
Abstract
In addition to the anti-infection response, neutrophils are linked to tumor progression through the secretion of inflammation components and neutrophil extracellular traps (NETs) formation. NET is a web-like structure constituted by a chromatin scaffold coated with specific nuclear and cytoplasmic proteins, such as histone and granule peptides. Increasing evidence has demonstrated that NETs are favorable factors to promote tumor growth, invasion, migration, and immunosuppression. However, the cell–cell interaction between NETs and other cells (tumor cells and immune cells) is complicated and poorly studied. This work is the first review to focus on the intercellular communication mediated by NETs in cancer. We summarized the complex cell–cell interaction between NETs and other cells in the tumor microenvironment. We also address the significance of NETs as both prognostic/predictive biomarkers and molecular targets for cancer therapy. Moreover, we presented a comprehensive landscape of cancer immunity, improving the therapeutic efficacy for advanced cancer in the future.
Similar content being viewed by others

Introduction
Neutrophils are the most abundant type of leukocytes in human peripheral blood serving as the front line in innate immunity [1]. Besides infections, ongoing efforts have expanded the roles of neutrophils in a wide range of human diseases, such as thrombosis, auto-immune diseases, pulmonary diseases, and cancer progression [2]. The contribution of tumor-associated neutrophils (TANs) in cancer remains elusive, as a result of the complex microenvironment of cancer [3]. Research using animal models has shown that TANs can be polarized into an anti-tumor (N1) or pro-tumor (N2) subtype, which is driven by the state of TGF-β [4].
In 2004, Brinkmann et al. discovered that neutrophil suicide could release web-like structures decorated with depolymerized chromatin and antimicrobial molecules, named as NETs. They elicited NETs using PMA and IL-8 in vitro and demonstrated that NETs could resist virulence factors and kill bacteria [5]. Apart from PMA and IL-8, additional stimuli were found to be responsible for the release of NETs, including protozoa, bacteria, fungi, IFN-α/IFN-γ/C5a, GM-CSF/C5a, lipopolysaccharide (LPS), antibody-antigen complexes, activated platelets, and calcium ionophores [6]. Within NETs, there are several proteins coated in the decondensed DNA, such as histones, neutrophil elastase (NE), myeloperoxidase (MPO) and cathepsin G [7,8,9].
Like the role of TANs, increasing evidence has recently revealed the role of NETs as a fundamental part in cancer progression [10,11,12]. The prior reviews mainly focused on the crucial role of NETs in contributing to the development and progression of cancer. Consistent with the previous studies, our research validated the role of NET as an unfavorable prognosis biomarker in most types of cancer, by developing a panel of genes signature as NET-score [13]. Nevertheless, this review first summarizes the intercellular communication between NETs, tumor cells, and immune cells in carcinogenesis. We aimed to figure out the cellular interplay and molecular pathway mechanism that regulate the innate immune and adaptive immune response in malignancies. The potential of NETs as diagnostic and prognostic markers and novel treatment targets is also discussed in this review.
The interplay between the tumor cell and NETs
The role of NETs in pro- and anti-tumorigenic functions remains unclear in different cancers. Very few articles now illustrated the in-vitro antitumor effect of NETs. An experiment in vitro showed that NETs could impede growth and induce apoptosis in colorectal cancer cells [14]. Consistently, Schedel F et al. found NETs inhibiting the migration and necrosis of melanoma cells in vitro [15]. However, NETs in the tumor microenvironment (TME) mostly exert promotion in the growth and progression of cancer, in the onset and spread of tumor metastasis, and in the poor response of anti-tumor therapy [12, 16,17,18,19,20,21,22,23]. The bond between cancer-cells and NETs was revealed in the Ewing sarcoma at first, in which the NETs were correlated with poor prognosis for patients [12]. Compared to control mice, the neutrophils in the tumor-bearing mice represented an increased ability to spontaneously form NETs [24, 25], which was associated with endothelial-to-mesenchymal transition (EMT) driving and cancer metastasis [25,26,27]. The cancer cell-derived factors supporting the NETosis include mainly IL-8, G-CSF, GROα/β, and CXCR1/2 chemokine receptor agonists [17, 28]. NETs also strengthen the metastatic potential of cancer cells by other mechanisms, including accumulation in the pre-metastatic niche or the circulation for entrapping the circulating tumor cells (CTCs) [17, 29].
Szczerba BM et al. manipulated single-cell RNA sequencing (scRNA-seq) and investigated the ligand/receptor pairs of the CTC–neutrophil cluster. They hypothesized that the expression of VCAM1 is a molecular feature possibly defining the CTC-neutrophil cluster formation in breast cancer [30]. Understanding the ligand/receptor pairing may explore new targets in cancer therapy.
Recently, several pieces of research demonstrated the molecular mechanism of the intracellular network of tumor cells and NETs. The most studied example of the ligand/receptor interaction was the TLR family in the tumor cell and NET-associated factors. The TLR family is characterized as the essential part of the innate immune and could recognize pathogen-associated molecular patterns (PAMPs) [31]. TLR receptors are ubiquitously expressed both in tumor and immune cells [32, 33], and exert a dual role in cancer [34,35,36,37]. To date, several TLR agonists have shown inspiring results for their survival benefits combined with immune vaccination, immune checkpoint inhibitors, and chemotherapy in clinical trials, especially for glioma [38, 39]. Nonetheless, TLR agonist-associated infection and the help of TLR overexpression in the carcinogenesis or tumor progression should be emphasized [33, 36, 40]. Higher levels of intratumor NETs and preoperative serum MPO-DNA as a marker of NETs were correlated with shorter survival in metastatic colorectal cancer. Mechanistically, NE as NETs-derived stimulatory factor directly activated the TLR4 pathway on tumor cells and subsequently upregulated Peroxisomes proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α), driving mitochondrial homeostasis and favoring the tumor growth [41]. In bladder cancer, Shinde-Jadhav S et al. found the level of NETs was increased after radiation therapy (RT), which contributed to tumor radiotherapy resistance. They further demonstrated that the activated formation of NETs was associated with HMGB1 via a TLR4-dependent manner, and inhibiting NETs or HMGB1 could improve radiation response [23]. For diffuse large B-cell lymphoma (DLBCL) patients, Nie M et al. investigated the mechanism of interleukin-8 (IL-8), secreted by lymphoma cells, binding to C-X-C Motif Chemokine Receptor 2 (CXCR2) on the cell-surface of neutrophil and inducing NET formation. Furthermore, an increased level of NETs activated TLR9 on the lymphoma cells, contributing to NFkB, STAT3, and p38 downstream pathways activation. The novel cross-talk as IL8-CXCR2-TLR9 axis augmented the tumor progression in DLBCL [21]. Tohme et al. proposed that HMGB1 released from NETs assisted in the TLR9 activation. TLR9 promoted colorectal cancer cell proliferation, migration, or invasion by activating the MAP kinase pathways [42]. The neutrophils derived from metastatic hepatocellular carcinoma (HCC) harbored an up-regulated capacity of producing NETs, compared with those in healthy adults. It was further investigated that NETs enhanced the invasion capacity of trapped tumor cells through the activation of the TLR4/9 receptor and the phosphorylation of P65 and cyclooxygenase-2 (COX2) overexpression. The direct inhibition of the TLR4/9-COX2 pathway wrecked the NET-driven metastatic potential [43]. Apart from the TLR ligand-associated pathway, CCDC25 is a transmembrane protein on the breast cancer cells, which could interact with the NET-DNA complex directly. As a result, it enhanced tumor cell motility and tumor metastasis by activating the downstream pathway including integrin-linked kinase (ILK) and β-Parvin. CCDC25-knockout cells abrogated the NET-mediated potential metastasis [11]. Cell-to-cell adhesion in cancer is complex and involved in each step of tumor progression. It enables tumor cells to loosen from the primary tumor mass and enhances cell attachment to the metastatic site [44, 45]. In the cell adhesion process, the integrin ligands (a combination of α and β subunits) determine the central role governing cancer cell migration [44]. In a panel of tumor cell lines, Monti M et al. revealed that NETs could adhere to tumor cells with high levels of integrin α5β1, αvβ3, and αvβ5 [46]. Tumor-derived integrin β1 promoted the co-localization of NETs and tumor cells in vivo and in vitro. Najmeh S et al. proposed the hypothesis that NETs captured CTC through the mediator integrin β1 [29]. Mechanism research demonstrated that NETs-related proteases, matrix metalloproteinase 9 (MMP9) and NE, resulted in the cleavage of laminin. The NET-remodeled laminin-111 subsequently activated the integrin a3β1 receptor on the tumor cell. The integrin a3β1 up-regulated the focal adhesion Kinase (FAK), extracellular signal-regulated kinase (ERK), and yes-associated Protein (YAP) and promoted the dormant tumor cell awaken [10].
