
Abstract
Genetic manipulation of bacteria often requires the joining together of more than one DNA segment to form a hybrid DNA molecule. This can be accomplished by PCR followed by restriction endonuclease digestions and ligations. However, this approach can often become laborious and expensive. Here is described a well-established method for using primer design and PCR to obtain hybrid products for use in cloning vectors, mutagenesis protocols, and other applications.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others

Keywords:
1 Introduction
Since the discovery of modern PCR (1, 2), the technique has been used for an extensive array of molecular biology applications. One example is splicing by overlap extension PCR or SOEing PCR (3, 4). SOEing PCR serves as a simple, yet powerful, method for manipulating DNA sequences without the need for laborious cloning protocols. The technique involves joining together individual segments of amplified DNA using homologous sequences added to the 5′-ends of specific primers. SOEing PCR is dependent on selection of fusion oligonucleotide primers which are a hybrid of the reverse primer for one segment and the forward primer for another segment. Initial DNA segments are amplified by standard PCR using combinations of regular primers and/or the hybrid fusion primers. Ends of PCR segments amplified using the fusion primers will contain a short sequence complementary to the segment to be fused. Following amplification of the individual segments, a final PCR reaction using the outermost primers generates a full length fusion PCR product combining all segments. The complementary ends between segments will serve like “internal primers” to generate a full length template comprised of all segments to be fused. If necessary, a filling reaction can be performed by combining all of the individual segments without primer and cycling with polymerase allowing the overlapping ends to serve as primers to generate some full length template for the final PCR reaction using the outermost primers. The entire process is simpler and cheaper than performing numerous digestion and ligation reactions.
2 Materials
Dissolve all buffer components in ultrapure water. Use only nuclease-free water for dilution of primers and for setting up PCR reactions. Keep DNA polymerase and PCR reactions on ice at all times prior to beginning PCR amplification run and follow manufacturer’s instructions for optimal activity. Oligonucleotide primer sequences can be selected manually or using the software program of your choice.
2.1 PCR Reaction
-
1.
10× amplification buffer (varies by manufacturer): generally consists of 500 mM KCl, 100 mM Tris–HCl, pH 8.0–8.3, 15–20 mM MgCl2.
-
2.
dNTP mixture: containing approximately 2.5 mM each of dCTP, dATP, dGTP, dTTP. Final concentration of each dNTP in the reaction should be ~200 μM.
-
3.
Stock oligonucleotide primers: dissolve each oligonucleotide primer to in TE (10 mM Tris pH 8.0, 1 mM ETDA) to make a 100 μM stock solution. Dilute from this stock 1:10 (in water or TE) to make a 10 μM working solution.
-
4.
Template DNA: <500 ng (see Note 1).
-
5.
Proofreading thermo-stable DNA polymerase: TaKaRa Ex Taq polymerase (5 units/μL) performs well in our hands.
-
6.
Nuclease-free water.
2.2 Agarose Gel Electrophoresis
-
1.
50× Tris-acetate (TAE): 242 g Tris base, 57.1 ml glacial acetic acid, 100 mL 0.5 M EDTA (pH 8.0). Alternatively Tris-borate (TBE) can be used from a 5× stock: 54 g Tris base, 27.5 g boric acid, 20 mL of 0.5 M EDTA (pH 8.0).
-
2.
Molecular biology grade, low-EEO agarose.
-
3.
Ethidium bromide: 1 % solution (see Note 2).
-
4.
6× DNA loading buffer: 30 % glycerol, 0.25 % bromophenol blue, and 0.5 % xylene cyanol in distilled water.
-
5.
DNA ladder.
2.3 DNA Isolation and Cloning Kits
-
1.
DNA Clean & Concentrator-5 (Zymo Research Corporation, Irvine, CA).
-
2.
TOPO-TA Cloning kit (Life Technologies Gaithersburg, MD) or other subcloning vector compatible with polymerase used (determined based on whether polymerase leaves an overhang or blunt end).
3 Methods
SOEing PCR can be used to fuse as many pieces of DNA as desired into a single product. Here we have simply described how to fuse three pieces together.
3.1 Primer Design
-
1.
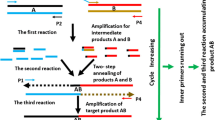
Design forward and reverse primers for each segment of DNA you wish to amplify. Here we refer to the segments as “upstream flanking,” “middle segment,” and “downstream flanking.” The first two primers, Primers 1 and 2, should serve as forward and reverse primers for your upstream flanking segment. Primers 3 and 4 should serve as forward and reverse primers for your downstream flanking segment and Primers 5 and 6 should amplify your middle segment.
