
Abstract
Considerable progress has been made in identifying genetic risk factors for idiosyncratic adverse drug reactions in the past 30 years. These reactions can affect various tissues and organs, including liver, skin, muscle and heart, in a drug-dependent manner. Using both candidate gene and genome-wide association studies, various genes that make contributions of varying extents to each of these forms of reactions have been identified. Many of the associations identified for reactions affecting the liver and skin involve human leukocyte antigen (HLA) genes and for reactions relating to the drugs abacavir and carbamazepine, HLA genotyping is now in routine use prior to drug prescription. Other HLA associations are not sufficiently specific for translation but are still of interest in relation to underlying mechanisms for the reactions. Progress on non-HLA genes affecting adverse drug reactions has been less, but some important associations, such as those of SLCO1B1 and statin myopathy, KCNE1 and drug-induced QT prolongation and NAT2 and isoniazid-induced liver injury, are considered. Future prospects for identification of additional genetic risk factors for the various adverse drug reactions are discussed.
Similar content being viewed by others
Introduction
Serious adverse drug reactions are a significant cause of death and serious illness in patients and an important cause of drug attrition in the pharmaceutical industry both during drug development and after licensing. These reactions are normally classed as idiosyncratic reactions that are not related directly to drug concentration but instead may be due to an unusual patient phenotype. Most serious adverse drug reactions can be classified as either type A, which are dose dependent, or type B (idiosyncratic), where the reaction is not predictable from normal drug pharmacology and is generally independent of dose [1]. Idiosyncratic adverse reactions are generally rarer than type A events, although frequencies vary with the type of reaction and the individual drug, with frequencies ranging from as high as 5% of users to as low as 1 in 10,000 to 100,000 users. Low frequencies mean the reactions are often seen only late in the drug development process or after the drug has been licensed.
Idiosyncratic adverse drug reactions can affect a number of different organs, including the liver, skin, kidney, heart and muscle, and, with some drugs, more generalized hypersensitivity reactions can occur. In terms of drug withdrawals from the market in recent years, the largest numbers of compounds were withdrawn because of either hepatotoxicity or toxicity affecting cardiac function. Adverse drug reactions affecting the liver show heterogeneity in their phenotypic effect but these reactions are collectively referred to as drug-induced liver injury (DILI); they are usually classified as hepatocellular when the injury mainly involves the hepatocyte, and cholestatic when the damage occurs at the hepatocyte canalicular membrane or within the biliary tree [2]. Up to 10% of these hepatotoxic adverse drug reactions can progress to liver failure, which can be fatal unless a liver transplant is performed. Cardiotoxic drugs can give rise to a delay in cardiac repolarization, which can be detected by prolongation of the QT interval on an electrocardiogram. QT prolongation is a risk factor for a form of ventricular tachycardia called torsade de pointes, which can lead to ventricular fibrillation and death.
Genetic susceptibility is an important feature of serious adverse drug reactions and there is considerable interest in the possibility that development of genetic tests to identify all those at risk of adverse events prior to prescription might lead to valuable drugs being retained. There are already two examples - abacavir hypersensitivity and HLA-B*57:01 and carbamazepine toxicity and HLA-B*15:02 - that have been translated to the clinic.
This article will consider progress to date in identifying pharmacogenomic risk factors for serious adverse drug reactions, including the different approaches that have been used, and prospects for further progress.
Pharmacogenomic approaches used to identify causative genes
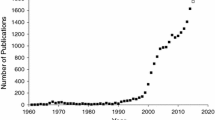
Pharmacogenomic studies to identify genes that contribute to susceptibility to adverse drug reactions have up to the present involved case-control association studies using either a candidate gene approach or genome-wide association (GWA) analysis. Though the development of GWA studies has led to considerable progress in the area of complex disease genomics and this would be generally considered the more appropriate approach to use currently to identify genes involved in adverse drug reactions, there are a several examples where candidate gene studies have been valuable in identifying causative genes. There are a number of reasons for this. Up to the present, most genetic risk factors identified have large effect sizes and are generally in biologically obvious genes. However, GWA studies have the advantage of their open approach where all genes and common variation are examined and there are now a few examples of entirely novel associations that would have been unlikely to have been predicted by candidate gene approaches. Generally, using GWA is particularly valuable in detecting small effects, but a limitation with most studies on adverse drug reactions is that the number of cases available for study is small, which limits power to detect significant effects. Recent international collaborative projects that aim to assemble large data sets are helpful in increasing sample numbers, but since genetic risk factors for adverse drug reactions tend to be drug specific and not simply end-organ specific, assembling large uniform cohorts is still challenging.
