
Abstract
Background
Interleukin (IL)-1β is a pro-inflammatory cytokine that plays a role in the pathogenesis of multiple sclerosis (MS) and experimental autoimmune encephalomyelitis (EAE), the animal model for MS. Yet, detailed studies on IL-1β expression in different stages of MS lesion development and a comparison of IL-1β expression in MS and EAE are lacking.
Methods
Here, we performed an extensive characterization of IL-1β expression in brain tissue of MS patients, which included different MS lesion types, and in brain tissue of rhesus macaques with EAE.
Results
In rhesus EAE brain tissue, we observed prominent IL-1β staining in MHC class II+ cells within perivascular infiltrates and at the edges of large demyelinating lesions. Surprisingly, staining was localized to resident microglia or differentiated macrophages rather than to infiltrating monocytes, suggesting that IL-1β expression is induced within the central nervous system (CNS). By contrast, IL-1β staining in MS brain tissue was much less pronounced. Staining was found in the parenchyma of active and chronic active MS lesions and in nodules of MHC class II+ microglia in otherwise normal appearing white matter. IL-1β expression was detected in a minority of the nodules only, which could not be distinguished by the expression of pro- and anti-inflammatory markers. These nodules were exclusively found in MS, and it remains to be determined whether IL-1β+ nodules are destined to progress into active lesions or whether they merely reflect a transient response to cellular stress.
Conclusions
Although the exact localization and relative intensity of IL-1β expression in EAE and MS is different, the staining pattern in both neuroinflammatory disorders is most consistent with the idea that the expression of IL-1β during lesion development is induced in the tissue rather than in the periphery.
Similar content being viewed by others


Background
IL-1β is a cytokine with potent pro-inflammatory characteristics. High levels of systemic IL-1β lead to a rise in body temperature by affecting the activity of the hypothalamus, to vasodilation, and to increased expression of adhesion factors on endothelial cells enabling transmigration of leukocytes [1, 2]. Furthermore, IL-1β orchestrates the innate immune response [3] and can induce skewing of T cells towards Th17 cells [4–7], thereby linking innate immune responses to activation of the adaptive immune system. The synthesis of IL-1β precursor protein is induced by IL-1α or by activation of receptors of the innate immune system such as Toll-like receptors (TLR) and NOD-like receptors (NLR) [8, 9]. Secretion of bioactive IL-1β requires additional cleavage of the precursor protein by a cysteine protease, which in turn requires activation [10]. Caspase 1 is the best-described cysteine protease that is activated by a protein complex called the inflammasome [10, 11].
Inflammasomes play a role in several neurodegenerative and neuroinflammatory diseases as well as in animal models for such diseases [12–18]. NLR-mediated activation is critically involved in inflammasome formation and is evoked by disturbances in cellular homeostasis, as caused by, e.g., pathogens, large protein aggregates, and neighboring cell death. Subsequently, NLR associate with inflammatory caspases, mostly via the adaptor protein ASC, leading to processing and secretion of pro-inflammatory cytokines such as IL-1β and IL-18 [19, 20].
The involvement of IL-1β and the inflammasome in experimental autoimmune encephalomyelitis (EAE), a commonly used animal model for MS, has been confirmed in different studies [21]. Inhibition of IL-1-induced signaling ameliorates the development of EAE in both rats and mice [22–25], and mice that are deficient in NLRP3, ASC, or caspase 1 expression are characterized by delayed onset of disease and less severe clinical symptoms [26–28]. Furthermore, expression levels of IL-1β [29–31], specific NLRs (e.g., NLRP1 and NLRP3) and caspase 1 are increased in the brain and spinal cord during disease [26, 32]. In addition, treatment with a caspase 1 inhibitor attenuates clinical signs of mouse EAE [33]. Treatment with interferon (IFN)β, a registered therapeutic biological for MS [34], decreases brain pathology by reducing serum IL-1β and caspase 1 activation levels [35].
In human macrophages, IFNβ inhibits inflammasome-mediated activation by inhibition of pro-IL1β transcription, by decreasing the availability of NLRP3-activating ligands, and by directly inhibiting NLRP3 and caspase-1 activation via post-translational modifications [35–37]. In line with this, monocytes derived from IFNβ-treated MS patients are characterized by decreased IL-1β production in response to inflammasome-activating stimuli [36]. More evidence for the involvement of IL-1β in MS pathogenesis comes from studies demonstrating that elevated IL-1β levels in cerebrospinal fluid (CSF) and blood of MS patients correlate with disease susceptibility, severity, and progression [38–43]. In addition, therapeutic approaches used for treatment of MS, i.e., IFNβ, Copaxone, or steroid treatment lead to increased levels of IL-1 receptor antagonist (IL-1RA), the natural inhibitor of the IL-1 receptor, in the blood [39, 44, 45].
Although these data suggest a role for IL-1β in both EAE and MS and provide a rationale for clinical trials that target the IL-1 axis [8], there are discordant results relating to the expression of IL-1β during the course of both diseases. While there is consensus on the abundant expression of IL-1β in the brain during EAE induced in rats and mice [29–31, 46], reports on IL-1β expression in MS lesions [47–49] are by no means unequivocal [50, 51]. Furthermore, it is unclear during which stages of pathogenesis IL-1β is produced and by which cells. We therefore characterized IL-1β expression in the brain tissue of MS patients, which included different types of MS lesions, side-by-side with brain tissue derived from rhesus macaques in which EAE was induced. We performed in depth analyses of the cellular sources of IL-1β expression and phenotyped these cells based on the expression of pro- and anti-inflammatory markers. Our results reveal distinct characteristics of either EAE or MS that might well reflect differences in pathogenesis. However, in both neuroinflammatory disorders, the expression of IL-1β during disease progression is mainly induced in the brain itself.
