
Abstract
Background
Multiple organellar RNA editing factor (MORF) genes play key roles in chloroplast developmental processes by mediating RNA editing of Cytosine-to-Uracil conversion. However, the function of MORF genes in peach (Prunus persica), a perennial horticultural crop species of Rosaceae, is still not well known, particularly the resistance to biotic and abiotic stresses that threaten peach yield seriously.
Results
In this study, to reveal the regulatory roles of RNA editing in plant immunity, we implemented genome-wide analysis of peach MORF (PpMORF) genes in response to biotic and abiotic stresses. The chromosomal and subcellular location analysis showed that the identified seven PpMORF genes distributed on three peach chromosomes were mainly localized in the mitochondria and chloroplast. All the PpMORF genes were classified into six groups and one pair of PpMORF genes was tandemly duplicated. Based on the meta-analysis of two types of public RNA-seq data under different treatments (biotic and abiotic stresses), we observed down-regulated expression of PpMORF genes and reduced chloroplast RNA editing, especially the different response of PpMORF2 and PpMORF9 to pathogens infection between resistant and susceptible peach varieties, indicating the roles of MORF genes in stress response by modulating the RNA editing extent in plant immunity. Three upstream transcription factors (MYB3R-1, ZAT10, HSFB3) were identified under both stresses, they may regulate resistance adaption by modulating the PpMORF gene expression.
Conclusion
These results provided the foundation for further analyses of the functions of MORF genes, in particular the roles of RNA editing in plant immunity. In addition, our findings will be conducive to clarifying the resistance mechanisms in peaches and open up avenues for breeding new cultivars with high resistance.
Similar content being viewed by others

Background
Peach (Prunus persica) is a deciduous tree or shrub in the rose family grown for its edible fruit with high minerals, vitamins, fiber, and antioxidant compounds, and it is native to China where is also the world’s largest producer and consuming country. Peach is usually used as the model species in Rosaceae with special characteristics such as self-pollinate ability, a short life cycle, and a small genome size of 265 Mb [1, 2]. However, peach is often subject to various stresses during the growing season, such as pathogens infection, light intensity, low or high temperature, and so on, limiting the growth and yield of peach. Being one of the biotic stresses, bacterial perforation disease caused by Xanthomonas arboricola pv. Pruni (Xap) is one of the most serious diseases in peach, which often causes leaf perforation, affects the normal growth of fruit or flower bud differentiation and development, and leads to flower drop and fruit quality deterioration, resulting in substantial economic losses worldwide [3]. Being one of abiotic stresses, ultraviolet radiation affect the growth of most plants including peach, which showed decreased plant height, leaf area, photosynthetic rate, and productivity when exposed to ultraviolet radiation [4]. These stresses are destructive and economically damaging for peaches. However, the resistance mechanism in response to multiple stresses remains unclear.
RNA editing is a type of post-transcriptional modification which is mainly manifested as nucleotide insertion/deletion or conversion, yielding genetic information on RNA products that are different from their DNA templates [5]. In flowering plants, the post-transcriptional modification includes C-to-U (Cytosine-to-Uracil), U-to-C (Uracil-to- Cytosine), and A-to-I (Adenosine-to-Inosine) editing, and there are about 400–500, 30–40 C-to-U editing in transcripts of mitochondria and chloroplast respectively [6,7,8]. RNA editing plays an indispensable role in plant organelle biogenesis, adaptation to environmental changes, and signal transduction. Many mutants with impaired editing of specific sites exhibited strong deleterious phenotypes, even lethality [9,10,11,12]. In our previous studies, we found that the RNA editing events in grapes were reduced in response to heat stress [11], the RNA editing events in kiwifruits were reduced in response to pathogens infection [13].
In flowering plants, RNA editing is mainly mediated by editing complexes involving multiple editing factors, including pentatricopeptide repeat (PPR), organelle zinc finger (OZ), organelle RNA recognition motif-containing protein (ORRM), protoporphyrinogen IX oxidase (PPO), and multiple organellar RNA editing factors (MORF) [9, 14,15,16,17]. A PPR protein utilizes its DYW domain to recognize the five cis-elements upstream of the edited cytosine, which is essential in the post-transcriptional regulation of mitochondrial and chloroplast RNA [18]. MORF protein binds to the DYW domain of PPR protein to modulate the RNA-binding activity. The loss of a MORF protein will abolish or lower editing at multiple sites, previous studies found that disruption of MORF8, MORF3 and MORF1 genes reduced 72%, 26% and 19% of mitochondria editing events respectively, whereas mutants of either MORF2 or MORF9 exhibited reduced editing at almost all sites in chloroplasts [8, 14].
The MORF gene family has been widely identified in plants, such as Arabidopsis with 9 members, Populus trichocarpa with 9 [9], O. sativa with 7 [19], Z. mays with 7 [20], Actinidia chinensis with 10 [13] and Nicotiana with 8 [21]. The crystal structures of MORF1/MORF9 protein complex in Arabidopsis were determined [17], which showed that they both adopt a novel globular fold, and validated the mechanism of MORF proteins multimerization. In Arabidopsis, MORF2 and MORF9 are targeted to the chloroplast, MORF5 and MORF8 are localized in mitochondria and chloroplast, and the other five members (MORF1, MORF3, MORF4, MORF6, and MORF7) are targeted to mitochondria, MORF8 can interact respectively with MORF1 and MORF2 in mitochondria and chloroplast [14, 22]. The chloroplast-located MORF2 and MORF9 proteins form homo- and heter- dimers and can affect the RNA editing of NADH dehydrogenase subunit 4 (ndhD) in chloroplasts [23, 24]. OsMORF9 plays a critical role in the biogenesis of chloroplast ribosomes, chloroplast development, and seedling survival [25]. It has been reported that the PtrMORF genes responded to drought in poplar [9]. OsMORF gene expression was proved to be affected by cold and salt stresses in rice [19]. In Nicotiana tabacum, NbMORF8 was reported to negatively regulate plant immunity to pathogens [21].
