
Abstract
Background
Maize (Zea mays L.) is a widely cultivated cereal and has been used as an optimum heavy metal phytoremediation crop. Metallothionein (MT) proteins are small, cysteine-rich, proteins that play important roles in plant growth and development, and the regulation of stress response to heavy metals. However, the MT genes for maize have not been fully analyzed so far.
Methods
The putative ZmMT genes were identified by HMMER.The heat map of ZmMT genes spatial expression analysis was generated by using R with the log2 (FPKM + 1).The expression profiles of ZmMT genes under three kinds of heavy metal stresses were quantified by using qRT-PCR. The metallothionein proteins was aligned using MAFFT and phylogenetic analysis were constructed by ClustalX 2.1. The protein theoretical molecular weight and pI, subcellular localization, TFs binding sites, were predicted using ProtParam, PSORT, PlantTFDB, respectively.
Results
A total of 9 ZmMT genes were identified in the whole genome of maize. The results showed that eight of the nine ZmMT proteins contained one highly conserved metallothio_2 domain, while ZmMT4 contained a Metallothio_PEC domain. All the ZmMT proteins could be classified into three major groups and located on five chromosomes. The ZmMT promoters contain a large number of hormone regulatory elements and hormone-related transcription factor binding sites. The ZmMT genes exhibited spatiotemporal specific expression patterns in 23 tissues of maize development stages and showed the different expression patterns in response to Cu, Cd, and Pb heavy metal stresses.
Conclusions
We identified the 9 ZmMT genes, and explored their conserved motif, tissue expression patterns, evolutionary relationship. The expression profiles of ZmMT genes under three kinds of heavy metal stresses (Cu, Cd, Pb) were analyzed. In summary, the expression of ZmMTs have poteintial to be regulated by hormones. The specific expression of ZmMTs in different tissues of maize and the response to different heavy metal stresses are revealed that the role of MT in plant growth and development, and stress resistance to heavy metals.
Similar content being viewed by others
Background
Excessive heavy metal ions, such as cadmium (Cd), copper (Cu), lead (Pb) often cause heavy metal stress in plants, resulting in metabolic dysfunction and growth inhibition [1]. Plants have evolved several mechanisms for detoxification of heavy metal, such as efflux of heavy metals from the cell, chelation, and sequestration heavy metal ions through specific transporters or ligands [1,2,3]. The glutathione (GSH), phytochelatins (PCs), and metallothionein (MT) are three well-known heavy metal-binding ligands in plant cells [4,5,6,7]. MT plays an important role in response to heavy metal stress and has been a research hotspot in the field of molecular biology [8].
MT proteins are small (7–8 kDa), cysteine-rich (20–30%) can keep the metal ion homeostasis by binding with heavy metal ions, and protect against heavy metal toxicity by sequestration [9,10,11,12]. The first plant MT protein was identified in wheat (EcMT) in 1987 [13], then more MT sequences have been reported in various species [14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. There are 7 and 14 MT genes in A. thaliana and O. sativa respectively, which can be classified into four subfamilies (type/class 1, 2, 3 and 4) [28, 33, 34]. The 6 MT genes in sugarcane (Saccharum officinarum L.) were found, which ScMT3-1 plays an active role in yeast (P. pastoris) response to Cd2+ and Cu2+ stress[35].Previous investigations showed that plant MT proteins acted as reactive oxygen species (ROS) scavenging enzymes [19, 36,37,38]. For example, rice OsMT1a and OsMT2b and cotton (Gossypium hirsutum) GhMT3a possessed of superoxide- and hydroxyl radical-scavenging activities in vitro [36,37,38]. Moreover, the lack of OsMT2b expression was found to promote epidermal cell death in stems and to accelerate H2O2-mediated aerenchyma formation in the internodes in rice [39, 40]. In maize, the expression level of MT1 in the root cortex was found to decrease during aerenchyma formation under waterlogged conditions [41]. These findings suggested that MT proteins had a role in determining the fate of cells in roots during inducible aerenchyma formation.
Maize (Zea mays L.) is a widely cultivated cereal and has been widely adopted for phytomanagement of Cd-contaminated soils due to its high biomass production and Cd accumulation capacity [42]. Recently, an increasing number of transcriptome studies screened out a series of candidate genes involved in the responses to heavy metal ions stress in various plant species [43,44,45,46,47,48,49,50,51]. Most transcriptome studies only provide fundamental information on the pathway involved in the responses to heavy metal ions stresses.
Although the MT genes have been studied in several plant species, however, they are rarely reported in maize. In this study, we identified 9 ZmMT genes in maize genome-wide, and their conserved motif and tissue expression patterns were analyzed. To understand their evolutionary relationship with other plants, a phylogenetic tree was constructed. Furthermore, the expression profiles of the ZmMT genes under three heavy metal stresses (Cu, Cd, Pb) were assessed by using qRT-PCR. The findings of our study will help to understand the roles of ZmMT genes in heavy metal ions response and to further identify the functions of this essential gene family in maize.