In conclusion, there is growing evidence from ligand/receptor pair analysis that NETs primarily promote the proliferation, adhesion, and metastatic capacity of tumor cells (shown in Table 1). Meanwhile, the molecular mechanism of its anti-tumorigenic effects requires further exploration. NETs directly or the NETs-associated factors interact with the receptors on the tumor cells and thus alter the tumor cell function. Nevertheless, there are few studies on the mechanism of interplay between tumor cells and NETs, especially for their ligand-receptor pairs. Furthermore, targeting the ligand-receptor pairing or specific kinases rather than neutrophils or tumor cells could be a potential strategy for anti-tumor treatment.
Potential interactions between the macrophage and NETs
Macrophages polarize to activated pro-inflammatory M1 and anti-inflammatory M2 phenotypes depending on the microenvironment stimuli [47, 48]. M1 macrophages exert pro-inflammatory effects through secreted cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor (TNF). In contrast to M1 phenotype macrophages, M2 macrophages are predominantly correlated with resolving inflammation and promoting tissue repair [49]. Tumor-associated macrophages (TAMs) are typically altered into M2 and mediate immune dysfunction in the TME [50]. Similar to neutrophils, macrophages also release web-like structures as extracellular traps (ETs) [51]. Macrophage extracellular traps (METs) exert tumor-promoting roles by assisting tumor growth, progression, and metastasis [51,52,53]. For pancreatic neuroendocrine tumors (pNETs), NETs and METs were deemed as the independent prognosis indicators for recurrence-free survival (RFS). However, no significant correlations were founded between NETs and METs in this research. It was postulated that NETs and METs were regulated by different mechanisms [54]. Zhang L et al. delineated the ability of NET to induce the migration and invasion of lung adenocarcinoma cells in vitro, which is partly dependent on macrophages [55]. Up to now, the molecular mechanism between NETs, macrophages, and METs is not fully described in cancer. To spur new ideas in cancer, we briefly introduced the recent studies, which uncovered the NETs-macrophage interaction in non-neoplastic diseases [56,57,58].
In response to infection, the caspase-1 dependent cell death pyroptosis is another regulated defending way in addition to generating ETs [59]. The novel mechanism was identified as NET-related HMGB1 activated the receptor for advanced glycation end products (RAGE) pathway signaling and subsequently trigger macrophage pyroptosis in sepsis [56]. Li H et al. indicated the interaction between NETs and macrophage pyroptosis, aggravating the inflammation of acute respiratory distress syndrome (ARDS) [60]. The inflammatory microenvironment plays an important role in all stages of tumor development and progression. It is necessary to further understand whether the interaction between NET and macrophages has the effect of amplifying the inflammatory response within the tumor and accelerating tumor progression.
A series of research investigated whether NETs could induce a cellular response in macrophage differentiation. After stimulation with low-density granulocyte (LDG)-derived NETs in the coronavirus disease 2019 (COVID-19), macrophages were characterized by supernatant proinflammatory cytokines secretion [61]. The role of NETs-related macrophage inflammatory phenotype polarization was similar in diabetic mice, contributing to atherosclerosis progression. Apart from the inflammasome-associated markers overexpression in NETs+ regions, the upregulation of the glycolytic pathway also symbolized a shift toward an M1-like phenotype in the atherosclerosis plaque area [62, 63]. EGF-like repeats and discoidin I-like domain 3 (EDIL3) were previously reported in the inflammatory regulation and neutrophil recruitment inhibition, through the interaction with lymphocyte function-associated antigen 1 (LFA1) or intercellular adhesion molecules (ICAMs) [64, 65]. EDIL3 was negatively associated with neutrophil recruitment and macrophage expansion in myocardial infarction (MI). The DNA moiety of NETs then licensed a switch towards M1-like macrophage polarization in the deficiency of EDIL3. Researchers further validated the mechanistic evidence that NETs induced inflammatory macrophage polarization via the TLR9 pathways, exerting DNA sensors to transduce NETs-macrophage interactive signals [66]. The evidence described above shows that the ubiquitously expressed TLR receptors are an important bridge for mediating NET and tumor cell interaction for cancer Therefore, it is crucial to understand whether TLR receptors are involved in mediating NET and macrophage interactions in tumor diseases. After inflammation declined in the late stage of wound healing, NETs-treated anti-inflammatory macrophages initiated the fibrotic cascades, resulting in postoperative epidural fibrosis formation [67]. Taken together, NETs may exert a dual role in the function switch of macrophages.
Based on the finding that the macrophage amount was negatively correlated with the localized NETs density for patients with abdominal aortic aneurysm, Haider P et al. hypothesized that macrophage may clear the NETs in vivo [68]. NET clearance refers to the phagocytosis of the NET by macrophage. Apart from the inflammatory macrophage phenotype polarization, aggregation of NETs with reduced clearance by macrophages may give rise to an ongoing inflammatory response. Mechanistically, MMP12 was validated as a key mediator for macrophages to remove NETs, preparing for inflammation degradation and restoring immune homeostasis [69]. Furthermore, AMPK-associated pathway activation is also an important mechanism for NET clearance. Chiang et al. explored the mechanism that the 13-series resolvins (RvTs) enhanced NETs clearance by macrophages through cyclic adenosine monophosphate (cAMP) / protein kinase A (PKA) / AMP-activated protein kinase (AMPK) axis, providing a molecular mechanism for inflammation resolution [58]. It was further illustrated that the restoration of AMPK in macrophages could recover the NET clearance ability [70].Additionally, the inhibitors preventing NETs formation abrogated the proinflammatory macrophage recruitment, macrophage pyroptosis, M1-like phenotype polarization, and macrophage-associated NET clearance [60, 62, 70,71,72,73].
This evidence identified that the crosstalk between NETs and macrophages was involved as a fundamental part of non-neoplastic disease progression. There is a lack of cell–cell interaction analysis between NET and macrophages in cancer. However, the above results of these studies in non-neoplastic disease provided potent interest or research ideas for the tumor in the future. Further questions remain as: Does NET-associated inflammation influences macrophage function in cancer? What is the mechanism for the cellular mechanism for NETs-macrophage interaction in cancers? Could NETs-macrophages molecular network be a potential novel treatment target for the tumor?
Cross-talk between NETs and lymphocytes
Lymphocytes including T lymphocytes, B lymphocytes, and natural killer (NK) cells, serve as a crucial mechanism to mediate the immune system hemostasis and regulate immune tolerance [74,75,76].
Recent findings in cancer demonstrated that tumor-specific lymphocytes primarily presented a dysfunctional state, shaped by the immunosuppressive tumor microenvironment, and thus promoted tumor escape and therapy resistance [75, 77, 78]. Especially T lymphocytes and NK cells exerted a fundamental part in tumor development and progression. There is increasing evidence that the complex interaction between NETs and lymphocytes may critically involve the immune function regulation in the tumor.
First, NETs exerted an inhibitory role in the amount and function of CD8+ T cells. The infiltrating rate of CD8+ T lymphocytes was inversely associated with the NETs density in human solid tumors including non-small cell lung cancer (NSCLC) and bladder cancer (BC) [79]. Teijeira Á et al. illustrated that the motility of CD8+ T cells migrating across the transwell was directly weakened by NETs in vitro[28]. Apart from the cell migration motility, Kaltenmeier C et al. investigated whether NETs could mediate the T cell dysfunction and exhaustion responses. In the NETs-rich TME, the tumor-infiltrating CD8+ T lymphocytes were characterized with functional exhausted phenotype, expressing high levels of exhaustion markers, such as PD-1, LAG-3, or TIM3. The direct modulatory role of NETs on CD8+ T cell's exhaustive differentiation was validated in the co-culture experiment. By co-culturing the NETs and CD8+ T cells in vivo, the exhausted phenotype changes of CD8+ T cells were as same as that in the NETs-rich TME. This phenotype shift was further reversed with the NETs inhibitor [80]. Second, NETs were illustrated for their positive correlation with CD4+ T cell exhausted phenotype differentiation and Foxp3+ regulatory T cells (Tregs) density [80, 81]. After the co-culture with NETs, the changes in naïve CD4+ T cells that could differentiate into Tregs, which presented with activated mitochondrial oxidative phosphorylation (OXPHOS) pathway. Naïve CD4+ T cells up-regulated the Treg-associated markers, including TGFB1, ID3, and DUSP4. Meanwhile, effector T cell-related genes in the program of effector T cell (Teff) differentiation were reduced [81]. It has been recognized that naïve CD4+ T cell differentiation depends on the balance of glycolysis and oxidative phosphorylation (OXPHOS) [82]. T cells in the NETs-rich area were presented with the down-regulated functioning mitochondria, reduced glucose but up-regulated fatty acid uptake [80]. The OXPHOS inhibitor reversed NETs-associated Treg differentiation [81]. Consistent with the previous study [83], the TLR4 on the naïve CD4+ T cell exerted an essential role in mediating the Treg activation and function. It was delineated that NETs directly contacted naïve CD4+ T cells mostly through TLR4, thus prompting Treg differentiation [81].