Methods
Brain tissue
We selected paraffin-embedded tissue blocks from three rhesus macaques without neurological disease, from eight rhesus macaques with EAE and from four immunized rhesus macaques that did not develop clinical disease (Table 1) from earlier studies [52–54] that were performed at the Biomedical Primate Research Centre (BPRC; Rijswijk, the Netherlands), and of which the tissue blocks were archived at the Department of Neuroimmunology from the Center of Brain Research (Vienna, Austria). As such no animals were sacrificed for the exclusive purpose of this study, thereby complying with the priority 3Rs program of the BPRC. EAE was induced by immunization with recombinant human (rh)MOG protein either in incomplete or complete Freund’s adjuvant (resp. IFA or CFA) [52–54]. All procedures were performed in compliance with guidelines of the Institutional Animal Care and Use Committee (IACUC) in accordance with Dutch law.
Human brain tissue samples were obtained from the Netherlands Brain Bank (NBB; coordinator Dr. Huitinga, Amsterdam, the Netherlands). NBB received permission to perform autopsies for the use of tissue and to access medical records for research purposes from the Medical Ethical Committee of the VU Medical Centre (Amsterdam, the Netherlands). All patients and controls, or their next of kin, had given informed consent for autopsy and the use of brain tissue for research purposes. Relevant clinical information was retrieved from the medical records and is summarized in Table 2. We selected a total of 45 tissue blocks (paraffin-embedded 22 blocks; frozen 23 blocks) from 28 MS patients (female-to-male ratio 4:3; average age 60.9 years; average post-mortem delay 8 h) and five tissue blocks from five donors without neurological disease (female-to-male ratio 2:3; average age 70.8 years; average post-mortem delay 6 h). This panel represented different types of MS, including relapsing remitting (RR), secondary progressive (SP), and primary progressive (PP) MS.
Immunohistochemistry
An overview of the used antibodies and their dilutions is given in Table 3. Isotype controls and omission of the primary antibodies were used to confirm specificity of the primary antibodies.
Five micrometer-thick paraffin sections were collected on Superfrost Plus glass slides (VWR international, Leuven, Belgium) and dried at 37 °C. Tissue sections were characterized for the presence of demyelination by staining for proteolipid protein (PLP) and for inflammation by staining for MHC class II. PLP and MHC class II were stained according to a previously described protocol [55] using mouse anti-human PLP or mouse anti-human HLA-DR antibodies (these are also cross reactive to the rhesus macaque equivalent Mamu-DR), EnVision horse radish peroxidase (HRP), and 3,3′-diaminobenzidine (DAB; both DAKO, Heverlee, Belgium). Consecutive sections were stained for IL-1β as described previously [56] using goat anti-human IL-1β antibodies, biotinylated anti-sheep/-goat antibodies, avidin-HRP, and DAB.
Five micrometer-thick cryosections were collected on Superfrost Plus glass slides and air-dried. For IL-1β stainings, sections were formalin-fixed for 10 min, endogenous peroxidase was quenched in 0.3 % H2O2 in phosphate-buffered saline (PBS) and sections were incubated with 10 % fetal calf serum (FCS) in wash buffer (DAKO) for 20 min at RT. Thereafter, sections were incubated with goat anti-human IL-1β antibodies overnight at 4 °C. After rinsing, the primary antibody was reapplied for 1 h at RT, followed by incubation with donkey anti-goat HRP and they were developed with DAB.
Immunohistochemical double staining for IL-1β with MHC class II, CD74, CD40, CD200R, CCL22, or MR were performed on cryosections. Slides were stained for IL-1β as described above and developed with DAB. Thereafter, slides were rinsed thoroughly and incubated with anti-human HLA-DR, CD74, CD40, CD200R, CCL22, or MR antibodies overnight at 4 °C. Then slides were incubated with either goat anti-mouse IgG alkaline phosphatase or goat anti-rabbit IgG alkaline phosphatase and further developed with Liquid Permanent Red solution (Dako) for 10 min at RT.
Sections were imaged using the Olympus BX50 microscope and Canvas X Pro (Canvas X software Inc, 2015, version 16, build 2115) was used for graphical representations.
Immunofluorescence
Immunofluorescent double staining for IL-1β with Iba-1 or MRP14 were performed on paraffin-embedded tissue sections. Antigen retrieval was performed by heating the slides in Tris-EDTA (pH 8.5). Thereafter, sections were incubated with anti-human IL-1β overnight at 4 °C. After rinsing the slides, the primary antibody was reapplied for 1 h at RT, followed by incubation with biotinylated anti-sheep/-goat antibodies and avidin-CY2 in the dark. Next, slides were incubated with anti-human Iba-1 or MRP14 antibodies for 1 h at RT. Then slides were incubated with either donkey anti-rabbit TRITC or donkey anti-mouse TRITC and embedded in Vectashield mounting medium containing DAPI (Brunswig chemie). Sections were imaged using the Nikon Microphot-FXA microscope and Canvas X Pro was used for graphical representations.
Results
Rhesus EAE
We studied brain tissue from three rhesus macaques without neurological disease and from 12 rhesus macaques that were immunized with rhMOG in either IFA or CFA, of which eight animals developed clinical EAE (Table 1). Brain tissue from control animals and from animals that did not develop clinical EAE did not contain detectable demyelination, inflammatory activity, or IL-1β. Tissue from animals that developed clinical EAE was characterized by perivascular infiltrates. We observed considerable inter-donor variability concerning the number and extent of the observed EAE lesions, probably attributable to the outbred nature of the model [54].
In animals immunized with rhMOG in IFA, we studied 30 perivascular lesions and three large areas with infiltrating cells and demyelination (Table 4). IL-1β staining was observed in 50 % of the perivascular infiltrates closely surrounding blood vessels (Fig. 1a, b) and in all large areas with extensive MHC class II expression and demyelination (Fig. 1c). Double immunofluorescent staining identified all IL-1β+ cells as Iba-1+ (Fig. 1d) and as MRP14− or MRP14low (Fig. 1e). As MRP14 is a marker that is strongly expressed on monocytes and neutrophils [57–59], this staining pattern is most consistent with microglia or differentiated macrophages as main sources of IL-1β.

