
Abstract
Background
Cardanol was previously reported to be an antiproliferative compound purified from Thai Apis mellifera propolis. By morphology, it could induce the cell death to many cancer cell lines but not the control (non-transformed human foreskin fibroblast cell line, Hs27). Here, it was aimed to evaluate the molecular effects of cardanol on breast cancer derived cell line (BT-474).
Methods
Morphological changes in BT-474 cells induced by cardanol compared to doxorubicin were evaluated by light microscopy, cytotoxicity by using the 3- (4, 5-dimethyl-thiazol-2-yl) 2, 5-diphenyl-tetrazolium bromide (MTT) assay, induction of cell cycle arrest and cell death by flow cytometric analysis of propidium iodide and annexin-V stained cells, and changes in the expression level of genes involved in the control of apoptosis and the cell cycle by quantitative reverse transcriptase-PCR (qRT-PCR) and western blot analyses.
Results
It revealed that cardanol induced a time- and dose-dependent cytotoxicity along with cell shrinkage and detachment from substratum. Cardanol caused cell cycle arrest at the G1 subphase (as opposed to at the G2/M subphase seen with doxorubicin) and cell death by late apoptosis, with both late apoptosis (27.2 ± 1.1 %) and necrosis (25.4 ± 1.4 %) being found in cardanol treated cells after 72 h, compared to a lower proportion of apoptosis (4.3 ± 0.4 %) and higher proportion of necrosis (35.8 ± 13.0 %) induced by doxorubicin. Moreover, cardanol changed the transcript expression levels of genes involved in the control of apoptosis (increased DR5 and Bcl-2 expression and decreased Mcl-1, MADD and c-FLIPP) and cell division (increased p21 and E2FI and decreased cyclin D1, cyclin E, CDK4 and CDK2 expression), as well as increasing the level of p21 p-ERK, p-JNK and p-p38 and decreasing cyclin D. This accounts for the failure to progress from the G1 to the S subphase.
Conclusion
Cardanol is a potential chemotherapeutic agent for breast cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Propolis, one of honeybee products, has mostly been used in traditional medicine. As known, the components and properties of propolis mainly depend on the geographic location and bee species. For example, the propolis derived from bees foraging on poplar (Populus nigra L.) was found to consist of plant resin (50 %), wax (30 %), oil (10 %), pollen (5 %) and other components (5 %) [1].
Propolis has been reported to show many bioactivities, including antibacterial, antiviral, anti-inflammatory and antioxidant activities [2–5]. These reports have typically presented the potential of propolis in the form of either crude or purified extracts/compounds and assayed in either an in vitro or an in vivo model. Also, it was linked to the regulation of gene expression in many types of cells like macrophages, spleenocyte cells and human monocytes [6, 7].
Interestingly, propoelix™, a water-soluble extract of propolis, has been used successfully in the treatment of patients with dengue hemorrhagic fever [8]. In addition, propolis has been reported to be a very rich source of polyphenolic compounds, flavonoids and fatty acids [9]. For example, baccharin isolated from Brazillian propolis and its analogs were able to inhibit aldo-keto reductase 1C3 (AKR1C3), which is involved in castration resistant prostate cancer [10]. The administration of caffeic acid phenethyl ester (CAPE) at 5 μM/kg in mice by intraperitoneal injection showed anti-depressant activity in mice receiving chronic unpredictable stress for 21 consecutive days. Downregulation of p38MAPK phosphorylation by CAPE, which contributed to enhance glucocorticoid receptor function, has also been reported [11].
The molecular mechanism of those active compounds has been revealed, or at least in part. For example, CAPE (25 μM) induces apoptosis in the HeLa cervical cancer cell line (ME 180) and induces cell cycle arrest at the S and G2/M subphases. The expression level of the E2F-1 target gene, cyclin A, cyclin E, apoptosis protease activating of factor-1 (Apaf-1) and myeloid leukemia cell differentiation protein (Mcl-1) were upregulated but cyclin B was down-regulated [12].
In addition, chrysin significantly reduced the serum levels of the pro-inflammatory cytokines IL-1β and IL-6 in high fat diet/streptozotocin -induced type 2 diabetic rats. Since these pro-inflammatory cytokines, along with especially TNF-α, have an important function in insulin resistance and inflammatory responses, chrysin could be a new target for the treatment of type 2 diabetes [13].
Cardanol, a phenolic compound found in members of the cashew tree (Anacardiaceae) family, has been associated with diverse biological effects, such as antiproliferative, antimicrobial and antioxidant activities [14–17]. However, the molecular mode of action of cardanol is unknown. In this research, the BT-474 cell line, as an in vitro breast cancer model, was focused because it is the leading cause of death in Thai women [18]. Here, the induction of cell cycle arrest as well as program cell death was reported. The change in the expression level of genes that control these functions was also investigated. Finally, a molecular mechanism of cardanol action on the BT-474 cell line is proposed.
Methods
Preparation of propolis
Propolis from Apis mellifera was collected from the hives at a bee farm in Pua district, Nan province, Thailand in January, 2012. It was wrapped in aluminum foil and kept in the dark at −20 °C until used. The extraction and enrichment to apparent homogeneity of cardanol from the propolis, along with the one-dimensional thin layer chromatography (1D-TLC), was performed as previously reported [14].