-
2.
Example primer sequences are shown in Fig. 1. Each primer number is located at its 5′-end. BamHI restriction sites have been added to the ends of Primers 1 and 4.
Fig. 1 
SOEing PCR diagram with example primer sequences. Primer sequences are numbered consecutively at their 5′-ends. Red and blue letters indicate overlapping fusion sequences. A BamHI restriction site (shown in italics) has been added to Primers 1 and 4 for cloning of the SOEing product
-
3.
Annealing temperatures of all primers should be as similar as possible, prior to adding additional sequences.
-
4.
Primer 2 (reverse primer for upstream segment) will need to have the inverse complement sequence of Primer 5 (forward primer for middle segment) added to its 5′ end. Additionally, Primer 3 will need the inverse complement sequence of primer 6 added to its 5′-end. These modifications will ensure overlap between the sequences during the later SOEing reaction (see Notes 3 and 4).
-
5.
Sequences complimentary to the upstream and downstream segments can also be added to the 5′-ends of Primers 5 and 6. However, the SOEing PCR will generally work without the added complimentary sequences and therefore is not shown here (see Note 5).

3.2 Initial PCR Reactions
-
1.
Set up individual PCR reactions to amplify the upstream, middle, and downstream segments. In a 50 μL reaction volume, add 5 μL of 10× amplification buffer, dNTPs (200 μM each), proofreading Taq DNA polymerase (1.25 units), forward and reverse primers (0.4 μM each), and template DNA (50–100 ng) (see Note 6).
-
2.
Run a standard 30-cycle PCR program: 1 cycle (4 min at 95 °C), 30 cycles (1 min at 95 °C, 1 min at 53 °C, X min at 72 °C where X = size in kilobases of product to be amplified), 1 cycle of 5 min at 72 °C (see Note 7).
3.3 Gel Imaging and PCR Cleanup
Pour a 0.8–1 % agarose gel by melting agarose in TAE buffer and pouring into cast. Immediately add a 10-well gel comb. Place gel into a gel electrophoresis apparatus fill with TAE to cover gel. If TBE is used to cast gel, fill apparatus with TBE instead of TAE.
-
1.
After the gel has solidified, add 1 μL of 6× loading dye to 5 μL of each PCR product, mix well, and load into wells.
-
2.
Apply current (90–100 V) for 35–40 min. Remove gel from apparatus and view on ultraviolet transilluminator.
-
3.
Example PCR products from three individual segments are shown in Fig. 2a. Individual band sizes are 500 bp (UP and DN) and 750 bp (Mid).
Fig. 2 
(a) Agarose gel UV image of products amplified using Primers 1 and 2 (UP), Primers 5 and 6 (Mid), and Primers 3 and 4 (DN). “L” indicates DNA ladder. 50 ng of template DNA was used for each reaction and 5 μL of PCR product was loaded into each well. (b) Agarose gel UV image of SOEing product following PCR fusion of all three segments from (a). Template for the SOEing PCR reaction consisted of a mixture of the three segments (100 ng each) from (a)
-
4.
After confirming amplification of each segment, perform a PCR cleanup using a commercially available kit to remove oligonucleotides and elute using the manufacturer’s recommended volume. Assess the concentration of each PCR product by a spectrophotometer.

3.4 SOEing PCR Reaction
-
1.
Add equal concentrations (~100 ng each) of the individual PCR segments to be fused to a PCR reaction mixture containing Primers 1 and 4 (see Note 8).
-
2.
Set the extension time minutes of the PCR program to equal the sum of all segments in kilobases. For instance, in the example used here the segments total 1.75 kb therefore an extension time of 1.45 min was used.
-
3.
Following PCR, run the SOEing product on a 0.8–1 % agarose gel as described above and check the gel on an UV transilluminator. A successful SOEing PCR should generate a product with a size totaling the sum of the individual segments. Fusion of the segments from Fig. 2a is shown in Fig. 2b.
3.5 Filling Reaction (Optional) (See Note 9)
-
1.
Combine equal concentrations (200 ng each) of the individual segments in a PCR reaction containing all components EXCEPT primers. The overlap sequences of the fusion primers (Primer 2 and 3) will serve as primers and the polymerase will fill the gaps to generate full length template (see Note 10).
-
2.
Run the standard PCR program as described above allowing an extension time in minutes that equals the length of desired full length product in kilobases.
