
Abstract
Both environmental and endogenous factors induce various forms of DNA damage. DNA double strand break (DSB) is the most deleterious DNA lesion. The swift initiation of a complexed network of interconnected pathways to repair the DNA lesion is essential for cell survival. In the past years, the roles of ubiquitin and ubiquitin-like proteins in DNA damage response and DNA repair has been explored. These findings help us better understand the complicated mechanism of DSB signaling pathways.
Similar content being viewed by others
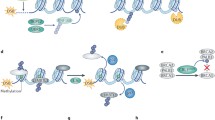
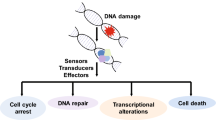

The human genome suffers either exogenous or endogenous damages all the time. Immediate and proper DNA damage response is essential for guarding genomic stability. Upon DNA damage, apical sensors are activated and transduce the signals to the downstream effectors through transducers. Ubiquitin and ubiquitin like proteins mediated posttranslational modifications play an important role in cellular response to these stresses to maintain genomic integrity.
The response and repair of the DNA double-strand breaks
Once DSB happens, the master kinase ATM is activated and initiates global signaling cascades and enriches the DNA damage signaling and repair factors at the DSB sites [1,2,3,4,5,6]. ATM kinase is recruited to DSB through interaction with the C-terminus of NBS1, which is a subunit of the DSB sensor- the MRN complex, in response to DSB [7,8,9]. It has been reported that MRN complex can stimulate ATM kinase activity directly in vitro [10, 11]. However, the exact mechanism by which MRN activates ATM is still not well understood. Activated ATM rapidly phosphorylates H2AX at serine 139, a variant of histone H2A, spreading in a bidirectional manner and spanning 1–2 Mb in mammals in the DSBs [12]. MDC1, the major reader for γH2AX, contains tandem BRCT domains at its C-terminus that specifically bind γH2AX at DSB sites [13, 14]. Oligomerization of MDC1 is critical for recruitment of MDC1 complex at DSBs [15,16,17]. Recruited MDC1 is phosphorylated by ATM and this phosphorylation is recognized by the FHA domain of the ubiquitin ligase RNF8 and recruits RNF8 to break sites [14, 18,19,20,21]. Meanwhile, MDC1 recruits more MRN complexes and ATM proteins, phosphorylating more H2AX at S139, spreading the assembly along chromatin and amplifying DDR signaling [14, 16, 17, 22,23,24,25]. RNF8, once recruited to the DSB, ubiquitinates the L3MBTL2 whose ubiquitination recruits another ubiquitin ligase, RNF168 to the damage site. RNA168 then ubiquitinates H2A at lysine 13/15 to promote engagement of 53BP1 and BRCA1 to DSBs [26,27,28,29]. DSBs are mainly repaired through two mutually exclusive pathways: homologous recombination (HR) and nonhomologous end joining (NHEJ) [23, 30]. Once a break is detected, 53BP1 and BRCA1 compete for directing the cell to commit to NHEJ or HR respectively [31]. CtIP, which is stimulated by BLM, MRN and RPA with enhanced endonuclease activity, cooperates with the nucleases EXO1 and BLM/DNA2 to resect the DNA [32,33,34,35,36]. This process creates extensive single-stranded (ss) 3′ DNA overhangs. Replication protein A (RPA) wraps these ssDNAs, thereby protecting it from breakage [32, 37]. Subsequently, BRCA1-PALB2-BRCA2 complex with DSS1 promotes the replacement of RPA and the loading of the recombinase RAD51 onto ssDNA to form the pre-synaptic filament [38, 39]. RAD51 nucleoprotein filaments search for a homologous sequence to invade and displace one strand of the homologous template to form a displacement loop, facilitating sister chromatid exchange [40,41,42,43]. To inhibit end resection induced homologous recombination, 53BP1 interacts with RIF1, the Shielding complex (REV7–SHLD1–SHLD2–SHLD3) and the CST/Pol α-Prim complex, and recruitment of these complexes shields DNA end from resection, and promote NHEJ [23, 44,45,46,47,48]. UHRF1, an E3 ligase, is also involved in this process. We discovered that UHRF1 mediates K63-linked polyubiquitination of RIF1, and leads to its dissociation from 53BP1 and DSBs, thereby regulating DSB repair choice [49]. In this review, we have summarized the role of ubiquitin and ubiquitin like modifiers (Table 1) in response to DNA double strand breaks (DSBs) in mammalian cells.
Ubiquitin dependent signaling response to DSB
Ubiquitin (Ub) is a highly conserved small protein, composed of 76 amino acids. Ubiquitination is the process of addition of ubiquitin protein to its substrate through a cascade of reactions. This process includes three steps reaction. First, E1 the Ub-activating enzyme activates Ub with adenosine triphosphate and transfers it to E2 Ub-conjugating enzyme. Second, C-terminal carboxyl group of Ub forms a thioester bond with active cysteine in E2 protein. Third, the E3 Ub ligases (E3s) catalyze the C-terminal carboxyl group of Ub, forming of an iso-peptide bond with the lysine ε-amino group of the substrate [50, 51]. More than 600 E3 ligases are predicted in human genome and these E3 ligase are categorized into two classes: the RING domain or U-box containing E3s (promoting direct Ub transfer from an E2 to a substrate), and the HECT (homologous to the E6AP carboxyl terminus) containing E3s (directly catalyze the covalent attachment of ubiquitin to substrate proteins) [52]. The substrate can be modified with one ubiquitin or multi-ubiquitin formed polyubiquitin chain at one or several lysine residues in the substrate protein. In the ubiquitin polypeptide, there are seven lysines: K6, K11, K27, K29, K33, K48 and K63. Ubiquitin moieties can be conjugated through one of the lysine residues or the N-terminal methionine residue to form specific polyubiquitin chain [52]. Different ubiquitin linkage could induce different functional outcomes. Usually, the substrate modification by Lys-48- or Lys-11-linked polyubiquitin chain is powerful degradation signal, which is recognized by proteasome, thus leading to substrate degradation. Whereas, the Lys-K63 or other different lysine linked polyubiquitin chain modification induces the change of the activity or cellular localization of its substrate [53]. In addition, single ubiquitin modified substrates are also frequently reported.