Although the stresses such as infections, high salt, low temperature, and draught are major limiting factors for peach production worldwide, the underlying response mechanisms particularly regarding the roles of RNA editing events remain unclear. Accordingly, we studied MORF genes as RNA editing factors in the peach genome based on public transcriptome data. In this tudy, we performed a meta-analysis to evaluate the expression pattern of these MORF genes and RNA editing profiles under pathogens infection and irradiation stresses. We observed apparent responses of RNA editing extent and MORF gene expression to both stresses and identified three candidate upstream transcription factors that may regulate plant immunity by modulating the MORF gene expression. The results provide novel insights into the biological functions of MORF genes in peaches and will help elucidate the roles of RNA editing in plant immunity.
Results
Characteristics and classification of PpMORF genes in peach
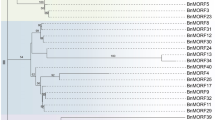
We searched the peach genome with known Arabidopsis MORF proteins as queries, BLASTP and HMM searches [26] were both performed against the entire protein sequences, thus, there were seven MORF genes identified in the peach genome (Table 1). All peach MORF genes were mapped to the peach reference genome (Fig. 1a), which indicates that these MORF genes are distributed in three peach chromosomes, including chromosomes 1, 3 and 4 (Fig. 1b). Based on the full-length amino acid sequences, a phylogenetic tree (Fig. 2) was constructed using the MEGA with maximum likelihood (ML) method [27]. Based on their phylogenetic relationship with MORF genes of Arabidopsis, we named them PpMORF1 (Prupe.1G574100), PpMORF2 (Prupe.1G045300), PpMORF3 (Prupe.1G130500), PpMORF7 (Prupe.3G039400), PpMORF8.1 (Prupe.4G168200), PpMORF8.2 (Prupe.4G168400) and PpMORF9 (Prupe.4G197100), accordingly. All the PpMORF genes were classified into six groups designated as A-F, nearly all the groups contain only one MORF copy except Group F, which contains two MORF copies including PpMORF8.1 and PpMORF8.2. The exon number of MORF genes range is mostly 4, except for PpMORF8.2, the encoded protein's length range from 187 to 537 amino acids (Table 1). Subcellular location prediction results showed that PpMORF2 and PpMORF9 were localized in the chloroplast, and PpMORF8.2 is localized in the nucleus, whereas the other five PpMORF genes were localized in the mitochondrion (Table 1), they shared the similar subcellular localization with their homologs in Arabidopsis.

Genomic structures and chromosomal locations of PpMORF genes. a The genomic structures of PpMORF genes. CDSs and UTRs are indicated by yellow and green boxes. The gene sizes are estimated using the length scale at the bottom. b The chromosomal locations of the PpMORF genes

Phylogenetic relationships of the MORF gene family from peach and Arabidopsis. The full-length amino acid sequences were used for phylogenetic tree construction by the maximum likelihood (ML) method [27]. All the MORF genes were classified into six groups and designated as Group A-F. Branches from different groups are indicated by different colors, the Bootstrap values are indicated on the branches
Identification of chloroplast RNA editing sites in peach
Two sets of public RNA-seq data from peaches were used to identify the chloroplast RNA editing sites. One set is about the samples of Xap infected leaves from two different peach varieties (‘Jh Hale’ and ‘Redkist’) with accession number SRP108345 [28], and the other set is about the samples of UVB irradiated peach fruits with accession number SRP103523 [29]. We used a combination of Bcftools 'mpileup' and GATK 'HaplotypeCaller' for variant calling, only the SNP sites that quantified the filter criterion of GATK were kept for the detection of RNA editing sites [30, 31]. Following the protocol in our previous study [11], a total of 79 chloroplast RNA editing sites that occurred in 35 genes were detected in peach leaves (Table S1), whereas 42 RNA editing sites that occurred in 26 genes were detected in peach fruits (Table S2). These results indicated the tissue specificity of RNA editing that fewer RNA editing sites in fruits than in leaves. Take the chloroplast RNA editing in leaves as an example, the average editing efficiency is 70.6%, nearly all of the editing types are C-to-U substitutions except for several mismatches, such as atpA_82 with A-to-C substitution type, which may result from sequencing error. We observed that the RNA editing efficiency varied among individual edited genes, ranging from 10 to 100%, such as the editing efficiency of rpl16_4 site is 13.4%, whereas it is nearly 100% for ndhB_50 site. In addition, several genes have more editing sites, especially NADH dehydrogenase subunit 2 (ndhB) gene and NADH dehydrogenase subunit 4 (ndhD) genes; ndhB has 10 editing sites while ndhD has 6 editing sites. All the sites of ndhB gene (ndhB_494, ndhB_246, ndhB_242, ndhB_204, ndhB_196, ndhB_181, ndhB_156, ndhB_50, ndhB_737, ndhB_149) were C-to-U editing type. We observed that the amino acid changes tend to be hydrophobic, such as proline-to-leucine and serine-to-leucine.