Materials and methods
Plant materials and stress treatments
Seeds of maize inbred line B73 were surface sterilized in 0.5% NaClO for 5 min, washed with distilled water, and then germinated on the filter paper moistened with distilled water and incubated at 26℃ in the dark. Seedlings in identical growing situations were selected, after three days, transplanted into an aerated complete nutrient solutions (Additional file 1: Table S1) and grew in a growth chamber as follows: kept for 3 days with a photoperiod of 14 h light/10 h dark at 26 ℃ and relative humidity of 70% [52]. After that, the maize seedlings were randomly divided into two groups, CK-grown seedlings, grown only in half-strength Hoagland solution were regarded as controls; Cd 200-grown (200 mg/L CdCl2·2.5H2O), Pb 1000-grown (1000 mg/L, Pb(NO3)2), Cu 200-grown (200 mg/L, CuCl2·2H2O) for heavy metal stresses. Both heavy metals (Cd, Pb, Cu)-grown and CK-grown root, stem, leaves of seedling three different tissues were separately sampling at 0 h, 24 h respectively after heavy metal treatment. All samples were harvested from each of the three maize seedlings, and three independent replicates were collected for each sample. Following harvested and immediately frozen in liquid nitrogen and stored at – 80 ℃ for extracting RNA and qRT-PCR analysis.
Identification of metallothionein genes in maize
The protein sequences of Zea mays (Zm, B73_RefGen_v4) were downloaded from Ensembl Plants (http://plants.ensembl.org/index.html). MT protein sequences of A. thaliana and O. sativa were downloaded from TAIR (https://www.arabidopsis.org/) and RGAP (http://rice.plantbiology.msu.edu/).
HMMER 3.0 software (http://hmmer.janelia.org/) was used to identify the putative MT genes by searching MT domain which was made by MT protein sequences of A. thaliana and O. sativa under default parameters. The putative MT genes were annotated by Pfam database. The protein theoretical molecular weight and pI were predicted using ProtParam (http://au.expasy.org/tools). The subcellular localization of ZmMT genes was predicted using the PSORT program (https://psort.hgc.jp/).
Phylogenetic analysis and conserved motif analysis
For the MT proteins phylogenetic analysis, all the MT proteins from Zea mays, A. thaliana and a outgroup sequence ScMT1 (S.cerevisiae metallothionein, accession No. AAA66061) were aligned using MAFFT software version 7 with L-INS-I [53], then a phylogenetic tree of Zea mays and A. thaliana MT proteins was constructed using ClustalX 2.1 software with 1000 bootstrap replicates [54]. The online program MEME Version 5.0.5 (http://meme.nbcr.net/meme/) was used to analyze conserved motifs for the 9 ZmMTs sequences with parameters as follows: maximum number, 5; site distribution, any number of repetitions; minimum width, 6; and maximum width, 50.
ZmMT genes distribution on Chromosomal and structure analysis
The chromosomal distribution of ZmMT genes was obtained from the Ensembl Plants (http://plants.ensembl.org/index.html), and the location images was drawn with TBtools software (https://github.com/CJ-Chen/TBtools/releases) [55]. Gff3 file of ZmMT genes was used for drawing schematic diagram gene structure.
Calculation of Ks and Ka of ZmMTs
Paralogous gene pairs of ZmMT genes were identified by Orthofinder v2.2.6 with the BLAST method under default parameters [56]. The paralogous gene pairs were used to calculate the synonymous (Ks) and nonsynonymous (Ka) values using the KaKs_Calculator2.0 [57]. Divergence time (T) was calculated using the formula T = Ks/2λ × 10 − 6(λ = 6.5 × 10−9 for grasses) million years ago (Mya) [58, 59].
Prediction of cis-responsive elements on the promoters of ZmMT genes
The 2000 bp genomic regions upstream of the initiation codon (ATG) of ZmMT genes were obtained and used to search for the cis-acting regulatory elements in PlantCARE database and PLACE database (http://www.dna.affrc.go.jp/PLACE/). TF binding sites were obtained by searching PlantTFDB (http://planttfdb.cbi.pku.edu.cn/).
Transcriptomic and quantitative real-time PCR (RT-qPCR) analysis
For spatial expression analysis ZmMT genes, transcriptomic sequencing (RNA-seq) data were collected from previous research [60]. These data represent 23 tissues of the spanning vegetative and reproductive stages of maize development. Gene transcript levels in various tissues were valued by fragments per kilobase (kb) of exon model per million mapped reads (FPKM) and the heat map was generated by using R (v3.4.0) with the log2 (FPKM + 1).
For the qRT-PCR assay, total RNA was extracted using the HiPure Universal RNA Kit (Biodata, Hefei, China) from respective tissues according to the manufacturer’s instructions. RNase-free DNase I (Biodata, Hefei, China) was used to remove genomic DNA. Total RNA (1 μg) was used to synthesize first-strand cDNAs using an equivalent amount of oligo-(dT)15 and random primers in 20 μL). volume with the GoScript™ Reverse Transcriptase 431 system (Promega, Madison, WI, USA) according to the manufacturer’s protocols. RT-qPCR was performed in 96-well plates in an ABI 7500 Real-time system (ABI, Alameda, CA, USA) using the SoAdvancedTM Universal SYBR® Green Supermix detection system (Bio-Rad, Hercules, CA, USA). The RT-qPCR reaction in a total volume of 10 μL consisted of 5 μL SYBR® Green Supermix, 0.5 μL of forward primer (10 μM), 0.5 μL of reverse primer (10 μM), 1 μL of cDNA, and 3 μL of ddH2O. The cycling conditions were 95 °C for 2 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. After 40 cycles, melting curve analysis was performed ranging from 60 to 95 °C. Maize 18S rRNA was used as the internal reference gene. The relative gene expression level was calculated using the 2−ΔΔCq method. All RT-qPCR experiments were carried out using three biological replicates of each sample. The gene-specific primers used for RT-qPCR are listed in Additional file 1: Table S2.