Ubiquitination of histone proteins is an important step in DNA damage response. The nucleosomes are composed of four core histones, H2A, H2B, H3 and H4, and linker histone H1. Following double strand break, H2A is ubiquitinated on K13 and K15 by RNF168. Both K127 and K129 can be ubiquitinated by BRCA1/BARD1 [27, 54, 55]. L3MBTL2 is tethered by MDC1 to the vicinity of the DNA lesion and K63-linked polyubiquitinated by RNF8. Ubiquitinated L3MBTL2 is subsequently recognized by RNF168 and recruited to DSBs. RNF168 then ubiquitinates proteins such as histone H2A and H2AX to further amplify the damage response and recruit repair proteins such as BRCA1 and 53BP1 [29]. The E3 ligase RNF168 primes mono-ubiquitination of H2A at lysine 15, and RNF8 extends the monoubiquitination on this site to help RNF168-mediated K63-linked polyubiquitination at lysine K15 subsequently once double strand breaks happen. RNF168 binds modified H2A by itself and elevates its concentration at the DSB and further amplifies H2A monoubiquitination signal [27, 56, 57]. This monoubiquitination signal and dimethylation of H4K20 is recognized by 53BP1 and recruitment of 53BP1 inhibits DNA end resection, thus suppressing HR and promotes NHEJ [58,59,60].
Protein ubiquitination is reversible and dynamic and the level of protein ubiquitination is balanced by E3 ligase and deubiquitinase. Deubiquitinase is responsible for removing ubiquitin from the substrate and regulating protein stability, activity or cellular localization. Till now, five DUB families have been discovered, including ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado–Joseph disease (MJD) protein domain proteases, and JAMM motif zinc metalloproteases [61].
The ubiquitination of H2A at lysine 13/15 is deubiquitinated by USP51 and USP3 and loss of USP51 causes increased 53BP1 foci formation, and boosted sensitivity to IR [62, 63]. Other deubiquitinases are also suggested to remove ubiquitin at both sites, for example, USP44, and USP11 [64, 65]. JAMM deubiquitinase family member BRCC36 complex with BRE or NBA1 opposes RNF8 mediated ubiquitination of H2AX and enhances radiosensitivity [66,67,68,69,70]. The ubiquitination of H2A at lysine 127/129 is performed by BRCA1/BARD1 E3 ligase complex and recruits SMARCAD1 for 53BP1 reposition, enhancing end resection and HR [55, 71]. PALB2 is also important for HR through interaction with BRCA1, forming complex with RAD51 and BRCA2 and facilitates RAD51 replacement of RPA [72,73,74,75]. The ubiquitination of BRCA1 interacting partners PALB2, which is mediated by KEAP1 E3 ligase complex, decreases its interaction with BRCA1 and suppresses HR in G1 phase [76]. The deubiquitinase, USP11, counteracts this process by removing polyubiquitin chain from PALB2 and sabotages the BRCA1-PALB2-BRCA2 complex assembly [76]. RNF169 is recruited to DSBs through its ubiquitin-binding MIU2 domain, competes with RNF168, removes 53BP1 and RAP80 bridging ubiquitin and histone surfaces, and promotes HR [77,78,79]. Ubiquitination of RAD51 also impairs RAD51-BRCA2 interaction, and UCHL3 mediated deubiquitination of RAD51 strengthens RAD51-BRCA2 binding and facilitates RAD51 recruitment to DSBs [80].
Another core histone H2B is globally monoubiquitinated at lysine 120 in response to DNA damage mediated by RNF20-RNF40 E3 ubiquitin ligase complex. Monoubiquitinated H2B leads to chromatin relaxation, thereby increasing the accessibility of DNA repair factors following DNA damage [81]. Interestingly, acetylation of H2BK120 is found at 1 KB window of DSB and monoubiquitination of H2BK120 is beyond that window [82]. The function of H2B ubiquitination is still not clear. It might be important for DNA damage checkpoint response and HR [83, 84]. USP22 and the SAGA complex are reported to remove the ubiquitin from H2B and regulate early stage of DNA damage response.
The recruitment of both 53BP1 and BRCA1 to DSB sites is through RNF8/RNF168 mediated ubiquitination, despite of their different functions in DNA repair. 53BP1 recognizes both H4K20me2 and H2A K15ub at DNA damage sites, whereas the recruitment of BRCA1 is depending on RAP80 mediated by its ubiquitin-binding modules (UIMs) binding K63-linked ubiquitin chains on chromatin [18, 19, 85]. The ubiquitination of RAP80 status affects its cellular localization in the cells. USP13, translocated to the DSBs after IR, deubiquitinates RAP80, enhances the recruitment of RPA80-BRCA1 complex to break sites [86]. BRCA1/BARD1 serves as an E3 for a range of substrates at DNA damage sites, including CtIP [87]. CtIP ubiquitination by BRCA1 does not mediate the degradation of CtIP protein. Actually, ubiquitinated CtIP facilitates its chromatin loading and plays an important role in regulating of G2/M checkpoint [87]. Recently, two groups reported that USP4 deubiquitinates CtIP, facilitates CtIP to the damage sites, and enhances HR [88, 89].Further studies are needed to explore the complicated relationship between both two proteins as well as the determinants for DSB repair pathway choices.
Sumoylation dependent signaling response to DSB
Small ubiquitin-like modifiers (SUMOs) mediated modification is essential for eukaryotes [90]. Four SUMO genes are discovered in vertebrate genomes, including SUMO1-4 [91]. SUMO2 and SUMO3 polypeptides are very similar, and they share 97% sequence similarity in human. However, SUMO1 is quite different (47% similarity shared by SUMO1 and SUMO2) [90]. Recently, SUMO4 is identified, sharing high similarity with SUMO2 and SUMO3. However, it contains a unique proline residue located at position 90, preventing its maturation by SUMO protease [92, 93]. With high similarity to ubiquitin, SUMO is also a small protein and can be conjugated to the target proteins through a serial enzymes, including E1 (SUMO-activating enzyme), E2 (SUMO-conjugating enzyme), and E3 (SUMO ligase) [94]. However, the number of these enzymes is much less than ubiquitin system. Till now, only 1 E1 and 1 E2 and around a dozen of known E3s. Like ubiquitination, sumoylation can be reversed by sentrin-specific proteases (SENPs) including SENP1, SENP2, SENP3, SENP5, SENP6 and SENP7 [91].