Response of PpMORF gene expression to Xap infection in peach
Based on RNA-seq data in resistant (‘Redkist’) and susceptible (‘Jh Hale’) peaches during Xap early infection (SRP108345), we examined the response of PpMORF genes in expression at four time points. We found that most PpMORF genes exhibited reduced expression tendency in both resistant and susceptible peach varieties under Xap stress, particularly in resistant peaches (Fig. 3 and Table S3). For ‘Redkist’, at the initial stage after Xap infection, most PpMORF genes were highly expressed, while the expressions of most PpMORF genes were down-regulated at three hours after pathogen infection (Fig. 3a), particularly for PpMORF9 and PpMORF2 (Fig. 3b). However, for the susceptible (‘Jh Hale’) peach, the number of down-regulated MORF genes was less than that of the resistant peach, although most PpMORF genes were down-regulated, but the down-regulation of PpMORF9 and PpMORF2 genes was insignificant. Whereas for other PpMORF genes, including PpMORF3, PpMORF8.1, PpMORF8.2, PpMORF1 and PpMORF7, they were generally down-regulated in both resistant and susceptible peach varieties, even at the initial stage of Xap infection (30 min). These results indicated that most PpMORF genes demonstrated down-regulated expression in response to Xap infection, especially in resistant peaches, and it’s speculated the changed expression of PpMORF genes improved the ability to regulate stress response. From the heatmap plotting (Fig. 3a), we also observed that PpMORF2 and PpMORF9 shared similar tissue expression patterns, indicating that PpMORF9, and PpMORF2 genes may work together and only play key roles in resistant peach variety. Interestingly, PpMORF9 and PpMORF2 only showed reduced expression in resistant peaches are both located in chloroplast, whereas the other PpMORF genes (PpMORF3, PpMORF8.1, PpMORF8.2, PpMORF1, and PpMORF7) that showed reduced expression in both peach varieties are localized in the mitochondrion, indicating the PpMORF genes in chloroplast rather than mitochondrion play a more important role in stress response and resistance.

Expression pattern of PpMORF genes between resistant (‘Redkist’) and susceptible (‘Jh Hale’) peaches in response to Xap infection. a Heat map of PpMORF gene expression between resistant and susceptible peaches after Xap infection. The x-axis represents hours after Xap infection (0, 30 min, 1 h, 3 h), and the y-axis represents PpMORF genes. The rows were clustered based on expression values. ‘Jh Hale’ and ‘Redkist’ represent susceptible and resistant peach, respectively; (b) Expression level of representative PpMORF genes (PpMORF2, PpMORF9, PpMORF3, and PpMORF8.1) between resistant and susceptible peaches in response to Xap infection. Asterisks denote significant differences: *p-value < 0.05
Response of RNA editing to Xap infection in peach
Considering the different expression patterns of PpMORF2 and PpMORF9 between two peach varieties in response to Xap infection (SRP108345), we further analyzed the corresponding chloroplast RNA editing events based on transcripts’ variants. As shown in Fig. 4a, ‘Jh Hale’ and ‘Redkist’ shared a comparable RNA editing pattern at 0 h, while with the Xap infection, RNA editing in ‘Redkist’ exhibited a more prominent reduction than that of ‘Jh Hale’. In ‘Redkist’, RNA editing in sites such as ndhB_246, ndhH_169, rpoC2_898, ycf1_859, rps2_83, ndhE_78, and rpoB_809 were completely lost at 30 min after infection, while editing in sites such as rpoC2_360, ycf1_641, psbF_26, and rpoC2_1242 were completely lost at 1 h after infection. However, these notable losses of RNA editing were not detected in ‘JH Hale’. In addition, the RNA editing level of all sites showed that the average RNA editing frequency of ‘Redkist’ was slightly lower than that of ‘Jh Hale’ (Fig. 4b). At 0 h, the average RNA editing frequency of ‘JH Hale’ was 0.68, and that of ‘Redkist’ was 0.61. After 30 min, the mean RNA editing frequency of both ‘JH Hale’ and ‘Redkist’ decreased, but the decrease of ‘Redkist’ was significantly greater. The RNA editing frequency of ‘Redkist’ dropped 18% from 0.61 to 0.50, and ‘JH Hale’ dropped 12% from 0.68 to 0.60. At 3 h after infection, the mean RNA editing frequency of ‘Redkist’ was significantly lower than the initial value (Decreased by 14.9%) with ~ 0.5, while the mean RNA editing frequency of ‘JH Hale’ was slightly higher than the initial value (increase by 0.8%). In addition, to rule out the influence of RNA-seq data abundance on the difference in RNA editing events, we also measured and compared gene expression levels of RNA editing genes. However, for RNA editing genes, no expression difference was detected under different treatments, suggesting that stress only affects the RNA editing events and has no influence on the expression level for those genes. Hence, compared with 'JH Hale', the resistant peach variety ‘Redkist’ demonstrated a sharper response to Xap infection in RNA editing levels than ‘JH Hale’, which is consistent with their different expression level of PpMORF genes in the chloroplast. Down-regulation of PpMORF2 and PpMORF9 may be in charge of the reduced chloroplast editing in ‘Redkist’. Under pathogen infection, the chloroplast PpMORF genes were prone to be down-regulated, thereby reducing the RNA editing level to trigger a series of defense responses and increase the resistance.

a Heat map of RNA editing efficiency in peach chloroplast genes between resistant (‘Redkist’) and susceptible (‘Jh Hale’) peaches in response to Xap infection. The x-axis represents infection time points, the y-axis represents chloroplast RNA editing sites, and the rows were clustered based on RNA editing efficiency. b RNA editing efficiency in peach chloroplast genes between resistant and susceptible peaches in response to Xap infection
Response of PpMORF gene expression and RNA editing to UVB irradiation in peach
We further examined the response of PpMORF gene expression under abiotic stress based on RNA-seq data in samples of UVB-irradiated peach fruits (SRP103523). In comparison with the control group, several PpMORF genes exhibited significantly reduced expression levels in UVB-irradiated peaches, especially for PpMORF2 and PpMORF9 (Fig. 5a and Table S3). At 6 h after UVB irradiation, the expression values of PpMORF2 and PpMORF9 decreased significantly, whereas, at 48 h after UVB irradiation, the expression values of PpMORF2 and PpMORF9 in UVB-irradiated peaches increased slightly compared with that of 6 h, but was still lower than those in the control peaches (Fig. 5b). The above observation indicated that peach PpMORF genes also exhibited down-regulated expression under UVB irradiation stress, especially for those chloroplast PpMORF genes, suggesting their roles in abiotic stress responses.