Results
Genome-wide identification of ZmMTs in maize
A total of 9 ZmMT genes termed ZmMT1 to ZmMT9 were identified (Table 1, Additional file 1: Table S3) by searching the Zea mays genome using known MT encoding genes and OsMt genes as queries. Most of all the identified ZmMT proteins contained one highly conserved Metallothio_2 domain, while ZmMT4 proteins contained a Metallothio_PEC domain. The length of the amino acid sequence in the ZmMT proteins (Table 1) ranged from 75 (ZmMT8) to 84 (ZmMT7), with an average of 7.78 kDa protein molecular weight. Subcellular localization prediction by PSORT shown that five ZmMTs were located in the chloroplast (chlo) and four were located at mitochondrion (mito).
Evolutionary relationships of ZmMTs
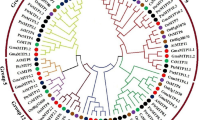
The phylogenetic relationship among the nine identified ZmMT proteins was examined based on OsMt proteins from the four groups, and a rooted tree with ScMT1 as outgroup was built by ClustalX 2.1 (Fig. 1). All the ZmMT proteins were classified into three major groups (I, II and IV) (Table 1, Fig. 1). Two members of ZmMTs (ZmMT3 and ZmMT9) belonged to group I, six members (ZmMT1, ZmMT2, ZmMT5, ZmMT6, ZmMT7 and ZmMT8) belonged to group II, and one member (ZmMT4) belonged to group IV (Fig. 1).

Classification of different groups of ZmMTs. Different color regions are used to distinguish different subgroups. The neighbor-joining (NJ) method was used to analyze the evolutionary trees of 9 ZmMTs and 14 OsMTs
Genomic structure, conserved domain and motif of ZmMT proteins
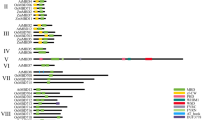
Through domain analysis, it can be seen that group I and group II members contain a Metallothio_2 domain and group IV members contain a Metallothio_PEC domain (Table 1). Genomic structure showed that ZmMT4 has one exon, ZmMT1 have 3 exons and other ZmMTs have 2 exons (Fig. 2).

Conserved motifs and gene structures of ZmMTs. MEME analysis showing distributions of 5 conserved motifs and coding sequences (CDS) regions in 9 ZmMTs that were demonstrated with a colorful box
The MEME program was used to predict the composition of the ZmMT proteins motifs. A total of five conserved motifs were detected (Fig. 2). Among them, motif 3 was conserved in all ZmMT proteins. Motif 1 and motif 5 were conserved in eight ZmMT proteins. Motif 2 and motif 4 were conserved in six and three ZmMT proteins. Except for motif 3 and motif 5, other motifs only exist in group I and group II. Interestingly, motif 3 exists in the 3' end of group I and group II, but in the middle of the sequence in group IV. In contrast, motif 5 exists in the 5' end of group I and group II, but in the 3' end in group IV (Fig. 2).
Chromosomal location and cis-elements
Analyses of the chromosomal distribution indicated that 9 ZmMTs were mapped on five chromosomes. ZmMT9 was anchored on chromosome 4. ZmMT1/ZmMT2, ZmMT3/ZmMT4, ZmMT5/ZmMT6, ZmMT7/ZmMT8 distributed on chromosome 8, 1, 6, and 3, respectively (Fig. 3). Based on the chromosomal distribution and paralogous analysis, the duplication events were proposed to occur in Zea Mays genome (Fig. 3 and Table 2). The substitution rate of nonsynonymous (Ka) and synonymous (Ks) is the basis for evaluating the positive selection pressure of duplication events, where Ka/Ks = 1 denoted neutral selection, Ka/Ks < 1 indicated purifying selection, and Ka/Ks > 1 referred to positive selection. KaKs Calculator 2.0 was used to calculate Ka/Ks ofZmMTs duplication event. Ka/Ks of paralogous ranged from 0.74 to 2.60. ZmMT5/ZmMT6, ZmMT2/ZmMT5, and ZmMT2/ ZmMT6 had Ka/Ks > 1, that is, 2.60, 1.43, and 1.23, respectively. These results suggested that thoseZmMTs were subjected to positive, negative, or balanced selection and functions of the duplicated genes became diverge along with the genome evolution after the duplication events of ZmMTs. The divergence time of the paralogous gene pairs of ZmMT gene pairs was estimated to be about range from 2.11 and 28.05 million years ago (Table 2).

The chromosomal distribution of ZmMT genes
To get an overview of the regulatory cis-acting elements involved in the responsiveness of abiotic and abiotic stresses, the 2 kb upstream sequences from each ZmMTs were programmed in PlantCARE and Plance server. As shown in Fig. 4A, potential environmental factor-related cis-regulatory elements were predicted to be correlated with ABA response, light response, and MeJA response which were most widely spread in promoters of ZmMTs. Stress-responsive elements were predicted to be mostly distributed in ZmMT4 and ZmMT8. Furthermore, some transcription factor (TF) binding sites, were also predicted as shown in Fig. 4B. In which, ERF binding sites are enriched in MzMT7 promoter, and MYB binding sites are enriched in MzMT1, MzMT2, and MzMT6 promoter. These results suggest that ERF and MYB may play an important role in regulating ZmMT gene expression.