SUMO plays important roles in the DNA damage response. Following DNA damage, numerous of DNA repair factors are sumoylated. SUMO1-3 aggregate at DSBs in human cells and SUMO ligase PIAS1 and PIAS4 are required for this accumulation of these SUMO proteins, which are important for subsequent robust ubiquitination of DSB-flanking chromatin mediated by RNF8, RNF168, and BRCA1 [95]. Later on, another study further found that sumoylation of HERC2 by PIAS4 increases its interaction with RNF8 in a DNA damage dependent manner and stabilizes the RNF8-Ubc13 complex [96]. These studies also suggest a more upstream role of SUMO in this response. RAP80 binding to SUMO at DNA damage sites via a SUMO-interacting motif (SIM) is also important for its recruitment to DSB sites [97, 98].
BRCA1 is also found to be sumoylated by PIAS SUMO E3 ligase in response to DSB, enhancing the ubiquitin ligase activity of BRCA1/BARD1complex and promoting HR [99]. CBX4 sumoylates BMI at lysine 88 and facilities the recruitment of BMI to the damage sites, mediating different DNA damage response pathways from PIAS1/PIAS4 [100]. Sumoylation is also critical for turning off DNA damage response after DNA repair is completed. Several labs independently reported that MDC1 is sumoylated at the damage site. Sumoylated MDC1 is recognized by the SUMO interacting motifs (SIM) of RNF4 and is further ubiquitinated by RNF4, inducing its degradation and removal of MDC1 and 53BP1 from DSBs [101,102,103]. In addition, RNF4 also interacts with other SUMOylated protein through its SIMs and ubiquitinates polysumoylated proteins in order to facilitate their degradation [103].
SUMO and ubiquitin can form hybrid chains. The hybrid chains are recognized by proteins which contain tandem SUMO- and ubiquitin-interacting motifs (tSIM-UIMs), such as RAP80. The tSIM-UIMs in RAP80 enable it to interact with greater affinity to hybrid SUMO-Ub chains compared with homotypic SUMO or ubiquitin chains. At damage sites, RNF4, a SUMO-targeted ubiquitin E3 ligase, catalyzes SUMO-Ub hybrid chains. These hybrid chains are recognized by RAP80, thus promoting BRCA1 accumulation and enhancing HR [97, 98]. The protease for removing the hybrid chains is still not clear.
Neddylation dependent signaling response to DSB
Neural Precursor Cell Expressed Developmentally Down-Regulated Protein 8 (NEDD8) is another ubiquitin like protein with 81 amino acids (NEDD8 precursor). DEN1 processes NEDD8 to its mature form by removing the last 5 amino acids from its C-terminal tail [104]. One family of the E3 ligases- Cullin-RING ligases (CRLs) requires covalent modification of its core cullin protein with NEDD8 to enhance their ligase activity [105]. NEDD8 is conjugated to its target proteins by an enzymatic neddylation cascade, including E1-activating enzyme (NAE) and E2-conjugating enzymes (UBC12, UBE2F) [106, 107]. NAE binds MgATP and NEDD8 and forms acyl adenylated NEDD8. Acyl adenylated NEDD8 subsequently reacts with the active thiol site of the enzyme to form a NEDD8-NAE thioester, coupled with the release of AMP. Another reaction generates a second acyl adenylated NEDD8, forming a ternary complex capable of transferring NEDD8 to E2. The last step is transferring NEDD8 from E2 to the respective cullins [108].
Neddylation functions critically in DNA damage response. CUL1 substrate receptor SKP2 modifies NBS1 with lysine 63 polyubiquitin chain and enhances its interaction with ATM, thus promoting ATM activation upon DNA damage happens [109]. RNF111/UBE2M-mediated neddylation inhibits BRCA1 and CtIP-mediated DNA end resection, and regulates the choice between NHEJ and HR and also the balances between different recombination sub-pathways [110]. Interestingly, RNF168 is reported to function as a NEDD8 E3 ligase after DNA damage [111]. It mediates both H2A ubiquitination and neddylation and the two modifications are against each other. RNF168 itself is also neddylated and this neddylation enhances its interaction with its E2 UBC13 [111]. CDC25A is a member of the CDC25 family of phosphatases and required for activation of cyclin-dependent kinases and cell cycle progression from G1 to the S phase. CDC25A is phosphorylated by CHEK2 following IR and ubiquitinated by CRL E3 ligase for degradation, leading to the cell cycle arrest, which leaves cells sufficient time to repair DNA damage [112]. Removal of NEDD8 from its substrate is also important. COP9 signalosome, a protein complex comprising 8 subunits, is in charge of deneddylation of CRLs [113]. NEDD8 is erased by the metalloprotease activity of a subunit COP9 signalosome-COPS5, thereby suppressing the CRLs [114]. COPS5 is found to be important for Rad51 protein stability. Suppression of COPS5 induces degradation of Rad51 protein and inhibition of HR due to enhanced CRL E3 ligase activity [115].
Ufmylation dependent signaling response to DSB
Ubiquitin fold modifier 1 (UFM1) is also a member of ubiquitin like proteins [116]. Similar to other modifiers, UFM1 protein needs further maturation by removing the last two amino acids in mammalian cells and exposing glycine residue for subsequent conjugating reactions. Parallel to ubiquitination system, ufmylation system also includes E1, the UFM1-activating enzyme (ubiquitin-like modifier-activating enzyme 5; UBA5), E2, the UFM1-conjugating enzyme 1 (UFC1); and E3, the UFM1-specific ligase 1 (UFL1). In contrast to numerous E3s for ubiquitination, only one E3 (UFL1) is identified so far. UFM1 can be conjugated to its substrates via these three enzymes: UFM1 is activated by UBA5 in the presence of ATP and undergoes thioester reaction with the Cysteine 250 of UBA5. Conjugated UBA5 interacts with UFC1 and transfer UFM1 to UFC1, forming a similar thioester linkage with Cysteine 116 in UFC1. The E3 ligase UFL1 transfers UFM1 from UFC1 to its target proteins [116]. The ufmylation is also a reversible process. UFM1 can be cut off from its substrate by the specific UFM1-specific proteases (UFSP). Again, in human, only one functional UFSP protein called UFSP2 has been identified so far [117]. Previous studies have suggested ufmylation system play important roles in hematopoiesis, reticulum homeostasis, liver development, mitosis, gene transcription, G-protein coupled receptor (GPCR) biogenesis, and fatty acid metabolism [118,119,120,121,122,123,124]. Genome-wide association studies also suggest UFM1 signaling is associated with a number of human diseases, including cancer, ischemic heart diseases, diabetes, atherosclerosis, hip dysplasia, and schizophrenia [125,126,127,128]. Recently, our group and others have discovered its new role in ATM activation following DNA damage [129, 130].