Expression patterns of PpMORF genes in peach fruits after UVB irradiation. a Heat map of PpMORF gene expression in peach fruits after UVB irradiation. The x-axis represents hours after UVB irradiation (6 h, 48 h), and the y-axis represents PpMORF genes. The rows were clustered based on expression values. b The expression level of PpMORF genes (PpMORF2 and PpMORF9) in peach fruits after UVB irradiation. Asterisks denote significant differences: *p-value < 0.05
Considering the reduced expression of PpMORF2 and PpMORF9 under UVB irradiation stress, we further analyzed the corresponding chloroplast RNA editing events (Fig. 6a). At 6 h after UVB irradiation, we observed that UVB-irradiated peaches exhibited a wide loss of editing sites, editing in sites ccsA_147, rpoC2_1242, ccsA_115, atpA_383, aptA_421, atpF_31, ndhA_358, petB_204, ndhA_321 were completely lost in comparison with the control group. While at 48 h after UVB irradiation, most of the lost RNA editing such as ccsA_115, atpA_383, aptA_421, atpF_31, ndhA_358, petB_204, and ndhA_321 returned to normal levels in the UVB-irradiated peaches. However, there were still several sites without editing including sites of ccsA_147, rpoC2_1242. A comparison of RNA editing frequency was further conducted. At 6 h after UVB radiation, the average RNA editing frequency in UVB-irradiated peaches was 33.3% lower than that of control peaches (Fig. 6b), while at 48 h after UVB radiation, the RNA editing frequency in UVB-irradiated peaches increased slightly, but was still 7.6% lowered than that of the control group. These observations revealed that chloroplast RNA editing exhibitted a reduction tendency under UVB irradiation stress, which is also consistent with the reduced expression level of chloroplast PpMORF genes. At the initial stage, the UVB irradiation stress elicits down-regulation of chloroplast PpMORF2 and PpMORF9, which further affect the normal RNA editing, the reduced RNA editing level may trigger a series of defense responses to stress. After some time, with the stress relieves, both the expression of PpMORF genes and RNA editing rebound to the normal state. Therefore, as the key elements of RNA editing, PpMORF genes may modulate the editing level and functions of chloroplast genes, thus providing a flexible strategy to increase stress tolerance.

Response of chloroplast RNA editing to UVB irradiation in peach fruits. a Heat map of RNA editing efficiency in peach chloroplast genes after UVB irradiation. The x-axis represents time points after UVB irradiation, the y-axis represents chloroplast RNA editing sites, and the rows were clustered based on RNA editing efficiency. b Comparison of RNA editing efficiency in peach chloroplast genes after UVB irradiation
Upstream transcription factors associated with PpMORF genes in peach
To investigate the underlying pathway that may regulate the PpMORF gene expression, we obtained 34 upstream transcription factors of PpMORF genes from the PlantRegMap database [32,33,34]. The regulatory interaction between transcription factors and PpMORF genes showed that the PpMORF3 gene had the most transcription factors with 11, followed by PpMORF9 with 10, PpMORF2 gene has the fewest transcription factors with only two (Fig. 7a). PpMORF9 shares one transcription factor with PpMORF1 and PpMORF3 respectively, and two copies of the PpMORF8 gene share four transcription factors. Based on two types of RNA-seq data under different treatments (biotic and abiotic stresses), differential expression analysis was conducted against these transcription factors, and a total of nine and six transcription factors showed differential expression under biotic (3 h after Xap infection for ‘Redkist’) and abiotic (6 h after UVB irradiation) stresses, respectively (Fig. 7b). There were three common transcription factors up-regulated in both conditions, including the myb 3R-1 gene (MYB3R-1, Pp.1G45000), zinc finger gene (ZAT10, Pp.1G424300), and heat stress transcription factor B-3 (HSFB3, Pp.7G056700). Interestingly, these three transcription factors only negatively regulate the expression of PpMORF2 and PpMORF9. These observations suggested that those differentially expressed transcription factors, especially the shared ones, may participate in the upstream regulation of chloroplast PpMORF gene expression and RNA editing in response to both biotic and abiotic stresses.

Upstream transcription factors associated with PpMORF genes in peach. a The regulatory network between PpMORF genes and transcription factors. The nodes of PpMORF genes and transcription factors are denoted by blue and red circles, respectively. The below red fonts represent the gene names of transcription factors. b Expression patterns of PpMORF genes’ upstream transcription factors in response to stress in peach. The left panel shows the response of expression to Xap infection (3 h after Xap infection in ‘Redkist’), while the right panel shows the response of expression to UVB irradiation (6 h after UVB irradiation). The transcription factors shared by both conditions are marked by the red dashed line
Discussion
Generally, plant RNA editing appears to act as an indirect repair mechanism to correct DNA mutations on the RNA level by restoration of conserved amino acids to guarantee proper protein function [35]. Previous studies showed plant RNA editing played multiple roles during plant developmental processes [36], organelle biogenesis [37], plant flowering [38], response to particular environmental conditions [39] and signal transduction [40]. However, the underlying mechanism still needs further clarification. MORF proteins have been identified as essential components of plant RNA editosome through interacting with other RNA editing factors, PPR proteins, organelle RNA recognition motif (ORRM) proteins, organelle zinc-finger (OZ) proteins, and protoporphyrinogen oxidase 1 (PPO1) [8, 14]. Recent evidence suggested that MORF genes played a critical role in plant development and stress response, such as seedling survival in rice, drought stress in poplar, and pathogen stress in tobacco and kiwifruit [9, 13, 19, 21]. It’s determined that NbMORF8 localized in mitochondrion negatively regulates plant immunity to Phytophthora pathogens [21] and indicated that nuclear gene regulation in plant enhanced resistance. Hence, the roles of RNA editing in response to stresses may be partly explained from the perspective of the function of MORFs.