IL-1β expression in brain tissue of rhesus macaques with EAE induced by rhMOG in IFA. Brain lesions were characterized based on the extent of myelin (PLP in brown, left panels) damage and activation of innate immune cells (MHC class II in brown, middle panels). In small perivascular lesions without signs of demyelination (a), IL-1β staining (in brown, right panels) was mainly localized in MHC class II+ cells at the edge of the lesion. In mid-sized MHC class II+ lesions with clear signs of demyelination (b), IL-1β staining was more pronounced. In large fulminating lesions with extensive demyelination and infiltration of MHC class II+ cells (c), IL-1β staining was less pronounced compared to the mid-sized lesions and observed at the edge of the demyelinated area. Double labeling of perivascular lesions for IL-1β (in green) and Iba-1 or MRP14 (in red) demonstrated that all IL-1β+ cells were Iba-1+ (d), whereas all IL-1β+ cells were MRP14− or MRP14low (e). Original magnifications ×10, scale bar represents 200 μm, insets ×100. Nuclei were counterstained with hematoxylin (blue)
As IFA does not contain mycobacteria that were previously shown to be involved in IL-1β production [60], we also studied the expression of IL1β in brain tissue of animals immunized with rhMOG in CFA. We studied 432 perivascular lesions and 10 large areas with strong MHC class II expression and demyelination. IL-1β staining was observed in 36 % of the perivascular infiltrates (Fig. 2a) and in 70 % of the large areas with strong MHC class II expression and demyelination (Fig. 2b). Although IL-1β+ cells and MRP14high cells were observed in close vicinity in the same lesions, all IL-1β+ cells were Iba-1+ (Fig. 2c) and MRP14− or MRP14low (Fig. 2d), similar to what was observed in animals immunized with rhMOG in IFA. In conclusion, IL-1β expression was associated mainly with MHC class II expressing cells present in perivascular infiltrates or at the edges of actively demyelinating lesions and not with infiltrating monocytes. Despite the fact that animals immunized with rhMOG in CFA were characterized by a much more rapid onset of clinical disease than those immunized with rhMOG in IFA, the IL-1β staining patterns were similar.

IL-1β expression in brain tissue of rhesus macaques with EAE induced by rhMOG in CFA. Brain lesions were characterized based on the extent of myelin (PLP in brown, left panels) damage and activation of innate immune cells (MHC class II in brown, middle panels). In small perivascular lesions without signs of demyelination (a), IL-1β staining (in brown, right panels) was mainly localized in MHC class II+ cells at the edge of the lesion. In large fulminating lesions with extensive demyelination and infiltration of MHC class II+ cells (b), IL-1β staining was more pronounced and mainly observed at the edges of the demyelinated area. Double labeling of perivascular lesions for IL-1β (in green) and Iba-1 or MRP14 (in red) demonstrated that all IL-1β+ cells were Iba-1+ (c), whereas all IL-1β+ cells were MRP14− or MRP14low (d). Original magnifications ×10, scale bar represents 200 μm, insets ×100
MS
We started our characterization of IL-1β in MS by examining well-characterized paraffin-embedded tissue blocks of five donors without neurological disease and of 17 MS patients. MS lesions were characterized for the presence of demyelination by staining for PLP and for inflammation by staining for MHC class II and categorized as active, chronic active, and inactive [55, 61, 62]. Most tissue blocks contained multiple lesions of different categories (Table 5).
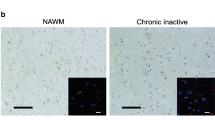
We did not observe IL-1β staining in healthy controls. In contrast to our expectation, we also did not detect IL-1β expression in active, chronic active, or in inactive MS lesions (Table 5, Fig. 3a–c). Surprisingly, examination of normal appearing white matter (NAWM) from MS patients revealed IL-1β expression in nodules of MHC class II+ microglia (Fig. 3d). These microglia nodules occurred without evident signs of demyelination or infiltration and were previously described by different research groups [55, 61–63]. Although their role in MS pathogenesis is unclear at present, some authors suggested that these nodules are preactive lesions [55, 61, 62]. In total, we studied 38 of such microglia nodules in nine patients. IL-1β staining was observed in eight of the 38 microglia nodules (21 %; Table 5, Fig. 3d). The number of IL-1β+ microglia nodules varied between patients. In one patient, we observed exclusively IL-1β+ microglia nodules. In two patients, we observed both IL-1β+ and IL-1β− microglia nodules, and in six patients, we observed exclusively IL-1β− microglia nodules. Formal confirmation of the identity of the IL-1β+ cells as microglia was obtained by colocalization with Iba-1 (Fig. 3e). Irrespective of the immunization protocol, no equivalent of these microglia nodules was found in rhesus macaques with clinical EAE.

IL-1β expression in different types of MS lesions. MS lesions in paraffin-embedded brain tissue sections were characterized based on the extent of myelin (PLP in brown, left panels) damage and activation of innate immune cells (MHC class II in brown, middle panels). Active MS lesions were classified as areas with ongoing demyelination and activation of MHC class II+ innate immune cells (a). Chronic active lesions were classified by the presence of a completely demyelinated (PLP−) center surrounded by a rim of MHC class II+ cells (b). Inactive lesions were classified by the presence of demyelinated areas where the immune response has resided (c). We did not detect IL-1β (in brown, right panels) in active, chronic active, and inactive MS lesions in paraffin-embedded tissue sections (a–c). In addition, we observed MHC class II+ microglia nodules in otherwise NAWM (d) in which IL-1β was expressed. Double labeling of these microglia nodules for IL-1β (in red) and Iba-1 (in green) implicated that all IL-1β+ cells were Iba-1+ (e). Original magnifications ×4, insets ×100 (a–d), scale bar represents 500 μm (a–d) or 10 μm (e). Nuclei were counterstained with hematoxylin (blue; a–d)
The existing literature on IL-1β expression in MS lesions contains contradicting observations. Whereas some studies reported a paucity of staining as we do [50, 51], others reported more extensive staining [47–49]. Differences in fixation procedures, tissue treatment and staining protocols may all have influenced the results. We therefore also characterized IL-1β staining in snap-frozen tissue blocks. For validation purposes, we included patients of which paraffin-embedded tissue sections had already been characterized by us.