-
3.
Following the filling PCR reaction, use 2–3 μL of the filling product as template in a SOEing reaction containing Primers 1 and 4. There is no need to perform a PCR cleanup of the filling reaction since it included no primers.
-
4.
Run the SOEing PCR product on a 0.8–1 % agarose gel and visualize on a UV transilluminator.
3.6 SOEing PCR Reaction Isolation and Cloning
-
1.
The SOEing PCR product can be isolated and concentrated using a kit as described for the individual segments (see Note 11).
-
2.
If more than one band appears on the gel, the annealing temperature may need to be adjusted. Run a gradient PCR to determine optimal conditions for production of a single full-length product.
-
3.
Clone the SOEing PCR product into a cloning vector such as pCR2.1-TOPO per manufacturer’s instructions. Confirm and isolate positive clones for freezer stocks. This prevents the need to repeat the SOEing PCR from scratch if the product is needed again in the future (see Note 12).
4 Notes
-
1.
Typically 20–100 ng of template DNA is sufficient to perform a standard PCR reaction.
-
2.
Ethidium bromide is a mutagen and should be handled with care. DNA gels can be stained following electrophoresis; however, we generally add ethidium bromide to our gels prior to casting. We typically add 2 μL 1 % ethidium bromide per 100 mL of gel.
-
3.
If possible, limit the length of the 3′ ends of primer 2 and primer 3 to approximately 15–20 nucleotides. Additionally, the 5′ additions (inverse complements should be approximately 15–20 nucleotides to help limit an extremely long fusion primer (i.e., final Primers 2 and 3).
-
4.
The additions to the 5′-ends of the primers will drastically increase the overall Tm of the primers. However, this should not be a concern since the entire primer is not adhering to the template.
-
5.
This prevents the need of ordering an additional set of fusion primers for each SOEing reaction you plan to perform. For example, if you are routinely fusing a common antibiotic cassette between segments of DNA, you can use your standard primers to amplify the cassette and will not require a second set of fusion primers.
-
6.
The concentration of template DNA can vary depending on the source. Empirical determination of optimal template concentration is recommended. We typically get strong amplification of DNA segments from 50 to 100 ng of chromosomal DNA. Less may be necessary if amplifying from plasmid templates.
-
7.
All annealing temperatures and extension times should be modified based on the products to be amplified as well as the polymerase used.
-
8.
The optimal ratio of segments for efficient amplification of the SOEing product can sometimes vary. Altering the ratio of the mid segment to the flanking pieces may improve the amplification efficiency and should be determined empirically.
-
9.
SOEing PCR occasionally results in multiple products or products that are not full length. For this reason, it is often beneficial to run a filling reaction prior to or in conjunction with the SOEing PCR reaction. Depending on the primers and sequences to be amplified, the filling reaction will sometimes generate a better full-length template than using the individual gene segments. If neither the standard SOEing PCR nor the filling reaction-as-template PCR leads to full length product, new primers may need to be designed as they may be incompatible.
-
10.
As described for the SOEing reaction, the optimal ratio of segments may need to be determined empirically if the filling reaction is not successful. Altering the ratio of mid-segment to flanking segments may improve the efficiency.
-
11.
A PCR purification column can be used to isolate the SOEing product only if a single band of the correct size is visualized following agarose gel electrophoresis.
-
12.
TOPO-TA cloning can also be used to isolate the correct SOEing PCR in the event that multiple bands are continually produced. If a band the size of all the combined segments is visualized, the band can be excised and gel purified prior to cloning. It is important to always sequence the TA clones prior to proceeding to ensure the SOEing product is correct.
References
Saiki RK, Scharf S, Faloona F et al (1985) Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 230:1350–1354
Mullis K, Faloona F, Scharf S et al (1986) Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51(Pt 1):263–273
Ho SN, Hunt HD, Horton RM et al (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59
Horton RM, Hunt HD, Ho SN et al (1989) Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this protocol
Cite this protocol
Thornton, J.A. (2014). Splicing by Overlap Extension PCR to Obtain Hybrid DNA Products. In: Bose, J. (eds) The Genetic Manipulation of Staphylococci. Methods in Molecular Biology, vol 1373. Humana Press, New York, NY. https://doi.org/10.1007/7651_2014_182
Download citation
DOI: https://doi.org/10.1007/7651_2014_182
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-3157-6
Online ISBN: 978-1-4939-3158-3
eBook Packages: Springer Protocols