Utilizing the recently released peach assembly, a total of seven members of PpMORF genes were identified in this study. Similar to Arabidopsis, PpMORF genes in peaches mostly were localized in mitochondria and a few in chloroplasts. PpMORF2 and PpMORF9 are exclusively localized in chloroplast, whereas PpMORF1, PpMORF7, PpMORF3, and PpMORF8.1 are localized in mitochondria, however, as a duplicated copy, PpMORF8.2 changed to be located in the nucleus. The similar expression patterns between PpMORF2 and PpMORF9 further confirmed their functional relevance and selective heteromer interactions. Based on two types of public RNA-seq data under different treatments (biotic and abiotic stresses), we conducted a meta-analysis to examine the roles of RNA editing in plant immunity, obvious response of PpMORF genes and varied chloroplast RNA editing profiles were observed, especially the reduced expression of PpMORF2 and PpMORF9. Their varied response to pathogens infection was also detected between resistant and susceptible peach cultivars, indicating the roles of PpMORF genes as controlling elements in stress response by modulating the chloroplasts RNA editing extent in plant immunity. The varied disease resistance capacity between resistant and susceptible peaches partly can be explained by their discrepancy of MORF gene expression in response to pathogen infection. In addition, for RNA editing genes in chloroplasts, no expression difference was detected under different treatments, suggesting that stress only affects the RNA editing events and has no influence on the expression level for those genes. Hence, it is reasoned that the response of RNA editing events may be regulated by the expression of RNA editing factors. Several transcription factors that regulate the expression of peach MORFs were also identified in our study, such as ZAT10, MYB3R-1, HSFB3, and so on. ZAT10 is a transcriptional repressor involved in abiotic stress responses, a previous study confirmed plants overexpressing ZAT10 showed growth retardation and enhanced tolerance to drought, salt, heat, and osmotic stresses [41]. MYB3R-1 is a transcription repressor that regulates organ growth, it specifically binds DNA sequence 5'-AGAAnnTTCT-3' known as heat shock promoter elements [42]. Those differentially expressed transcription factors may participate in the upstream regulation of chloroplast MORF gene expression and RNA editing in response to both biotic and abiotic stresses.
Chloroplasts play key roles in plant-pathogen interactions and are important for reactive oxygen species (ROS) that act as key defense molecules in plant immune responses [43]. However, it remains largely unclear how chloroplast proteins achieve modulation of the plant immune system. Recently, nuclear gene expression has been acknowledged to be involved in the post-transcriptional regulation of chloroplast function in response to external stimuli, and RNA editing is one such control mechanism [39]. It’s confirmed that overexpressor of cationic peroxidase3 (ocp3) which is targeted to chloroplasts contributes to control over the extent of ndhB transcripts editing and proposed that ocp3 mediated chloroplast RNA editing in plant immunity. ndhB encodes the B subunit of the chloroplast NADH dehydrogenase-like complex (NDH) involved in cyclic electron flow (CEF) around photosystem I. Ocp3-silenced mutants lead to ndhB editing efficiency decays, thereby impairing CEF and enhancing disease resistance to pathogens substantially [39]. In our study, the affected chloroplast genes with reduced RNA editing in response to stresses mostly function in DNA-RNA transcription and RNA splicing and photosystem, such as ndhB, ndhH, ndhD, rpoC, ndhE, and rpoB. ndhB encodes the B subunit of the chloroplast NADH dehydrogenase-like complex (NDH) involved in CEF around photosystem I. NDH complex activity and plant immunity appear as interlinked processes. MORF genes, similar to ocp3, may modulate the plant-pathogen interaction by controlling the extent of chloroplast RNA editing, especially components of the NDH complex. Hence, we speculated the decays of editing efficiency in these genes might trigger the impaired CEF, thereby leading to the activation of ROS-mediated retrograde signaling, and the disease resistance to pathogens or other stresses substantially enhanced (Fig. 8). However, we also found that this regulatory strategy is flexible from the result of response to UVB irradiation. If the stress relieves, both the expression of MORF genes and RNA editing return to the normal state. There is a ‘trade-off’ between resistance and homeostasis.

Schematic model for the role of MORF genes in plant immunity. The MORF-regulated ROS burst is likely achieved through its effect on the functionality of Photosystem I and II. Upstream transcription factors regulate the expression of MORF genes. MORF genes participate in the RNA editing of chloroplast photosystem genes and subsequently affect the cyclic electron flow activities. Stress such as pathogen infection and UVB irradiation leads to the down-regulation of MORF genes and reduced RNA editing efficiency, thereby impairing CEF, and up-regulating ROS levels, Calcium influx, which enhances the immunity to stress
Conclusions
The present study is a comprehensive meta-analysis of the PpMORF gene family in peaches, particularly for their roles in response to biotic and abiotic stresses. We identified seven PpMORF genes in total and performed a series of analyses of their basic structures, classification, chromosomal, subcellular localization, and expression. The findings revealed that most PpMORF genes were localized in mitochondria or chloroplast, with one in the nucleus. In response to different stresses, including pathogen infection and UVB radiation, chloroplast localized PpMORF genes exhibited down-regulated expression, accompanied by reduced chloroplast RNA editing. In addition, different expressions of MORF genes and RNA editing profiles in chloroplasts between resistant and susceptible peaches after pathogen infection were also observed, indicating the contributions of PpMORF genes to the disease resistance of different peach varieties. Finally, some transcription factors that regulated PpMORF gene expression were predicted, they may play an essential role in the MORFs-mediated stress adaption pathway. This study will be highly useful for further molecular elucidation of plant immunity and the breeding of resistant peaches.
Methods
Genome-wide identification of PpMORF genes in peach
The peach (Prunus persica) genome and annotation files were downloaded from Genome Database for Rosaceae (GDR) (https://www.Rosaceae.org/). We used two searching strategies to obtain peach MORF genes. First, using the previously identified MORF genes in Arabidopsis as queries [14], we implemented BLASTP searches against the entire protein database of peaches with an E-value cut-off of 0.00001 to reduce false positives. Second, Hidden Markove Model (HMM) profiles of MORF genes in Arabidopsis were constructed and used to search against the peach protein database by using HMMER software [26], nine MORF genes in Arabidopsis were aligned and used to build HMM profiles using ‘‘hmmbuild’’ command, thus the resulting HMM profiles were used to search against peach protein sequences with an E-value cut-off of 0.001 using ‘hmmsearch’ command. Finally, all the candidate peach MORF genes were named based on their phylogenetic relationship with that of Arabidopsis accordingly. The phylogenetic tree from full-length amino acid sequences was constructed using the MEGA with maximum likelihood (ML) method [27].
