pathophenotype. More recently, the D2.B10-Dmdmdx/J (D2/mdx ) mouse has been utilized, which presents a more severe pathology and therefore more closely mimics the human pathophenotype, particularly in the heart. Here, we outline important considerations when utilizing the D2/mdx model by highlighting the differences between these models in addition to describing histological and immunohistochemical methods utilized in Kennedy et al. (Mol Ther Methods Clin Dev 11:92–105, 2018) for both cardiac and skeletal muscle, which can quantify these differences. These considerations are particularly important when investigating treatment strategies that may be affected by regeneration; such is the case for upregulation of the dystrophin paralogue, utrophin.
Access this chapter
Tax calculation will be finalised at checkout
Purchases are for personal use only
Similar content being viewed by others
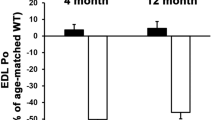
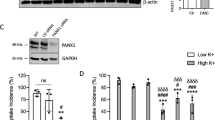
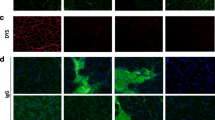
Change history
02 March 2023
The corresponding author of the chapter informed that he realised the reference to Kennedy (2018) paper was not included in Figure 1 legend, which needs to be added for potential copyright reasons.
References
Kennedy TL et al (2018) Micro-utrophin improves cardiac and skeletal muscle function of severely affected D2/mdx mice. Mol Ther Methods Clin Dev 11:92–105
Mendell JR et al (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 71(3):304–313
Mah JK et al (2014) A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 24(6):482–491
Forcina L et al (2017) Insights into the pathogenic secondary symptoms caused by the primary loss of dystrophin. J Funct Morphol Kinesiol 2(4):44
Head S, Williams D, Stephenson G (1994) Increased susceptibility of EDL muscles from mdx mice to damage induced by contraction with stretch. J Muscle Res Cell Motil 15(4):490–492
Bach JR et al (1987) Management of end stage respiratory failure in Duchenne muscular dystrophy. Muscle Nerve 10(2):177–182
Rideau Y et al (1983) Prolongation of life in Duchenne’s muscular dystrophy. Acta Neurol 5(2):118–124
Birnkrant DJ, Ararat E, Mhanna MJ (2016) Cardiac phenotype determines survival in Duchenne muscular dystrophy. Pediatr Pulmonol 51(1):70–76
Love DR et al (1989) An autosomal transcript in skeletal muscle with homology to dystrophin. Nature 339(6219):55–58
Lin S, Burgunder J-M (2000) Utrophin may be a precursor of dystrophin during skeletal muscle development. Dev Brain Res 119(2):289–295
Tinsley J et al (1998) Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 4(12):1441–1444
Krag TO et al (2004) Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc Natl Acad Sci 101(38):13856–13860
Tinsley JM et al (2011) Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 6(5):e19189. https://doi.org/10.1371/journal.pone.0019189. PMID: 21573153; PMCID: PMC3089598
Coley WD et al (2016) Effect of genetic background on the dystrophic phenotype in mdx mice. Hum Mol Genet 25(1):130–145
Vohra R et al (2017) Magnetic resonance monitoring of disease progression in mdx mice on different genetic backgrounds. Am J Pathol 187(9):2060–2070
Finsterer J, Stöllberger C (2008) Cardiac involvement in Becker muscular dystrophy. Can J Cardiol 24(10):786–792
van Putten M et al (2019) Natural disease history of the D2-mdx mouse model for Duchenne muscular dystrophy. FASEB J 33(7):8110–8124
Fukada S-I et al (2010) Genetic background affects properties of satellite cells and mdx phenotypes. Am J Pathol 176(5):2414–2424
Hammers DW et al (2020) The D2. mdx mouse as a preclinical model of the skeletal muscle pathology associated with Duchenne muscular dystrophy. Sci Rep 10(1):1–12
Rodrigues M et al (2016) Impaired regenerative capacity and lower revertant fibre expansion in dystrophin-deficient mdx muscles on DBA/2 background. Sci Rep 6(1):1–9
Luz MA, Marques MJ, Santo Neto H (2002) Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz J Med Biol Res 35(6):691–695
Clerk A et al (1993) Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J 25(8):554–561
Lin S, Gaschen F, Burgunder J-M (1998) Utrophin is a regeneration-associated protein transiently present at the sarcolemma of regenerating skeletal muscle fibers in dystrophin-deficient hypertrophic feline muscular dystrophy. J Neuropathol Exp Neurol 57(8):780–790
Weir AP, Burton EA, Harrod G, Davies KE (2002) A- and B-utrophin have different expression patterns and are differentially up-regulated in mdx muscle*. J Biol Chem 277:45285–45290
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Science+Business Media, LLC, part of Springer Nature
About this protocol

Cite this protocol
Kennedy, T.L., Dugdale, H.F. (2023). Cardiac and Skeletal Muscle Pathology in the D2/mdx Mouse Model and Caveats Associated with the Quantification of Utrophin. In: Maruyama, R., Yokota, T. (eds) Muscular Dystrophy Therapeutics. Methods in Molecular Biology, vol 2587. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-2772-3_4
Download citation
DOI: https://doi.org/10.1007/978-1-0716-2772-3_4
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-2771-6
Online ISBN: 978-1-0716-2772-3
eBook Packages: Springer Protocols