MRN complex serves as an initiator of DNA damage response and DNA repair. It is responsible for ATM recruitment to DSBs. One study suggest that MRE11 is ufmylated, and this modification is important for the stability of MRN complex and ATM activation [130]. Our group also found that ufmylation signaling is important for ATM activation. We found that UFL1 aggregated at break sites quickly after double-strand breaks in an MRN complex dependent manner [129]. Recruited UFL1 mono-ufmylates histone H4 at Lysine 31, which helps recruiting Suv39H1-KAP1-HP1 complex to the DSBs, resulting in H3K9 trimethylation and the activation of Tip60-ATM pathway [129]. Interestingly, activated ATM phosphorylates UFL1 at serine 462 and elevates UFL1 activity, forming a positive feedback loop and amplifying ATM activation signal. These studies suggest a tight interplay between the DDR and the UFM1 pathway [129].
Isgylation dependent signaling response to DSB
Interferon-stimulated gene 15 (ISG15) is a 17 kd protein and becomes mature until 8 amino acids are removed from its C terminus [131]. Type I interferon and virus infection stimulate its expression [132]. Like ubiquitination and other ubiquitin like modification, Isgylation also requires cascades of enzymes: an E1 (ubiquitin-activating enzyme E1-like protein, Ube1L; also named as UBA7), an E2 (ubiquitin-carrier protein H8, UbcH8), and an E3 [HECT domain and RCC1-like domain-containing protein 5 (HERC5), and TRIM25 in humans] [133]. ISG15 is activated by E1 enzyme UBE1L, forming a high-energy thioester intermediate. Then it is transferred to the active-site cysteine of E2 UbcH8. E3 ligases HERC5 subsequently transfers the activated ISG15 to the substrates [134, 135]. USP18, USP2, USP5, USP13 and USP14 are the ISG15-specific proteases and unconjugate ISG15 from its substrates [136,137,138,139]. DNA damage inducers induce Isgylation of p53 at K291 and K292 by the E3 ligase EFP and greatly enhances p53 transcriptional activity; hence the transcription of p53 target genes (CDKN1, BAX, MDM2 and ISG15) and Isgylation factors are elevated. Isgylated p53 also facilitates its phosphorylation and acetylation, thus suppression of cell growth and tumorigenesis [140]. Another DNA damage response protein, PCNA also can be modified by ISG15 [141].
Fatylation dependent signaling response to DSB
Ubiquitin like protein HLA-F adjacent transcript 10 (FAT10) is encoded in the major histocompatibility complex class I locus and is synergistically inducible with interferon-γ and tumor necrosis factor α. FAT10 is different from other members. Its protein contains two ubiquitin like domain [142] and a free GG motif located at its C terminal tail [143]. Due to its unique structure, it is immediately available for activation and conjugation. In FAT10 conjugation cascade, FAT10 binds to its E1 UBA6 and forms UBA6-FAT10 thioester. E2 protein USE1, also named UBE2Z, transfers the thioestered FAT10 to its lysine to form a stable isopeptide linkage [144]. The E3 ligase for FAT10 and the deconjugating enzymes have not been discovered yet.
The function of FAT10 has previously been suggested. Depletion of FAT10 in mice prolongs lifespan and reduces adiposity, thus suggesting that FAT10 has a role in aging [145]. Its aberrant expression have been investigated in various cancer types, such as gastrointestinal cancer, hepatocellular carcinoma (HCC), pancreatic ductal adenocarcinoma, human glioma and cervical cancer [146]. The role of FAT10 in DNA damage response is not very clear. Only limited studies have been performed. Proteomic analyses of Fatylation have identified many DNA damage response proteins as FAT10 substrates, including Ku70, RECQ1, FUS, RAD51C, PCNA, DNAJA1, H2AX, KAP-1 [147, 148]. Recently, it is confirmed that IR induces the increase of Fatylated PCNA and leads to degradation of PCNA [149]. More studies are required to better understand the role of FAT10 in DNA damage response.
Conclusions
Impressive studies of the principle and mechanism of ubiquitination, and ubiquitination- like modifications in DSB induced DNA damage response and DNA repair have been published. Here we listed the targets of ubiquitination, sumoylation, neddylation, Isgylation, and ufmylation, which are involved in the DSB signaling. We expect that the list of the substrates of those modifications will keep growing. Many challenges are still present. Even though ubiquitination and sumoylation are extensively studied, new question is emerging. For example, what is the function of branched Ub chain and Ub chain with hybrid linkage? Recent studies also suggest that Ub itself can be modified by PTMs, such as phosphorylation, and how these PTMs affecting Ub reaction and substrate function is not entirely clear. In addition, the exact function of ISG15 and FAT10 is still not clear. For some other Ub like molecules, such as ubiquitin-related modifier-1 (URM1), fan ubiquitin-like protein 1 (FUB1) and histone mono-ubiquitination 1 (HUB1), their functions in DNA damage have not yet been explored. Further studies are therefore needed to explore new principles and mechanism of these new ubiquitin like proteins in maintenance of genome stability. Further efforts are also likely to study the crosstalk among these modifiers and their contribution to DSB induced DNA damage response. A deep understanding of the principle and mechanism of these posttranslational modifications can also potentially provide new therapeutics for the cancer patients. The inhibitor of Neddylation E1 enzyme NAE—MLN4924 has been developed and displays dramatic inhibition of hematological malignancies and solid tumors [150,151,152,153,154,155,156,157,158,159,160,161]. Multiple Phase I clinical study of MLN4924 are undergoing [162,163,164,165,166]. Better understanding of the complexity of these signaling will help develop more small molecular inhibitors to target the factors in uibiquitination and ubiquitination like modification pathways for treating cancer or other diseases.
Abbreviations
- DSB:
-
DNA double strand break
- HR:
-
Homologous recombination
- NHEJ:
-
Nonhomologous end joining
- Ub:
-
Ubiquitin
- SUMO:
-
Small ubiquitin-like modifier
- NEDD8:
-
Neural precursor cell expressed developmentally down-regulated protein 8
- UFM1:
-
Ubiquitin fold modifier 1
- ISG15:
-
Interferon-stimulated gene 15
- FAT10:
-
HLA-F adjacent transcript 10
- URM1:
-
Ubiquitin-related modifier-1
- FUB1:
-
Fan ubiquitin-like protein 1
- HUB1:
-
Histone mono-ubiquitination 1
References
Bekker-Jensen S, Mailand N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair (Amst). 2010;9(12):1219–28.
Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316(5828):1202–5.
Morris JR, Solomon E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet. 2004;13(8):807–17.
Panier S, Durocher D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amst). 2009;8(4):436–43.
Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316(5828):1198–202.
Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316(5828):1194–8.
Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G. Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem. 2001;276(41):38224–30.
Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173(2):195–206.
Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature. 2005;434(7033):605–11.
Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308(5721):551–4.
Lee JH, Paull TT. Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science. 2004;304(5667):93–6.
Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010;29(8):1446–57.
Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–26.
Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21(2):187–200.
Luo K, Yuan J, Lou Z. Oligomerization of MDC1 protein is important for proper DNA damage response. J Biol Chem. 2011;286(32):28192–9.
Liu J, Luo S, Zhao H, Liao J, Li J, Yang C, Xu B, Stern DF, Xu X, Ye K. Structural mechanism of the phosphorylation-dependent dimerization of the MDC1 forkhead-associated domain. Nucleic Acids Res. 2012;40(9):3898–912.
Melander F, Bekker-Jensen S, Falck J, Bartek J, Mailand N, Lukas J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J Cell Biol. 2008;181(2):213–26.
Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131(5):901–14.
Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318(5856):1637–40.
Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131(5):887–900.
Lou Z, Chini CC, Minter-Dykhouse K, Chen J. Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J Biol Chem. 2003;278(16):13599–602.
Jungmichel S, Clapperton JA, Lloyd J, Hari FJ, Spycher C, Pavic L, Li J, Haire LF, Bonalli M, Larsen DH, et al. The molecular basis of ATM-dependent dimerization of the Mdc1 DNA damage checkpoint mediator. Nucleic Acids Res. 2012;40(9):3913–28.
Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci. 2012;125(Pt 15):3529–34.
Spycher C, Miller ES, Townsend K, Pavic L, Morrice NA, Janscak P, Stewart GS, Stucki M. Constitutive phosphorylation of MDC1 physically links the MRE11-RAD50-NBS1 complex to damaged chromatin. J Cell Biol. 2008;181(2):227–40.
Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci USA. 2008;105(32):11200–5.
Bohgaki M, Bohgaki T, El Ghamrasni S, Srikumar T, Maire G, Panier S, Fradet-Turcotte A, Stewart GS, Raught B, Hakem A, et al. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc Natl Acad Sci USA. 2013;110(52):20982–7.
Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150(6):1182–95.
Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, Narita T, Sixma TK, Choudhary C, Bekker-Jensen S, et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;527(7578):389–93.
Nowsheen S, Aziz K, Aziz A, Deng M, Qin B, Luo K, Jeganathan KB, Zhang H, Liu T, Yu J, et al. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat Cell Biol. 2018;20(4):455–64.
Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204.
Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54.
Eid W, Steger M, El-Shemerly M, Ferretti LP, Pena-Diaz J, Konig C, Valtorta E, Sartori AA, Ferrari S. DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 2010;11(12):962–8.
Marrero VA, Symington LS. Extensive DNA end processing by exo1 and sgs1 inhibits break-induced replication. PLoS Genet. 2010;6(7):e1001007.
Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25(4):350–62.
Liao S, Guay C, Toczylowski T, Yan H. Analysis of MRE11′s function in the 5′→3′ processing of DNA double-strand breaks. Nucleic Acids Res. 2012;40(10):4496–506.
Makharashvili N, Tubbs AT, Yang SH, Wang H, Barton O, Zhou Y, Deshpande RA, Lee JH, Lobrich M, Sleckman BP, et al. Catalytic and noncatalytic roles of the CtIP endonuclease in double-strand break end resection. Mol Cell. 2014;54(6):1022–33.
Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450(7169):509–14.
Zhao W, Vaithiyalingam S, San Filippo J, Maranon DG, Jimenez-Sainz J, Fontenay GV, Kwon Y, Leung SG, Lu L, Jensen RB, et al. Promotion of BRCA2-dependent homologous recombination by DSS1 via RPA targeting and DNA mimicry. Mol Cell. 2015;59(2):176–87.
Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22(6):719–29.
Carreira A, Hilario J, Amitani I, Baskin RJ, Shivji MK, Venkitaraman AR, Kowalczykowski SC. The BRC repeats of BRCA2 modulate the DNA-binding selectivity of RAD51. Cell. 2009;136(6):1032–43.
Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467(7316):678–83.
Liu J, Doty T, Gibson B, Heyer WD. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol. 2010;17(10):1260–2.
Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC. The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol. 2010;17(10):1263–5.
Wilson MD, Benlekbir S, Fradet-Turcotte A, Sherker A, Julien JP, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, Durocher D. The structural basis of modified nucleosome recognition by 53BP1. Nature. 2016;536(7614):100–3.
Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Alvarez-Quilon A, Moatti N, Zimmermann M, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560(7716):117–21.
Mirman Z, Lottersberger F, Takai H, Kibe T, Gong Y, Takai K, Bianchi A, Zimmermann M, Durocher D, de Lange T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature. 2018;560(7716):112–6.
Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar-Lele M, Sanchiz-Calvo M, Fueyo-Marcos E, et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560(7716):122–7.
Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173(4):972–88.
Zhang H, Liu H, Chen Y, Yang X, Wang P, Liu T, Deng M, Qin B, Correia C, Lee S, et al. A cell cycle-dependent BRCA1-UHRF1 cascade regulates DNA double-strand break repair pathway choice. Nat Commun. 2016;7:10201.
Di Fiore PP, Polo S, Hofmann K. When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol. 2003;4(6):491–7.
Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–33.
Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86:129–57.
Akutsu M, Dikic I, Bremm A. Ubiquitin chain diversity at a glance. J Cell Sci. 2016;129(5):875–80.
Gatti M, Pinato S, Maspero E, Soffientini P, Polo S, Penengo L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle. 2012;11(13):2538–44.
Kalb R, Mallery DL, Larkin C, Huang JT, Hiom K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014;8(4):999–1005.
Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136(3):435–46.
Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136(3):420–34.
Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127(7):1361–73.
Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510.
Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470(7332):124–8.
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. A genomic and functional inventory of deubiquitinating enzymes. Cell. 2005;123(5):773–86.
Wang Z, Zhang H, Liu J, Cheruiyot A, Lee JH, Ordog T, Lou Z, You Z, Zhang Z. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016;30(8):946–59.