Gene structure analysis, subcellular and physical localization
TargetP [44] and LOCALIZER (http://localizer.csiro.au/) were used for predicting the putative subcellular localization of peach MORF genes. The gene structure and positional information of peach MORF genes on the genome were obtained from the annotation documents, and we utilized TBtools [45] to draw the sketch map of gene structure and physical location.
Transcriptome data collection and preprocessing
Two types of transcriptome data of peaches under different stresses (biotic and abiotic stresses) were downloaded from the Short Read Archive (SRA) database of the National Center for Biotechnology Information (NCBI). The first transcriptome data was collected from resistant ‘Redkist’ and susceptible ‘Jh Hale’ peach leaf samples in response to Xap during early infection with accession number SRP108345 [28]. Both cultivars are yellow melting flesh peaches, ‘Redkist’ was obtained from a mutation of ‘Redskin’ and is highly resistant to bacterial spot; ‘JH Hale’ was obtained from self-pollination of ‘Elberta’ and moderately susceptible to Xap [28]. The early infection consists of 30 min, 1 and 3 h-post-infection (hpi) after inoculation with Xap, and each condition consists of two replicates. The second data is peach fruit samples under different treatments (control; 6 h, 48 h after UVB irradiation) with accession number SRP103523 [29], melting flesh peach (Prunus persica L. Batsch cv. Hujingmilu) fruits were harvested in Ningbo, China. Eeach treatment consists of three biological replicates, an average of ~ 11 million clean reads (Q30 > 94.87%) were obtained per sample, with ~ 155 million clean reads in a total of 12 samples with 91.92% mapped to the peach genome. Before analysis of RNA-Seq data, we utilized the FastQC tool to check the quality of the transcriptome data first [46], and trimmed the adapter and low-quality bases (phred score < 33) with Trimmomatic (v0.39) [47], only reads > 40 bp were kept.
Expression analysis of PpMORF genes in peach in response to stress
The clean reads of RNA-seq data from each sample were mapped against the peach genome reference with HISAT2 [48], and each SAM file was converted into a BAM file and sorted with SAMtools [49, 50]. Further transcript assembly and quantification of the read alignments were performed with Stringtie [51]. Gene expression levels were measured by FPKM (fragments per kilobase of transcript per million mapped reads). The differential expressed genes were determined by using EdgeR [52]. Cluster analysis was also performed using the HeatMap function implemented in TBtools [45] based on the matrix of MORF gene expression, which was initially normalized by subtracting the row-wise mean from the values in each row of data and divided by the standard deviation value of each row.
Identification of RNA editing sites
For RNA editing site detection, we retrieved the genome sequences (NC_014697.1) of peach chloroplast as well as their annotation files from the nucleotide database of NCBI. The transcriptome data were mapped to chloroplast genome reference by using HISAT2 software with default parameters [48]. Afterward, each SAM file was converted into a BAM file, sorted with SAMtools [49, 50]. We used a combination of Bcftools 'mpileup' and GATK 'HaplotypeCaller' for variant calling. Firstly, The variant calling process was conducted by SAMtools ‘mpileup' command, and the single nucleotide polymorphisms (SNPs) were identified by BCFtools ‘call’ command [31]. Secondly, to validate the SNPs, we also employed Genome Analysis Toolkit (GATK, v4.0) to detect SNPs [30], we mapped the clean reads to the reference using BWA-MEM with default parameters [53]; the multiple tools (‘MarkDuplicates’, ‘HaplotypeCaller’ and ‘VariantFiltration’, etc.,) implemented in GATK [30] were used to obtain high-quality SNPs, with strict filter settings “QD < 2.0 || MQ < 40.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < -12.5 || 218 ReadPosRankSum < -8.0”. For chloroplast, based on their SNP-calling results and gene annotation files, RNA editing sites were identified by using the REDO tool [54]. A series of comprehensive rule-dependent and statistical filters implemented in the REDO tool were used to reduce the false positives. Afterward, we further minimize false-positive sites by manually examining all mismatches. To rule out the influence of RNA-seq data abundance on the difference in RNA editing events, we excluded sites with no RNA-seq data in a certain sample. For each site, RNA editing efficiency was quantified by the proportion of edited transcripts in total covered transcripts. The matrix of RNA editing efficiency was initially normalized by subtracting the row-wise mean from the values in each row of data and divided by the standard deviation value of each row, for comparison between conditions, cluster analysis was subsequently performed by using the HeatMap function implemented in TBtools [45].
Identification of upstream regulatory transcription factors of PpMORF genes
The transcriptional regulatory map of peach was retrieved from the PlantRegMap database [32,33,34]. Transcriptional regulations in PlantRegMap were identified from the literature and ChIP-seq data, or inferred by combining transcript factors (TF) binding motifs and regulatory elements data, this tool was used to infer potential regulatory interactions between TF and input genes, and found the TFs which possess over-represented targets in the input gene set. We submitted the gene symbols of PpMORF genes in the website tools (http://plantregmap.gao-lab.org/network.php) to retrieve corresponding regulations and upstream regulatory transcription factors under default settings. Finally, the regulatory transcription factors were annotated, and the regulatory interactions were further mapped using Cytoscape [55].
Availability of data and materials
The datasets analyzed during the current study are available in the NCBI repository (https://www.ncbi.nlm.nih.gov/) with accession SRP103523 and SRP108345.