GWA studies are unlikely to identify all genetic risk factors for adverse drug reactions. There may also be a contribution from rare variants, which can be detected only by sequencing studies. Good progress is being made in some diseases by use of exome sequencing where all coding areas of genes are sequenced. Exome sequencing has tended to be of most value in detecting variants involved in rare diseases showing Mendelian inheritance (for example, [3, 4]) rather than complex diseases, though there are some recent exceptions to this reported in the fields of infectious disease and type II diabetes [5, 6]. Whole genome sequencing where regulatory sequences are also determined may be necessary to provide sufficient sensitivity to detect rare variants relevant to adverse drug reactions.
Both candidate gene and GWA studies on adverse drug reactions of several types have provided strong evidence for a role for human leukocyte antigen (HLA) genes in susceptibility. In view of this, the next section will consider HLA genes as a general risk factor for adverse drug reactions and describe some specific HLA associations in detail. It should be noted that HLA genes may not be the sole genetic risk factor for these reactions and are not relevant at all to some types of adverse drug reaction, including cardiotoxicity and muscle toxicity.
HLA associations in drug-induced liver injury, hypersensitivity reactions and skin rash
It has been believed for over 30 years that HLA type is a predictor of risk for certain adverse drug reactions, and well-established and replicated associations have now been described for both DILI, including some reactions that do not show obvious features of a hypersensivity reaction, and hypersensitivity reactions affecting the skin.
HLA and drug-induced liver injury
Many different drugs in current use can cause DILI, though the incidence of this adverse drug reaction will typically be very low, in the order of 1 in 10,000 patients treated (for review, see [7]). The underlying mechanism may involve direct toxic effects by the drug, for example involving oxidative stress or cellular damage, and formation of reactive intermediates resulting in either direct toxicity or an inappropriate immune response [8].
For DILI, the first reports linking HLA and genetic susceptibility involved the anesthetic halothane, which was used widely up to the 1980s and was also an important cause of idiosyncratic hepatitis up to that time. An association between the HLA class II serotype DR2 was reported by a study based in Japan [9], though this was not found in two smaller studies in Europe [10, 11]. In a study of DILI associated with a range of different drugs, a small but not statistically significant increase in incidence in frequency of both HLA-DR2 and another serotype, HLA-DR6, was seen [12]. A larger study of a number of different drugs found a trend towards significance for the class I serotype HLA-A11 for DILI induced by tricyclic antidepressants and diclofenac, and for the class II serotype HLA-DR6 in relation to DILI due to chlorpromazine [13].
More recently, HLA associations with DILI have been studied directly by genotyping rather than serotype determination. The first HLA genotyping studies were candidate gene association studies on amoxicillin-clavulanate-related DILI. Though this form of DILI does not generally show classical immune-related features, two independent candidate gene association studies reported an identical association with the HLA-DRB1*15:01 allele, which corresponds to the DR2 serotype mentioned above [14, 15]. This form of DILI has been suggested to relate predominantly to the clavulanic acid component of the drug [16], though this has still not been demonstrated directly. Subsequent genetic studies on DILI using both candidate gene and GWA methods have resulted in the identification of a number of different HLA class I and II associations (Table 1). The effect sizes observed vary considerably, with odds ratios of between 2 and 80 reported for different drugs. The strongest HLA association reported up to the present for DILI relates to reactions to the antimicrobial flucloxacillin. A GWA study showed a very strong association (odds ratio 80) with the class I HLA allele B*57:01 [17], which had been previously demonstrated to be a strong risk factor for hypersensitivity reactions to abacavir (see below). A role for HLA in reactions to drugs other than those listed in Table 1 seems less likely in view of a recent GWA covering DILI due to a wide range of drugs, which failed to show any signal for the HLA region when cases due to drugs known to show a HLA association were excluded [18]. The observed HLA associations point to a role for T-cell responses in DILI reactions and possible mechanisms are discussed in more detail below.
