
Abstract
Eosinophil apoptosis (programmed cell death) plays an important role in several inflammatory and allergic conditions. Apoptosis triggers various mechanisms including activation of cysteine-aspartic proteases (caspases) and is characterized by morphological and biochemical changes. These include cellular condensation, nuclear fragmentation, increased mitochondrial permeability with loss of membrane potential, and exposure of phosphatidylserine on the cell membrane. A greater understanding of apoptotic mechanisms, subsequent phagocytosis (efferocytosis), and regulation of these processes is critical to understanding disease pathogenesis and development of potential novel therapeutic agents. Release of soluble factors and alterations to surface marker expression by eosinophils undergoing apoptosis aid them in signaling their presence to the immediate environment, and their subsequent recognition by phagocytic cells such as macrophages. Uptake of apoptotic cells usually suppresses inflammation by restricting proinflammatory responses and promoting anti-inflammatory and tissue repair responses. This, in turn, promotes resolution of inflammation. Defects in the apoptotic or efferocytosis mechanisms perpetuate inflammation, resulting in chronic inflammation and enhanced disease severity. This can be due to increased eosinophil life span or cell necrosis characterized by loss of cell membrane integrity and release of toxic intracellular mediators. In this chapter, we detail some of the key assays that are used to assess eosinophil apoptosis, as well as the intracellular signaling pathways involved and phagocytic clearance of these cells.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others

Key words
1 Introduction
Increased eosinophil accumulation at sites of inflammation is evident in a number of allergic diseases including asthma, eczema, and rhinitis [1]. Recruited as part of a process including Th2 lymphocyte infiltration and immunoglobulin E (IgE)-mediated mast cell activation, eosinophils are central to disease pathogenesis [2]. The subsequent release of reactive oxygen species, proteases, and inflammatory mediators including cytokines can result in increased inflammation, tissue damage, and organ dysfunction. In order to attenuate this inflammatory process eosinophil apoptosis and subsequent non-phlogistic clearance of apoptotic cells are important for ensuring efficient resolution of the inflammatory process. Dysregulation of apoptosis or efferocytosis mechanisms result in sustained inflammation and may contribute to tissue injury and chronic inflammation correlated with various inflammatory diseases [3,4,5,6].
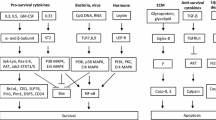
The circulating life span of eosinophils is short with an intravascular presence of around 18–25 h prior to migration into tissues, with the thymus and gastrointestinal tract the usual destination of eosinophils in health [7]. Here their life span is thought to be several days. Eosinophils isolated from peripheral blood undergo constitutive apoptosis, although at a much slower rate than the closely related neutrophil granulocyte . The eosinophil half-life in vitro is approximately 48 h following isolation [8,9,10]. The process of eosinophil apoptosis is dependent upon the activation of cysteine-aspartic proteases (caspases) which are contained as inactive zymogens within the cell with eosinophils containing caspases 3, 6, 7, 8, and 9. Caspase cleavage occurs due to activation of either the extrinsic or the intrinsic pathways of apoptosis [3]. The extrinsic pathway relies on the ligation of “death receptors” such as the tumor necrosis factor receptor (TNF-R), the Fas receptor (FasR), and the TNF-related apoptosis-inducing ligand receptor (TRAIL-R) [11]. Cross-linking of these receptors causes clustering and, through associations with internal adaptor proteins, allows the formation of pro-caspase complexes, caspase-8 cleavage, and subsequent apoptosis. The intrinsic pathway is activated when the cell faces withdrawal of survival factors, genotoxic stress, and exposure to ultraviolet radiation or chemotherapeutic agents allowing pro-apoptotic members of the B-cell lymphoma 2 (Bcl-2) family to dissociate from their anti-apoptotic regulators and translocate to the mitochondria. Resultant increased mitochondrial membrane permeability and pore formation lead to pro-apoptotic factors such as cytochrome c to be released into the cytosol with cleavage of pro-caspase-9 to active caspase-9, thereby committing the cell to caspase-3-mediated apoptosis. The process of apoptosis results in morphological and biochemical changes including cell shrinkage, nuclear condensation, and apoptotic body formation; increased mitochondrial permeability with loss of membrane potential; DNA fragmentation; and caspase activation and externalization of phosphatidylserine on the plasma membrane.
Eosinophil life span can be modulated through alterations in the balance of pro-survival and pro-apoptotic signals and proteins. There is evidence suggesting that Bc1-xL (an anti-apoptotic Bcl-2 family member) plays a significant role in eosinophil survival, independent of Mcl-1, a protein which is crucial in the regulation of neutrophil life span. This may, in part, explain why eosinophils undergo constitutive apoptosis at a slower rate compared to neutrophils [12]. Apoptosis can be delayed by a variety of factors including cytokines (interleukin-5 (IL-5), GM-CSF , eotaxin, and interferon-γ), hypoxia, and bacterial exotoxins [13] and DNA which also serve to induce eosinophil migration and activation into areas of inflammation.
Conversely apoptosis can be accelerated by IL-4 , FAS ligand, ligation of CD69, CD45 and CD30 cell surface receptors, and intracellular oxidant production with pharmacological agents including corticosteroids and theophyllines also driving apoptosis [14]. Glucocorticoids (a class of corticosteroids) increase eosinophil apoptosis via the intrinsic pathway, with the mechanism involving changes in the phosphorylation state of Bcl-2 family members as well as inhibition of cytokine-dependent survival [5, 15]. Additionally, the cyclin-dependent kinase inhibitor (CDKi) R-roscovitine induces eosinophil apoptosis by mitochondrial membrane potential loss and downregulation of the key survival protein Mcl-1 [8, 16]. The CDKi-driven apoptosis of eosinophils in vitro is a time-, concentration-, and caspase-dependent effect [8]. Similarly, flavones (polyphenolic plant-derived compounds) such as wogonin also induce eosinophil apoptosis in a time-, concentration-, and caspase-dependent manner due to augmented loss of eosinophil mitochondrial membrane potential [5].