Nevertheless, these studies mentioned above merely considered the interrelationship between T cells and NETs in cancer. The relationship between NETs and B cells is not well understood. Most of the studies in this area are concerned about autoimmune diseases. Recent research reported that B cells were drivers of chronic inflammation in Rheumatoid arthritis (RA). Activated B cells were capable to release IL-8 recruiting neutrophils to the synovium, and produce autoantibodies activating the complement pathway and promoting NETs formation [84]. Additionally, citrullinated histones in NETs acted as a continuous source of fresh antigens to B cells [85]. In Systemic Lupus Erythematosus (SLE), uptake of NETs in Lupus Nephritis (LN-NETs) by B cells was found, and LN-NETs could also stimulate Naïve B Cells to produce IgG2 in SLE [86]. Correlating with disease severity, spontaneous NETs formation was enhanced by circulating neutrophils in Bullous pemphigoid (BP) patients [87]. Mechanism research showed that NETs formation could be abrogated by blocking Fcγ receptor and/ or NADPH pathway. Additionally, elevated levels of NETs in BP patients triggered B cells differentiation into plasma cells, producing a large amount of autoantibody, and this procedure was mediated by the activation of MAPK P38 cascade [87].
The situation becomes more complicated when considering the relationship between NET, lymphocytes, tumor cells, and others. It was postulated that the anti-tumor effect of lymphocytes was compromised by the reduced contact with NETs-shielding tumor cells [28], yet, the relevant molecular interaction mechanisms need to be further explored. In conclusion, the existing studies mostly focused on T lymphocytes, and NETs had a regulatory mechanism on the T lymphocytes infiltrating and functioning in cancer. The effects of NETs on NK cells and B cells are poorly understood and deserve further investigation.
A positive feedback cycle between NETs and platelets
In addition to their well-known role in coagulation and hemostasis, accumulating evidence shows that platelets also exert a regulative role in the immune system [88,89,90]. The level of NETs is elevated in different cancers, including colorectal cancer (CRC), gastric cancer (GC), oral squamous cell carcinoma (OSCC), as well as pancreatic tumors, and the inhibition of NETs diminishes the hypercoagulability in cancer [91,92,93,94].
Increasing evidence has illustrated the role of platelet in NETs formation. Clark et al. first described platelet involved in DNA extracellular trap formation in a mouse model of sepsis. They found that platelets, activated by LPS through TLR4, could bind neutrophils and lead to their activation and NETs formation [95]. One potent mechanism of platelets-induced NETs formation seems to be the combination of P-selectin to its receptor P-selectin glycoprotein ligand-1 (PSGL-1) on their surface [96]. Animal studies demonstrated that platelets from mice with overexpressed P-selectin were more prone to generate NETs when co-incubated with neutrophils, while platelets from P-selectin knock-out mice failed to induce NETs [97]. Moreover, using anti‐P‐selectin and PSGL‐1 antibodies to abrogate the interaction between neutrophils and platelets could remarkably decrease NETs formation in the plasma of glioma patients [98]. The pro-inflammatory molecule platelet-derived high mobility group box 1 (HMGB1), secreted from activated platelets has also been shown to facilitate NET formation [99]. According to this study, platelets from colorectal cancer patients stimulated neutrophils to release NETs, which could be abolished by the absence of HMGB1 [92]. Meanwhile, platelets acted as carriers of tumor-derived exosomes, which in turn contributed to the generation of NETs [90].
It was shown that platelets promote neutrophils to generate NET and its components,which in turn activate platelets as wel l[100]. NETs could function in procoagulant response by providing a scaffold for platelets, red blood cells, extracellular vesicles, and pro-coagulant molecules [101,102,103]. NETs could convert platelets to a procoagulant phenotype and stimulate the activation and aggregation of platelets by upregulating phosphatidylserine and P-selectin expression on its membrane [98, 104]. Brian A. Boone et al. found that DNA and its receptor for advanced glycation end products (RAGE) were necessary for NETs-relevant platelet aggregation and RAGE KO tumor-bearing mice exhibited decreased platelet aggregation [93]. Another study showed that DNase I treatment could attenuate platelet aggregation, while some platelets still adhered to the glass slides. Histones that are the most abundant proteins in NETs or NE are sufficient to induce platelet aggregation [104]. Co-culture platelets with histones H3 and H4 promote its aggregation, whereas histones 1H, H2A, and H2B had no such effect [102]. More specifically, histone-enhanced platelet aggregation by recruiting fibrinogen and histone-dependent platelet activation seems to be mediated by the signaling pathway of TLR2 and TLR4 receptors, via the transcription factor NF-κB [105].
Holistically, activated platelets simultaneously interplay with neutrophils, promoting NETs formation. NETs provide a scaffold for platelets and induce activation and aggregation of platelet via their complex components, thus generating a positive feedback cycle to each other.
NETs as a valuable marker in cancer from the clinical perspective
Several techniques for the detection of NETs showed promising clinical applications for diagnosis, therapeutic response, and prognosis. ELISA technique was the most commonly acknowledged to detect the circulating NETs-associated complexes, allowing the quantitative assessment of NETs. In certain studies, the circulating level of NET-derived DNA was measured as MPO-DNA, NE-DNA, or circulating DNA [11, 106,107,108]. Apart from NET-related DNA complex, the circulating H3Cit level was also identified as the number of NETs [109, 110]. Meanwhile, circulating MPO-DNA, NE-DNA, and H3Cit were more specific for NETs quantification than circulating DNA alone [107]. Based on the evidence of a cohort of 283 gastric adenocarcinoma (GAC) patients, it seemed that both the serum and plasma of blood samples could all be employed for NETs detection [108]. The immunohistochemical (IHC) technique was also used to measure NET formation in the primary tumor lesion or metastatic site of the tumor tissue sample [79, 111]. The NET formation was identified as the neutrophils positive for the H3Cit signal [112, 113]. In some cases, the NETs level, measured as other NETs-specific proteins like MPO, NE, and so on, has been applied as a surrogated marker of NETs [10, 13, 103].
The increment of circulating DNA in plasma, considered a specific marker of NETs in this research, was founded in cancer-related stroke patients [114]. Nevertheless, this data should be cautiously interpreted due to the circulating DNA also involving in the apoptotic, necrotic, and so on [115]. NET-derived proteins like MPO or NE could bind to DNA in the circulating system. For both esophagogastric and lung adenocarcinoma, the level of circulating MPO-DNA was elevated compared to healthy people [106]. According to the analysis of pancreatic adenocarcinoma patients, the level of circulating MPO-DNA before treatment was correlated positively with the clinical stage [19]. The expression of serum MPO-DNA was validated as a predictor of liver metastasis for early-stage breast cancer patients [11]. Tohme S et al. revealed that metastatic colorectal cancer patients with elevated levels of serum MPO-DNA after liver resection surgery were more likely to have a reduction in disease-free survival (DFS) [42]. Yazdani HO et al. also investigated that the pre-operatively serum MPO-DNA complexes levels increased in proportion to the clinical outcome, observing the added NETs level in patients with shorter DFS and overall survival [41]. In addition, the serum MPO-DNA complex level was confirmed to monitor the HER2 inhibitor-associated vasculitis activity from a prospective cohort of breast cancer [116]. Compared with localized breast cancer, the levels of plasma NE-DNA complexes were higher in regional and distant stages [117]. As a specific NETs biomarker, serum NE-DNA showed better diagnostic efficiency compared with other common clinical biomarkers in gastric Adenocarcinoma, like carbohydrate antigen 19–9 (CA19-9) and carcinoembryonic antigen (CEA). It was identified that serum NE-DNA increased along with the existence of lymph node metastasis. The baseline serum level of NETs was inversely correlated with PFS for GC patients with negative HER2 status. Prompted by the above evidence, NETs quantified by NE-DNA complexes could be identified as an effective diagnostic and prognostic risk factor value in GC [108].
Citrullinated histone H3 (H3Cit) is a representative marker of chromatin decondensation during the NET formation process. It was reported that a significant increment in circulating H3Cit was directly associated with poor clinical outcomes in a panel of tumors [109]. Grilz E et al. collected the venous blood samples of 957 patients with cancer and performed a median of 666 days follow-up, revealing that the increased plasma H3Cit level was in an independent correlation with higher cancer mortality (HR = 1.1, P < 0.001) [110]. In a cohort enrolling 317 pancreatic ductal adenocarcinoma patients, the amounts of tumor-infiltrating NETs which were quantified by the IHC staining for H3Cit was correlated with RFS and OS, regardless of the state of neutrophil infiltration in the tumor. What’s more, combined with NETs, the diagnostic accuracy was improved by the TNM staging system in pancreatic cancer [113].