HLA and hypersensitivity reactions affecting the skin
Adverse drug reactions affecting the skin involving hypersensitivity can be divided into early and delayed responses (for review, see [19]). Early or immediate-type responses involve IgE and their underlying mechanism is well understood, though genetic risk factors are still unclear and this type of reaction will not be discussed further here. Delayed-type hypersensitivity reactions involving the skin show considerable heterogeneity, ranging from very mild forms, where the skin is the only organ affected and drug withdrawal leads to rapid improvement, to drug-induced hypersensitivity syndrome (sometimes referred to as DRESS), where other organs and tissues may be affected and where there is fever and eosinophilia. In addition, some patients may show an unusually severe skin rash, which involves blistering in the conditions known as Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN).
There is a considerable body of data showing that T-cell responses to drugs are a key event in delayed immune-mediated reactions affecting the skin [19]. Since HLA genes code for proteins involved in antigen presentation to T cells, the possibility that HLA genotype is a predictor of delayed hypersensitivity reactions has been investigated widely. Before the more recent studies showing a role for T-cell reactions in drug-induced skin rash, HLA associations with these reactions had been reported. TEN and SJS were found to be weakly associated with the HLA class I serotype B12 [20]. Among patients reacting to a particular drug, stronger associations were seen, especially for A29-B12-DR7 haplotype and sulfonamide-induced toxicity.
Carbamazepine-induced skin reactions
Further progress on HLA associations in relation to skin reactions was slower than that for liver reactions until a candidate gene study involving genotyping for HLA alleles and a range of polymorphisms in cytochromes P450 in Taiwanese cases of carbamazepine-induced SJS found a very strong association of this adverse drug reaction with the class I allele B*15:02 (Table 1) [21]. Genotyping for B*15:02 is now recommended in individuals of Han Chinese, Thai, Malaysian, Indonesian, Philippino and south Indian ethnicity prior to carbamazepine prescription in a number of countries (see, for example, [22]), but the association does not extend to most other ethnic groups, probably because the frequency of B*15:02 is lower. HLA allele B*15:02 does not appear to be a risk factor for more common mild skin rash reactions induced by carbamazepine. An association involving another HLA allele, A*31:01, and carbamazepine-induced skin rash of varying severity has now been shown for both European and Japanese individuals in GWA studies [23, 24].
Abacavir hypersensitivity
A severe hypersensitivity reaction to the anti-HIV drug abacavir is characterized by a skin rash, and also gastrointestinal and respiratory symptoms. Though it may be relatively mild initially and alleviated by drug withdrawal, a subsequent re-exposure will result in more severe symptoms, which are potentially fatal. An association between abacavir hypersensitivity and a haplotype including HLA-B*57:01, HLA-DR7 and HLA-DQ3 was initially demonstrated by Mallal and colleagues using a candidate gene approach [25] and then replicated in other cohorts [26, 27]. These findings were confirmed in a large randomized controlled trial [28], which has led to widespread adoption of genetic testing for B*57:01 prior to initiation of abacavir treatment.
Other adverse drug reactions affecting the skin
TEN and SJS, together with milder hypersensitivity reactions induced by the drug allopurinol, have been shown to associate with HLA-B*58:01 in Taiwanese using a candidate gene approach [29]. This association was later shown to extend to other ethnic groups [30]. Nevirapine, another widely used anti-HIV drug, is also associated with a skin rash, which varies in severity. Several HLA associations have been reported for this adverse drug reaction with the risk allele varying according to ethnic group. An association with the HLA class I allele Cw*8 was reported in a Sardinian population [31] and subsequently in Japanese [32]. However, in Thais, there is a clear association with B*35:05 [33], which was recently confirmed in a GWA study [34]. A role for B*35 in this reaction in Asians has been confirmed in a multiethnic study [35] that also reports an association with Cw*04 for Europeans, Asians and African-Americans.