Prediction of cis-responsive elements (A) and transcription factor (B) binding sites in the 2-k upstream regulatory regions of ZmMT genes. A Prediction of cis-responsive elements in the 2-k upstream regulatory regions of ZmMT genes. Different cis-responsive elements are represented by different colored boxes. B Prediction of TF-binding sites in the 2-k upstream regulatory regions of ZmMT genes. Different TF-binding sites are represented by different colored boxes
Expression profiling of ZmMT genes in different tissues
To study the spatiotemporal specific expression of ZmMT genes, we used public available RNA-seq data of 23 tissues to investigate the expression profiles of the ZmMT genes. These data represent 23 tissues of the spanning vegetative and reproductive stages of maize development in Fig. 5. The cluster heatmap shows that ZmMT1 and ZmMT8 are highly expressed in all tissues. ZmMT9 is highly expressed in germination kernels, pericarp, aleurone, mature leaf, primary root, root cortex, root elongation, root meristem zone, second root 7–8 days, female spikelet, silk. Both ZmMT5 and ZmMT6 are highly expressed in kernels. ZmMT4 and ZmMT7 are mainly expressed in seeds after fertilization, especially in the 38DAP embryo. ZmMT2 and ZmMT3 are expressed in low amounts in most tissues, among which ZmMT2 is expressed in internodes and meristems higher than other tissues, and ZmMT3 is only expressed relatively high in the root cortex.

Hierarchical clustering of ZmMT genes expression levels in 23 tissues of the spanning vegetative and reproductive stages of maize development
Response to heavy metal stresses
To verify the response expression of ZmMTs, the qRT-PCR assay was performed under three heavy metal stresses (Cu, Cd, and Pb). The expression of ZmMT2, ZmM4, and ZmMT5 is not presented due to its extremely low signal (Fig. 6). In young roots, compared with CK, ZmMT1, ZmMT3, ZmMT8, ZmMT9 are significantly up-regulated, while ZmMT7 is significantly down-regulated under Cu stress. ZmMT6 and ZmMT9 are significantly up-regulated, while ZmMT3 and ZmMT7 are significantly down-regulated under Cd stress. ZmMT3, ZmMT7, and ZmMT8 are significantly down-regulated under Pb stress. In young stems, compared with the control, ZmMT1, ZmMT3, ZmMT6, ZmMT7, ZmMT8, and ZmMT9 are significantly down-regulated under Cu stress. ZmMT3 is significantly up-regulated, while ZmMT1, ZmMT7, and ZmMT8 are significantly down-regulated under Cd stress. ZmMT1, ZmMT3, ZmMT6, ZmMT7, ZmMT8 are significantly down-regulated under Pb stress. In young leaves, compared with the control, ZmMT3, ZmMT7, ZmMT9 are significantly up-regulated, while ZmMT1 is significantly down-regulated under Cu stress. ZmMT3, ZmMT7, and ZmMT9 are significantly up-regulated, while ZmMT1 and ZmMT8 are significantly down-regulated under Cd stress. ZmMT3 and ZmMT7 are significantly up-regulated, while CK, ZmMT1, ZmMT6, and ZmMT8 are significantly down-regulated under Pb stress.

Expression profilings of ZmMT genes in different tissues
Discussion
Identification and structural conservation of ZmMT proteins
Maize is a widely cultivated cereal and tolerant to heavy metal ions-contaminated soils [61]. Maize has been used as an optimum plant for heavy metal ions phytoremediation in contaminated soils [62, 63]. Even if the entire genome sequencing of maize has been released for a decade, a thorough survey of ZmMTs has not been reported hitherto. We aimed to gain novel insights into the molecular aspects of the ZmMT gene family in maize. An initial comprehensive genome-wide analysis demonstrated that a total of 9 ZmMTs were identified from the recently released maize genome (Zm, B73_RefGen_v4) in Ensembl Plants.
The members of MT genes range from 4 (Hevea brasiliensis) to 14 (Oryza sativa) in higher plants in the previous reports [15, 16, 20, 23, 29, 33, 34, 64, 65]. It was showed that the number of ZmMT genes is not correlated with genome size but with genomic ploidy. For example, only four MT genes were identified in H. brasiliensis, which has a genome of 1460 megabases (Mb), while O. sativa has 14 members of MT genes and a moderate 420 Mb genome, and arabidopsis has 7 members of MT genes and a compact 135 Mb genome. In Brassica, three diploid species, B. rapa (AA, 2n = 20), B. oleracea (CC, 2n = 18), and B. nigra (BB, 2n = 20) have 8, 9 and 7 MT genes while in two allotetraploid species, B. napus (AACC, 2n = 38) and B. juncea (AABB, 2n = 36) have 16 and 12 MT genes. The number of ZmMT genes was smaller when considering its much larger genome size (~ 2500 Mb) [48] as compared with those of arabidopsis (125 Mb) [64] and rice (420 Mb) [65]. All ZmMTs proteins contain 75 to 84 amino acids, similar in length to the MTs of arabidopsis and rice [48, 64, 65].