Numerous changes in cell surface marker expression and secretion of soluble factors that occurs during apoptosis facilitate the uptake of these dying cells by surrounding phagocytes including macrophages, dendritic cells, and nonprofessional phagocytes such as epithelial cells [17, 18]. Important alterations to the cell membrane include phosphatidylserine exposure, changes in ICAM-1 epitopes, modification in glycosylation patterns, and charge and expression of calreticulin. Phagocytosis is a key event in macrophage phenotypic switching from a pro-inflammatory to a pro-resolving phenotype with release of anti-inflammatory cytokines and lipids (including IL-10, transforming growth factor-β (TGF-β), and resolvins) [19]. Failure in clearance of apoptotic cells results in eventual disintegration of the cell membrane (termed secondary necrosis) with release of toxic intracellular contents, tissue damage, and perpetuation of the inflammatory response.
Eosinophils form approximately 1–3% of the granulocyte population in the peripheral blood of nonatopic humans; therefore enrichment following conventional granulocyte isolation methods is essential [20, 21]. Negative selection (anti-CD16) is most widely used although some care should be taken as eosinophils also have low levels of CD16 expression. Eosinophil life span can be influenced by several factors, therefore ensuring that eosinophil activation is prevented during isolation and culture is vital for in vitro study. Factors which may alter constitutive eosinophil life span and affect interpretation of results include the method of isolation, serum presence, temperature, pH level, oxygen tension, and cell density [3].
As described, the molecular and morphological changes that occur during the process of apoptosis allow in vitro study through a variety of different assays and approaches ranging from assessment of externalization of phosphatidylserine by flow cytometry and nuclear condensation by light microscopy to assessment of DNA fragmentation by hypodiploid peak analysis and mitochondrial membrane permeability with chromogenic dyes. This chapter therefore updates our earlier chapter on the methodologies used to examine components of the apoptotic process and subsequent phagocytic clearance of eosinophils.
2 Materials
2.1 Culture of Human Eosinophils In Vitro
-
1.
Iscove’s modified Dulbecco’s medium (IMDM ).
-
2.
Penicillin/streptomycin 100×.
-
3.
10% Autologous serum (see Note 1).
-
4.
96-Well flat-bottomed plate.
2.2 Cytocentrifuge Preparations of Eosinophils for Light Microscopy
-
1.
Cytocentrifuge chambers, filter cards, glass slides, and coverslips.
-
2.
Methanol, Diff-Quik™ stains.
-
3.
DPX mounting medium.
2.3 Preparation for Electron Microscopy
-
1.
3% Glutaraldehyde, 25% stock solution diluted in 0.1 M sodium cacodylate buffer (pH 7.2).
-
2.
1% Osmium tetroxide in 0.1 M sodium cacodylate.
-
3.
100, 90, 70, and 50% normal-grade acetones and analar acetone.
-
4.
Araldite resin.
-
5.
Reichert OmU4 ultramicrotome.
2.4 Annexin V/Propidium Iodide Staining
-
1.
96-Well flat-bottomed plate.
-
2.
Fluorophore-conjugated annexin V, e.g., fluorescein isothiocyanate (FITC).
-
3.
Annexin-binding buffer (Hanks’ balanced salt solution (HBSS) with 2.5 mM Ca2+). Store at 4 °C.
-
4.
Stock solution of 1 mg/mL propidium iodide in sterile ddH2O.
2.5 Alterations in Mitochondrial Permeability
-
1.
MitoCapture™ Mitochondrial Apoptosis Detection Fluorometric Kit (Biovision, Milpitas, CA 95035 USA) contains MitoCapture™ reagent (store at −20 °C), incubation buffer (store at 4 °C).
2.6 Western Blotting for Intracellular Proteins That Regulate Apoptosis
-
1.
Tris-buffered saline (TBS 10×): NaCl (87.66 g), Tris base (24.22 g), distilled water (ddH2O, 800 mL), pH adjusted to 7.4 with HCl and then made to 1 L with ddH20, dilute 1:10 with ddH20 prior to use for 1×TBS.
-
2.
Protease inhibitor buffer (see Note 2): 780 μL 1×TBS added to 20 μL protease inhibitor cocktail, supplemented with 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF; 20 μL–400 mM stock in H2O), aprotinin (20 μL–0.15 μM stock in H2O), leupeptin (20 μL–20 mM stock in H2O), pepstatin A (40 μL–0.75 μM stock in methanol), sodium vanadate (20 μL–1 M stock in H2O, pH 10, boiled), benzamidine (20 μL–0.5 M stock in H2O), levamisole (20 μL–2 M stock in H2O), β-glycerophosphate (60 μL of 3.33 M stock in H2O).
-
3.
10% Nonidet P-40 (NP-40) detergent in 1× TBS.
-
4.
BCA protein assay.
-
5.
Sample buffer (for 4×): 50% Glycerol (4 mL), 20% SDS (4 mL), Tris–HCl (2.5 mL 1 M, pH 6.8), 1% (w/v in ethanol) bromophenol blue (20 μL), β-mercaptoethanol (400 μL—add in fume hood).
-
6.
Benchmark™ pre-stained molecular weight standards (Invitrogen).
-
7.
12% SDS gel.
-
8.
Running buffer (10×): Tris base (121 g), SDS (10 g), Hepes (238 g), ddH20 (800 mL), once dissolved make up 1 L (ddH2O); diluted 1:10 (ddH20) for 1× solution prior to use.
-
9.
Transfer buffer (10×): Tris base (30.3 g), glycine (144.12 g), ddH20 (800 mL), once dissolved make up 1 L (ddH2O).
-
10.
Transfer buffer (1×): 10× transfer buffer (100 mL), methanol (200 mL), ddH2O (700 mL).
-
11.
Polyvinylidene difluoride (PVDF) membrane.
-
12.
Blocking buffer: 1×TBS: 0.1% Tween®20 (polysorbat 20), 5% dried milk powder.
-
13.
Primary antibodies: Mcl-1 (1:500; Santa-Cruz, Biotechnology, CA, USA), GAPDH (1,10,000; Sigma), cleaved caspase-3 (11,000, Cell Signaling, Danvers, MA, USA), cleaved caspase-9 (11,000; Cell Signaling).
-
14.
Secondary antibodies: corresponding horseradish peroxidase-conjugated antibodies (1:2500, Dako, Cambridgeshire, UK).