Sharma N, Zhu Q, Wani G, He J, Wang QE, Wani AA. USP3 counteracts RNF168 via deubiquitinating H2A and gammaH2AX at lysine 13 and 15. Cell Cycle. 2014;13(1):106–14.
Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem. 2013;288(23):16579–87.
Yu M, Liu K, Mao Z, Luo J, Gu W, Zhao W. USP11 Is a negative regulator to gammaH2AX ubiquitylation by RNF8/RNF168. J Biol Chem. 2016;291(2):959–67.
Shao G, Lilli DR, Patterson-Fortin J, Coleman KA, Morrissey DE, Greenberg RA. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc Natl Acad Sci USA. 2009;106(9):3166–71.
Cooper EM, Cutcliffe C, Kristiansen TZ, Pandey A, Pickart CM, Cohen RE. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009;28(6):621–31.
Shao G, Patterson-Fortin J, Messick TE, Feng D, Shanbhag N, Wang Y, Greenberg RA. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23(6):740–54.
Feng L, Huang J, Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009;23(6):719–28.
Wang B, Hurov K, Hofmann K, Elledge SJ. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23(6):729–39.
Densham RM, Garvin AJ, Stone HR, Strachan J, Baldock RA, Daza-Martin M, Fletcher A, Blair-Reid S, Beesley J, Johal B, et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat Struct Mol Biol. 2016;23(7):647–55.
Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7(7):1110–8.
Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA. 2009;106(17):7155–60.
Zhang F, Bick G, Park JY, Andreassen PR. MDC1 and RNF8 function in a pathway that directs BRCA1-dependent localization of PALB2 required for homologous recombination. J Cell Sci. 2012;125(Pt 24):6049–57.
Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. 2009;19(6):524–9.
Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528(7582):422–6.
Chen J, Feng W, Jiang J, Deng Y, Huen MS. Ring finger protein RNF169 antagonizes the ubiquitin-dependent signaling cascade at sites of DNA damage. J Biol Chem. 2012;287(33):27715–22.
Kitevski-LeBlanc J, Fradet-Turcotte A, Kukic P, Wilson MD, Portella G, Yuwen T, Panier S, Duan S, Canny MD, van Ingen H, et al. The RNF168 paralog RNF169 defines a new class of ubiquitylated histone reader involved in the response to DNA damage. Elife. 2017;6:e23872.
Hu Q, Botuyan MV, Cui G, Zhao D, Mer G. Mechanisms of ubiquitin-nucleosome recognition and regulation of 53BP1 chromatin recruitment by RNF168/169 and RAD18. Mol Cell. 2017;66(4):473–87.
Luo K, Li L, Li Y, Wu C, Yin Y, Chen Y, Deng M, Nowsheen S, Yuan J, Lou Z. A phosphorylation-deubiquitination cascade regulates the BRCA2-RAD51 axis in homologous recombination. Genes Dev. 2016;30(23):2581–95.
Ramadan K, Dikic I. Editorial: ubiquitin and ubiquitin-relative SUMO in DNA damage response. Front Genet. 2017;8:188.
Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 2011;7(2):113–9.
Moyal L, Lerenthal Y, Gana-Weisz M, Mass G, So S, Wang SY, Eppink B, Chung YM, Shalev G, Shema E, et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol Cell. 2011;41(5):529–42.
Giannattasio M, Lazzaro F, Plevani P, Muzi-Falconi M. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem. 2005;280(11):9879–86.
Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25(7):685–700.
Li Y, Luo K, Yin Y, Wu C, Deng M, Li L, Chen Y, Nowsheen S, Lou Z, Yuan J. USP13 regulates the RAP80-BRCA1 complex dependent DNA damage response. Nat Commun. 2017;8:15752.
Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006;20(13):1721–6.
Liu H, Zhang H, Wang X, Tian Q, Hu Z, Peng C, Jiang P, Wang T, Guo W, Chen Y, et al. The deubiquitylating enzyme USP4 cooperates with CtIP in DNA double-strand break end resection. Cell Rep. 2015;13(1):93–107.
Wijnhoven P, Konietzny R, Blackford AN, Travers J, Kessler BM, Nishi R, Jackson SP. USP4 auto-deubiquitylation promotes homologous recombination. Mol Cell. 2015;60(3):362–73.
Flotho A, Melchior F. Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem. 2013;82:357–85.
Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol. 2010;11(12):861–71.
Guo D, Li M, Zhang Y, Yang P, Eckenrode S, Hopkins D, Zheng W, Purohit S, Podolsky RH, Muir A, et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet. 2004;36(8):837–41.
Owerbach D, McKay EM, Yeh ET, Gabbay KH, Bohren KM. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem Biophys Res Commun. 2005;337(2):517–20.
van der Veen AG, Ploegh HL. Ubiquitin-like proteins. Annu Rev Biochem. 2012;81:323–57.
Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462(7275):935–9.
Danielsen JR, Povlsen LK, Villumsen BH, Streicher W, Nilsson J, Wikstrom M, Bekker-Jensen S, Mailand N. DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. J Cell Biol. 2012;197(2):179–87.
Guzzo CM, Berndsen CE, Zhu J, Gupta V, Datta A, Greenberg RA, Wolberger C, Matunis MJ. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci Signal. 2012;5(253):ra88.
Hu X, Paul A, Wang B. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J Biol Chem. 2012;287(30):25510–9.
Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature. 2009;462(7275):886–90.
Ismail IH, Gagne JP, Caron MC, McDonald D, Xu Z, Masson JY, Poirier GG, Hendzel MJ. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012;40(12):5497–510.
Luo K, Zhang H, Wang L, Yuan J, Lou Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012;31(13):3008–19.
Yin Y, Seifert A, Chua JS, Maure JF, Golebiowski F, Hay RT. SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 2012;26(11):1196–208.
Galanty Y, Belotserkovskaya R, Coates J, Jackson SP. RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 2012;26(11):1179–95.
Gan-Erdene T, Nagamalleswari K, Yin L, Wu K, Pan ZQ, Wilkinson KD. Identification and characterization of DEN1, a deneddylase of the ULP family. J Biol Chem. 2003;278(31):28892–900.
Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6(1):9–20.
Huang DT, Schulman BA. Expression, purification, and characterization of the E1 for human NEDD8, the heterodimeric APPBP1-UBA3 complex. Methods Enzymol. 2005;398:9–20.