As described in the results, the ZmMT genes in maize can be divided into three main groups based on phylogenetic analysis. There is no group III type gene in the maize genome, which is different from rice (Fig. 1). The motif-based sequence analysis tools (MEME) results showed that there are two conserved C-enriched motifs in the ZmMT protein sequences, namely motif 3 and motif 5. Motif 3 exists in all sequences, and motif 5 is deleted in the ZmMT7 protein sequence (Fig. 2). More interestingly, in most ZmMT proteins, motif 3 and motif 5 are present at the 3' and 5' ends of the protein, respectively. In ZmMT4 protein, motif 5 is present in the 5' segment, and motif 3 is present in the middle of the protein. The deletion of motif 5 and the misalignment of the two motifs may be caused by the rich transposon elements in the maize genome during genome replication evolution. In addition, motif1 and motif 2 exist in group I and group II, and motif4 only exists in group II.
Cis-Elements of ZmMT genes
To analyze the regulatory elements of ZmMT genes, we extracted a 2 Kb candidate promoter sequence upstream of ATG and used the components of the PlantCARE database and the PLACE database to predict the regulatory elements on the promoter. The statistical results show that the promoter region of ZmMTgenes contains a large number of hormone-related elements, such as ABA response, ethylene response, GA response, MeJA response components, light response components, low-temperature response components, and wound response components. Furthermore, using the PlantRegMap to analyze the binding elements on the promoter, the results showed that the promoters of ZmMT2, ZmMT7, and ZmMT9 have a large number of ERF binding components, the promoters of ZmMT2 and ZmMT6 have a large number of MYB or MYB-related TF binding components, while the promoter of ZmMT9 has a large number of NAC TF binding components, the promoter of ZmMT7 has a large number of LBD binding components, the promoters of ZmMT3, ZmMT5 and ZmMT9 has a large number of WRKY binding elements. Hormones are able to decreasing levels of ROS or peroxidation for plants to against heavy metal stress[66]. Moreover, studies reported that hormone can enhance plant resistance to heavy metal stress[66]. For example, exogenous melatonin, epibrassinolide and jasmonic acid could enhance the antioxidant capacity of rice by inducing antioxidant enzyme activity, remove excess ROS, and thereby alleviate the toxicity of Cd and As to rice[67]. Thus, expressions of ZmMTs may regulate by hormones. These gave us a strong hint that molecular regulation of ZmMTs highly relies on the crosstalk among hormones, stress, and TFs (Fig. 3 and Additional file 1: Table S3). It also indicated that the regulation of ZmMTs by hormones may directly affect their promoters, or regulation is initiated indirectly by ERF, MYB, WRKY, or other TFs.
Spatio-temporal expression and responses to heavy metal ions of ZmMTs
RNA-seq data of 23 tissues of the spanning vegetative and reproductive stages of maize development showed that ZmMTs have five expression patterns. Among them, ZmMT1 and ZmMT8 have higher expression in all tissues, indicating that these two genes may have important regulatory roles in the whole plant growth and development cycle of maize. ZmMT9 is highly expressed in germination kernels, pericarp, aleurone, mature leaf, primary root, root cortex, root elongation, root meristem zone, second root 7–8 Days, spikelet, silk, indicating that ZmMT9 may play an important regulatory role in the development of female organs, the development of root tissues and seed germination. Both ZmMT5 and ZmMT6 are highly expressed in kernels. ZmMT4 and ZmMT7 are mainly expressed in the seeds after fertilization, especially in the 38 DAP embryo, indicating that these two genes may be involved in the regulation of fertilization and seed maturity. ZmMT2 and ZmMT3 are expressed in low amounts in most tissues. ZmMT2 is expressed in internodes and meristems higher than other tissues, indicating that ZmMT2 may regulate the growth of aerial parts. ZmMT3 is only expressed relatively high in root cortex, which shows that ZmMT3 may be involved in regulating the growth and development of the underground part. Under different tissues and different heavy metal stress conditions, each gene has a different response pattern, indicating that plants have different regulatory mechanisms for different heavy metal stresses. In root, ZmMT1, ZmMT6, ZmMT8 are all activated by Cu stress, however, ZmMT6 is also sensitive to Cd stress. In stem, ZmMT1 and ZmMT8 are not activated by metal ion stressed. While in leaf, expression of ZmMT1, ZmMT6, and ZmMT8 are suppressed by stress. But ZmMT3, ZmMT7, and ZmMT9 are up-regulated in leaf under Cu stress. It is showed that most of ZmMTs in root are important for responding to heavy metal stresses, whereas the situation in stem and leaf are totally differed.
Conclusions
MT proteins play an important role in the growth and development of plants and the regulation of stress response to heavy metals. The abundant regulatory elements and TF binding sites on the MT gene promoters result in a wide range of spatiotemporal expressions and different responses to heavy metal signals. Although the MT family genes have been identified and studied in different plants over the past two decades, the MT genes for maize has not been fully analyzed so far. Here, we provide bioinformatic analysis and quantitative analysis of expression level by genome-wide analysis of maize MT family. In summary, a large number of hormone regulatory elements and hormone-related transcription factor binding sites are resided on the ZmMT promoters. The specific expression of ZmMTs in different tissues of maize and the response to different heavy metal stresses are revealed that the role of MT in plant growth and development, and stress resistance to heavy metals.
Availability of data and materials
Please contact author for data requests.