Cell culture
The BT-474 cells (ATCC no. HTB 20) was cultured in complete medium (CM) comprised of Roswell Park Memorial Institute (RPMI) 1640 medium containing 5 % (v/v) fetal calf serum. Cells were seeded at 1 × 105 cells/5 ml CM/ 25-cm2 flask and incubated at 37 °C with 5 % (v/v) CO2. Cells were re-passaged when they reached 70–80 % confluency.
Cytotoxicity
Cytotoxicity was evaluated indirectly from MTT assay. Thus, the results are influenced by changes in the average cell proliferation rate and/or cell viability, and the reduction in the total number of viable cells is herein referred to as the cytotoxicity without delineation of these two components. BT-474 cells (5 103 cells in 198 μl) were seeded in each well of a 96 well plate, and incubated at 37 °C with 5 % (v/v) CO2 for 24 h. Then 2 μl of cardanol or doxorubicin, dissolved in dimethylsulfoxide (DMSO) to a concentration of 10000, 1000, 100, 10, 1 and 0.1 μg/ml for cardanol and 50 μg/ml for doxorubicin, was added to the wells in triplicate, along with DMSO only (2 μl/well) as the solvent (no treatment) control. The cells were then incubated for 72 h before 10 μl of 5 mg/ml of MTT solution was added to each well and incubated for another 4 h. After that, the media was removed and replaced with 150 μl of DMSO and 25 μl of 0.1 M glycine and gently aspirated to lyse the cells and dissolve the formazan crystals. The absorbance was then measured at 540 nm (A540) by a microplate reader. Setting the total number of viable cells in the control culture to be 100 %, the relative percentage of viable cells was calculated from Eq. (1):
The concentration of the test compound that caused a 50 % maximal inhibition of the viable cell number (IC50) was derived from the graphical plot of the relative number of viable cells vs. test compound concentration.
Growth curve of BT-474 cells
BT-474 cells treated with solvent only (control) or with cardanol at the IC50 value (15.6 ± 1.76 μg/ml) were assayed for the relative number of viable cells using the MTT assay after 1, 2, 3, 5 and 7 d of culture. The graph of relative number of viable cells vs. time was drawn, where the trend line was compared to the control cell line.
Cell morphology
BT-474 cells (2 × 105 cells/ml) were cultured in CM with the addition of (i) the DMSO solvent only (Control), (ii) 30 μg/ml of cardanol and (iii) 0.5 μg/ml of doxorubicin (positive control). The morphology of the cells was observed after 0, 24, 48, 72 and 96 h incubation using inverted light microscope (Ziess, Jena) connected to a digital camera (Canon EOS 7D, Tokyo).
Detection of apoptosis and necrosis
BT-474 cells (3–5 × 106 cells/ml) were cultured in CM with the addition of (i) the DMSO solvent only (Control), (ii) 30 μg/ml of cardanol and (iii) 0.5 μg/ml of doxorubicin (positive control). After the indicated time in culture (24–72 h) the cells were harvested by centrifugation (3000 × g, 4 °C for 10 min), washed in 1 ml of cold 1 x phosphate buffer saline (PBS) and harvested as before. The pellet was resuspended in 50 μl of 1 × binding buffer pH 7.4 (10 mM Hepes, 140 mM NaCl and 2.5 mM CaCl2) and stained with the addition 1 μl of annexin V (Alexa Fluor 488 conjugate, Life Technologies, Carlsbad, CA) and 5 μl of 1 mg/ml propidium iodide (PI) solution (Sigma Aldrich, St. Louis, MO) in the dark at room temperature (RT) for 30 min. Cells were then analyzed by flow cytometry on a FC 500 MPL cytometer (Beckman Coulter, Brea, CA) recording 2 events (cells).
Detection of cell cycle arrest
BT-474 cells (1–100 × 106 cells/ml) were cultured in CM with the addition of (i) the DMSO solvent only (Control), (ii) 30 μg/ml of cardanol and (iii) 0.5 μg/ml of doxorubicin (positive control) for 24, 48 and 72 h and then harvested and washed as above. The cell pellet was resuspended and fixed in 500 μl of cold PBS and 200 μl of 70 % (v/v) ethanol at −20 °C overnight or on ice for 4 h. Cells were then harvested and washed as above and the pellet resuspended in 250 μl of PBS with 0.1 mg/ml RNAse and incubated at 37 °C for 30 min. After harvesting, the cells were resuspended in 12.5 μl of 1 mg/ml PI and incubated at RT in the dark for 30 min before being analyzed by flow cytometry on a FC 500 MPL cytometer (Beckman Coulter) recording 2 events per sample. The obtained linear fluorescence profile was interpreted in terms of the (1) sub G1 phase (apoptotic cells), (ii) G1 phase (diploid chromosome content), (iii) S phase (DNA synthesis) and (iv) G2/M subphase (double diploid).
Change in gene expression levels
Transcript expression levels
BT474 cells were cultured in CM with the addition of (i) the DMSO solvent only (Control), (ii) 30 μg/ml of cardanol and (iii) 0.5 μg/ml of doxorubicin (positive control) for 72 h, and then harvested. Total RNA was then extracted from them using the RNeasy Plus Mini Kit (Qiagen, Hilden) as per the suppliers protocol. The extracted RNA was eluted in 20 μl of RNase-free H2O and the absorbance at 260 and 280 nm (A260 and A280, respectively) was measured. The concentration of RNA was calculated from Eq. (2),
The purity of the extracted RNA was estimated from the A260/A280 ratio. The RNA samples were stored at −20 °C until use.