Abbreviations
- C-to-U:
-
Cytosine-to-uracil
- PPR:
-
Pentatrico peptide repeat
- ndhB:
-
Subunit of the NADH dehydrogenase-like complex
- MORF:
-
Multiple organelle RNA editing factors
- atpA:
-
ATP synthase alpha-subunit
- Xap:
-
Xanthomonas arboricola pv. Pruni
- MYB3R-1:
-
Myb 3R-1 gene
- ZAT10:
-
Zinc finger gene
- HSFB3:
-
Heat stress transcription factor B-3
- CEF:
-
Cyclic electron flow
- ROS:
-
Reactive oxygen species
- TF:
-
Transcript factors
References
International Peach Genome Initiative, Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45(5):487–94.
Verde I, Jenkins J, Dondini L, Micali S, Pagliarani G, Vendramin E, Paris R, Aramini V, Gazza L, Rossini L, et al. The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genomics. 2017;18(1):225.
Stefani E. Economic significance and control of bacterial spot/canker of stone fruits caused by xanthomonas arboricola pv PRUNI. J Plant Pathol. 2010;92(1):S99–103.
Biggs RH, Webb PG, Biggs RH, Webb PG. Effects of enhanced ultraviolet-B radiation on yield, and disease incidence and severity for wheat under field conditions. In 'Stratospheric ozone reduction, solar ultraviolet radiation and plant life'. Ecological Sci. 1986;8:303–11.
Petriccione M, Di Cecco I, Arena S, Scaloni A, Scortichini M. Proteomic changes in Actinidia chinensis shoot during systemic infection with a pandemic Pseudomonas syringae pv. actinidiae strain. J Proteomics. 2013;78:461–76.
Okudaira C, Sakari M, Tsukahara T. Genome-wide identification of U-To-C RNA editing events for nuclear genes in Arabidopsis thaliana. Cells. 2021;10(3):635.
Giege P, Brennicke A. RNA editing in arabidopsis mitochondria effects 441 C to U changes in ORFs. Proc Natl Acad Sci U S A. 1999;96(26):15324–9.
Yan J, Zhang Q, Yin P. RNA editing machinery in plant organelles. Sci China Life Sci. 2018;61(2):162–9.
Wang D, Meng S, Su W, Bao Y, Lu Y, Yin W, Liu C, Xia X. Genome-wide analysis of multiple organellar RNA editing factor family in poplar reveals evolution and roles in drought stress. Int J Mol Sci. 2019;20(6):1425.
Chateigner-Boutin AL, Small I. Plant RNA editing. RNA Biol. 2010;7(2):213–9.
Zhang A, Jiang X, Zhang F, Wang T, Zhang X. Dynamic response of RNA editing to temperature in grape by RNA deep sequencing. Funct Integr Genomics. 2020;20(3):421–32.
Fang J, Jiang XH, Wang TF, Zhang XJ, Zhang AD. Tissue-specificity of RNA editing in plant: analysis of transcripts from three tobacco (Nicotiana tabacum) varieties. Plant Biotechnol Rep. 2021;15(4):471–82.
Xiong Y, Fang J, Jiang X, Wang T, Liu K, Peng H, Zhang X, Zhang A. Genome-wide analysis of multiple organellar RNA editing factor (MORF) family in kiwifruit (actinidia chinensis) reveals its roles in chloroplast RNA editing and pathogens stress. Plants (Basel). 2022;11(2):146.
Takenaka M, Zehrmann A, Verbitskiy D, Kugelmann M, Hartel B, Brennicke A. Multiple organellar RNA editing factor (MORF) family proteins are required for RNA editing in mitochondria and plastids of plants. Proc Natl Acad Sci U S A. 2012;109(13):5104–9.
Zhang F, Tang W, Hedtke B, Zhong L, Liu L, Peng L, Lu C, Grimm B, Lin R. Tetrapyrrole biosynthetic enzyme protoporphyrinogen IX oxidase 1 is required for plastid RNA editing. Proc Natl Acad Sci U S A. 2014;111(5):2023–8.
Tian F, Yu J, Zhang Y, Xie Y, Wu B, Miao Y. MORF9 functions in plastid RNA editing with tissue specificity. Int J Mol Sci. 2019;20(18):4635.
Haag S, Schindler M, Berndt L, Brennicke A, Takenaka M, Weber G. Crystal structures of the arabidopsis thaliana organellar RNA editing factors MORF1 and MORF9. Nucleic Acids Res. 2017;45(8):4915–28.
Barkan A, Small I. Pentatricopeptide repeat proteins in plants. Annu Rev Plant Biol. 2014;65:415–42.
Zhang Q, Shen L, Ren D, Hu J, Chen G, Zhu L, Gao Z, Zhang G, Guo L, Zeng D, et al. Characterization, expression, and interaction analyses of OsMORF gene family in rice. Genes. 2019;10(9):694.
Luo M, Cai M, Zhang J, Li Y, Zhang R, Song W, Zhang K, Xiao H, Yue B, Zheng Y, et al. Functional divergence and origin of the DAG-like gene family in plants. Sci Rep. 2017;7(1):5688.
Yang Y, Fan G, Zhao Y, Wen Q, Wu P, Meng Y, Shan W. Cytidine-to-uridine RNA editing factor NbMORF8 negatively regulates plant immunity to phytophthora pathogens. Plant Physiol. 2020;184(4):2182–98.
Bentolila S, Heller WP, Sun T, Babina AM, Friso G, van Wijk KJ, Hanson MR. RIP1, a member of an Arabidopsis protein family, interacts with the protein RARE1 and broadly affects RNA editing. Proc Natl Acad Sci U S A. 2012;109(22):E1453-1461.
Zehrmann A, Hartel B, Glass F, Bayer-Csaszar E, Obata T, Meyer E, Brennicke A, Takenaka M. Selective homo- and heteromer interactions between the multiple organellar RNA editing factor (MORF) proteins in Arabidopsis thaliana. J Biol Chem. 2015;290(10):6445–56.
Huang C, Li ZR, Yu QB, Ye LS, Cui YL, Molloy DP, Yang ZN. MORF2 tightly associates with MORF9 to regulate chloroplast RNA editing in arabidopsis. Plant Sci. 2019;278:64–9.