In total, we studied 25 active lesions in 15 patients and six chronic active lesions in two patients. IL-1β staining in cryosections was more extensive than in paraffin-embedded sections, now also revealing expression in active and chronic active lesions. IL-1β staining was observed in 52 % of active lesions (Table 6, Fig. 4a). In nine patients, we observed IL-1β staining in ramified MHC class II+ cells in the parenchyma, whereas in six patients, we could not detect IL-1β in any of the active lesions. Similarly, IL-1β staining was observed in ramified MHC class II+ cells in the rim of all chronic active lesions (Table 6, Fig. 4b). In line with our earlier results, we also observed IL-1β staining in MHC class II+ microglia nodules in otherwise NAWM. In total, we studied 106 microglia nodules in seven patients. IL-1β staining was observed in 52 of these microglia nodules (49 %; Table 6, Fig. 4c). The number of IL-1β+ microglia nodules varied between patients. In one patient, all microglia nodules were IL-1β+, in three patients, we observed both IL-1β+ and IL-1β− microglia nodules, and in three patients, we observed only IL-1β− microglia nodules.

IL-1β expression in different types of MS lesions. MS lesions in frozen brain tissue sections were characterized based on the extent of myelin (PLP in brown, left panels) damage and activation of innate immune cells (MHC class II in brown, middle panels). Active MS lesions were classified as areas with ongoing demyelination and activation of MHC class II+ innate immune cells (a). IL-1β expression (in brown, right panels) was mainly observed in ramified MHC class II+ cells in the parenchyma, which were mainly localized at the edges of active lesions. Chronic active lesions were classified by the presence of a completely demyelinated (PLP−) center surrounded by a rim of MHC class II+ cells (b). IL-1β expression was observed in MHC class II+ cells in the rim of the lesion. Again, we observed MHC class II+ microglia nodules in otherwise NAWM (c) in which IL-1β was expressed. Original magnifications ×10, insets ×40 (a–c), scale bar represents 200 μm
To further characterize the IL-1β+ cells, microglia nodules were stained for molecules associated with pro- and anti-inflammatory phenotypes [64]. IL-1β staining in MHC class II+ microglia nodules (Fig. 5a) colocalized with the pro-inflammatory markers CD74 (Fig. 5b) and CD40 (Fig. 5c) as well as with the anti-inflammatory marker CD200R (Fig. 5d). In most microglia nodules, IL-1β staining also colocalized with the anti-inflammatory marker CCL22, although some microglia nodules contained IL-1β+/CCL22− cells (Fig. 5e). By contrast, IL-1β+ microglia did not stain for mannose receptor (MR; Fig. 5f). As previously described [64, 65], MR staining was predominantly observed in perivascular spaces and not in microglia nodules. IL-1β+ and IL-1β− microglia nodules could not be distinguished based on the expression of these markers. In conclusion, IL-1β+ nodular microglia expressed a mix of pro-inflammatory and anti-inflammatory markers, in line with other reports [64].

Activation status of IL-1β+ cells in microglia nodules. Microglia nodules were classified as clusters of MHC class II+ cells in otherwise NAWM. These clusters of activated microglia were double stained for IL-1β (in brown) and cell surface markers associated with pro-inflammatory or anti-inflammatory cellular phenotypes (in red). IL-1β staining colocalized with MHC class II (a) and cell surface markers CD74 (b) and CD40 (c) as well as with CD200R (d). In most microglia nodules, IL-1β staining also colocalized with CCL22 (e), although some microglia nodules contained IL-1β+/CCL22− cells. IL-1β+ microglia did not express MR (f). Original magnifications: ×40, scale bar represents 50 μm. Nuclei were counterstained with hematoxylin (blue)
Microglia in the rim of chronic active lesions expressed a similar mix of pro- and anti-inflammatory markers as the microglia in the nodules (Additional file 1), except that not all IL-1β+ cells in the rim of chronic active lesions were CD74+ and that more colocalization was found with CCL22, which may be indicative of a slightly less pro-inflammatory profile.

IL-1β expression in perivascular infiltrates in active MS lesions. Within the active lesions of two patients, we observed perivascular infiltrates in areas with ongoing demyelination (PLP, in brown; a). These perivascular cells were strongly MHC class II+ (in brown; b). We observed IL-1β expression (in brown) in cells associated with these perivascular infiltrates, mainly at the edges of the infiltrate and the parenchyma (c). These cells were double stained for IL-1β (in brown) and cell surface markers associated with pro-inflammatory or anti-inflammatory cellular phenotypes (in red). IL-1β staining colocalized with MHC class II (d) and CD74, although we also observed multiple IL-1β+/CD74− cells (e). Furthermore, IL-1β staining colocalized with CD40 (f), CD200R (g), and CCL22, although we also observed some IL-1β+/CCL22− cells (h). IL-1β and MR staining were both observed in the same perivascular infiltrates, but all IL-1β+ cells were MR− (i). In addition, within one active lesion, we observed MHC class II+ cells (in red) with a foamy appearance (j). These MHC class II+ foamy cells did not express detectable levels of IL-1β. In addition, we observed some IL-1β staining (in brown) in reactive astrocytes in the same active lesion (k). Original magnifications ×40, scale bar represents 50 μm. Nuclei were counterstained with hematoxylin (blue)
Since IL-1β expression in EAE was mainly localized to perivascular infiltrates, we screened our available patient material for such lesions. In two patients, very active lesions were found that were associated with large perivascular infiltrates. Here, we also observed IL-1β staining in cells associated with the perivascular infiltrates (Fig. 6a–c), mainly at the edges of the infiltrates. These cells were MHC class II+ (Fig. 6d) and again expressed a mix of pro- and anti-inflammatory markers (Fig. 6e-h). Although IL-1β and MR staining were observed in the same perivascular infiltrates, IL-1β staining never colocalized with MR staining (Fig. 6i), nor with MHC class II+ cells with a foamy appearance (Fig. 6j). Although this may suggest that myelin ingestion inhibits the production of IL-1β, as was shown previously for other pro-inflammatory cytokines [50], we could not confirm this in vitro (data not shown). We did also observe some IL-1β immunoreactivity in reactive astrocytes (Fig. 6k), as reported by other authors [47]. In both patients that showed these IL-1β+ perivascular lesions, microglia nodules and ramified cells within the rim of chronic active lesions were also IL-1β+.