-
15.
ECL prime, light-sensitive film, X-ray developer.
2.7 Fluorimetric Caspase Kit
-
1.
Homogeneous Caspases Assay Kit (Roche Diagnostics Ltd., Lewes, UK): 1× Incubation buffer, stock caspase substrate solution (500 μM DEVD-R110 in DMSO), positive control (e.g., lysate from apoptotic camptothecin-treated U937 cells), and R110 standard for calibration curve construction (1 mM in DMSO).
2.8 Caspase Profiling Plate
ApoAlert™ Caspase Profiling Plate (Clontech, Saint-Germain-en-Laye, France) contains 96-well microplate with immobilized substrates for caspase-2 (VDVAD-AMC), caspase-3 (DEVD-AMC), caspase-8 (IETD-AMC), and caspase-9 (LEHD-AMC) in 24 wells each, lysis buffer, 2× reaction buffer, 100× DTT solution, and inhibitors of caspases 2, 3, 8, and 9.
2.9 Gel Electrophoresis for DNA Fragmentation
-
1.
Wizard® genomic DNA purification kit.
-
2.
5× TBE running buffer: Tris base (54 g), boric acid (27.5 g) EDTA (20 mL −0.5 M pH 8.0). ddH2O (800 mL), pH adjusted to 8.3, then made to 1 L with ddH20, 0.5×TBE.
-
3.
SeaKem LE agarose for DNA electrophoresis.
-
4.
GelRedTM Nucleic Acid Gel Stain.
-
5.
6× Blue/orange loading dye.
2.10 Propidium Iodide Staining for Hyplodiploid Nuclei
-
1.
96-Well flat-bottom plate.
-
2.
PI solution: Propidium iodide (250 μL–10 mg/mL in ddH2O), sodium citrate (2.2 mL–2.2 g in 10 mL ddH2O), Triton X-100 (50 μL), ddH2O (made up to 50 mL total volume); store solution at 4 °C in the dark.
2.11 TUNEL Staining
-
1.
96-Well flat-bottom flexible plate.
-
2.
In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics Ltd.) contains 10× enzyme solution (TdT) in storage buffer and 1× labeled nucleotide mixture in reaction buffer. This protocol also requires PBS (wash buffer), 3% H2O2 in methanol (blocking solution), 4% paraformaldehyde in PBS at pH 7.4 (fixation buffer; freshly prepared), and 0.1% Triton-X100 in 0.1% sodium citrate (permeabilization buffer; freshly prepared).
2.12 Flow Cytometry-Based Phagocytosis Assay
-
1.
5-Chloromethylfluorescein diacetate (CMFDA; CellTracker™ Green, Invitrogen).
-
2.
pHrodo™ Red succinimidyl ester (Invitrogen).
-
3.
0.25% Trypsin/1 mM ethylenediaminetetraacetic acid solution.
3 Methods
3.1 Assessing Morphological Changes of Apoptosis Using Light Microscopy
Apoptotic eosinophils are distinguishable from viable cells, under light microscopy, by their characteristic morphological appearances of nuclear condensation and cell shrinkage (Fig. 1a, c).
-
1.
Suspend eosinophils (of at least 97% purity—determined by cytocentrifuge preparation as described below) at 4 × 106 cells/mL in IMDM supplemented with 10% autologous serum and penicillin/streptomycin (1×) (see Note 1).
-
2.
In a 96-well flat-bottomed plate add 75 μL of eosinophil suspension. Add 60 μL IMDM with 10% autologous serum to each well and 15 μL of apoptosis-modifying agents (10× concentration) or vehicle control. (NB: If two agents are used only 45 μL of IMDM is required for a total volume of 150 μL.)
-
3.
Cover the plate with a lid and incubate at 37 °C in a 5% CO2 incubator for the required amount of time.
-
4.
Gently pipette the cell suspension in the well to resuspend adherent cells and load 200 μL (150 μL cells and 50 μL IMDM) into a cytocentrifuge chamber.
-
5.
Cytocentrifuge at 300 rpm for 3 min.
-
6.
Air-dry for 5 min.
-
7.
Fix in methanol for 2 min.
-
8.
Stain in Diff Quik™ solution 1 or equivalent acid dye for 2 min.
-
9.
Stain in Diff Quik™ solution 2 or equivalent basic dye for 2 min.
-
10.
Rinse with distilled water.
-
11.
Air-dry slides before mounting with a drop of DPX and coverslip. View slides using a light microscope with a 40× or 100× (oil) objective and count >300 cells per slide (see Note 3).

Determining eosinophil apoptosis by light microscopy and flow cytometry. Viable eosinophils isolated from peripheral venous human blood have eosinophilic cytoplasmic staining, abundant granules, and bilobed nuclei (white arrow) (a). Flow cytometric analysis of cell death immediately following isolation demonstrates that the majority of cells are viable (AnnV−ve/PI−ve) (b). Apoptotic eosinophils in vitro display characteristic morphological changes of cell shrinkage, membrane blebbing, nuclear condensation, darkening of cytoplasmic staining, and nuclear condensation (black arrows) (c). Similarly increased apoptotic (AnnV+ve/PI−ve) and necrotic (AnnV+ve/PI+ve) staining is seen with flow cytometry (d). All images 1000× magnification
3.2 Analysis of Eosinophil Morphology by Electron Microscopy
Although conventional light microscopy provides information regarding the general morphological changes that occur during apoptosis it is electron microscopy that allows detailed structural analysis of these processes.
-
1.
Suspend eosinophils (at least 97% purity) at 4 × 106 cells/mL in IMDM supplemented with 10% autologous serum and penicillin/streptomycin (1×) (see Note 1).
-
2.
In a 96-well flat-bottomed plate add 75 μL of cell suspension, 15 μL of apoptosis-modifying agents (10× concentration) or vehicle control, and 60 μL IMDM with 10% autologous serum to each well. (NB: If two agents are used only 45 μL of IMDM is required for a total volume of 150 μL.)
-
3.
Cover and incubate at 37 °C in a 5% CO2 incubator for the duration of the experiment.
-
4.