In our previous analysis, we first constructed a panel of genes signature as NET-score using the RNA-sequencing data of The Cancer Genome Atlas (TCGA) pan-cancer cohort and measured the NETs density in tumor tissue through IHC validation. NET was correlated with unfavorable prognosis in most types of cancer, like Kidney renal clear cell carcinoma (KIRC), Lung adenocarcinoma (LUAD), and Colon adenocarcinoma (COAD) [13]. In summary, NETs exert a fundamental role as a marker from the clinical perspective (shown in Table 2). The above studies provide the value of NETs as a biomarker in cancer with several NET-associated metrics. From a technical point of view, the main difficulty in the clinical application of NETs was the lack of unified detection metrics and standard quantification threshold.
The synergistic effect of NETs inhibition combined with immunotherapy
There is a growing study focusing on the mechanism of the immunosuppressive environment in cancer, especially after the clinical application of immune checkpoint inhibitors (ICIs) with promising therapeutic effects. The goal of ICIs is to reactivate the immunity response and rescue the anti-tumor effects in cancer. Although a certain percentage of patients showed a favorable benefit from the ICIs treatment, how to improve the response rate and relieve the adverse effects of treatment is always a challenge for clinicians. It was founded that the inhibition of NETs maybe has a synergistic effect with ICIs for cancer treatment [118]. Zhang Y et al. further investigated that increased CD8+ T cells were recruited in the mouse deficient of PAD4. And the pancreatic implantation tumor showed a significant reduction with the additional employment of PD-1 blockade in the mouse deficient in PAD4 (PAD4-KO) [119]. The PAD4 inhibition of NETosis has also been described with the potential synergistic effect with ICIs through the modulatory role for lymphocyte function, like T and NK cells [28]. The implantation of melanoma cells in mice resulted in tumor regression with anti-PD1 or anti-CTLA4 immunotherapy, and the increment of NETs during treatment was speculated in association with adverse reactions [118]. In addition, the current research on the impact of NETs on immunotherapy is mainly carried out in vitro or in vivo. There is a lack of relevant evidence for the role of NETs on immunotherapy in humans. We also intend to explore the relationship between NET and immunotherapy efficacy and adverse effects in the patient cohort of tumor immunotherapy. Whether the use of NETs inhibitors can improve the response rate of immunotherapy and relief adverse reactions deserves further exploration. Therapeutic strategies targeting NETs combined with immunotherapy treatment may be a promising regimen with improved treatment benefits in the future.
Potential treatment strategies for inhibiting NETs
Since plenty of shreds of evidence have indicated that NETs might play vital roles in different diseases, including inflammation injury, sepsis, auto-immune disease, and cancer, targeting NETs is now expected to be a potential treatment strategy. Several works which are under experimental research or preclinically used include degrading already formed NETs and blocking the aberrant formation of NETs (shown in Table 3).
DNase I is one of the endonucleases that cleave DNA, resulting in the collapse of the web-like structure, and is commonly used as a NETs inhibitor [140]. Previous studies reported that Dnase I could improve diabetic wound healing through the clearance of NETs [141] and mechanistically research revealed that Dnase I exerted its function mainly by improving inflammation resolution, reactivating epithelial regeneration-related signaling pathways, and attenuating the cumulation of reactive oxygen species (ROS) [142]. Timely elimination of excessive NETs is crucial for tissue homeostasis and avoiding the presentation of self-antigens [123]. Evidence showed that inhibiting NET formation by the treatment of Dnase I significantly protected hepatocytes and reduced inflammation after liver ischemia/reperfusion (I/R) injury [121], lessened lung injury, and improved survival both in mouse models and in humans with ARDS from pneumonia or sepsis [122]. Impairing the function of Dnase I failed to remove NETs in time might result in the pathogenesis of lupus nephritis in systemic lupus erythematosus (SLE) patients [124]. Treatment with DNase I might be a therapeutic target in SLE, which still demanded further research [123, 124]. In a mouse spine operation model, the authors proved that NETs promoted scar formation in post-epidural fibrosis, which was remarkably decreased with the administration of DNase I [67]. Moreover, recombinant Dnase I is used for the treatment of cystic fibrosis currently due to its inhibitory function in NETs [125, 126]. Emerging evidence revealed that NET formation favors tumor cell proliferation, metastasis, as well as immunosuppression [115, 143]. Degrading NETs by Dnase I not only reduced circulating NET levels, but also suppressed tumor cell growth and metastasis in colorectal cancer, hepatocellular carcinoma, breast cancer, and pancreatic cancer [19, 127]. PAD4 catalyzes the citrullination of histones and promotes chromatin decondensation during the formation of NETs [144, 145]. Inhibition of PAD4 is an effective method for NETs suppression and is under research in different diseases [144]. Inhibiting PAD4 by GSK484 reduced NETs formation, and was proved to improve renal function via apoptosis limitation in a mouse IRI model [130], and could attenuate the swelling of the alveolar interstitium caused by a subarachnoid hemorrhage in mouse [131]. Moreover, GSK484 decreased tumor lung metastasis in hepatocellular carcinoma and breast cancer mouse models due to NETs inhibition [28, 129, 132]. Besides GSK484, Cl-amidine and BMS-P5 are also PAD4 inhibitors that are under investigation [135, 146]. In an LPS-induced mouse mastitis model, Cl-amidine reduced NETs release and pathological injury, which might be relevant to inhibiting NF-κB, MAPK, and NLRP3 signaling pathways [133]. It was reported that Cl-amidine could also weaken the inflammatory response of LPS-induced endometritis in rats by decreasing the formation of NETs [134]. BMS-P5 is a novel PAD4-specific inhibitor. A recent study reported that BMS-P5 blocked citrullination of histone H3 and NETs formation in human multiple myeloma (MM) cells, and treatment with BMS-P5 to MM-bearing mice attenuated symptoms and disease progression [135].
The protease NE and MPO are vital components of NETs, and they work synergistically to decondense chromatin in NETs [147]. Some studies reported that NE or MPO deficiency fails to produce NETs both in vitro and in vivo [148, 149]. Inhibition of these two enzymes would likely serve as therapeutic targets in NET-associated diseases [149]. Sivelestat is a NE inhibitor that has been approved to treat ARDS in Japan and South Korea [115]. The generation of NETs proves to be beneficial for pro-migratory tumor behavior. In a three-dimensional (3D) model that mimics a tumor-immune microenvironment, the authors demonstrated that inhibiting NETs formation by sivelestat prevents ovarian tumor cells from acquiring an invasive phenotype [136]. A recent study reported that CXCR2 favored NETs formation by enhancing the recruitment of TANs towards brain metastasis in breast cancer cells, and it could be impeded through sivelestat administration [150]. Reparixin as a CXCR1/2 inhibitor could impede NETosis [28]. GW311616 is another NE inhibitor that could attenuate the proliferation and migration of diffuse large B-cell lymphoma cells induced by NETs [21]. Minerals-organic particles in the human body were shown to promote pro-inflammatory responses via NETs which could also be blocked by GW311616 [137]. Strategies to impede NETs by the inhibitors of MPO are also effective. Excessive release of NETs in septic patients and high levels of NETs-MPO in septic patients were relevant to the severity of organ dysfunction. AZD5904, an inhibitor of MPO could reduce the formation of NETs and therefore may be a new therapeutic option for multiple organ dysfunction in sepsis [138].
Fortunately, phase I (NCT04409925) and phase II (NCT03250689) clinical trials inhibiting NETs are being carried out in non-neoplastic diseases. Once successful, it will lay the foundation for clinical trials in cancer.