The transcript expression levels of the selected genes were then assayed by single-stage qRT-PCR. Two groups of genes were selected for screening. The first group were the death receptor group of the apoptosis regulated genes b-cell lymphoma-2 (Bcl-2), Mcl-1, mitogen activating protein-kinase activating death domain (MADD), cellular FLICE-like inhibitory protein (c-FLIP) and human death receptor 5 (DR5). The second group were the cell cycle regulating genes of p21, cyclin D1, cyclin E, cyclin A, cyclin-dependent kinase 4 (CDK4), CDK6 and CDK2.
The reaction mixture was prepared using the One Step SYBR PrimeScript RT-PCR Kit II (Takara, Tokyo) as per the manufacturer’s protocol. Each qRT-PCR reaction mixture (20 μl final volume) contained total RNA (10 ng), 10 μl of 2x one step SYBR RT-PCR buffer, 1 μl of Prime Script 1 step enzyme mix, 0.5 μl of each gene fragment specific forward and reverse PCR primer (20 μM stock) and RNase-free d-H2O. The respective forward and reverse primers are listed in Table 1. The PCR thermocycling was performed at 95 °C for 15 min, followed by 40 cycles of 94 °C for 15 s, x °C for 30 s and 72 °C for 30 s, where x is gene specific and is given in Table 1. The relative expression level of each gene was normalized to the expression level of the ß-actin gene as an internal control. The crossing point (Cp) was used to calculated the relative gene expression level as per Eq. (3),
The Cp value is correlated to the amount of the initial template and so indicates the expression of the target mRNA [19].
Protein expression levels
Changes in the expression level of selected proteins were evaluated by western blot analysis following the protocol of Lirdprapamongkol et al. [20] with slight modification. BT-474 cancer cells (2 × 105 cells/ml) were cultured in CM with the addition of (i) the DMSO solvent only (Control), (ii) cardanol at the 2x IC50 concentration (30 μg/ml) and (iii) 0.5 μg/ml of doxorubicin (positive control) for 24 h. Cells were then harvested and lysed in 150 μl of radioimmunoprecipitation assay buffer, which contained 1x halt protease phosphatase and phosphatase inhibitor cocktail with EDTA (Thermo Scientific, Waltham, MA), on ice. The concentration of protein in the lysate was measured by the Bradford assay.
Twenty μg of protein was loaded per well of a sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) with a 7 and 4 % (w/v) acrylamide separating and stacking gel, respectively. After electrophoresis at 15 mA for 105 min, the protein was transferred to immobilon-P nylon membrane (Millipore, Billerica, MA) by electroblotting at 100 V for 90 min. The membrane was later blocked with 3 % (w/v) bovine serum albumin (BSA) for 1 h with gentle shaking at RT. After that, the membrane was cut and probed with the primary antibodies (Cell Signaling Technology, Danvers, MA) diluted in 3 % (w/v) BSA to 1: 1000 (all except for anti-pERK that was 1: 5000) overnight at 4 °C in the dark. The membrane was washed in 1x TBS/T pH 7.6 (20 mM Tris and 137 mM NaCl) and incubated with the diluted horseradish peroxidase-conjugated secondary antibody (Promega, Fitchburg, WI) in TBS/T containing 5 % (w/v) skim milk (1: 10000 mouse, 1: 5000 rabbit) with gentle shaking at RT for 1 h. The bound secondary antibodies were then visualized using western bright ECL reagents (Advansta, Menlo Park, CA) as per the supplier’s protocol and the image was captured using an Image Quant LAS 4000 mini instrument (GE Healthcare Life Sciences, Little Chalfont).
Statistical analysis
Data are expressed as the mean ± one standard deviation (1 S.D.), derived from triplicate replications in each experiment. The data were analyzed by one way analysis of variance (ANOVA) followed by Tukey’s test of multiple comparisons to test for the significance of differences in the means. Significance was accepted at the p < 0.05 level. All analyses were performed using the SPSS program version 19.0.
Results
Cardanol isolation
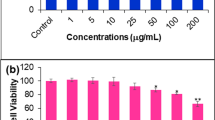
Starting with 90 g of Apis mellifera propolis, 1.54 g of crude dichloromethane extract (CDE) was obtained and then further fractionated by successive quick column and adsorption column chromatography (CC). The obtained cardanol (0.52 mg) was confirmed from its 1D-TLC derived Rf value [14] and from its mass spectrometry derived spectrum. The IC50 value for the cytotoxicity against BT-474 cells was calculated to be 15.6 ± 1.76 μg/ml (Fig. 1), which was close to the 14.0 ± 1.0 μg/ml previously reported on this cell line [14].

Growth curve of the solvent only (Control) and cardanol treated (0.001–199 μg/ml) BT-474 cell line. Data are shown as the mean ± 1SD, derived from three independent repeats
In order to confirm that the BT-474 cells were healthy under these culture conditions and were responsive to cardanol, the growth of BT-474 cells in CM with either DMSO only (solvent control) or with various concentrations of cardanol (0.001–100 μg/ml) was evaluated, and is shown in terms of relative to the initial amount as 100 % (Fig. 1). Among the four phases of a usual growth curve, the total number of viable cells was recorded from the lag phase to the log (exponential growth) phase because treated cells at any cardanol concentration started to die at the end of the log phase. Overall, the inhibition by cardanol was time- and dose-dependent manner.