Gong L, Yeh ET. Identification of the activating and conjugating enzymes of the NEDD8 conjugation pathway. J Biol Chem. 1999;274(17):12036–42.
Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 conjugation pathway and its relevance in cancer biology and therapy. Genes Cancer. 2010;1(7):708–16.
Wu J, Zhang X, Zhang L, Wu CY, Rezaeian AH, Chan CH, Li JM, Wang J, Gao Y, Han F, et al. Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol Cell. 2012;46(3):351–61.
Jimeno S, Fernandez-Avila MJ, Cruz-Garcia A, Cepeda-Garcia C, Gomez-Cabello D, Huertas P. Neddylation inhibits CtIP-mediated resection and regulates DNA double strand break repair pathway choice. Nucleic Acids Res. 2015;43(2):987–99.
Li T, Guan J, Huang Z, Hu X, Zheng X. RNF168-mediated H2A neddylation antagonizes ubiquitylation of H2A and regulates DNA damage repair. J Cell Sci. 2014;127(Pt 10):2238–48.
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85.
Wei N, Serino G, Deng XW. The COP9 signalosome: more than a protease. Trends Biochem Sci. 2008;33(12):592–600.
Wu JT, Lin HC, Hu YC, Chien CT. Neddylation and deneddylation regulate Cul1 and Cul3 protein accumulation. Nat Cell Biol. 2005;7(10):1014–20.
Pan Y, Zhang Q, Atsaves V, Yang H, Claret FX. Suppression of Jab1/CSN5 induces radio- and chemo-sensitivity in nasopharyngeal carcinoma through changes to the DNA damage and repair pathways. Oncogene. 2013;32(22):2756–66.
Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, Tanaka K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004;23(9):1977–86.
Kang SH, Kim GR, Seong M, Baek SH, Seol JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K, et al. Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases, UfSP1 and UfSP2. J Biol Chem. 2007;282(8):5256–62.
Cai Y, Pi W, Sivaprakasam S, Zhu X, Zhang M, Chen J, Makala L, Lu C, Wu J, Teng Y, et al. UFBP1, a key component of the Ufm1 conjugation system, is essential for ufmylation-mediated regulation of erythroid development. PLoS Genet. 2015;11(11):e1005643.
Wu J, Lei G, Mei M, Tang Y, Li H. A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-kappaB signaling. J Biol Chem. 2010;285(20):15126–36.
Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW, Lee SW, Ka SH, Sou YS, Komatsu M, Tanaka K, et al. Modification of ASC1 by UFM1 is crucial for ERalpha transactivation and breast cancer development. Mol Cell. 2014;56(2):261–74.
Zhang M, Zhu X, Zhang Y, Cai Y, Chen J, Sivaprakasam S, Gurav A, Pi W, Makala L, Wu J, et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015;22(12):1922–34.
Xie Z, Fang Z, Pan Z. Ufl1/RCAD, a Ufm1 E3 ligase, has an intricate connection with ER stress. Int J Biol Macromol. 2019;135:760–7.
Chen C, Itakura E, Weber KP, Hegde RS, de Bono M. An ER complex of ODR-4 and ODR-8/Ufm1 specific protease 2 promotes GPCR maturation by a Ufm1-independent mechanism. PLoS Genet. 2014;10(3):e1004082.
Homrich M, Wobst H, Laurini C, Sabrowski J, Schmitz B, Diestel S. Cytoplasmic domain of NCAM140 interacts with ubiquitin-fold modifier-conjugating enzyme-1 (Ufc1). Exp Cell Res. 2014;324(2):192–9.
Colin E, Daniel J, Ziegler A, Wakim J, Scrivo A, Haack TB, Khiati S, Denomme AS, Amati-Bonneau P, Charif M, et al. Biallelic variants in UBA5 reveal that disruption of the UFM1 cascade can result in early-onset encephalopathy. Am J Hum Genet. 2016;99(3):695–703.
Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol. 2006;291(3):H1411–20.
Watson CM, Crinnion LA, Gleghorn L, Newman WG, Ramesar R, Beighton P, Wallis GA. Identification of a mutation in the ubiquitin-fold modifier 1-specific peptidase 2 gene, UFSP2, in an extended South African family with Beukes hip dysplasia. S Afr Med J. 2015;105(7):558–63.
Rubio MD, Wood K, Haroutunian V, Meador-Woodruff JH. Dysfunction of the ubiquitin proteasome and ubiquitin-like systems in schizophrenia. Neuropsychopharmacology. 2013;38(10):1910–20.
Qin B, Yu J, Nowsheen S, Wang M, Tu X, Liu T, Li H, Wang L, Lou Z. UFL1 promotes histone H4 ufmylation and ATM activation. Nat Commun. 2019;10(1):1242.
Wang Z, Gong Y, Peng B, Shi R, Fan D, Zhao H, Zhu M, Zhang H, Lou Z, Zhou J, et al. MRE11 UFMylation promotes ATM activation. Nucleic Acids Res. 2019;47(8):4124–35.
Potter JL, Narasimhan J, Mende-Mueller L, Haas AL. Precursor processing of pro-ISG15/UCRP, an interferon-beta-induced ubiquitin-like protein. J Biol Chem. 1999;274(35):25061–8.
Haas AL, Ahrens P, Bright PM, Ankel H. Interferon induces a 15-kilodalton protein exhibiting marked homology to ubiquitin. J Biol Chem. 1987;262(23):11315–23.
Skaug B, Chen ZJ. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143(2):187–90.
Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20(3):362–71.
Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol. 2004;24(21):9592–600.
Malakhova O, Malakhov M, Hetherington C, Zhang DE. Lipopolysaccharide activates the expression of ISG15-specific protease UBP43 via interferon regulatory factor 3. J Biol Chem. 2002;277(17):14703–11.
Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem. 2002;277(12):9976–81.
Catic A, Fiebiger E, Korbel GA, Blom D, Galardy PJ, Ploegh HL. Screen for ISG15-crossreactive deubiquitinases. PLoS ONE. 2007;2(7):e679.
Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–97.
Park JH, Yang SW, Park JM, Ka SH, Kim JH, Kong YY, Jeon YJ, Seol JH, Chung CH. Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nat Commun. 2016;7:12513.
Park JM, Yang SW, Yu KR, Ka SH, Lee SW, Seol JH, Jeon YJ, Chung CH. Modification of PCNA by ISG15 plays a crucial role in termination of error-prone translesion DNA synthesis. Mol Cell. 2014;54(4):626–38.