Conclusion and the future perspective
Given that neutrophils are core innate immune signaling hubs that could transmit or activate most of the pathways indispensable for adaptive immune activities, there is a growing interest in the research of neutrophils, especially for NETs. The prior reviews mainly discussed the crucial role of NETs in contributing to the development and progression of cancer [115, 143, 151,152,153]. Our review illustrated the altered immune networks mediated by NETs in cancer. Based on the available data, we focused on the intercellular communication between NETs, cancer cells, and immune cells (Fig. 1). This information is the key issue to figuring out the specific molecular mechanism of NET-cell contact for targeted therapy and therapeutic intervention. Meanwhile, it remains much left to delineate the NETs-primed molecular networks in cancer. Previously, the focus in the field of tumor immunology was mostly on lymphocytes, but now there is increasing evidence that the role of neutrophils is indispensable. For example, the neutrophil-to-lymphocyte ratio (NLR) can serve as an independent prognostic factor and predict the efficacy of immunotherapy. Therefore, it is necessary to further understand neutrophils. One of the key limitations is that primary human neutrophils have a short survival time with only several days in vitro. Neutrophils could not be transfected to inhibit the specific signaling pathway in association with NETs formation and function. Most advanced technologies, like sc-RNA sequencing or labeling system, have been highly adaptable to ligand/receptor interactions. Nevertheless, whether these technologies could be a technology in the application for deeply resolving cellular interaction of NETs deserves further validation. We highlighted the recent research in our review that the inhibition of NETs exerted a synergistic part combined with ICIs immunotherapy in preclinical studies. Due to their connection between innate and adaptive immune response, we aspired to supply a novel framework regarding NETs targeted inhibitors combined with other immunotherapy strategies to improve clinical treatment benefits for cancer.

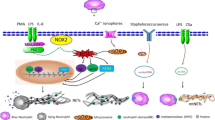
Intercellular communication between NETs and tumor cells or immune cells. a NET-DNA served as a chemotactic factor, which was sensor by cell-surface receptor CCDC25 on the metastatic tumor cell, and subsequently activated the downstream ILK–β-Parvin signaling to accelerate the metastasis. b NETs functioning as a substrate for the integrins expressed on the tumor cell, could enhance the cell adhesion ability and sequester the CTC. c NET-remodeled laminin-111 contributed to tumor proliferation by activating the integrin a3β1 signaling of the dormant cancer cell. d NET clearance refers to the process by which macrophage phagocytoses the NET. NET clearance is often impaired in cases of abnormal immunity. Specific molecules could enhance the ability of NET clearance through the cAMP/PKA/AMPK axis. e The impact of NETs on the immunomodulatory function may be due in part to the phenomenon of NETs-shielding tumor cells, reducing the direct contact between effector cytotoxic lymphocytes and tumor cells. f NET-primed naïve CD4+ T cell was more inclined to the differential of Treg cell through increasing the mitochondrial OCR and OXPHOS. Genes essential for Treg differentiation and activity were up-regulated. Meanwhile, Teff programming genes were significantly down-regulated. g Activated platelets bind to the receptor P-selective (P-sel) glycoprotein ligand-1 (PSGL-1) on the surface of the neutrophil via P-selective protein and promote the production of NETs. And NETs such as the DNA backbone and adherent thrombosis-related enzymes can activate platelets in turn
Availability of data and materials
Not applicable.
Abbreviations
- NETs:
-
Neutrophil extracellular traps
- TANs:
-
Tumor-associated neutrophils
- MPO:
-
Myeloperoxidase
- TLRs:
-
Toll-like receptors
- PKC:
-
Protein kinase C
- NE:
-
Neutrophil elastase
- PAD4:
-
Peptidyl arginine deiminase 4
- TME:
-
Tumor microenvironment
- EMT:
-
Endothelial-to-mesenchymal transition
- CTCs:
-
Circulating tumor cells
- scRNA-seq:
-
Single-cell RNA sequencing
- PAMP:
-
Pathogen-associated molecular pattern
- PGC1-α:
-
Peroxisomes proliferator-activated receptor gamma coactivator 1-alpha
- DLBCL:
-
Diffuse large B-cell lymphoma
- IL-8:
-
Interleukin-8
- CXCR2:
-
C-X-C Motif Chemokine Receptor 2
- HCC:
-
Hepatocellular carcinoma
- COX2:
-
Cyclooxygenase-2
- ILK:
-
Integrin-linked kinase
- MMP9:
-
Matrix metalloproteinase 9
- FAK:
-
Focal adhesion Kinase
- ERK:
-
Extracellular signal regulated kinase
- YAP:
-
Yes-associated Protein
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- TNF:
-
Tumor necrosis factor
- TAMs:
-
Tumor associated macrophages
- ETs:
-
Extracellular traps
- MET:
-
Macrophage extracellular trap
- pNET:
-
Pancreatic neuroendocrine tumor
- RFS:
-
Recurrence-free survival
- ARDS:
-
Acute respiratory distress syndrome
- LDG:
-
Low density granulocyte
- COVID-19:
-
Coronavirus disease 2019
- EDIL3:
-
EGF-like repeats and discoidin I-like domain 3
- LFA1:
-
Lymphocyte function-associated antigen 1
- ICAM:
-
Intercellular adhesion molecule
- Mertk−MHC-IIlo − int :
-
Myeloid-epithelial-reproductive tyrosine kinase/major histocompatibility complex II
- MI:
-
Myocardial infarction
- RvTs:
-
13-Series resolvins
- cAMP:
-
Cyclic adenosine monophosphate
- PKA:
-
Protein kinase A
- AMPK:
-
AMP-activated protein kinase
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- BC:
-
Bladder cancer
- Tregs:
-
Regulatory T cells
- OXPHOS:
-
Oxidative phosphorylation
- Teff:
-
Effector T cell
- GC:
-
Gastric adenocarcinoma
- IHC:
-
Immunohistochemical
- DFS:
-
Disease-free survival
- CA19-9:
-
Carbohydrate antigen 19–9
- CEA:
-
Carcinoembryonic antigen
- H3Cit:
-
Citrullinated histone H3
- TCGA:
-
The Cancer Genome Atlas
- KIRC:
-
Kidney renal clear cell carcinoma
- LUAD:
-
Lung adenocarcinoma
- COAD:
-
Colon adenocarcinoma
- ICIs:
-
Immune checkpoint inhibitors
- ROS:
-
Reactive oxygen species
- I/R:
-
Ischemia/reperfusion
- SLE:
-
Systemic lupus erythematosus
- MM:
-
Multiple myeloma
- NLR:
-
Neutrophil-to-lymphocyte ratio
References
Hedrick CC, Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat Rev Immunol. 2022;22(3):173–87.
Nemeth T, Sperandio M, Mocsai A. Neutrophils as emerging therapeutic targets. Nat Rev Drug Discov. 2020;19(4):253–75.
Mollinedo F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019;40(3):228–42.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, Cichon I, Clancy DM, Desai J, Dumych T, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019;26(3):395–408.
Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. 2018;15(4):206–21.
Tan C, Aziz M, Wang P. The vitals of NETs. J Leukoc Biol. 2021;110(4):797–808.
Pinegin B, Vorobjeva N, Pinegin V. Neutrophil extracellular traps and their role in the development of chronic inflammation and autoimmunity. Autoimmun Rev. 2015;14(7):633–40.
Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409):eaao4227.
Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133–8.
Berger-Achituv S, Brinkmann V, Abed UA, Kuhn LI, Ben-Ezra J, Elhasid R, Zychlinsky A. A proposed role for neutrophil extracellular traps in cancer immunoediting. Front Immunol. 2013;4:48.
Zhang Y, Guo L, Dai Q, Shang B, Xiao T, Di X, Zhang K, Feng L, Shou J, Wang Y. A signature for pan-cancer prognosis based on neutrophil extracellular traps. J Immunother Cancer. 2022;10(6):e004210.
Arelaki S, Arampatzioglou A, Kambas K, Papagoras C, Miltiades P, Angelidou I, Mitsios A, Kotsianidis I, Skendros P, Sivridis E, et al. Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PLoS ONE. 2016;11(5):e0154484.
Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, Loser K, Sunderkotter C, Luger TA, Weishaupt C. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020;33(1):63–73.
Cools-Lartigue J, Spicer J, Najmeh S, Ferri L. Neutrophil extracellular traps in cancer progression. Cell Mol Life Sci. 2014;71(21):4179–94.
Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216(1):176–94.
Boone BA, Orlichenko L, Schapiro NE, Loughran P, Gianfrate GC, Ellis JT, Singhi AD, Kang R, Tang D, Lotze MT, et al. The receptor for advanced glycation end products (RAGE) enhances autophagy and neutrophil extracellular traps in pancreatic cancer. Cancer Gene Ther. 2015;22(6):326–34.
Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D, Kang R, Lotze MT, Zeh HJ 3rd. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology. 2019;8(9):e1605822.
Demers M, Wong SL, Martinod K, Gallant M, Cabral JE, Wang Y, Wagner DD. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology. 2016;5(5):e1134073.
Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, Liu P, Li Z, Xia Y, Jiang W. Neutrophil extracellular traps induced by il8 promote diffuse large b-cell lymphoma progression via the tlr9 signaling. Clin Cancer Res. 2019;25(6):1867–79.
Yang L, Liu L, Zhang R, Hong J, Wang Y, Wang J, Zuo J, Zhang J, Chen J, Hao H. IL-8 mediates a positive loop connecting increased neutrophil extracellular traps (NETs) and colorectal cancer liver metastasis. J Cancer. 2020;11(15):4384–96.