Discussion
Different lines of evidence suggest that IL-1β has a pathogenic role in MS and in the animal model for MS, EAE [26, 28, 32, 35]. Here, we characterized the expression of IL-1β in brain tissue from rhesus macaques with EAE and in different MS lesion types. Contrary to our expectations, we observed that IL-1β expression was mainly restricted to glia cells, most importantly microglia, both in EAE as well as in MS. In rhesus EAE, IL-1β expression was most abundant in perivascular lesions and in active demyelinating lesions with large infiltrates, whereas in MS IL-1β expression was much less abundant and mainly observed in parenchymal nodules of activated microglia.
Although the perivascular localization of IL-1β in rhesus EAE was largely in line with previous studies in rodents [29, 30, 46] and in accordance with the peripheral induction of disease, we did not detect IL-1β in MRP14high monocytes that had recently infiltrated the CNS or in T lymphocytes [31, 66]. Especially in animals immunized with rhMOG in CFA, the enhanced immunogenicity caused by the presence of mycobacteria in the adjuvant has been linked to their ability to directly cause IL-1β expression, inflammasome activation, and IL-1β secretion in monocytes and macrophages [60, 67–69]. The induced expression of pro-IL-1β by immunization is however local and most likely of a transient nature. Recently, we described that in vitro pro-IL-1β expression can be potently induced in rhesus macaque primary microglia and peripheral macrophages, but that expression is subject to strong and rapid negative regulation [70]. As the last immunization was performed at least 12 days before euthanasia, it is unlikely that the immunization-induced expression of IL-1β is responsible for the staining pattern observed in the CNS. Our results suggest that IL-1β expression is induced within the CNS and reflects a tissue response to stress that is associated with infiltration of peripheral immune cells. This would also be in line with the IL-1β+ microglia in brain tissue of animals immunized with rhMOG in IFA. We are not the first to report on this phenomenon, as previous studies demonstrated that IL-1β expression in microglia-like cells is increased by infiltration of immune cells into the CNS [71] and that NLRP3 inflammasome activation is induced in rodents where EAE was passively induced [35]. Although different studies have reported on the expression of IL-1β in infiltrating T lymphocytes in rodent EAE [31, 66], we did not detect IL-1β+ T cells in rhesus EAE tissue. This discrepancy may be attributable to differences in immunization protocols or to differences between species. In this context, it is noteworthy that the rhesus EAE model is characterized by a hyperacute development of clinical symptoms, rendering the model less suitable to study more chronic features of the neuroinflammatory process. The marmoset EAE model might provide a suitable alternative for further studies on this topic as it is characterized by a more chronic development of clinical symptoms [72].
IL-1β expression in MS was much less prominent as in rhesus EAE and the staining pattern was markedly different. In MS, IL-1β expression was mainly localized in the parenchyma, especially in parenchymal nodules of activated microglia. We observed that only a portion of these nodules was IL-1β+. Characterization of the IL-1β+ microglia in these nodules using markers for anti- and pro-inflammatory phenotypes showed that these cells express a mix of both markers, which is in line with other studies [64, 65]. It has been proposed that most of these nodules might resolve spontaneously while other might progress into an active lesion [61], and previous studies have demonstrated that IL-1β can initiate the demyelination process [73, 74]. Whether the expression of IL-1β is a discriminating factor regarding the fate of the nodules remains to be determined as it is also well possible that the microglial expression of IL-1β merely reflects a transient response to cellular stress or to neuronal degeneration [75]. Various molecules associated with acute cellular stress induce IL-1β expression, including IL-1α, TNFα, the small stress protein alphaB-crystallin (HspB5), and high mobility group box 1 (HMGB1) [9, 76, 77]. Interestingly, HspB5 and TNFα are expressed in microglia nodules in MS [55, 76] and may contribute to the IL-1β expression as described here. However, whether these factors are specifically associated with the IL-1β+ microglia nodules remains to be investigated.
Interestingly, IL-1β has recently been demonstrated to play a role in neuronal degeneration via a p53-mediated apoptotic cascade [78]. In addition, IL-1β might affect cortical excitability in MS patients [43] and can be detected in the gray matter of rats in which chronic-relapsing EAE was induced [29]. We have therefore also analyzed IL-1β expression in five leukocortical lesions that were present in four patients. In the limited number of lesions we studied, IL-1β expression was barely detectable and almost exclusively restricted to the white matter (data not shown). A possible explanation might be that most cortical demyelination is thought to occur early during MS pathogenesis [79], and inflammatory activity might already have resolved in the lesions we studied. This topic warrants further investigations, both in MS and in EAE. Again, the rhesus EAE model is not suitable for such a study, as gray matter lesions are not present.
The etiology of MS is still debated, and both infectious and non-infectious factors have been proposed as inducers or precipitators of the disease [80–82]. NLR activation has been reported in response to infectious and sterile inflammation, and inflammasome-induced IL-1β might represent an a-specific hallmark of disrupted brain homeostasis, both in EAE and in MS. However, in contrast to MS, we did not observe IL-1β+ microglia nodules in rhesus EAE, which is most probably due to the acute nature of the model. Whether IL-1β expression as observed in MS can also be observed in more chronic EAE models requires further study.
Conclusions
In conclusion, the expression pattern of IL-1β in EAE and MS is consistent with a response that is initiated in the tissue rather than with the infiltration of IL-1β-producing monocytes. Whether this response plays a role in the exacerbation of the disease remains to be demonstrated. Most importantly, we here describe that a subpopulation of parenchymal IL-1β+ microglial nodules can be distinguished exclusively in MS with an as yet unknown role in lesion initiation or progression.
References
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: back to the future. Immunity. 2013;39:1003–18.