Morphological changes in BT-474 cells
The morphology of the control BT-474 cells (Fig. 2a) showed no major changes over the 96 h culture period (except for the increased cell density) with most cells being alive, attached to the substratum and flat in appearance. However, the morphology of the cardanol treated cells (IC50 concentration of 15.6 ± 1.76 μg/ml) was different, with unattached cells being observed after 48 h incubation, whilst the attached cells had started to shrink and large clumps of cells were observed at 72 h after treatment with markedly lower cell numbers being visible after 96 h (Fig. 2b).

Morphology of the a control BT-474 cells and those treated with b cardanol at the IC50 concentration (15.6 ± 1.76 μg/ml) and c doxorubicin (0.5 μg/ml) for (I) 24 h, (II) 48 h, (III) 72 h and (IV) 96 h of incubation. The scale bar represents 50 μM. The arrow indicates custard apple shaped cells. All images were magnified at 200 x and are representative of at least 3 such fields of view per sample and three independent repeats
Doxorubicin treatment (0.5 μg/ml) induced broadly similar changes in the BT-474 morphology as those induced by cardanol, except smaller clumps of custard apple shaped cells and a lower number of viable cells was observed (Fig. 2c).
Induction of apoptosis and necrosis
The induction of apoptosis and necrosis in BT-474 cells was determined by the distribution of annexin V and PI stained cells using flow cytometry. Representative flow cytometry dot plots are shown in Additional file 1: Figure S1 in the supplementary information (SI), whilst the analysis of all three replications is summarized in Fig. 3. The control cells remained largely viable (98 % at 24 h to 78 % at 72 h) with very few apoptotic cells. In contrast, the cardanol (30 μg/ml) treated cells were dead by late apoptosis at 72 h of incubation (27.2 ± 1.1 %), whereas the doxorubicin (0.5 μg/ml) treated cells had mostly died by necrosis from 48 h of incubation (29.9 ± 2.9 %) and this proportion was higher (35.8 ± 13.0 %) after 72 h of incubation. Significant difference between the control and treated cells in both groups could be noticed after 48 h of exposure.

The percentage of cell death. Three groups of cells which were stained untreated cells, cardanol treated cells and doxorubicin treated cells were used. The percentage of livable cells, early apoptosis, late apoptosis, and necrosis was shown. a, b and c represented 24, 48 and 72 h of incubation. A symbol of “**” represented significant difference between the control and treated cells at p < 0.01
Cell cycle arrest
The cell cycle position, in terms of the interphase subphases G1, S and G2/M, were identified by the DNA content as determined by flow cytometric analysis of PI stained cells. Representative histograms (PI fluorescence vs. number of cells) are shown in Additional file 2: Figure S2 (SI) and a summary of all the data is shown in Table 2.
For the control cells after 24–72 h culture, around 17–19.3 % of the cells were in the G2/M phase and 66–71.5 % in the G1 phase of the cell cycle. Cardanol (30 μg/ml) treatment increased the proportion of cells in the G1 subphase of the cell cycle compared to the control cells at all three time points, from 66.2 to 72.9 %, 67.2 to 74.6 % and 71.5 to 80.7 % at 24, 48 and 72 h, respectively. Thus, cardanol appeared to induce the cell cycle arrest of BT-474 cells at the G1 subphase. Furthermore, 0.5 μg/ml doxorubicin increased the proportion of cells in the G2/M subphase of the cell cycle compared to the control at all three time points, from 19.3 to 20.6 %, 20.1 to 30.0 %, and 17.0 to 41.3 % at 24, 48 and 72 h, respectively. Thus, doxorubicin induced cell cycle arrest of BT-474 cells at the G2/M subphase.
Changes in gene transcript expression levels
Since 30 μg/ml cardanol induced the late apoptosis of BT-474 cells after 72 h of incubation, total RNA was extracted from BT-474 cells at this period and the transcript level of genes in the apoptosis and cell cycle regulating groups were evaluated by single stage qRT-PCR.
Within the apoptosis regulating genes evaluated, cardanol (30 μg/ml) treatment increased the transcript expression level of DR5 and Bcl-2 significantly at p < 0.01 but decreased significantly that of Mcl-1 (p < 0.01), MADD (p < 0.01) and c-FLIP (p < 0.05). Doxorubicin (0.5 μg/ml) up-regulated the transcript expression level of Bcl-2 significantly at p < 0.01 but down-regulated significantly that of Mcl-1 (p < 0.01), MADD (p < 0.01), c-FLIP (p < 0.05) and DR5 (p < 0.05) (Fig. 4a). A significant difference at either p < 0.01 or p < 0.05 levels was compared between the control and treated cells.

The change in transcript expression levels of genes in the a death receptor group (apoptosis regulating) and b cell cycle regulating genes (transcription factors important for the cell cycle). BT-474 cells were cultured for 72 h in CM with the addition of DMSO only (Control) or 30 μg/ml cardanol or 0.5 μg/ml doxorubicin. Data are shown as the mean ± 1 SD, derived from three independent repeats. Significant difference between the control and treated cells are shown at the (**) p < 0.01 and (*) p < 0.05 levels
For the cell cycle regulation group of genes evaluated, 30 μg/ml cardanol increased the transcript expression level of p21 and E2F1 but decreased that of cyclin D1, cyclin E, CDK4 and CDK2 significantly at p < 0.01, whilst 0.5 μg/ml doxorubicin up-regulated significantly E2F1, p21, cyclin A, CDK6 and CDK2 transcript expression levels (Fig. 4b). A significant difference at p < 0.01 level was compared between the control and treated cells.