Shinde-Jadhav S, Mansure JJ, Rayes RF, Marcq G, Ayoub M, Skowronski R, Kool R, Bourdeau F, Brimo F, Spicer J, et al. Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat Commun. 2021;12(1):2776.
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, Scadden DT, Wagner DD. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci U S A. 2012;109(32):13076–81.
Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, Schott AF, Kinugasa-Katayama Y, Lee Y, Won NH, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138.
Stehr AM, Wang G, Demmler R, Stemmler MP, Krug J, Tripal P, Schmid B, Geppert CI, Hartmann A, Munoz LE, et al. Neutrophil extracellular traps drive epithelial-mesenchymal transition of human colon cancer. J Pathol. 2022;256(4):455–67.
Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, Konig S, Monteiro RQ. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial-Mesenchymal Transition. Cancers (Basel). 2020;12(6):1542.
Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, de Andrea C, Ochoa MC, Otano I, Etxeberria I, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-871 e858.
Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, Giannias B, Berube J, Rousseau S, Ferri LE, et al. Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int J Cancer. 2017;140(10):2321–30.
Szczerba BM, Castro-Giner F, Vetter M, Krol I, Gkountela S, Landin J, Scheidmann MC, Donato C, Scherrer R, Singer J, et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature. 2019;566(7745):553–7.
Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13(5):816–25.
Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. 2014;5:461.
Urban-Wojciuk Z, Khan MM, Oyler BL, Fahraeus R, Marek-Trzonkowska N, Nita-Lazar A, Hupp TR, Goodlett DR. The Role of TLRs in Anti-cancer Immunity and Tumor Rejection. Front Immunol. 2019;10:2388.
Li C, Ma L, Liu Y, Li Z, Wang Q, Chen Z, Geng X, Han X, Sun J, Li Z. TLR2 promotes development and progression of human glioma via enhancing autophagy. Gene. 2019;700:52–9.
Vinnakota K, Hu F, Ku MC, Georgieva PB, Szulzewsky F, Pohlmann A, Waiczies S, Waiczies H, Niendorf T, Lehnardt S, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol. 2013;15(11):1457–68.
Gowing SD, Chow SC, Cools-Lartigue JJ, Chen CB, Najmeh S, Goodwin-Wilson M, Jiang HY, Bourdeau F, Beauchamp A, Angers I, et al. Gram-negative pneumonia augments non-small cell lung cancer metastasis through host toll-like receptor 4 activation. J Thorac Oncol. 2019;14(12):2097–108.
Chang CY, Jeon SB, Yoon HJ, Choi BK, Kim SS, Oshima M, Park EJ. Glial TLR2-driven innate immune responses and CD8(+) T cell activation against brain tumor. Glia. 2019;67(6):1179–95.
Lee SN, Jin SM, Shin HS, Lim YT. Chemical strategies to enhance the therapeutic efficacy of toll-like receptor agonist based cancer immunotherapy. Acc Chem Res. 2020;53(10):2081–93.
Xun Y, Yang H, Kaminska B, You H. Toll-like receptors and toll-like receptor-targeted immunotherapy against glioma. J Hematol Oncol. 2021;14(1):176.
Khan AA, Khan Z, Warnakulasuriya S. Cancer-associated toll-like receptor modulation and insinuation in infection susceptibility: association or coincidence? Ann Oncol. 2016;27(6):984–97.
Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, Loughran P, Shiva S, Geller DA, Bartlett DL, et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. 2019;79(21):5626–39.
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, Wang Y, Simmons RL, Huang H, Tsung A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76(6):1367–80.
Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, Lin ZF, Wang XY, Wang CQ, Lu M, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. 2020;13(1):3.
Sokeland G, Schumacher U. The functional role of integrins during intra- and extravasation within the metastatic cascade. Mol Cancer. 2019;18(1):12.
Hamidi H, Ivaska J. Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer. 2018;18(9):533–48.
Monti M, De Rosa V, Iommelli F, Carriero MV, Terlizzi C, Camerlingo R, Belli S, Fonti R, Di Minno G, Del Vecchio S. Neutrophil Extracellular Traps as an Adhesion Substrate for Different Tumor Cells Expressing RGD-Binding Integrins. Int J Mol Sci. 2018;19(8).
Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering Macrophages for Cancer Immunotherapy and Drug Delivery. Adv Mater. 2020;32(40):e2002054.
Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090.
Meli VS, Veerasubramanian PK, Atcha H, Reitz Z, Downing TL, Liu WF. Biophysical regulation of macrophages in health and disease. J Leukoc Biol. 2019;106(2):283–99.
Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117(5):1155–66.
Doster RS, Rogers LM, Gaddy JA, Aronoff DM. Macrophage extracellular traps: a scoping review. J Innate Immun. 2018;10(1):3–13.
Wang H, Yang L, Wang D, Zhang Q, Zhang L. Pro-tumor activities of macrophages in the progression of melanoma. Hum Vaccin Immunother. 2017;13(7):1556–62.
Chen T, Wang Y, Nan Z, Wu J, Li A, Zhang T, Qu X, Li C. Interaction between macrophage extracellular traps and colon cancer cells promotes colon cancer invasion and correlates with unfavorable prognosis. Front Immunol. 2021;12:779325.
Xu SS, Li H, Li TJ, Li S, Xia HY, Long J, Wu CT, Wang WQ, Zhang WH, Gao HL, et al. Neutrophil extracellular traps and macrophage extracellular traps predict postoperative recurrence in resectable nonfunctional pancreatic neuroendocrine tumors. Front Immunol. 2021;12:577517.
Zhang L, Yi H, Chen J, Li H, Luo Y, Cheng T, Yang H, Jiang Z, Pan C. Neutrophil extracellular traps facilitate a549 cell invasion and migration in a macrophage-maintained inflammatory microenvironment. Biomed Res Int. 2022;2022:8316525.
Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, Fei K, Jiang G, Fan J. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. 2018;9(6):597.
Wei X, Zou S, Xie Z, Wang Z, Huang N, Cen Z, Hao Y, Zhang C, Chen Z, Zhao F, et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc Res. 2022;118(9):2179–95.
Chiang N, Sakuma M, Rodriguez AR, Spur BW, Irimia D, Serhan CN. Resolvin T-series reduce neutrophil extracellular traps. Blood. 2022;139(8):1222–33.
Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352(6281):aaf2154.
Li H, Li Y, Song C, Hu Y, Dai M, Liu B, Pan P. Neutrophil extracellular traps augmented alveolar macrophage pyroptosis via AIM2 inflammasome activation in LPS-Induced ALI/ARDS. J Inflamm Res. 2021;14:4839–58.
Torres-Ruiz J, Absalon-Aguilar A, Nunez-Aguirre M, Perez-Fragoso A, Carrillo-Vazquez DA, Maravillas-Montero JL, Mejia-Dominguez NR, Llorente L, Alcala-Carmona B, Lira-Luna J, et al. Neutrophil Extracellular Traps Contribute to COVID-19 Hyperinflammation and Humoral Autoimmunity. Cells. 2021;10(10).
Josefs T, Barrett TJ, Brown EJ, Quezada A, Wu X, Voisin M, Amengual J, Fisher EA. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight. 2020;5(7):e134796.
Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169(4):570–86.
Hajishengallis G, Chavakis T. Endogenous modulators of inflammatory cell recruitment. Trends Immunol. 2013;34(1):1–6.
Shin J, Maekawa T, Abe T, Hajishengallis E, Hosur K, Pyaram K, Mitroulis I, Chavakis T, Hajishengallis G. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med. 2015;7(307):307ra155.
Wei X, Zou S, Xie Z, Wang Z, Huang N, Cen Z, Hao Y, Zhang C, Chen Z, Zhao F, et al. EDIL3 deficiency ameliorates adverse cardiac remodeling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc Res. 2021;118(9):2179–95.
Jin Z, Sun J, Song Z, Chen K, Nicolas YSM, Kc R, Ma Q, Liu J, Zhang M. Neutrophil extracellular traps promote scar formation in post-epidural fibrosis. NPJ Regen Med. 2020;5(1):19.
Haider P, Kral-Pointner JB, Mayer J, Richter M, Kaun C, Brostjan C, Eilenberg W, Fischer MB, Speidl WS, Hengstenberg C, et al. Neutrophil extracellular trap degradation by differently polarized macrophage subsets. Arterioscler Thromb Vasc Biol. 2020;40(9):2265–78.
Bellac CL, Dufour A, Krisinger MJ, Loonchanta A, Starr A, Auf dem Keller U, Lange PF, Goebeler V, Kappelhoff R, Butler GS, et al. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 2014;9(2):618–32.