Sims JE, Smith DE. The IL-1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89–102.
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–9.
Duan J, Chung H, Troy E, Kasper DL. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe. 2010;7:140–50.
Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–91.
Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–41.
Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–52.
Hanamsagar R, Hanke ML, Kielian T. Toll-like receptor (TLR) and inflammasome actions in the central nervous system. Trends Immunol. 2012;33:333–42.
Dinarello CA. Interleukin-1 beta, interleukin-18, and the interleukin-1 beta converting enzyme. Ann N Y Acad Sci. 1998;856:1–11.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8.
Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med. 2008;12:2255–62.
Hafner-Bratkovic I, Bencina M, Fitzgerald KA, Golenbock D, Jerala R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1beta and neuronal toxicity. Cell Mol Life Sci. 2012;69:4215–28.
Hanamsagar R, Torres V, Kielian T. Inflammasome activation and IL-1beta/IL-18 processing are influenced by distinct pathways in microglia. J Neurochem. 2011;119:736–48.
Jamilloux Y, Pierini R, Querenet M, Juruj C, Fauchais AL, Jauberteau MO, Jarraud S, Lina G, Etienne J, Roy CR, et al. Inflammasome activation restricts Legionella pneumophila replication in primary microglial cells through flagellin detection. Glia. 2013;61:539–49.
Shi F, Yang L, Kouadir M, Yang Y, Wang J, Zhou X, Yin X, Zhao D. The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J Neuroinflammation. 2012;9:73.
Latz E. The inflammasomes: mechanisms of activation and function. Curr Opin Immunol. 2010;22:28–33.
Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65.
Inoue M, Shinohara ML. The role of interferon-beta in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis—in the perspective of inflammasomes. Immunology. 2013;139:11–8.
Furlan R, Bergami A, Brambilla E, Butti E, De Simoni MG, Campagnoli M, Marconi P, Comi G, Martino G. HSV-1-mediated IL-1 receptor antagonist gene therapy ameliorates MOG(35-55)-induced experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther. 2007;14:93–8.
Martin D, Near SL. Protective effect of the interleukin-1 receptor antagonist (IL-1ra) on experimental allergic encephalomyelitis in rats. J Neuroimmunol. 1995;61:241–5.
Matsuki T, Nakae S, Sudo K, Horai R, Iwakura Y. Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol. 2006;18:399–407.
Wiemann B, Van GY, Danilenko DM, Yan Q, Matheson C, Munyakazi L, Ogenstad S, Starnes CO. Combined treatment of acute EAE in Lewis rats with TNF-binding protein and interleukin-1 receptor antagonist. Exp Neurol. 1998;149:455–63.
Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol. 2010;185:974–81.
Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:10480–5.
Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4610–4.
Prins M, Eriksson C, Wierinckx A, Bol JG, Binnekade R, Tilders FJ, Van Dam AM. Interleukin-1beta and interleukin-1 receptor antagonist appear in grey matter additionally to white matter lesions during experimental multiple sclerosis. PLoS One. 2013;8:e83835.
Vainchtein ID, Vinet J, Brouwer N, Brendecke S, Biagini G, Biber K, Boddeke HW, Eggen BJ. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia. 2014;62:1724–35.
Mandolesi G, Musella A, Gentile A, Grasselli G, Haji N, Sepman H, Fresegna D, Bullitta S, De Vito F, Musumeci G. Interleukin-1beta alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J Neurosci. 2013;33:12105–21.
Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis. J Neurosci. 2009;29:14965–79.
Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol. 2011;186:5738–48.
Gajofatto A, Benedetti MD. Treatment strategies for multiple sclerosis: when to start, when to change, when to stop? World J Clin Cases. 2015;3:545–55.
Inoue M, Williams KL, Oliver T, Vandenabeele P, Rajan JV, Miao EA, Shinohara ML. Interferon-beta therapy against EAE is effective only when development of the disease depends on the NLRP3 inflammasome. Sci Signal. 2012;5:ra38.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23.
Hernandez-Cuellar E, Tsuchiya K, Hara H, Fang R, Sakai S, Kawamura I, Akira S, Mitsuyama M. Cutting edge: nitric oxide inhibits the NLRP3 inflammasome. J Immunol. 2012;189:5113–7.
de Jong BA, Huizinga TW, Bollen EL, Uitdehaag BM, Bosma GP, van Buchem MA, Remarque EJ, Burgmans AC, Kalkers NF, Polman CH, Westendorp RG. Production of IL-1beta and IL-1Ra as risk factors for susceptibility and progression of relapse-onset multiple sclerosis. J Neuroimmunol. 2002;126:172–9.
Dujmovic I, Mangano K, Pekmezovic T, Quattrocchi C, Mesaros S, Stojsavljevic N, Nicoletti F, Drulovic J. The analysis of IL-1 beta and its naturally occurring inhibitors in multiple sclerosis: the elevation of IL-1 receptor antagonist and IL-1 receptor type II after steroid therapy. J Neuroimmunol. 2009;207:101–6.
Reale M, de Angelis F, di Nicola M, Capello E, di Ioia M, Luca G, Lugaresi A, Tata AM. Relation between pro-inflammatory cytokines and acetylcholine levels in relapsing-remitting multiple sclerosis patients. Int J Mol Sci. 2012;13:12656–64.
Rossi S, Studer V, Motta C, Germani G, Macchiarulo G, Buttari F, Mancino R, Castelli M, De Chiara V, Weiss S, et al. Cerebrospinal fluid detection of interleukin-1beta in phase of remission predicts disease progression in multiple sclerosis. J Neuroinflammation. 2014;11:32.
Seppi D, Puthenparampil M, Federle L, Ruggero S, Toffanin E, Rinaldi F, Perini P, Gallo P. Cerebrospinal fluid IL-1beta correlates with cortical pathology load in multiple sclerosis at clinical onset. J Neuroimmunol. 2014;270:56–60.
Rossi S, Furlan R, De Chiara V, Motta C, Studer V, Mori F, Musella A, Bergami A, Muzio L, Bernardi G, et al. Interleukin-1beta causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol. 2012;71:76–83.