Gently pipette the cell suspension in the well to resuspend adherent cells, combine the contents of 5 replicate wells together into a 500 μL Eppendorf tube, and centrifuge for 5 min at 300 × g.
-
5.
Resuspend in 3% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.3, for 2 h.
-
6.
Centrifuge at 300 × g for 5 min and resuspend in 0.1 M cacodylate. Incubate for 10 min (repeat three times).
-
7.
Postfix in 1% osmium tetroxide in 0.1 M sodium cacodylate for 45 min.
-
8.
Centrifuge for 5 min at 300 × g and resuspend in 0.1 M cacodylate. Incubate for 10 min (repeat three times).
-
9.
Dehydrate sequentially in 50%, 70%, 90%, and 100% normal-grade acetones (10 min each) and then for 10 min in analar acetone (repeat twice).
-
10.
Embed in araldite resin.
-
11.
Cut 1 μm sections on Reichert OmU4 ultramicrotome, and stain with toluidine blue.
-
12.
Select appropriate areas for further study using a light microscope.
-
13.
From those areas cut ultrathin (60 nm) sections and stain with uranyl acetate and lead citrate.
-
14.
View section with a Philips CM120 transmission electron microscope.
3.3 Annexin V/Propidium Iodide Staining and Flow Cytometric Analysis of Apoptosis
Externalization of phosphatidylserine to the outer surface of the cell membrane is a key component in the apoptotic process, allowing the recognition of apoptotic cells by surrounding phagocytes. Annexin V (AnnV), in the presence of Ca2+, binds phosphatidylserine and when fluorescently conjugated (commonly AnnV-FITC) it can be used to identify apoptotic cells. Discrimination between viable, apoptotic, and necrotic cells is possible by using AnnV together with a nucleophilic dye such as propidium iodide (PI) in a simple flow cytometry assay. PI is excluded from cells with an intact cell membrane; however when membrane integrity is lost PI enters the cell and binds nuclear material with a consequent increase in fluorescence. Numerous other viability dyes exist that can be used in place of PI including (but not limited to) DAPI , 7AAD, and SYTOX™ dyes.
-
1.
Suspend eosinophils (of at least 97% purity) at 4 × 106 cells/mL in IMDM supplemented with 10% autologous serum and penicillin/streptomycin (1×) (see Note 1).
-
2.
In a 96-well flat-bottomed plate add 75 μL of cell suspension, 15 μL of apoptosis-modifying agents (10× concentration) or vehicle control, and 60 μL IMDM with 10% autologous serum to each well. (NB: If two agents are used only 45 μL of IMDM is required for a total volume of 150 μL.).
-
3.
Cover and incubate at 37 °C in a 5% CO2 incubator for the duration of the experiment.
-
4.
Gently pipette the cell suspension in the well to resuspend adherent cells and pipette 50 μL of the cell suspension into a flow tube with 250 μL AnnV buffer (see Note 4).
-
5.
Incubate on ice for 5 min.
-
6.
Add PI (1 μL of 1 mg/mL solution) to each sample immediately prior to running the sample on a flow cytometer.
-
7.
Analyze on a flow cytometer using FL-1/FL-2 channel analysis. Viable cells are dual AnnV/PI negative; apoptotic cells are AnnV positive, and PI negative; necrotic cells are dual AnnV/PI positive (Fig. 1 b, d).
3.4 Measuring Mitochondrial Membrane Potential Using MitoCapture™
The formation of pores within the mitochondrial membrane that occurs during the intrinsic apoptotic process results in loss of mitochondrial membrane potential (ΔΨM) and facilitates the movement of proteins into the cytoplasm, in particular cytochrome c, with resultant caspase activation. Changes in the mitochondrial membrane potential of eosinophils can be measured using MitoCapture™, a cationic dye which in viable cells accumulates and polymerizes within mitochondria and fluoresces in the red (FL-2) channel—indicated by a fluorescence emission shift from green (535 nm) to red (590 nm). During apoptosis, when ΔΨM is compromised, the dye remains monomeric within the cytoplasm and fluoresces in the green (FL-1) channel. This mitochondrial depolarization can be quantified by flow cytometry as an increase in FL-1 fluorescence (Fig. 2a), or by plate-based fluorometric assay as a decrease in the red/green fluorescence intensity ratio.
-
1.
For each sample dilute 0.5 μL MitoCapture™ reagent in 500 μL pre-warmed (37 °C) MitoCapture™ incubation buffer in a 1.5 mL Eppendorf tube. (NB: This protocol relies on the use of a MitoCapture™ mitochondria permeability detection kit).
-
2.
Suspend eosinophils (at least 97% purity) at 4 × 106 cells/mL in IMDM (10% autologous serum).
-
3.
In a 96-well flat-bottomed plate add 75 μL of cell suspension, 15 μL of apoptosis-modifying agents (10× concentration) or vehicle control, and 60 μL IMDM with 10% autologous serum to each well. (NB: If two agents are used only 45 μL of IMDM is required for a total volume of 150 μL).
-
4.
Cover and incubate at 37 °C in a 5% CO2 incubator for the duration of the experiment.
-
5.
Add 150 μL of cell suspension to 500 μL diluted MitoCapture™ reagent.
-
6.
Incubate on shaking heat block at 37 °C, 300 rpm, for 15 min.
-
7.
Centrifuge at 300 × g for 5 min and discard supernatant.
-
8.
Resuspend cells in 300 μL MitoCapture™ incubation buffer.
-
9.
Analyze using a flow cytometer with increased fluorescence in FL-1 channel indicating loss of ΔΨM and increased apoptosis.