Gregoire M, Uhel F, Lesouhaitier M, Gacouin A, Guirriec M, Mourcin F, Dumontet E, Chalin A, Samson M, Berthelot LL, et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur Respir J. 2018;52(2):1702590.
Carai P, Florit Gonzalez L, Van Bruggen S, Spalart V, De Giorgio D, Geuens N, Martinod K, Jones EAV, Heymans S. Neutrophil Inhibition Improves Acute Inflammation in a Murine Model of Viral Myocarditis. Cardiovasc Res. 2022;118(17):3331–45.
Zhang XL, Wang TY, Chen Z, Wang HW, Yin Y, Wang L, Wang Y, Xu B, Xu W. HMGB1-promoted neutrophil extracellular traps contribute to cardiac diastolic dysfunction in mice. J Am Heart Assoc. 2022;11(4):e023800.
Abaricia JO, Shah AH, Musselman RM, Olivares-Navarrete R. Hydrophilic titanium surfaces reduce neutrophil inflammatory response and NETosis. Biomater Sci. 2020;8(8):2289–99.
Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33(4):547–62.
Cozar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-infiltrating natural killer cells. Cancer Discov. 2021;11(1):34–44.
Wei Y, Huang CX, Xiao X, Chen DP, Shan H, He H, Kuang DM. B cell heterogeneity, plasticity, and functional diversity in cancer microenvironments. Oncogene. 2021;40(29):4737–45.
Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19(1):116.
Toker A, Ohashi PS. Expression of costimulatory and inhibitory receptors in FoxP3(+) regulatory T cells within the tumor microenvironment: Implications for combination immunotherapy approaches. Adv Cancer Res. 2019;144:193–261.
de Andrea CE, Ochoa MC, Villalba-Esparza M, Teijeira A, Schalper KA, Abengozar-Muela M, Eguren-Santamaria I, Sainz C, Sanchez-Gregorio S, Garasa S, et al. Heterogenous presence of neutrophil extracellular traps in human solid tumours is partially dependent on IL-8. J Pathol. 2021;255(2):190–201.
Kaltenmeier C, Yazdani HO, Morder K, Geller DA, Simmons RL, Tohme S. Neutrophil Extracellular Traps Promote T Cell Exhaustion in the Tumor Microenvironment. Front Immunol. 2021;12:785222.
Wang H, Zhang H, Wang Y, Brown ZJ, Xia Y, Huang Z, Shen C, Hu Z, Beane J, Ansa-Addo EA, et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J Hepatol. 2021;75(6):1271–83.
Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125(1):194–207.
Gonzalez-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, Corr M, Katakura K, Eckman L, Lee J, et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. 2010;120(2):570–81.
Kristyanto H, Blomberg NJ, Slot LM, van der Voort EIH, Kerkman PF, Bakker A, Burgers LE, Ten Brinck RM, van der Helm-van Mil AHM, Spits H, et al. Persistently activated, proliferative memory autoreactive B cells promote inflammation in rheumatoid arthritis. Sci Transl Med. 2020;12(570):eaaz5327.
Karmakar U, Vermeren S. Crosstalk between B cells and neutrophils in rheumatoid arthritis. Immunology. 2021;164(4):689–700.
Bertelli R, Schena F, Antonini F, Reverberi D, Signa S, Pedemonte N, Consolaro A, Gattorno M, Negrini S, Pupo F, et al. Neutrophil extracellular traps in systemic lupus erythematosus stimulate IgG2 production from B lymphocytes. Front Med (Lausanne). 2021;8:635436.
Fang H, Shao S, Xue K, Yuan X, Qiao P, Zhang J, Cao T, Luo Y, Bai X, Li W, et al. Neutrophil extracellular traps contribute to immune dysregulation in bullous pemphigoid via inducing B-cell differentiation and antibody production. FASEB J. 2021;35(7):e21746.
Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. 2016;7:271.
McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, Jenne CN. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129(10):1357–67.
Cedervall J, Hamidi A, Olsson AK. Platelets, NETs and cancer. Thromb Res. 2018;164(Suppl 1):S148–52.
Li B, Liu Y, Hu T, Zhang Y, Zhang C, Li T, Wang C, Dong Z, Novakovic VA, Hu T, et al. Neutrophil extracellular traps enhance procoagulant activity in patients with oral squamous cell carcinoma. J Cancer Res Clin Oncol. 2019;145(7):1695–707.
Zhang Y, Wang C, Yu M, Zhao X, Du J, Li Y, Jing H, Dong Z, Kou J, Bi Y, et al. Neutrophil extracellular traps induced by activated platelets contribute to procoagulant activity in patients with colorectal cancer. Thromb Res. 2019;180:87–97.
Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, Ross MA, Wallace CT, Sperry JL, Lotze MT, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 2018;18(1):678.
Yang C, Sun W, Cui W, Li X, Yao J, Jia X, Li C, Wu H, Hu Z, Zou X. Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int J Clin Exp Pathol. 2015;8(11):14075–86.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–9.
Snoderly HT, Boone BA, Bennewitz MF. Neutrophil extracellular traps in breast cancer and beyond: current perspectives on NET stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res. 2019;21(1):145.
Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. 2015;126(2):242–6.
Zhang S, Guo M, Liu Q, Liu J, Cui Y. Neutrophil extracellular traps induce a hypercoagulable state in glioma. Immun Inflamm Dis. 2021;9(4):1383–93.
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12(12):2074–88.
Olsson AK, Cedervall J. NETosis in Cancer - Platelet-Neutrophil Crosstalk Promotes Tumor-Associated Pathology. Front Immunol. 2016;7:373.
Ferrer-Marin F, Cuenca-Zamora EJ, Guijarro-Carrillo PJ, Teruel-Montoya R. Emerging Role of Neutrophils in the Thrombosis of Chronic Myeloproliferative Neoplasms. Int J Mol Sci. 2021;22(3):1143.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107(36):15880–5.
Thalin C, Hisada Y, Lundstrom S, Mackman N, Wallen H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(9):1724–38.
Li JC, Zou XM, Yang SF, Jin JQ, Zhu L, Li CJ, Yang H, Zhang AG, Zhao TQ, Chen CY. Neutrophil extracellular traps participate in the development of cancer-associated thrombosis in patients with gastric cancer. World J Gastroenterol. 2022;28(26):3132–49.
Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood. 2011;118(7):1952–61.
Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, Quail D, Walsh L, Sangwan V, Bertos N, et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5(16):e128008.
Yoo DG, Floyd M, Winn M, Moskowitz SM, Rada B. NET formation induced by pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol Lett. 2014;160(2):186–94.
Zhang Y, Hu Y, Ma C, Sun H, Wei X, Li M, Wei W, Zhang F, Yang F, Wang H, et al. Diagnostic, therapeutic predictive, and prognostic value of neutrophil extracellular traps in patients with gastric adenocarcinoma. Front Oncol. 2020;10:1036.
Thalin C, Lundstrom S, Seignez C, Daleskog M, Lundstrom A, Henriksson P, Helleday T, Phillipson M, Wallen H, Demers M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE. 2018;13(1):e0191231.
Grilz E, Mauracher LM, Posch F, Konigsbrugge O, Zochbauer-Muller S, Marosi C, Lang I, Pabinger I, Ay C. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br J Haematol. 2019;186(2):311–20.
Shang B, Guo L, Shen R, Cao C, Xie R, Jiang W, Wen L, Bi X, Shi H, Zheng S, et al. Prognostic significance of NLR about NETosis and lymphocytes perturbations in localized renal cell carcinoma with tumor thrombus. Front Oncol. 2021;11:771545.
Arpinati L, Shaul ME, Kaisar-Iluz N, Mali S, Mahroum S, Fridlender ZG. NETosis in cancer: a critical analysis of the impact of cancer on neutrophil extracellular trap (NET) release in lung cancer patients vs. mice. Cancer Immunol Immunother. 2020;69(2):199–213.
Jin W, Xu HX, Zhang SR, Li H, Wang WQ, Gao HL, Wu CT, Xu JZ, Qi ZH, Li S, et al. Tumor-Infiltrating NETs Predict Postsurgical Survival in Patients with Pancreatic Ductal Adenocarcinoma. Ann Surg Oncol. 2019;26(2):635–43.
Bang OY, Chung JW, Cho YH, Oh MJ, Seo WK, Kim GM, Ahn MJ. Circulating DNAs, a Marker of Neutrophil Extracellular Traposis and Cancer-Related Stroke: The OASIS-Cancer Study. Stroke. 2019;50(10):2944–7.
Cristinziano L, Modestino L, Antonelli A, Marone G, Simon HU, Varricchi G, Galdiero MR. Neutrophil extracellular traps in cancer. Semin Cancer Biol. 2022;79:91–104.