Abbreviations
- MT:
-
Metallothionein
- TF:
-
Transcription factor
- chlo:
-
Chloroplast
- mito:
-
Mitochondrion
- Cd:
-
Cadmium
- Cu:
-
Copper
- Pb:
-
Lead
- GSH:
-
Glutathione
- PCs:
-
Phytochelatins
References
Dubey S, Shri M, Gupta A, Rani V, Chakrabarty D. Toxicity and detoxification of heavy metals during plant growth and metabolism. Environ Chem Lett. 2018;16:1169–92.
Hall JL. Cellular mechanisms for heavy metal detoxification and tolerance. J Exp Bot. 2002;53:1–11.
Zhao FJ, Mcgrath SP, Meharg AA. Arsenic as a food chain contaminant: mechanisms of plant uptake and metabolism and mitigation strategies. Annu Rev Plant Biol. 2010;61:535–59.
Xu WZ, Dai WT, Yan HL, Li S, Shen Yan HL, Chen YH, Xu H. Arabidopsis NIP3;1 plays an important role in arsenic uptake and root-to-shoot translocation under arsenite stress conditions. Mol Plant. 2015;8:722–33.
He ZY, Yan HL, Chen YS, Shen HL, Xu WX, Zhang HY, Shi L, Zhu YG, Ma M. An aquaporin PvTIP4;1 from pterisvittata may mediate arsenite uptake. New Phytol. 2016;209:746–61.
Shruti M, Dubey RS. Heavy metal uptake and detoxification mechanisms in plants. Int J Agr Res. 2006;1:122–41.
Gautam N, Verma PK, Verma S, Tripathi RD, Trivedi PK, Adhikari B, Chakrabarty D. Genome-wide identification of rice class I metallothionein gene: tissue expression patterns and induction in response to heavy metal stress. Funct Integ Genomi. 2012;12:635–47.
Chi CN, Ding GH. Research progress of the molecular biology in heavy metal tolerance of plants. Biotechnol Bull. 2017;33:6–11 (in Chinese).
Margoshes M, Vallee BL. A cadmium protein from equine kidney cortex. J Am Chem Soc. 1957;79:4813–4.
Leszczyszyn OI, Imam HT, Blindauer CA. Diversity and distribution of plant metallothioneins: a review of structure, properties and functions. Metallomics Integr Biometal Sci. 2013;5:1146–69.
Domènech J, Mir G, Huguet G, Capdevila M, Molinas M, Atrian S. Plant metallothionein domains: functional insight into physiological metal binding and protein folding. Biochimie. 2006;88:583–93.
Prasad MNV. Metallothioneins, metal binding complexes and metal sequestration in plants[M]. Heavy metal stress in plants: From biomolecules to ecosystems, Prasad MNV, Berlin, Heidelberg,Springer Berlin Heidelberg. 2004; 47–83.
Lane B, Kajioka R, Kennedy T. The wheat-germ Ec protein is a zinc-containing metallothionein. Biochem Cell Biol. 1987;65:1001–5.
Kohler A, Blaudez D, Chalot M, Francis M. Cloning and expression of multiple metallothioneins from hybrid poplar. New Phytol. 2010;164:83–93.
Hegelund JN, Schiller M, Kichey T, Hansen TH, Pedas P, Husted S, Schjoerring JK. Barley metallothioneins: MT3 and MT4 are localized in the grain aleurone layer and show differential zinc binding. Plant Physiol. 2012;159:1125–37.
Hassinen VH, Tuomainen M, Peraniemi S, Schat H, Karenlampi SO, Tervahauta AI. Metallothioneins 2 and 3 contribute to the metal-adapted phenotype but are not directly linked to Zn accumulation in the metal hyperaccumulator, Thlaspicaerulescens. J Exp Bot. 2009;60:187–96.
Giritch A, Ganal M, Stephan UW, Bäumlein H. Structure, expression and chromosomal localisation of the metallothionein-like gene family of tomato. Plant Mol Biol. 1998;37:701–14.
Pagani MA, Tomas M, Carrillo J, Bofill R, Capdevila M, Atrian S, Andreo CS. The response of the different soybean metallothionein isoforms to cadmium intoxication. J Inorg Biochem. 2012;117:306–15.
Zhou Y, Chu P, Chen H, Li Y, Liu J, Ding Y, Tsang EWT, Jiang LW, Wu KQ, Huang SZ. Overexpression of Nelumbo nucifera metallothioneins 2a and 3 enhances seed germination vigor in Arabidopsis. Planta. 2012;235:523–37.
Roosens NH, Leplae R, Bernard C, Verbruggen N. Variations in plant metallothioneins: the heavy metal hyperaccumulator Thlaspicaerulescensas a study case. Planta. 2005;222:716–29.
Schor-Fumbarov T, Goldsbrough PB, Adam Z, Elisha TO. Characterization and expression of a metallothionein gene in the aquatic fern Azolla filiculoides under heavy metal stress. Planta. 2005;223:69–76.
Liu P, Goh CJ, Loh CS, Pua EC. Differential expression and characterization of three metallothionein-like genes in Cavendish banana (Musa acuminata). Physiol Plantarum. 2002;114:241–50.
Zhou GK, Xu YF, Liu JY. Characterization of a rice class II metallothionein gene: tissue expression patterns and induction in response to abiotic factors. J Plant Physiol. 2005;162:686–96.
Zhang J, Zhang M, Tian S, Lu LL, Shohag MJI, Yang XE. Metallothionein 2 (SaMT2) from Sedum alfredii Hance confers increased Cd tolerance and accumulation in yeast and tobacco. PLoS ONE. 2014;9:e102750.