Assessing intracellular events during apoptosis and phagocytic clearance of apoptotic cells. Loss of mitochondrial membrane potential due to increased membrane permeability during apoptosis is measured by increased fluorescence of Mitotracker™ dye. Representative histogram (a) of control (blue)- and dexamethasone (red)-treated eosinophils after 20-h in vitro culture—apoptotic cells indicated by gate. Changes in the expression of intracellular regulators of apoptosis can be assessed by western blot (b). Caspase inhibitor zVAD delays apoptosis, maintains Mcl-1 expression, and prevents caspase-3 cleavage while pro-apoptotic cyclin-dependent kinase inhibitors (CDKi) cause Mcl-1 downregulation and caspase-3 cleavage. Measurement of phagocytosis of apoptotic cells by monocyte-derived macrophages (gated) is demonstrated by increased CellTracker™-green fluorescence from stained apoptotic cells in dexamethasone-treated cells (d) relative to control (c) (black arrows). Overlay histogram demonstrates population of macrophages containing apoptotic cells (e, gated)
3.5 Western Blotting for Caspases and Apoptotic Proteins
Caspases are essential throughout the apoptotic process in both the initiation and execution of the cell death process. The detection of the cleaved active forms of these proteins, or the disappearance of their inactive forms, alongside changes in the expression of other pro- and anti-apoptotic proteins in response to extrinsic and intrinsic modulators of eosinophil life span is possible through a variety of assays described below (Fig. 2b).
-
1.
Suspend eosinophils (at least 97% purity) at 4 × 106 cells/mL in IMDM (10% autologous serum). Pipette 750 μL cells into a 2 mL Eppendorf tube and incubate with 150 μL apoptosis-modifying agents and 600 μL IMDM (10% autologous serum). (NB: If two agents are used only 450 μL of IMDM is required for a total volume of 1500 μL.)
-
2.
Incubate at 37 °C in a shaking heat block for the duration of the experiment.
-
3.
Centrifuge the Eppendorf tube at 13,000 rpm for 1 min. Discard the supernatants.
-
4.
Resuspend cell pellets in 90 μL protease inhibitor buffer and incubate on ice for 10 min (see Note 2).
-
5.
Add 10 μL 10% NP-40 (diluted in TBS), vortex thoroughly, and incubate for a further 10 min on ice.
-
6.
Centrifuge at 16,000 × g for 20 min at 4 °C, and transfer the protein-rich supernatant into a 500 μL Eppendorf tube. Freeze samples (−20 °C) until use.
-
7.
Calculate protein concentration of each sample using BCA protein assay as per the manufacturer’s instructions.
-
8.
Transfer volume equivalent to 30 μg protein into fresh Eppendorf tubes and make up to total volume of 30 μL with PBS (without cations) and 8 μL of 4× sample buffer.
-
9.
Heat at 95 °C for 5 min.
-
10.
Load samples onto a 12% polyacrylamide (or equivalent) gel including molecular weight standards. Run at 110 V until the dye front reaches the bottom of the gel.
-
11.
Transfer proteins onto the PVDF membrane at 80 V for 1 h at 4 °C.
-
12.
Wash the membrane in TBS/0.1% Tween®20 for 5 min on a rocking platform.
-
13.
Block the membrane for 1 h with 10 mL of 5% dried milk powder in TBS/0.1% Tween®20 at room temperature on a rocking platform.
-
14.
Wash the membrane in TBS/0.1% Tween®20 for 5 min (repeat three times).
-
15.
Incubate with primary antibody overnight at 4 °C—concentrations as per 2.4.1. in TBS/0.1% Tween®20 containing 5% dried milk powder (5 mL).
-
16.
Wash membrane in TBS/0.1% Tween®20 each for 5 min (repeat three times).
-
17.
Incubate with the corresponding secondary antibody diluted 1:2500 in TBS/0.1% Tween®20 containing 5% dried milk powder (5 mL) for 2 h.
-
18.
Wash membrane in TBS/0.1% Tween®20 for 5 min (repeat three times).
-
19.
Develop using enhanced chemiluminescence according to the manufacturer’s instructions.
-
20.
Strip and re-probe blot with β-actin or GAPDH as a loading control.
3.6 Fluorometric Homogeneous Caspase Assay
As discussed previously apoptosis is a caspase-dependent process; therefore assessment of caspase activity can be used as a marker of apoptotic cell death. Quantification of total caspase activity is possible using commercially available assays (homogeneous caspase assay) but their use is limited given the inability to discriminate between individual caspases. Cleavage of a fluorescently conjugated caspase substrate (e.g., VAD-fmk) to produce a fluorescent product (e.g., FITC, rhodamine 110) enables fluorescent intensity to be measured as a marker of total caspase activity.
-
1.
These instructions are based on the use of Homogeneous Caspases Assay Kit.
-
2.
Suspend eosinophils (at least 97% purity) at 1 × 106 cells/mL in IMDM (10% autologous serum).
-
3.
In a black 96-well microplate load 100 μL of cell suspension (1 × 105 cells per well), with apoptosis-modifying agents and incubate at 37 °C for the duration of the experiment.
-
4.
Dilute stock caspase substrate 1:10 in incubation buffer and add 100 μL freshly prepared 1× caspase substrate to each well. Include negative and positive controls (media alone and cell lysate).
-
5.
Incubate at 37 °C in the dark for at least 1 h.
-
6.
Use a plate reader to measure fluorescence (excitation: 470–500 nm, emission: 500–560 nm).
3.7 Caspase Profiling Assay
Fluorometric assays for specific caspases are similar to the homogeneous assays described above but have a greater degree of specificity between the substrates of individual caspases or groups of caspases. Fluorescently conjugated substrates specific for certain caspases are immobilized in a 96-well plate. When cell lysates are added to the wells, the level of fluorescence emitted is an indicator of the activity of that particular caspase allowing delineation of specific pathways of the apoptotic process.
-
1.
These instructions assume the use of the ApoAlert™ Caspase Profiling Plate (see Note 5).
-
2.
Suspend eosinophils (at least 97% purity) at 2 × 106 cells/mL in IMDM (10% autologous serum). In 2 mL Eppendorf tubes pipette 1 mL of cell suspension (2 × 106 cells) and apoptosis-modifying agents and incubate at 37 °C for the desired length of time.
-
3.
Centrifuge at 220 × g for 5 min at 4 °C and aspirate the supernatant.
-
4.
Resuspend the cell pellet in 400 μL ice-cold 1× lysis buffer; incubate on ice for 10 min.
-
5.
While cells are incubating add 10 μL DTT per 1 mL 2× reaction buffer, and then pipette 50 μL to each well of the 96-well caspase profiling plate.
-
6.
Cover the plate and incubate for 5 min at 37 °C.
-
7.
Vortex the cell lysate and add 50 μL of lysate to duplicate wells of each caspase substrate.