Chang CH, Jung CJ, Huang YM, Chiao L, Chang YL, Hsieh SC, Lin CH, Kuo YM. The first reported case of trastuzumab induced interstitial lung disease associated with anti-neutrophil cytoplasmic antibody vasculitis - A case report and a prospective cohort study on the usefulness of neutrophil derived biomarkers in monitoring vasculitis disease activity during follow-up. Breast. 2022;61:35–42.
Rivera-Franco MM, Leon-Rodriguez E, Torres-Ruiz JJ, Gomez-Martin D, Angles-Cano E, de la Luz S-G. neutrophil extracellular traps associate with clinical stages in breast cancer. Pathol Oncol Res. 2020;26(3):1781–5.
Blenman KRM, Wang J, Cowper S, Bosenberg M. Pathology of spontaneous and immunotherapy-induced tumor regression in a murine model of melanoma. Pigment Cell Melanoma Res. 2019;32(3):448–57.
Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, Zoltan M, Arora N, Baydogan S, Horne W, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. 2020;217(12):e20190354.
Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–9.
Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–14.
Lefrancais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogeninduced lung injury. JCI Insight. 2018;3(3):e98178.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci U S A. 2010;107(21):9813–8.
Martinez Valle F, Balada E, Ordi-Ros J, Vilardell-Tarres M. DNase 1 and systemic lupus erythematosus. Autoimmun Rev. 2008;7(5):359–63.
Gray RD, McCullagh BN, McCray PB. NETs and CF lung disease: current status and future prospects. Antibiotics (Basel). 2015;4(1):62–75.
Thomson AH. Human recombinant DNase in cystic fibrosis. J R Soc Med. 1995;88(Suppl 25):24–9.
Xia Y, He J, Zhang H, Wang H, Tetz G, Maguire CA, Wang Y, Onuma A, Genkin D, Tetz V, et al. AAV-mediated gene transfer of DNase I in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol Oncol. 2020;14(11):2920–35.
Guan X, Lu Y, Zhu H, Yu S, Zhao W, Chi X, Xie C, Yin Z. The Crosstalk Between Cancer Cells and Neutrophils Enhances Hepatocellular Carcinoma Metastasis via Neutrophil Extracellular Traps-Associated Cathepsin G Component: A Potential Therapeutic Target. J Hepatocell Carcinoma. 2021;8:451–65.
Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, Wang Y, Yang S, Liang C, Liang Y, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423-437 e427.
Wu X, You D, Cui J, Yang L, Lin L, Chen Y, Xu C, Lian G, Wan J. Reduced neutrophil extracellular trap formation during ischemia reperfusion injury in C3 KO mice: C3 requirement for NETs release. Front Immunol. 2022;13:781273.
Zeng H, Fu X, Cai J, Sun C, Yu M, Peng Y, Zhuang J, Chen J, Chen H, Yu Q, et al. Neutrophil extracellular traps may be a potential target for treating early brain injury in subarachnoid hemorrhage. Transl Stroke Res. 2022;13(1):112–31.
Jiang ZZ, Peng ZP, Liu XC, Guo HF, Zhou MM, Jiang D, Ning WR, Huang YF, Zheng L, Wu Y. Neutrophil extracellular traps induce tumor metastasis through dual effects on cancer and endothelial cells. Oncoimmunology. 2022;11(1):2052418.
Wang C, Wang J, Liu X, Han Z, Aimin J, Wei Z, Yang Z. Cl-amidine attenuates lipopolysaccharide-induced mouse mastitis by inhibiting NF-kappaB, MAPK, NLRP3 signaling pathway and neutrophils extracellular traps release. Microb Pathog. 2020;149:104530.
Shen W, Oladejo AO, Ma X, Jiang W, Zheng J, Imam BH, Wang S, Wu X, Ding X, Ma B, et al. Inhibition of Neutrophil Extracellular Traps Formation by Cl-Amidine Alleviates Lipopolysaccharide-Induced Endometritis and Uterine Tissue Damage. Animals (Basel). 2022;12(9):1151.
Li M, Lin C, Deng H, Strnad J, Bernabei L, Vogl DT, Burke JJ, Nefedova Y. A novel Peptidylarginine Deiminase 4 (PAD4) inhibitor BMS-P5 blocks formation of neutrophil extracellular traps and delays progression of multiple myeloma. Mol Cancer Ther. 2020;19(7):1530–8.
Surendran V, Rutledge D, Colmon R, Chandrasekaran A. A novel tumor-immune microenvironment (TIME)-on-Chip mimics three dimensional neutrophil-tumor dynamics and neutrophil extracellular traps (NETs)-mediated collective tumor invasion. Biofabrication. 2021;13(3):035029.
Peng HH, Liu YJ, Ojcius DM, Lee CM, Chen RH, Huang PR, Martel J, Young JD. Mineral particles stimulate innate immunity through neutrophil extracellular traps containing HMGB1. Sci Rep. 2017;7(1):16628.
Shao Y, Li L, Liu L, Yang Y, Huang J, Ji D, Zhou Y, Chen Y, Zhu Z, Sun B. CD44/ERM/F-actin complex mediates targeted nuclear degranulation and excessive neutrophil extracellular trap formation during sepsis. J Cell Mol Med. 2022;26(7):2089–103.
Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil extracellular trap mitochondrial DNA and Its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015;67(12):3190–200.
Hosseinnejad A, Ludwig N, Wienkamp AK, Rimal R, Bleilevens C, Rossaint R, Rossaint J, Singh S. DNase I functional microgels for neutrophil extracellular trap disruption. Biomater Sci. 2021;10(1):85–99.
Fadini GP, Menegazzo L, Rigato M, Scattolini V, Poncina N, Bruttocao A, Ciciliot S, Mammano F, Ciubotaru CD, Brocco E, et al. NETosis delays diabetic wound healing in mice and humans. Diabetes. 2016;65(4):1061–71.
Zhang J, Dai Y, Wei C, Zhao X, Zhou Q, Xie L. DNase I improves corneal epithelial and nerve regeneration in diabetic mice. J Cell Mol Med. 2020;24(8):4547–56.
Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of Neutrophil Extracellular Traps (NETs) in tumor progression and metastasis. Front Immunol. 2020;11:1749.
Liu X, Arfman T, Wichapong K, Reutelingsperger CPM, Voorberg J, Nicolaes GAF. PAD4 takes charge during neutrophil activation: impact of PAD4 mediated NET formation on immune-mediated disease. J Thromb Haemost. 2021;19(7):1607–17.
Thiam HR, Wong SL, Wagner DD, Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. 2020;36:191–218.
Biron BM, Chung CS, O’Brien XM, Chen Y, Reichner JS, Ayala A. Cl-Amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. 2017;9(1):22–32.
Burgener SS, Schroder K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb Perspect Biol. 2020;12(7):a037028.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91.
Metzler KD, Fuchs TA, Nauseef WM, Reumaux D, Roesler J, Schulze I, Wahn V, Papayannopoulos V, Zychlinsky A. Myeloperoxidase is required for neutrophil extracellular trap formation: implications for innate immunity. Blood. 2011;117(3):953–9.
Safarulla S, Madan A, Xing F, Chandrasekaran A. CXCR2 Mediates Distinct Neutrophil Behavior in Brain Metastatic Breast Tumor. Cancers (Basel). 2022;14(3):515.
Ronchetti L, Boubaker NS, Barba M, Vici P, Gurtner A, Piaggio G. Neutrophil extracellular traps in cancer: not only catching microbes. J Exp Clin Cancer Res. 2021;40(1):231.
Yang D, Liu J. Neutrophil extracellular traps: a new player in cancer metastasis and therapeutic target. J Exp Clin Cancer Res. 2021;40(1):233.
De Meo ML, Spicer JD. The role of neutrophil extracellular traps in cancer progression and metastasis. Semin Immunol. 2021;57:101595.
Acknowledgements
Thanks for the drawing platform of “Figdraw”. The figures in our manuscript were made by “Figdraw”.
Funding
This work was supported by The National Natural Science Foundation of China (ID Number: 82072837). This study was also funded by Beijing Municipal Natural Science Foundation (ID Number: 7212083) and CAMS Initiative for Innovative Medicine (CAMS-I2M) [2016-I2M-1–007].
Author information
Authors and Affiliations
Contributions
JZS and LF organized the review schedule and provided funding support. BQS and HLC have written the review and completed the figures. RYX, JW, HZS and XGB have collected the references cited in the review. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shang, B., Cui, H., Xie, R. et al. Neutrophil extracellular traps primed intercellular communication in cancer progression as a promising therapeutic target. Biomark Res 11, 24 (2023). https://doi.org/10.1186/s40364-023-00463-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00463-y