Changes in protein expression levels by western blot analysis
The protein expression levels of ERK, JNK and p38 MAPK plus their phosphorylated (active) forms (p-ERK, p-JNK and p-P38), as well as p21 and cyclin D1 in BT-474 cells was evaluated after a 24 h incubation with or without 30 μg/ml cardanol or 0.5 μg/ml doxorubicin (Fig. 5). Cardanol activated ERK, JNK and p38 MAPK, as seen by the increased expression levels of the phosphorylated forms of these three proteins. The increased phosphorylation of ERK, JNK and p38 MAPK, and so their active enzyme levels, is likely to have caused the increased the p21 and cyclin D1 expression levels. Overall, the results strongly suggested that the G1 subphase arrest induced by cardanol was mediated by activation of the MAPK-p21 pathway.

Western blot analysis of protein expression levels in BT-474 cells after incubation in CM with the addition of DMSO only (Control) or with 30 μg/ml cardanol or 0.5 μg/ml doxorubicin for 24 h. Unphosphorylated and phosphorylated forms (p-) of the ERK, p38 and JNK proteins are shown. The blot shown is representative of those seen from 3 independent repeats
Discussion
Bees collect nectar, bee pollen and resin from different plants and so many compounds can found in bee products. For example, α-pinene was reported to be the major compound in European propolis, and it originated from many plant species, including coniferous species like Cupressus sempervirens [21]. Moreover, galangin, chrysin and pinocembrin were found to be the main compounds in Serbian propolis, and were similar to the composition of the resin in poplar trees that are widely distributed in Europe [22]. The propolis from Hungary, Bulgaria, France and Northern Italy were all found to contain resin from poplar trees as well, although the major compounds found in those propolis types were the non-terpenic compounds of benzyl alcohol and benzyl benzoate [23]. Benzyl benzoate was not detected in the volatile oils of poplar buds, although this might reflect differences in the volatiles of different poplar subspecies. Thus, the bud exudates of even of the same species can demonstrate quantitative variability.
Cardanol inhibited the growth of BT-474 cells in a time- and dose-dependent manner (Fig. 1), which is broadly consistent with other reports. For example, the CEE of propolis harvested from many regions in Korea inhibited the angiogenesis, as in tube formation of human umbilical vein endothelial cells (HUVECs), in a dose-dependent manner (6.25–25 μg/ml) [24]. In addition, the CEE of propolis from the Uijeongbu and Pyoseon regions significantly suppressed the proliferation of HUVECs in a dose dependent manner (3.13–25 μg/ml) [24].
Considering the induction of apoptosis by cardanol (Fig. 3 and Additional file 1: Figure S1), cardanol killed BT-474 cells at the late apoptosis (apoptosis and necrosis) stage, which was somewhat similar to doxorubicin, a currently used chemotherapeutic drug, although the later had a higher proportion of necrotic cells. The induction of apoptosis like this is commonly found in compounds purified from natural products, including chemotherapeutic drugs. In addition to propolis, Tualang honey induced late apoptosis in human breast adenocarcinoma (MCF-7 and MDA-MB-231) and cervical (HeLa) cancer cell lines with an EC50 value of 2.4–2.8 % (v/v) [25]. MDA-MB-231 cells treated with Tualang honey at 24 h showed the highest percentage of late apoptosis at 37.8 %, while for MCF-7 and Hela cells it was 55.6 and 56.2 %, respectively [25].
The mechanism of induction of apoptotic cells has many pathways. In this study, cardanol increased the transcript expression level of the Bcl-2 and DR5 apoptosis-related genes but decreased that of Mcl-1, c-FLIP and MADD (Figs. 4 and 5), somewhat similar to doxorubicin (positive control). The DR5 protein is an apoptosis inducing membrane receptor for TNF-related apoptosis-inducing ligand, where apoptosis in human renal cancer cells is induced by up-regulation of DR5 and down-regulation of c-FLIP [26]. In addition, the combined treatment with rosiglitazone and TNF-α-related apoptosis inducing ligand (TRAIL) could induce apoptosis in renal cancer cells via induction of Bcl-2 overexpression [26]. Similarly acrolein can effectively sensitize human renal Caki cells to TRAIL-induced apoptosis through down-regulating Bcl-2 and up-regulating DR5, mediated via generation of reactive oxygen species and induction of the C/EBP homologous protein [27]. Thus, lowering the TRAIL resistance or increasing the damage of tumor cells could help in cancer therapy. Moreover, the knockdown of MADD and c-FLIP reduced the resistance to TRAIL-induced apoptosis in SKOV-3 ovarian cancer cells to 64.2 ± 3.0 % [27].
The Mcl-1 protein is an anti-apoptotic member in the Bcl-2 family of apoptosis regulating proteins. Benzyl isothiocyanate, an anti-cancer agent, causes G2/M cell cycle arrest and apoptosis in human leukemia cell lines via the down-regulation of Mcl-1 [28].