Fan W, Cai W, Parimoo S, Schwarz DC, Lennon GG, Weissman SM. Identification of seven new human MHC class I region genes around the HLA-F locus. Immunogenetics. 1996;44(2):97–103.
Theng SS, Wang W, Mah WC, Chan C, Zhuo J, Gao Y, Qin H, Lim L, Chong SS, Song J, et al. Disruption of FAT10-MAD2 binding inhibits tumor progression. Proc Natl Acad Sci USA. 2014;111(49):E5282–91.
Schmidtke G, Aichem A, Groettrup M. FAT10ylation as a signal for proteasomal degradation. Biochim Biophys Acta. 2014;1843(1):97–102.
Canaan A, DeFuria J, Perelman E, Schultz V, Seay M, Tuck D, Flavell RA, Snyder MP, Obin MS, Weissman SM. Extended lifespan and reduced adiposity in mice lacking the FAT10 gene. Proc Natl Acad Sci USA. 2014;111(14):5313–8.
Aichem A, Groettrup M. The ubiquitin-like modifier FAT10 in cancer development. Int J Biochem Cell Biol. 2016;79:451–61.
Aichem A, Kalveram B, Spinnenhirn V, Kluge K, Catone N, Johansen T, Groettrup M. The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J Cell Sci. 2012;125(Pt 19):4576–85.
Leng L, Xu C, Wei C, Zhang J, Liu B, Ma J, Li N, Qin W, Zhang W, Zhang C, et al. A proteomics strategy for the identification of FAT10-modified sites by mass spectrometry. J Proteome Res. 2014;13(1):268–76.
Chen Z, Zhang W, Yun Z, Zhang X, Gong F, Wang Y, Ji S, Leng L. Ubiquitinlike protein FAT10 regulates DNA damage repair via modification of proliferating cell nuclear antigen. Mol Med Rep. 2018;17(6):7487–96.
Zeng Y, Iv YS, Pan QH, Zhou YG, Li H. An overactive neddylation pathway serves as a therapeutic target and MLN4924 enhances the anticancer activity of cisplatin in pancreatic cancer. Oncol Lett. 2019;18(3):2724–32.
Hu L, Bai ZG, Ma XM, Bai N, Zhang ZT. MRFAP1 plays a protective role in neddylation inhibitor MLN4924-mediated gastric cancer cell death. Eur Rev Med Pharmacol Sci. 2018;22(23):8273–80.
Lv Y, Li B, Han K, Xiao Y, Yu X, Ma Y, Jiao Z, Gao J. The Nedd8-activating enzyme inhibitor MLN4924 suppresses colon cancer cell growth via triggering autophagy. Korean J Physiol Pharmacol. 2018;22(6):617–25.
Chen X, Cui D, Bi Y, Shu J, Xiong X, Zhao Y. AKT inhibitor MK-2206 sensitizes breast cancer cells to MLN4924, a first-in-class NEDD8-activating enzyme (NAE) inhibitor. Cell Cycle. 2018;17(16):2069–79.
Oladghaffari M, Shabestani Monfared A, Farajollahi A, Baradaran B, Mohammadi M, Shanehbandi D, Asghari Jafar Abadi M, Pirayesh Islamian J. MLN4924 and 2DG combined treatment enhances the efficiency of radiotherapy in breast cancer cells. Int J Radiat Biol. 2017;93(6):590–9.
Lan H, Tang Z, Jin H, Sun Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci Rep. 2016;6:24218.
Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, Pan Y, Ding C, Qian J, Wu L, et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72(13):3360–71.
Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, Danilov AV. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20(6):1576–89.
Han K, Wang Q, Cao H, Qiu G, Cao J, Li X, Wang J, Shen B, Zhang J. The NEDD8-activating enzyme inhibitor MLN4924 induces G2 arrest and apoptosis in T-cell acute lymphoblastic leukemia. Oncotarget. 2016;7(17):23812–24.
Khalife J, Radomska HS, Santhanam R, Huang X, Neviani P, Saultz J, Wang H, Wu YZ, Alachkar H, Anghelina M, et al. Pharmacological targeting of miR-155 via the NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia. 2015;29(10):1981–92.
Knorr KL, Schneider PA, Meng XW, Dai H, Smith BD, Hess AD, Karp JE, Kaufmann SH. MLN4924 induces Noxa upregulation in acute myelogenous leukemia and synergizes with Bcl-2 inhibitors. Cell Death Differ. 2015;22(12):2133–42.
Visconte V, Nawrocki ST, Espitia CM, Kelly KR, Possemato A, Beausoleil SA, Han Y, Carraway HE, Nazha A, Advani AS, et al. Comprehensive quantitative proteomic profiling of the pharmacodynamic changes induced by MLN4924 in acute myeloid leukemia cells establishes rationale for its combination with azacitidine. Leukemia. 2016;30(5):1190–4.
Lockhart AC, Bauer TM, Aggarwal C, Lee CB, Harvey RD, Cohen RB, Sedarati F, Nip TK, Faessel H, Dash AB, et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs. 2019;37(1):87–97.
Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Mulligan G, Pickard MD, Faessel H, Dezube BJ, Hamid O. A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Invest New Drugs. 2016;34(4):439–49.
Shah JJ, Jakubowiak AJ, O’Connor OA, Orlowski RZ, Harvey RD, Smith MR, Lebovic D, Diefenbach C, Kelly K, Hua Z, et al. Phase I Study of the novel investigational NEDD8-activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22(1):34–43.
Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ, Cleary JM, Mahalingam D, Pickard MD, Faessel HM, et al. Phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res. 2016;22(4):847–57.
Swords RT, Erba HP, DeAngelo DJ, Bixby DL, Altman JK, Maris M, Hua Z, Blakemore SJ, Faessel H, Sedarati F, et al. Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169(4):534–43.
Acknowledgements
We gratefully acknowledge funding from National Institutes of Health Grants (CA217183, CA203561 and CA130996).
Author information
Authors and Affiliations
Contributions
Each author substantially contributed to the review. JY and BQ: conception and design, drafting the review; ZL: conception and design, revising it critically for important intellectual content, and final approval of the version to be published. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yu, J., Qin, B. & Lou, Z. Ubiquitin and ubiquitin-like molecules in DNA double strand break repair. Cell Biosci 10, 13 (2020). https://doi.org/10.1186/s13578-020-0380-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-020-0380-1