-
8.
Cover the plate and incubate for 2 h at 37 °C.
-
9.
Use a plate reader to measure fluorescence (excitation: 380 nm, emission 460 nm).
3.8 Assessing Nuclear Changes During Apoptosis: Gel Electrophoresis of DNA
Activation of the apoptotic process results in endonuclease-mediated cleavage of DNA. After early large-scale degradation of DNA (50–200 kbp) endonuclease activity generates single-nucleosome or oligonucleosomal fragments of around 180 bp (or multiples thereof). This cleavage process creates discrete sized lengths of DNA which, when run through gel electrophoresis, produce a characteristic “laddering” effect that is distinct from the “smear” generated by the random DNA cleavage that occurs during cell necrosis.
-
1.
Suspend eosinophils (at least 97% purity) at 5 × 106 cells/mL in IMDM (10% autologous serum). In 2 mL Eppendorf tubes pipette 1 mL of cell suspension (2 × 106 cells) and apoptosis-modifying agents and incubate at 37 °C for the desired length of time.
-
2.
Extract genomic DNA using Wizard® Genomic DNA Purification Kit.
-
3.
Run the DNA (23 μL DNA mixed with 7 μL loading dye) on a 2% agarose gel containing GelRed (5 μL in 50 mL) in 1× TBE buffer at 110 V.
-
4.
Run until the dye front reaches the end of the gel and visualize the gel under ultraviolet illumination.
3.9 Hypodiploid DNA Content
Endonuclease-mediated cleavage of nuclear DNA during apoptosis causes an apparent decrease in DNA content of triton-permeabilized cells. Nuclear staining using propidium iodide allows detection of this “hypodiploid” cell population. This technique works well with eosinophils, as they are terminally differentiated and do not undergo proliferation meaning only two peaks are visible when DNA content is measured: diploid (viable) cells and hypodiploid (apoptotic) cells.
-
1.
Suspend eosinophils (at least 97% purity) at 4 × 106 cells/mL in IMDM (10% autologous serum).
-
2.
In a 96-well flat-bottomed plate add 75 μL of cell suspension, 15 μL of apoptosis-modifying agents (10× concentration) or vehicle control, and 60 μL IMDM with 10% autologous serum to each well. (NB: If two agents are used only 45 μL of IMDM is required for a total volume of 150 μL.)
-
3.
Cover and incubate at 37 °C in a 5% CO2 incubator for the duration of the experiment.
-
4.
Gently pipette the well to resuspend adherent cells and add 50 μL to a flow tube containing 250 μL of PI solution.
-
5.
Incubate in the dark at 4 °C for 15 min.
-
6.
Analyze by flow cytometry (FL-2 channel) to determine the percentage of cells with hypodiploid DNA content.
3.10 TUNEL Staining for DNA Breaks
DNA cleavage can be measured enzymatically as DNA breaks create acceptor sites for enzymes such as terminal deoxyribonucleotidyltransferase (TdT). TdT together with fluorescein-12-2′deoxyuridine-5′-triphosphate is used to identify DNA fragmentation in terminal uridine nucleotide end-labeling (TUNEL) staining.
-
1.
These instructions assume the use of in situ cell death detection kit, fluorescein.
-
2.
Suspend eosinophils (at least 97% purity) at 2 × 107 cells/mL in IMDM (10% autologous serum).
-
3.
To a 96-well flat-bottom plate add 90 μL of cell suspension and 10 μL of apoptosis-modifying agents (10× concentration) or vehicle control.
-
4.
Cover and incubate at 37 °C in a 5% CO2 incubator for the duration of the experiment.
-
5.
To a 96-well U-bottom flexible plate pipette 100 μL of cell suspension and centrifuge at 200 × g for 2 min at 4 °C. Discard the supernatants.
-
6.
Wash the cells three times. Adding 100 μL PBS per well. Spin the plate at 200 × g for 3 min at 4 °C, discarding the supernatants, and vortex for 5 s.
-
7.
Add 100 μL of fixation solution to each well.
-
8.
Incubate on a shaking heat block for 60 min at 300 rpm at room temperature.
-
9.
Add 200 μL PBS to each well, then spin the plate at 200 × g for 10 min at 4 °C, and discard the supernatant.
-
10.
Resuspend the cells in permeabilization solution and incubate for 2 min on ice.
-
11.
Add 50 μL of nucleotide mixture to two negative control wells.
-
12.
Make the TUNEL reaction mixture by adding the enzyme solution (50 μL) to 450 μL nucleotide mixture.
-
13.
Treat the two positive control wells with DNase I for 10 min at room temperature to introduce DNA strand breaks.
-
14.
Wash the plate twice in PBS (200 μL per well) and then resuspend in TUNEL reaction mixture (50 μL per well).
-
15.
Cover the plate and incubate at 37 °C for 60 min.
-
16.
Wash twice in PBS (200 μL per well) and then transfer to flow cytometry tubes for analysis of fluorescence levels (FL-1).
3.11 Assessing Phagocytic Uptake of Apoptotic Cells: Culture of Monocyte-Derived Macrophages
To assess monocyte-derived macrophage phagocytosis of apoptotic eosinophils it is necessary to differentiate blood-derived human monocytes into macrophages in in vitro culture. A variety of methods exist in order to, as closely as possible, recapitulate the phenotype of tissue macrophages. Isolation by adherence utilizes monocyte ability to rapidly attach to tissue culture plastic in preference to neutrophils and lymphocytes; washing off non-adherent cells after 1 h leaves a relatively homogenous cell population for subsequent culture. Alternatively, use of a pan-monocyte isolation kit by negative selection with anti-CD14-coated magnetic beads yields a highly pure monocyte population (see Note 6). Immortalized macrophage cell lines or primary macrophages isolated from animal tissue can also be used.
-
1.
Resuspend peripheral blood mononuclear cells at 4 × 106 cells/mL in IMDM with 10% autologous serum, add 500 μL/well in a 48-well plate, and incubate for 60 min at 37 °C.
-
2.
Wash adherent cells 3–4 times with IMDM and incubate in 500 μL IMDM with 10% autologous serum at 37 °C (see Note 7).
-
3.