Cardanol appears to arrest BT-474 cells at the G1 subphase of the cell cycle, somewhat similar to the effect of propolin H from Taiwanese propolis that arrested the human lung carcinoma H460 cell line in the G1 subphase. Treatment of H460 cells with 40 μM of propolin H increased the proportion of cells in the G1 subphase from 57.8 to 75.1 % [29].
Cancer cells display an uncontrolled growth and present abnormal gene expression profiles. The expression level of regulating genes, such as cyclin and CDKs, are typically higher and so induce the cell cycle to move to the next phase. With respect to the effect of cardanol on BT-474 cells, it affected the expression of many genes important for the cell cycle, such as decreasing p21, cyclin D1, cyclin E, CDK2 and CDK4 expression levels and increasing that for p21 and E2F1. These results are in accord with those for CAPE at a concentration of 2.5–80 mg/l that increased the proportion of cells in the G1 subphase in a dose-dependent manner, and also increased the expression of beta-catenin and decreased the expression of cyclin D1 and c-myc [30]. In addition, a 24 h exposure to CAPE (50 μg/ml) inhibited the growth of C6 glioma cells, inducing cell cycle arrest at the G1 subphase after a 24 h incubation, decreasing the CDK2/cyclin E and CDK4/cyclin D activity and inhibiting Rb phosphorylation by increasing p21, p27 and p16 expression [31].
The potential induction of the G1 cell cycle arrest by cardanol via increasing p21, p-p38 MAPK, p-JNK and p-ERK protein levels was similar to isothiocyanate sulforaphane, a chemotherapeutic drug. The mechanism of SFN on human colon carcinoma HT-29 cells was reported to be mediated via inducing expression of p21CIP1 and cyclin D1 through activating the MAPK pathways, including ERK, JNK and p38 [32].
In this report, doxorubicin was used as positive control since its action is already well reported [33]. It is an anthracyline drug extracted from Streptomyces peucetius var caesivs and has been used for treatment of diverse cancers, including breast, lung, gastric, ovarian, thyroid, non-Hodgkin’s and Hodgkin’s lymphoma. The mechanism of action of this drug on cancer inhibition has been described in two pathways. First, it binds to DNA and disrupts topoisomerase II-mediated DNA repair. Second, it produces free radicals and damages the cell membrane, DNA and protein leading to cell death. The data of this research supported the effect of doxorubicin on the BT-474 cell line with a cell cycle arrest at the G2/M subphase, up-regulated transcript expression levels of Bcl-2, E2F1, p21, cyclin A, CDK6 and CDK2 and down-regulated expression of Mcl-1, MADD, c-FLIP and cyclin D1. Thus, doxorubicin is likely to act via inhibiting DNA synthesis through increased p21 and cyclin D1 activities. Moreover, doxorubicin decreased the expression level of the anti-apoptotic genes Mcl-1 and c-FLIP.
In summary, the proposed mechanism of how cardanol could inhibit the growth of BT-474 cells is shown in Fig. 6. In this model, cardanol increases the phosphorylation of ERK, JNK and p38 MAPK leading to p21 activation. Then, the active p21 suppressed CDK4/cyclin D and cyclin E/CDK2 and so prevented the hyperphosphorylation of the retinoblastoma protein. This led to the obstruction of DNA synthesis and prevented the movement of cells into and from the S subphase, causing the G1 subphase arrest.

A model of the mechanism of action of cardanol to induce cell cycle arrest at the G1 subphase and cell death in BT-474 cancer cells
However, the data mentioned above were from in vitro only. In the future, primary normal cell culture and animal models must be performed before going forward to human testing. As known, in vitro cultured cells can not represent the whole organism due to lack of precise control of physicochemical surrounding, physiological conditions and so on [34].
In addition, it should be aware of an agent with both anticancer and antioxidant activities. Recently, it has been contradictorily reported whether such compound could be applied to the treatment of cancer [35].
Conclusion
Cardanol, purified from Apis mellifera propolis from Nan province, Thailand, had a cytotoxic activity (IC50 value of 15.6 ± 1.76 μg/ml) against the BT-474 cell line. The inhibition by cardanol was time- and dose-dependent manner. Morphologically, cardanol treated cells revealed a loss of adhesion and cell shrinking with the formation of large clumps and a reduced number of viable cells. After 72 h, significant numbers of cells were dead by late apoptosis. The change in transcript and protein expression levels of genes involved in apoptosis induction and cell proliferation strongly suggested that the MAPK regulated p21-mediated G1 phase cell cycle arrest was a mechanism underlying the growth inhibitory effect of cardanol on BT-474 cells.
References
Burdock GA. Review of the biological properties and toxicity of bee propolis (propolis). Food Chem Toxicol. 1998;36:347–63.
Popova M, Dimitrova R, Al-Lawati HT, Tsvetkova I, Najdenski H, Bankova V. Omani propolis: chemical profiling, antibacterial activity and new propolis plant sources. Chem Cent J. 2013;7:158.
Diaz-Carballo D, Ueberl K, Kleff V, Ergun S, Malak S, Freistuehler M, et al. Antiretroviral activity of two polyisoprenylated acylphloroglucinols, 7-epi-nemorosone and plukenetione A, isolated from Caribbean propolis. Int J Clin Pharm Th. 2010;48:670–7.
Hu F, Hepburn HR, Li Y, Chen M, Radloff SE, Daya S. Effects of ethanol and water extracts of propolis (bee glue) on acute inflammatory animal models. J Ethnopharmacol. 2005;100:276–83.