Singh RK, Anandhan S, Singh S, Patade VY, Ahmed Z, Pande V. Metallothionein-like gene from Cicer microphyllum is regulated by multiple abiotic stresses. Protoplasma. 2011;248:839–47.
Chyan CL, Lee TT, Liu CP, Yang YC, Tzen JTC, Chou WM. Cloning and expression of a seed-specific metallothionein-like protein from sesame. Biosci Biotech Bioch. 2005;69:2319–25.
Zhou J, Goldsbrough PB. Structure, organization and expression of the metallothionein gene family in Arabidopsis. Mol Gen Genet. 1995;248:318–28.
Guo W, Bundithya W, Goldsbrough PB. Characterization of the Arabidopsis metallothionein gene family: tissue-specific expression and induction during senescence and in response to copper. New Phytol. 2003;159:369–81.
Pan Y, Zhu M, Wang S, Ma GQ, Huang XH, Qiao CL, Wang R, Xu XF, Liang Y, Lu K, Li JN, Qu CM. Genome-wide characterization and analysis of metallothionein family genes that function in metal stress tolerance in Brassica napus L. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19082181.
Huang Y, Fang Y, Long X, Liu LY, Wang J, Zhu JH, Ma YY, Qin YX, Qi JY, Hu XW, Tang CR. Characterization of the rubber tree metallothionein family reveals a role in mitigating the effects of reactive oxygen species associated with physiological stress. Tree Physiol. 2018;38:911–24.
Pakdee O, Songnuan W, Panvisavas N, Pokethitiyook P, Yokthongwattana K, Meetam M. Functional characterization of metallothionein-like genes from Physcomitrella patens: expression profiling, yeast heterologous expression, and disruption of PpMT1.2a gene. Planta. 2019;250:427–43.
Bulgarelli RG, Araujo P, Tezotto T, Mazzafera P, Andrade SAL. Expression of metallothionein genes in coffee leaves in response to the absence or excess of Cu and Zn. Theor Exp Plant Phys. 2016;28:371–83.
Kim YO, Kang H. Comparative expression analysis of genes encoding metallothioneins in response to heavy metals and abiotic stresses in rice (Oryza sativa) and Arabidopsis thaliana. Biosci Biotech Bioch. 2018;82:1656–65.
Zhou G, Xu Y, Li J, Yang LY, Liu JY. Molecular analyses of the metallothionein gene family in rice (Oryza sativa L.). J Biochem Mol Biol. 2006;39:595–606.
Chen Yun. Cloning and functional identification of sugarcane metallothionein family genes [D]. Fujian Agriculture and Forestry University, 2016.
Kumar G, Kushwaha HR, Panjabi-Sabharwal V, Kumari S, Joshi R, Karan R, Mittal S, Pareek SLS, Pareek A. Clustered metallothionein genes are co-regulated in rice and ectopic expression of OsMT1e-P confers multiple abiotic stress tolerance in tobacco via ROS scavenging. BMC Plant Biol. 2012;12:107.
Wong HL, Sakamoto T, Kawasaki T, Umemura K, Shimamoto K. Down-regulation of metallothionein, a reactive oxygen scavenger, by the small GTPase OsRac1 in rice. Plant Physiol. 2004;135:1447–56.
Xue T, Li X, Zhu W, Wu C, Yang G, Zheng C. Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J Exp Bot. 2008;60:339–49.
Steffens B, Sauter M. Epidermal cell death in rice is confined to cells with a distinct molecular identity and is mediated by ethylene and H2O2 through an autoamplified signal pathway. Plant Cell. 2009;21:184–96.
Steffens B, Geske T, Sauter M. Aerenchyma formation in the rice stem and its promotion by H2O2. New Phytol. 2011;190:369–78.
Rajhi I, Yamauchi T, Takahashi H, Nishiuchi S, Shiono K, Watanabe R, Mliki A, Nagamura Y, Tsutsumi N, Nishizawa NK, Nakazono M. Identification of genes expressed in maize root cortical cells during lysigenous aerenchyma formation using laser microdissection and microarray analyses. New Phytol. 2011;190:351–68.
Rizwan M, Ali S, Qayyum MF, Ok YS, Zia-ur-Rehman M, Abbas Z, Hannan F. Use of Maize (Zea mays L.) for phytomanagement of Cd-contaminated soils: a critical review. Environ Geochem Health. 2017;39:259–77.
Xu J, Sun JH, Du LG, Liu XJ. Comparative transcriptome analysis of cadmium responses in Solanum nigrum and Solanum torvum. New Phytol. 2012;196:110–24.
Zhou Q, Guo JJ, He CT, Shen C, Huang YY, Chen JX, Guo JH, Yuan JG, Yang ZY. Comparative transcriptome analysis between low- and high-cadmium-accumulating genotypes of Pakchoi (Brassica chinensis L.) in Response to cadmium stress. Environ Sci Technol. 2016;50:6485–94.
Weber M, Trampczynska A, Clemens S. Comparative transcriptome analysis of toxic metal responses in Arabidopsis thaliana and the Cd2+-hypertolerant facultative metallophyte Arabidopsis halleri. Plant Cell Environ. 2006;29:950–63.
He F, Liu QQ, Zheng L, Cui YQ, Shen ZG, Zheng LQ. RNA-Seq analysis of rice roots reveals the involvement of post-transcriptional regulation in response to cadmium stress. Front Plant Sci. 2016;6:1136.