Culture monocytes for 5–7 days with media changed after day 3 in culture prior to use in subsequent experiments.
3.12 Flow Cytometry-Based Phagocytosis Assay
This assay uses a fluorescent chloromethyl dye that diffuses across cell membranes to label the cytoplasm of live eosinophils without altering their functional activity (Fig. 2c–e). These pH-sensitive dyes such as pHrodo™ or CypHer5E are minimally fluorescent at a neutral pH but fluoresce brightly in acidic conditions (i.e., within the phagolysosome). This allows distinguishing of internalized apoptotic cells that are acidified within the phagolysosome versus apoptotic cells that are adherent to the phagocyte cell surface. Alone, this is a valid method for assessment of phagocytic uptake of apoptotic cells; however CellTracker™-green can also be used alongside in order to stain macrophages and identify the dual-positive population indicating ingested apoptotic cells. Efferocytosis probes can also be used to measure phagocytosed apoptotic cells by fluorescence microscopy and flow cytometry, for example a anxA5-pHrodo probe; these techniques may also have the added benefit of use in vivo [22].
-
1.
This method assumes the use of adherent monocyte-derived macrophages in Costar® 24-well TC-treated microplates.
-
2.
Suspend eosinophils (at least 97% purity) at 2 × 107 cells/mL in IMDM (10% autologous serum) in a 15 mL Falcon® conical polypropylene tube. Add 2 μg/mL of 10 mM 5-chloromethylfluorescein diacetate (CellTracker™ Green), pipette gently, and incubate at 37 °C for 30 min.
-
3.
Centrifuge at 220 × g for 5 min, wash cell pellet in PBS, and spin again at 220 × g for 5 min.
-
4.
Resuspend cells at 4 × 106 cells/mL in IMDM (10% autologous serum). Transfer the eosinophil suspension into Costar® 75 cm2 cell culture flask and incubate for 20 h at 37 °C (5% CO2).
-
5.
Transfer the eosinophils into a 50 mL Falcon® conical polypropylene tube, wash twice in warm IMDM (50 mL volume per wash) (220 g for 5 min), and discard the supernatant. Following each wash, resuspend the eosinophil pellet in 1 mL of warm IMDM to avoid cell clumping. Resuspend the aged eosinophils at 1 × 106 cells/mL in warm HBSS (37 °C).
-
6.
Incubate cells with 20 ng/mL pHrodo™ (30 min, room temperature).
-
7.
Centrifuge at 220 × g for 5 min, wash cell pellet in PBS, and spin again at 220 × g for 5 min.
-
8.
Resuspend cells at 4 × 106/mL in warm IMDM (serum free).
-
9.
Rinse the macrophages with warm IMDM to wash off non-adherent cells.
-
10.
Add 2 μg/mL CellTrace™ Far Red to IMDM, pipette gently, and incubate macrophage monolayer at 37 °C for 30 min.
-
11.
Pipette 500 μL (2 × 106 cells) of labeled aged eosinophils in IMDM (serum free) atop the macrophage monolayer. Incubate for 60 min at 37 °C in a 5% CO2 atmosphere (see Note 8).
-
12.
Remove the eosinophil suspension from the plate and wash macrophages with PBS three times.
-
13.
Incubate the macrophages with 300 μL 0.25% trypsin/1 mM ethylenediaminetetraacetic acid solution for 15 min at 37 °C followed by 5 min at 4 °C (see Note 9).
-
14.
Collect the detached macrophages by pipetting vigorously and place in a flow cytometer tube on ice.
-
15.
Analyze samples immediately by flow cytometry.
-
16.
Apoptotic cells and macrophage populations are identified by their distinct forward and side scatter characteristics. By dividing the number of dual CellTracker/pHrodo-positive events in the macrophage gate by the total macrophage number the percentage of macrophages that have internalized apoptotic cells can be calculated.
4 Notes
-
1.
Reference to autologous serum used in the culture of eosinophils denotes autologous plasma-derived serum. It is made by adding 20 mM CaCl2 to platelet-rich plasma (harvested after centrifugation of citrate-anticoagulated blood) and incubated for 1 h at 37 °C in glass tubes. Alternatively, fetal calf serum can be used or eosinophils may be cultured without serum but with a small amount of supplemental protein (e.g., 0.5% (w/v) serum albumin)—the latter will however accelerate the rate of apoptosis.
-
2.
Eosinophil granules are rich in proteases; therefore, in order to prevent the protein of interest being degraded, care should be taken to keep all samples on ice during the preparation of lysates. In addition, higher protease inhibitor concentrations are necessary than for lysis of other cell types.
-
3.
Supplemental serum can be added to cells in the cytocentrifuge chamber to prevent artifacts caused by cell breakage during centrifugation. This however reduces the effectiveness of visualizing secondary necrotic eosinophils, thereby underestimating the rate of eosinophil apoptosis. Secondary necrotic eosinophils appear as cell ghosts with little or no evidence of nuclear staining, having undergone “nuclear evanescence,” and as such may, incorrectly, not be included in the quantification of morphological changes.
-
4.
Annexin-binding buffer should always be used in the preparation of annexin V as the absence of supplemental Ca2+ causes rapid dissociation of annexin V from phosphatidylserine on the apoptotic cell surface.
-
5.
Colorimetric assays for single-caspase activity can also be used which are similar to the fluorometric assays and follow a similar protocol. However, caspase activation is measured by the cleavage of a chromophore (e.g., p-nitroanilide) from caspase substrates. A spectrophotometer or microplate reader (405 nm) records color development and activity quantified by comparison with a calibration curve constructed using known standards. Caspase activity assays can be applied to any cell type, as caspase activation is a general event of apoptosis. However, to select the most appropriate assay to use in eosinophils, differences between the expression profiles of the various caspases in different cell types should be considered. Caspases 3, 6, 7, 8, and 9 are contained as inactive zymogens within eosinophils [2].
-
6.
Purification of monocytes from the mononuclear cell population by adherence is frequently used as described. Isolation is also possible using negative selection of contaminating lymphocytes and neutrophils with magnetic beads (Pan Monocyte Isolation Kit, Miltenyi Biotec, Surrey, UK), according to the manufacturer’s instructions [15]. Caution should be taken using anti-CD16-negative selection to remove neutrophils as this may also remove CD16hi inflammatory monocytes. Isolated monocytes are plated out at 4 × 105/well in a 24-well plate.