Sulaiman GM, Al Sammarrae KW, Ad’hiah AH, Zucchetti M, Frapolli R, Bello E, et al. Chemical characterization of Iraqi propolis samples and assessing their antioxidant potentials. Food Chem Toxicol. 2011;49:2415–21.
Orsatti CL, Missima F, Pagliarone AC, Sforcin JM. Th1/Th2 cytokines’ expression and production by propolis-treated mice. J Ethnopharmacol. 2010;129:314–8.
Bufalo MC, Bordon-Graciani AP, Conti BJ, De Assis GM, Sforcin JM. The immunomodulatory effect of propolis on receptors expression, cytokine production and fungicidal activity of human monocytes. J Pharm Pharmacol. 2014;66:1497–504.
Soroy L, Bagus S, Yongkie IP, Djoko W. The effect of a unique propolis compound (Propoelix™) on clinical outcomes inpatients with dengue hemorrhagic fever. Infect Drug Resist. 2014;7:323–9.
Lotfy M. Biological activity of bee propolis in health and disease. Asian Pac J Cancer Prev. 2006;7:22–31.
Zang T, Verma K, Chen M, Jin Y, Trippier PC, Penning TM. Screening baccharin analogs as selective inhibitors against type 5 17β-hydroxysteroid dehydrogenase (AKR1C3). Chem Biol Interact. 2014; doi:10.1016/j.cbi.2014.12.015.
Lee MS, Kim YH, Lee BR, Kwon SH, Moon WJ, Hong KS, et al. Novel antidepressant-like activity of caffeic acid phenethyl ester is mediated by enhanced glucocorticoid receptor function in the hippocampus. Evid-Based Compl Alt Med. 2014; doi:10.1155/2014/646039.
Hsu TH, Chu CC, Hung MW, Lee HJ, Hsu HJ, Chang TC. Caffeic acid phenethyl ester induces E2F-1-mediated growth inhibition and cell-cycle arrest in human cervical cancer cells. FEBS J. 2013;280:2581–93.
Ahad A, Ganai AA, Mujeeb M, Siddiqui WA. Chrysin, an anti-inflammatory molecule, abrogates renal dysfunction in type 2 diabetic rats. Toxicol Appl Pharm. 2014; doi:10.1016/j.taap.2014.05.007.
Teerasripreecha D, Puthong S, Kimura K, Okuyama M, Mori H, Kimura A, et al. In vitro antiproliferative/cytotoxic activity on cancer cell lines of a cardanol and a cardol enriched from Thai Apis mellifera propolis. BMC Complement Altern Med. 2012;12:27.
Ola A. Molecular identification and anticancer activity of alkylphenol from cashew nut shell oil (Anacardium occidantale) grown in Timor Island. Indonesian J Pharm. 2008;19:137–44.
Gopalakrishnan S, Nevaditha NT, Mythili CV. Antibacterial activity of azo compounds synthesized from the natural renewable source, cardanol. J Chem Pharm Res. 2011;3:490–7.
Trevisan MT, Pfundstein B, Haubner R, Wurtele G, Spiegelhalder B, Bartsch H, et al. Characterization of alkyl phenols in cashew (Anacardium occidentale) products and assay of their antioxidant capacity. Food Chem Toxicol. 2006;44:188–97.
Suwisith N, Hanucharurnkul S, Dodd M, Vorapongsathorn T, Pongthavorakamol K, Asavametha N. Symptom clusters and functional status of women with breast cancer. Thai J Nurs Res. 2008;12:153–65.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8.
Lirdprapamongkol K, Sakurai H, Abdelhamed S, Yokoyama S, Athikomkulchai S, Viriyaroj A, et al. Chrysin overcomes TRAIL resistance of cancer cells through Mcl-1 downregulation by inhibiting STAT3 phosphorylation. Int J Oncol. 2013;43:329–37.
Milos M, Radonic A, Mastelic J. Seasonal variation in essential oil compositions of Cupressus sempervirens L. J Essent Oil Res. 2002;14:222–3.
Ristivojevic P, Trifkovic J, Gasic U, Andric F, Nedic N, Tesic Z, et al. Ultra high-performance liquid chromatography and mass spectrometry (UHPLC–LTQ/Orbitrap/MS/MS) study of phenolic profile of Serbian poplar type propolis. Phytochem Anal. 2015;26:127–36.
Bankova V, Popova M, Trusheva B. Propolis volatile compounds: chemical diversity and biological activity: a review. Chem Cent J. 2014;8:28.
Park SI, Ohta T, Kumazawa S, Jun M, Ahn MR. Korean propolis suppresses angiogenesis through inhibition of tube formation and endothelial cell proliferation. Nat Prod Commun. 2014;9:555–60.
Fauzi AN, Norazmi MN, Yaacob NS. Tualang honey induces apoptosis and disrupts the mitochondrial membrane potential of human breast and cervical cancer cell lines. Food Chem Toxicol. 2011;49:871–8.
Kim YH, Jung EM, Lee TJ, Kim SH, Choi YH, Park JW, et al. Rosiglitazone promotes tumor necrosis factor-related apoptosis-Inducing ligand-induced apoptosis by reactive oxygen species-mediated up- regulation of death receptor 5 and down-regulation of c-FLIP. Free Radical Biol Med. 2008;44:1055–68.