Zhang Q, Wang Y, Xie W, Chen C, Ren D, Hu J, Zhu L, Zhang G, Gao Z, Guo L, et al. OsMORF9 is necessary for chloroplast development and seedling survival in rice. Plant Sci. 2021;307:110907.
Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, Finn RD. HMMER web server: 2018 update. Nucleic Acids Res. 2018;46(W1):W200-w204.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Gervasi F, Ferrante P, Dettori MT, Scortichini M, Verde I. Transcriptome reprogramming of resistant and susceptible peach genotypes during Xanthomonas arboricola pv. pruni early leaf infection. Plos One. 2018;13(4):e0196590.
Liu H, Cao X, Liu X, Xin R, Wang J, Gao J, Wu B, Gao L, Xu C, Zhang B, et al. UV-B irradiation differentially regulates terpene synthases and terpene content of peach. Plant, Cell Environ. 2017;40(10):2261–75.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491.
Danecek P, McCarthy SA. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 2017;33(13):2037–9.
Tian F, Yang D-C, Meng Y-Q, Jin J, Gao G. PlantRegMap: charting functional regulatory maps in plants. Nucleic Acids Res. 2019;48(D1):D1104–13.
Jin J, Tian F, Yang DC, Meng YQ, Kong L, Luo J, Gao G. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45(D1):D1040-d1045.
Jin J, He K, Tang X, Li Z, Lv L, Zhao Y, Luo J, Gao G. An arabidopsis transcriptional regulatory map reveals distinct functional and evolutionary features of novel transcription factors. Mol Biol Evol. 2015;32(7):1767–73.
Bentolila S, Oh J, Hanson MR, Bukowski R. Comprehensive high-resolution analysis of the role of an arabidopsis gene family in RNA editing. PLoS Genet. 2013;9(6):e1003584.
Takenaka M, Zehrmann A, Verbitskiy D, Härtel B, Brennicke A. RNA editing in plants and its evolution. Annu Rev Genet. 2013;47:335–52.
Lurin C, Andrés C, Aubourg S, Bellaoui M, Bitton F, Bruyère C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al. Genome-wide analysis of arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell. 2004;16(8):2089–103.
Shi X, Germain A, Hanson MR, Bentolila S. RNA recognition motif-containing protein ORRM4 broadly affects mitochondrial rna editing and impacts plant development and flowering. Plant Physiol. 2016;170(1):294–309.
Garcia-Andrade J, Ramirez V, Lopez A, Vera P. Mediated plastid RNA editing in plant immunity. Plos Pathog. 2013;9(10):e1003713.
Tang J, Kobayashi K, Suzuki M, Matsumoto S, Muranaka T. The mitochondrial PPR protein LOVASTATIN INSENSITIVE 1 plays regulatory roles in cytosolic and plastidial isoprenoid biosynthesis through RNA editing. Plant J. 2010;61(3):456–66.
Mittler R, Kim Y, Song L, Coutu J, Coutu A, Ciftci-Yilmaz S, Lee H, Stevenson B, Zhu JK. Gain- and loss-of-function mutations in Zat10 enhance the tolerance of plants to abiotic stress. FEBS Lett. 2006;580(28–29):6537–42.
Bourbousse C, Vegesna N, Law JA. SOG1 activator and MYB3R repressors regulate a complex DNA damage network in arabidopsis. P Natl Acad Sci USA. 2018;115(52):E12453–62.
Amirsadeghi S, Robson CA, Vanlerberghe GC. The role of the mitochondrion in plant responses to biotic stress. Physiol Plantarum. 2007;129(1):253–66.
Boos F, Muhlhaus T, Herrmann JM. Detection of Internal Matrix Targeting Signal-like Sequences (iMTS-Ls) in mitochondrial precursor proteins using the targetp prediction tool. Bio Protoc. 2018;8(17):e2474.
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–202.
Brown J, Pirrung M, McCue LA. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics (Oxford, England). 2017;33(19):3137–9.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. genome project data processing s: the sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, et al. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10(2):008.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England). 2010;26(1):139–40.
Jung Y, Han D. BWA-MEME: BWA-MEM emulated with a machine learning approach. Bioinformatics. 2022;38(9):2404–13.
Wu S, Liu W, Aljohi HA, Alromaih SA, Alanazi IO, Lin Q, Yu J, Hu S. REDO: RNA Editing Detection in Plant Organelles Based on Variant Calling Results. J Comput Biol. 2018;25(5):509–16.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Acknowledgements
We would like to thank the members of the Bioinformatics Group of Wuhan Botanical Garden, Chinese Academy of Sciences, China for the discussion and suggestions to improve the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32070682), the Key Research and Development Program of Hubei Province (2022BBA0076), the National Science & Technology Innovation Zone Project (1816315XJ00100216), and CAS Pioneer Hundred Talents Program.
Author information
Authors and Affiliations
Contributions
ADZ and XJZ designed the experiments. XJZ supervised the project. ADZ, YHX, and JF performed statistical analyses of gene expression data. KCL and HXP performed statistical analyses. ADZ drafted the manuscript. XJZ and ADZ approved the final manuscript. All authors have read the manuscript and agreed with the current version of the manuscript for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All methods and materials were in compliance with relevant institutional, national, and international guidelines and legislation.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Detailed information of chloroplast RNA editing sites identified in peach leaves.
Additional file 2: Table S2.
Detailed information of chloroplast RNA editing sites identified in peach fruits.
Additional file 3: Table S3.
Detailed information of differential expressed PpMORF genes under Xap infection and UVB irradiation treatments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, A., Xiong, Y., Fang, J. et al. Genome-wide identification and expression analysis of peach multiple organellar RNA editing factors reveals the roles of RNA editing in plant immunity. BMC Plant Biol 22, 583 (2022). https://doi.org/10.1186/s12870-022-03982-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-022-03982-2