Burger D, Molnarfi N, Weber MS, Brandt KJ, Benkhoucha M, Gruaz L, Chofflon M, Zamvil SS, Lalive PH. Glatiramer acetate increases IL-1 receptor antagonist but decreases T cell-induced IL-1beta in human monocytes and multiple sclerosis. Proc Natl Acad Sci U S A. 2009;106:4355–9.
Comabella M, Julia E, Tintore M, Brieva L, Tellez N, Rio J, Lopez C, Rovira A, Montalban X. Induction of serum soluble tumor necrosis factor receptor II (sTNF-RII) and interleukin-1 receptor antagonist (IL-1ra) by interferon beta-1b in patients with progressive multiple sclerosis. J Neurol. 2008;255:1136–41.
Bauer J, Berkenbosch F, Van Dam AM, Dijkstra CD. Demonstration of interleukin-1 beta in Lewis rat brain during experimental allergic encephalomyelitis by immunocytochemistry at the light and ultrastructural level. J Neuroimmunol. 1993;48:13–21.
Kawana N, Yamamoto Y, Tsuyoshi I, Saito Y, Konno H, Arima K, Satoh JI. Reactive astrocytes and perivascular macrophages express NLRP3 inflammasome in active demyelinating lesions of multiple sclerosis and necrotic lesions of neuromyelitis optica and cerebral infarction. Clin Exp Neuroimmunol. 2013;4:296–304.
Brosnan CF, Cannella B, Battistini L, Raine CS. Cytokine localization in multiple sclerosis lesions: correlation with adhesion molecule expression and reactive nitrogen species. Neurology. 1995;45:S16–21.
Cannella B, Raine CS. The adhesion molecule and cytokine profile of multiple sclerosis lesions. Ann Neurol. 1995;37:424–35.
Boven LA, Van Meurs M, Van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, Aerts JM, Amor S, Nieuwenhuis EE, Laman JD. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain. 2006;129:517–26.
Kitic M, Hochmeister S, Wimmer I, Bauer J, Misu T, Mader S, Reindl M, Fujihara K, Lassmann H, Bradl M. Intrastriatal injection of interleukin-1 beta triggers the formation of neuromyelitis optica-like lesions in NMO-IgG seropositive rats. Acta Neuropathol Commun. 2013;1:5.
Haanstra KG, Hofman SO, Lopes Estevao DM, Blezer EL, Bauer J, Yang LL, Wyant T, Csizmadia V, t Hart BA, Fedyk ER. Antagonizing the alpha4beta1 integrin, but not alpha4beta7, inhibits leukocytic infiltration of the central nervous system in rhesus monkey experimental autoimmune encephalomyelitis. J Immunol. 2013;190:1961–73.
Haanstra KG, Jagessar SA, Bauchet AL, Doussau M, Fovet CM, Heijmans N, Hofman SO, van Lubeek-Veth J, Bajramovic JJ, Kap YS, et al. Induction of experimental autoimmune encephalomyelitis with recombinant human myelin oligodendrocyte glycoprotein in incomplete Freund’s adjuvant in three non-human primate species. J Neuroimmune Pharmacol. 2013;8:1251–64.
Haanstra KG, Dijkman K, Bashir N, Bauer J, Mary C, Poirier N, Baker P, Scobie L, t Hart BA, Vanhove B. Selective blockade of CD28-mediated T cell costimulation protects rhesus monkeys against acute fatal experimental autoimmune encephalomyelitis. J Immunol. 2015;194:1454–66.
van Horssen J, Singh S, van der Pol S, Kipp M, Lim JL, Peferoen L, Gerritsen W, Kooi EJ, Witte ME, Geurts JJ, et al. Clusters of activated microglia in normal-appearing white matter show signs of innate immune activation. J Neuroinflammation. 2012;9:156.
Bauer J, Lassmann H. Neuropathological techniques to investigate central nervous system sections in multiple sclerosis. Methods Mol Biol. 2016;1304:211–29.
Lagasse E, Clerc RG. Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol. 1988;8:2402–10.
Lominadze G, Rane MJ, Merchant M, Cai J, Ward RA, McLeish KR. Myeloid-related protein-14 is a p38 MAPK substrate in human neutrophils. J Immunol. 2005;174:7257–67.
Odink K, Cerletti N, Bruggen J, Clerc RG, Tarcsay L, Zwadlo G, Gerhards G, Schlegel R, Sorg C. Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature. 1987;330:80–2.
Kleinnijenhuis J, Joosten LA, van de Veerdonk FL, Savage N, van Crevel R, Kullberg BJ, van der Ven A, Ottenhoff TH, Dinarello CA, van der Meer JW, Netea MG. Transcriptional and inflammasome-mediated pathways for the induction of IL-1beta production by Mycobacterium tuberculosis. Eur J Immunol. 2009;39:1914–22.
van der Valk P, Amor S. Preactive lesions in multiple sclerosis. Curr Opin Neurol. 2009;22:207–13.
van der Valk P, De Groot CJ. Staging of multiple sclerosis (MS) lesions: pathology of the time frame of MS. Neuropathol Appl Neurobiol. 2000;26:2–10.
De Groot CJ, Bergers E, Kamphorst W, Ravid R, Polman CH, Barkhof F, van der Valk P. Post-mortem MRI-guided sampling of multiple sclerosis brain lesions: increased yield of active demyelinating and (p)reactive lesions. Brain. 2001;124:1635–45.
Peferoen LA, Vogel DY, Ummenthum K, Breur M, Heijnen PD, Gerritsen WH, Peferoen-Baert RM, van der Valk P, Dijkstra CD, Amor S. Activation status of human microglia is dependent on lesion formation stage and remyelination in multiple sclerosis. J Neuropathol Exp Neurol. 2015;74:48–63.
Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, van der Valk P, Amor S, Teunissen CE, van Horssen J, Dijkstra CD. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation. 2013;10:35.