-
7.
Differentiation of monocytes in in vitro culture can also be performed by culturing a combination of IL-4, IL-6, and GM-CSF (as described in ref. 8).
-
8.
This is an excess number of apoptotic eosinophils in order to determine macrophage phagocytic capacity and not a surrogate marker of eosinophil apoptosis.
-
9.
Treatment with trypsin-EDTA may lead to clumping of cells leading to blockage of the flow cytometer’s sample intake nozzle. Clumping may be minimized by adding 50 μL of bovine serum to each well following incubation with trypsin-EDTA.
Change history
27 August 2021
Chapter 10 was previously published non-open access. It has now been changed to open access under a CC BY 4.0 license and the copyright holder has been updated to ‘The Author(s)’. The book has been updated with these changes.
References
Rosenberg HF, Dyer KD, Foster PS (2013) Eosinophils: changing perspectives in health and disease. Nat Rev Immunol 13(1):9–22
Felton JM, Lucas CD, Rossi AG, Dransfield I (2014) Eosinophils in the lung – modulating apoptosis and efferocytosis in airway inflammation. Front Immunol 5:302
Leitch AE, Duffin R, Haslett C, Rossi AG (2008) Relevance of granulocyte apoptosis to resolution of inflammation at the respiratory mucosa. Mucosal Immunol 1(5):350–363
IPoon IK, Lucas CD, Rossi AG, Ravichandran KS (2014) Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14(3):166–180
Lucas CD, Dorward DA, Sharma S et al (2015) Wogonin induces eosinophil apoptosis and attenuates allergic airway inflammation. Am J Respir Crit Care Med 191(6):626–636
Felton JM, Lucas CD, Dorward DA et al (2018) Mer-mediated eosinophil efferocytosis regulates resolution of allergic airway inflammation. J Allergy Clin Immunol 142(6):1884–1893. e6
Farahi N, Singh NR, S. Heard S, et al. (2012) Use of 111-Indium-labeled autologous eosinophils to establish the in vivo kinetics of human eosinophils in healthy subjects. Blood 120(19):4068–4071
Duffin R, Leitch AE, Sheldrake TA (2009) The CDK inhibitor, R-roscovitine, promotes eosinophil apoptosis by down-regulation of Mcl-1. FEBS Lett 583(15):2540–2546
Park YM, Bochner BS (2010) Eosinophil survival and apoptosis in health and disease. Allergy Asthma Immunol Res 2(2):87–101
Tai PC, Sun L, Spry CJ (1991) Effects of IL-5, granulocyte/macrophage colony-stimulating factor (GM-CSF) and IL-3 on the survival of human blood eosinophils in vitro. Clin Exp Immunol 85(2):312–316
Robertson NM, Zangrilli JG, Steplewski A (2002) Differential expression of TRAIL and TRAIL receptors in allergic asthmatics following segmental antigen challenge: evidence for a role of TRAIL in eosinophil survival. J Immunol 169(10):5986–5996
Duffin R, Leitch AE, Fox S, Haslett C, Rossi AG (2010) Targeting granulocyte apoptosis: mechanisms, models, and therapies. Immunol Rev 236(1):28–40
Wedi B, Wieczorek D, Stunkel T, Breuer K, Kapp A (2002) Staphylococcal exotoxins exert proinflammatory effects through inhibition of eosinophil apoptosis, increased surface antigen expression (CD11b, CD45, CD54, and CD69), and enhanced cytokine-activated oxidative burst, thereby triggering allergic inflammatory reactions. J Allergy Clin Immunol 109(3):477–484
Akakura S, Singh S, Spataro M et al (2004) The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res 292(2):403–416
Baiula M, Bedini A, Carbonari G, Dattoli S, Spampinato S (2012) Therapeutic targeting of eosinophil adhesion and accumulation in allergic conjunctivitis. Front Pharmacol 3:203
Cartwright JA, Lucas CD, Rossi AG (2019) Inflammation resolution and the induction of granulocyte apoptosis by cyclin-dependent kinase inhibitor drugs. Front Pharmacol 10:55
Ravichandran KS (2010) Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med 207(9):1807–1817
Walsh GM, Sexton DW, Blaylock MG, Convery CM (1999) Resting and cytokine-stimulated human small airway epithelial cells recognize and engulf apoptotic eosinophils. Blood 94(8):2827–2835
Serhan CN, Savill J (2005) Resolution of inflammation: the beginning programs the end. Nat Immunol 6(12):1191–1197
Alessandri AL, Duffin R, Leitch AE et al (2011) Induction of eosinophil apoptosis by the cyclin-dependent kinase inhibitor AT7519 promotes the resolution of eosinophil-dominant allergic inflammation. PLoS One 6(9):e25683
Dorward DA, Lucas CD, Alessandri AL et al (2013) Technical advance: autofluorescence-based sorting: rapid and nonperturbing isolation of ultrapure neutrophils to determine cytokine production. J Leukoc Biol 94(1):193–202
Stöhr R, Deckers N, Schurgers L, Marx N, Reutelingsperger C (2018) AnnexinA5-pHrodo: a new molecular probe for measuring efferocytosis. Sci Rep 8:17731
Acknowledgments
The authors would like to acknowledge funding from the Wellcome Trust [206566/Z/17/Z, CDL], the UK Medical Research Council [MR/K013386/1: AGR], and the Pathological Society [#2219, NG].
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2021 The Author(s)
About this protocol

Cite this protocol
Gachanja, N.N., Dorward, D.A., Rossi, A.G., Lucas, C.D. (2021). Assays of Eosinophil Apoptosis and Phagocytic Uptake. In: Walsh, G.M. (eds) Eosinophils. Methods in Molecular Biology, vol 2241. Humana, New York, NY. https://doi.org/10.1007/978-1-0716-1095-4_10
Download citation
DOI: https://doi.org/10.1007/978-1-0716-1095-4_10
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-0716-1094-7
Online ISBN: 978-1-0716-1095-4
eBook Packages: Springer Protocols