Xie Y, Ye S, Wang Y, Xu L, Zhu XW, Yang JL, Feng HY, Yu RG, Karanja B, Gong YQ, Liu LW. Transcriptome-based gene profiling provides novel insights into the characteristics of radish root response to Cr stress with next-generation sequencing. Front Plant Sci. 2015;6:202.
Tang MF, Mao DH, Xu LW, Li DY, Song SH, Chen CY. Integrated analysis of miRNA and mRNA expression profiles in response to Cd exposure in rice seedlings[J]. BMC Genomics. 2014;15:835.
Rui HY, Zhang XX, Shinwari KI, Zheng LQ, Shen ZG. Comparative transcriptomic analysis of two Vicia sativa L. varieties with contrasting responses to cadmium stress reveals the important role of metal transporters in cadmium tolerance. Plant Soil. 2018;423:241–55.
Yao YN, Xiao XL, Ou YB, Wu XL, Xu G. Root transcriptome analysis on the grape genotypes with contrast translocation pattern of excess manganese from root to shoot. Plant Soil. 2015;387:49–67.
Chen YK, Zhi JK, Zhang H, Li J, Zhao QH, Xu JH. Transcriptome analysis of Phytolacca americana L. in response to cadmium stress. PLoS ONE. 2017;12:e184681.
Peng H, He XJ, Gao J, Ma HX, Zhang ZM, Shen YO, Pan GT, Lin HJ. Transcriptomic changes during maize roots development responsive to Cadmium (Cd) pollution using comparative RNAseq-based approach. Biochem Bioph Res Com. 2015;464:1040–7.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13:1194–202.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157.
Wang DP, Zhang YB, Zhang Z, Zhu J, Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genom Proteom Bioinf. 2010;8:77–80.
Morton BR, Gaut BS, Clegg MT. Evolution of alcohol dehydrogenase genes in the palm and grass families. Proc Natl Acad Sci USA. 1996;93:11735–9.
Quraishi UM, Abrouk M, Murat F, Pont C, Foucrier S, Desmaizieres G, Confolent C, Rivière N, Charmet G, Paux E, Murigneux A, Guerreiro L, Lafarge S, Gouis JL, Feuillet C, Salse J. Cross-genome map based dissection of a nitrogen use efficiency ortho-meta QTL in bread wheat unravels concerted cereal genome evolution. Plant J. 2011;65:745–56.
Walley JW, Sartor RC, Shen ZX, Schmitz RJ, Wu KJ, Urich MA, Nery JR, Smith LG, Schnable JC, Ecker JR, Briggs SP. Integration of omic networks in a developmental atlas of maize. Science. 2016;353:814–8.
Van SS, Witters N, Meers E, Peene A, Michels E, Adriaensen K, Ruttens A, Vangronsveld J, Du LG, Wierinck I, Dael MV, Passel SV, Tack FMG. Safe use of metal-contaminated agricultural land by cultivation of energy maize (Zea mays). Environ Pollut. 2013;178:375–80.
Meers E, Van SS, Adriaensen K, Ruttens A, Vangronsveld J, Du LG, Witters N, Thewys T, Tack FM. The use of bio-energy crops (Zea mays) for “phytoattenuation” of heavy metals on moderately contaminated soils: a field experiment. Chemosphere. 2010;78:35–41.
Xu XH, Liu CY, Zhao XY, Li RY, Deng WJ. Involvement of an antioxidant defense system in the adaptive response to cadmium in maize seedlings (Zea mays L.). Bull Environ Contam Toxicol. 2014;93:618–24.
Kaul S, Koo HL, Jenkins J, Rizzo M, Rooney T, Tallon LJ, Feldblyum T, Nierman W, Benito MI, Lin XY, et al. Analysis of the genome sequence of the flowering plant, Arabidopsis thaliana. Nature. 2000;408:796–815.
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 2002;296:92–100.
Masood A, Khan MI, Fatma M, Asgher M, Per TS, Khan NA. Involvement of ethylene in gibberellic acid-induced sulfur assimilation, photosynthetic responses, and alleviation of cadmium stress in mustard. Plant Physiol Biochem. 2016;104:1–10.
Zhang SN. Effects of exogenous plant hormones on tolerance of rice and rape to cadmium and arsenic stress and its physiological mechanism. Chinese Academy of Agricultural Sciences; 2021.
Acknowledgements
The authors are grateful to Mingzhi Li (Bio&Data Biotechnologies Co. Ltd., Guangzhou) for providing technical assistance.
Funding
This research was funded by Natural Science Foundation of Anhui Province, China(No. 1908085MC69), National Natural Science Foundation of China (No. 31971895).
Author information
Authors and Affiliations
Contributions
CHG. and QQC. initiated, designed, conducting and monitored the experiment. CHG., KG., ZLT, and LXZ performed the RT-qPCR. HXY, TDG and JYZ analyzed the data. CHG. and KG wrote the first draft of the manuscript. CHG and QQC revised and edited the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The detailed components of the nutrient solution. Table S2. Primers for qRT-PCR. Table S3. Identified ZmMTgenes from maize and their related information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gao, C., Gao, K., Yang, H. et al. Genome-wide analysis of metallothionein gene family in maize to reveal its role in development and stress resistance to heavy metal. Biol Res 55, 1 (2022). https://doi.org/10.1186/s40659-021-00368-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40659-021-00368-w