Martin BN, Wang C, Zhang CJ, Kang Z, Gulen MF, Zepp JA, Zhao J, Bian G, Do JS, Min B, et al. T cell-intrinsic ASC critically promotes TH17-mediated experimental autoimmune encephalomyelitis. Nat Immunol. 2016;17:583–92.
Mishra BB, Moura-Alves P, Sonawane A, Hacohen N, Griffiths G, Moita LF, Anes E. Mycobacterium tuberculosis protein ESAT-6 is a potent activator of the NLRP3/ASC inflammasome. Cell Microbiol. 2010;12:1046–63.
Welin A, Eklund D, Stendahl O, Lerm M. Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PLoS One. 2011;6:e20302.
Lee HM, Kang J, Lee SJ, Jo EK. Microglial activation of the NLRP3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia. 2013;61:441–52.
Burm SM, Zuiderwijk-Sick EA, t Jong AE, van der Putten C, Veth J, Kondova I, Bajramovic JJ. Inflammasome-induced IL-1beta secretion in microglia is characterized by delayed kinetics and is only partially dependent on inflammatory caspases. J Neurosci. 2015;35:678–87.
Grebing M, Nielsen HH, Fenger CD, K T Jensen, Von Linstow CU, Clausen BH, Soderman M, Lambertsen KL, Thomassen M, Kruse TA, Finsen B. Myelin-specific T cells induce interleukin-1beta expression in lesion-reactive microglial-like cells in zones of axonal degeneration. Glia. 2016;64:407–24.
t Hart BA, Bauer J, Brok HP, Amor S. Non-human primate models of experimental autoimmune encephalomyelitis: variations on a theme. J Neuroimmunol. 2005;168:1–12.
Ferrari CC, Depino AM, Prada F, Muraro N, Campbell S, Podhajcer O, Perry VH, Anthony DC, Pitossi FJ. Reversible demyelination, blood-brain barrier breakdown, and pronounced neutrophil recruitment induced by chronic IL-1 expression in the brain. Am J Pathol. 2004;165:1827–37.
Jana M, Pahan K. Redox regulation of cytokine-mediated inhibition of myelin gene expression in human primary oligodendrocytes. Free Radic Biol Med. 2005;39:823–31.
Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Bruck W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol. 2013;125:595–608.
Bsibsi M, Holtman IR, Gerritsen WH, Eggen BJ, Boddeke E, van der Valk P, van Noort JM, Amor S. Alpha-B-crystallin induces an immune-regulatory and antiviral microglial response in preactive multiple sclerosis lesions. J Neuropathol Exp Neurol. 2013;72:970–9.
Yang H, Wang H, Chavan SS, Andersson U. High mobility group box protein 1 (HMGB1): the prototypical endogenous danger molecule. Mol Med. 2015;21 Suppl 1:S6–S12.
Rossi S, Motta C, Studer V, Macchiarulo G, Volpe E, Barbieri F, Ruocco G, Buttari F, Finardi A, Mancino R, et al. Interleukin-1beta causes excitotoxic neurodegeneration and multiple sclerosis disease progression by activating the apoptotic protein p53. Mol Neurodegener. 2014;9:56.
Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H, Bruck W, Parisi JE, Scheithauer BW, Giannini C, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med. 2011;365:2188–97.
Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis, Part I: the role of infection. Ann Neurol. 2007;61:288–99.
Ascherio A, Munger KL. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors Ann Neurol. 2007;61:504–13.
Gilden DH. Infectious causes of multiple sclerosis. Lancet Neurol. 2005;4:195–202.
Acknowledgements
The authors thank U. Köck, I. Kondova, W. Collignon, W. Gerritsen, R. Peferoen, M. Breur, and K. Ummenthum for expert technical assistance.
Funding
This work was financially supported by the Dutch MS Research Foundation (MS12-805). The Dutch MS Research Foundation had no role in the design of the study or in the collection, analysis and interpretation of the data.
Availability of data and materials
All data and materials represented in this article are freely and fully available upon request.
Authors’ contributions
SB, SA, PV, JaB, JeB were involved in conception and design of the study. KH, BH, JaB and PV provided rhesus and human CNS material. SB, LP and EZ acquired the data for the study. SB, LP, SA, JaB and JeB analyzed and interpreted the data. SB, LP, SA and JeB wrote the manuscript. All authors were involved in revising and providing intellectual contribution for the manuscript.
Competing interests
The authors have no competing interests to disclose.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The studies were performed on post-mortem human brain tissue obtained according to the protocol of the Netherlands Brain Bank (coordinator Dr. I. Huitinga, Amsterdam, the Netherlands), with the approval of the Brains Bank and Institute’s Ethical Committee’s (Amsterdam, the Netherlands). Patients and controls, or their next of kin, had given informed consent for the use of their brain tissue and clinical details for research purposes. The post-mortem rhesus monkey brain tissue was obtained from studies which were reviewed and approved by the Institutional Animal Care and Use Committee of the Biomedical Primate Research Centre, in accordance with Dutch legislation on animal experimentation.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Figure S1
Activation status of IL-1β+ cells in the rim of chronic active lesions. IL-1β+ cells in the rim of chronic active lesions were double stained for IL-1β (in brown) and cell surface markers associated with pro-inflammatory or anti-inflammatory cellular phenotypes (in red). IL-1β staining colocalized with MHC class II (a) and with CD74 (b), although we also observed some IL-1β+/CD74− cells. Furthermore, IL-1β staining colocalized with CD40 (c), CD200R (d), and CCL22 (e), although we also observed some IL-1β+/CCL22− cells. IL-1β and MR staining were both observed in rim of chronic active lesions (f), but all IL-1β+ cells were MR−. Original magnifications ×40, scale bar represents 50 μm. Nuclei were counterstained with hematoxylin (blue). (TIF 7190 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Burm, S.M., Peferoen, L.A.N., Zuiderwijk-Sick, E.A. et al. Expression of IL-1β in rhesus EAE and MS lesions is mainly induced in the CNS itself. J Neuroinflammation 13, 138 (2016). https://doi.org/10.1186/s12974-016-0605-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-016-0605-8