Yang ES, Woo SM, Choi KS, Kwon TK. Acrolein sensitizes human renal cancer Caki cells to TRAIL-induced apoptosis via ROS-mediated up-regulation of death receptor-5 (DR5) and down-regulation of Bcl-2. Exp Cell Res. 2011;317:2592–601.
Zhou T, Li G, Cao B, Liu L, Cheng Q, Kong H, et al. Downregulation of Mcl-1 through inhibition of translation contributes to benzyl isothiocyanate induced cell cycle arrest and apoptosis in human leukemia cells. Cell Death Dis. 2013;4:1–11.
Weng MS, Liao CH, Chen CN, Wu CL, Lin JK. Propolin H from Taiwanese propolis induces G1 arrest in human lung carcinoma cells. J Agric Food Chem. 2007;55:5289–98.
He YJ, Liu BH, Xiang DB, Qiao ZY, Fu T, He YH. Inhibitory effect of caffeic acid phenethyl ester on the growth of SW480 colorectal tumor cells involves beta-catenin associated signaling pathway down-regulation. World J Gastroentero. 2006;12:4981–5.
Kuo HC, Kuo WH, Lee YJ, Lin WL, Chou FP, Tseng TH. Inhibitory effect of caffeic acid phenethyl ester on the growth of C6 glioma cells in vitro and in vivo. Cancer Lett. 2006;234:199–208.
Shen G, Xu C, Chen C, Hebbar V, Kong AN. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother Pharmacol. 2006;57:317–27.
Thorn CF, Oshiro C, Marsh S, Hernandez-Boussard T, McLeod H, Klein TE, et al. Doxorubicin pathways: pharmacodynamics and adverse effects. Pharmacogenet Genomics. 2011;21:440–6.
Shetab-Boushehri SV, Abdollahi M. Current concerns on the validity of in vitro models that use transformed neoplastic cells in pharmacology and toxicology. Int J Pharmacol. 2012;8:594–5.
Abdollahi M, Shetab-Boushehri SV. Is it right to look for anti-cancer drugs amongst compounds having antioxidant effect? DARU. 2012;20:61.
Li LC, Jayaram S, Ganesh L, Qian L, Rotmensch J, Maker AV, et al. Knockdown of MADD and c-FLIP overcomes resistance to TRAIL-induced apoptosis in ovarian cancer cells. Am J Obstet Gynecol. 2011; doi:10.1016/j.ajog.2011.05.035.
Pillai MR, Collison LW, Wang X, Finkelstein D, Rehg JE, Boyd K, et al. On the plasticity of regulatory T cell function. J Immunol. 2011;187:4987–97.
Weglarz L, Molin I, Orchel A, Parfiniewicz B, Dzierzewicz Z. Quantitative analysis of the level of p53 and p21(WAF1) mRNA in human colon cancer HT-29 cells treated with inositol hexaphosphate. Acta Biochim Pol. 2006;53:349–56.
Galanti G, Fisher T, Kventsel I, Shoham J, Gallily R, Mechoulam R, et al. Delta 9-tetrahydrocannabinol inhibits cell cycle progression by downregulation of E2F1 in human glioblastoma multiforme cells. Acta Oncol. 2008;47:1062–70.
Ullmannova V, Stockbauer P, Hradcova M, Soucek J, Haskovec C. Relationship between cyclin D1 and p21Waf1/Cip1 during differentiation of human myeloid leukemia cell lines. Leuk Res. 2003;27:1115–23.
Potemski P, Pluciennik E, Bednarek AK, Kusinska R, Dorota JK, Grazyna PW, et al. Cyclin E expression in operable breast cancer quantified using real-time RT–PCR: a comparative study with immunostaining. Jpn J Clin Oncol. 2006;33:142–9.
Chiang PCL, Su CP, Shiow LK, Ching HT, Lin K, Mao TW, et al. Antroquinonol displays anticancer potential against human hepatocellular carcinoma cells: a crucial role of AMPK and mTOR pathways. Biochem Pharmacol. 2010;79:162–71.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SB conducted the experiments. SP helped in cell culture. TP provided the convenience in using real-time PCR machine and flow cytometry. KL provided advice in western blot analysis. PP provided overall advice in chemistry. JS gave valuable comments and suggestions on the manuscript. CC designed the experiments, provided overall advice in biology and wrote the manuscript. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Program cell death of BT-474 cells. A, B and C represented untreated cells as control, 30 μg/ml cardanol treated cells and 0.5 μg/ml doxorubicin treated cells while I, II and III represented 24, 48 and 72 h of incubation, respectively. Duplication of experiments was done. This figure was from one replication only. (DOCX 91 kb)
Additional file 2:
The cell cycle arrest of BT-474 cells. (A) Control, (B) 30 μg/ml cardanol treated and (C) 0.5 μg/ml doxorubicin treated cells after (I) 24 h, (II) 48 h and (III) 72 h of incubation. Histograms shown are derived from 2 events (cells) and are representative of three independent repeats. (DOCX 235 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Buahorm, S., Puthong, S., Palaga, T. et al. Cardanol isolated from Thai Apis mellifera propolis induces cell cycle arrest and apoptosis of BT-474 breast cancer cells via p21 upregulation. DARU J Pharm Sci 23, 55 (2015). https://doi.org/10.1186/s40199-015-0138-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40199-015-0138-1