HLA and adverse drug reactions affecting the skin: summary
A combination of candidate gene and GWA studies has led to the identification of a number of HLA associations for adverse drug reactions involving specific drugs affecting skin, as summarized in Table 1. There is evidence suggesting that particular HLA alleles may be risk factors for skin reactions to additional drugs, but problems with small numbers of cases for individual drugs have limited the ability to obtain statistically significant associations in some recent candidate gene and GWA studies [36–38].
Underlying mechanism for HLA associations with adverse drug reactions
Up to recently, two main mechanisms for the observed HLA associations with adverse drug reactions affecting skin and liver were postulated. One involved formation of a covalent complex between the drug or a metabolite and cellular proteins [39]. This complex could then be presented to T cells by particular HLA molecules, resulting in an inappropriate local T-cell response and cellular damage. An alternative mechanism proposed that drugs interact directly with HLA molecules, resulting in a T-cell response without the need to form a covalent complex (p-I concept) [40]. However, recent data on T-cell responses to abacavir are more consistent with a third mechanism. Using several different approaches, three independent groups of investigators have suggested that abacavir binds to the B*57:01 gene product and induces a conformational change. This results in incorrect recognition of self-peptides as foreign by the immune system, which triggers an inappropriate immune response [41–43]. However, flucloxacillin, which may give rise to DILI in B*57:01-positive individuals, did not induce inappropriate recognition of self-peptides [42]. Instead, flucloxacillin appears to induce cell proliferation in T cells that are B*57:01-positive when covalently bound to peptides [44]. Similarly, carbamazepine also appears to interact covalently with peptides with the B*15:02 gene product [45]. It has also been proposed recently that the available T-cell repertoire, which could also be genetically determined, may be an additional factor to HLA genotype in determining whether an adverse drug reaction occurs [46]. There are currently no data showing an association between susceptibility to HLA-associated adverse drug reactions and the T-cell receptor gene, but it would be of interest to investigate this further.
Non-HLA genetic associations in adverse drug reactions
In addition to HLA, a number of genetic risk factors for idiosyncratic adverse drug reactions have been identified, though only a few of these have been well replicated. Idiosyncratic adverse drug reactions are often considered to be concentration-independent, but genetic factors that affect drug concentration by their role in drug disposition also make a contribution to susceptibility to some adverse drug reactions. Other genetic risk factors identified include polymorphisms affecting the innate immune system and in genes that protect the cell against oxidative stress. Cardiotoxicity reactions are different to other forms of adverse drug reaction in that polymorphisms in cardiac ion channels are the best described genetic risk factors currently and there appears to be no overlap with genetic risk factors for other adverse drug reactions. As the area of non-HLA genetic associations in adverse reactions covers a wide range of different types of genes, this section will consider in separate subsections the contribution of genes affecting drug disposition to adverse drug reactions, the contribution of innate immune system and oxidative stress genes, and finally the role of cardiac ion channel polymorphisms in cardiotoxicity reactions.
Idiosyncratic adverse drug reactions and genes affecting drug disposition
Well-replicated associations have been described for SLCO1B1 with statin myopathy and for NAT2 with isoniazid-induced DILI. There are also a number of other more poorly replicated associations particularly involving the transporter ABCC2 and various UGT isoforms. Table 2 summarizes current data in this area.
Though they are very effective drugs, statins can cause muscle toxicity in some individuals. This is usually seen as an asymptomatic rise in creatine phosphokinase levels, which is reversible by drug discontinuation but can be more serious on rare occasions with a more severe form of disease resulting in rhabdomyolysis followed by possible death (for review, see [47]). A GWA study involving 85 cases of simvastatin-induced myopathy found a significant signal for a single SNP in the gene SLCO1B1, which encodes a transporter expressed at high levels in hepatocytes [48]. The transporter is located on the sinusoidal membrane and transports statins and various other drugs into hepatocytes from the general circulation. The SNP giving the positive signal in the GWA study was in complete linkage disequilibrium with a non-synonymous polymorphism in the SLCO1B1*5 and *15 alleles that had already been shown to be associated with higher plasma levels of certain statins due to impaired transport [49]. The association of statin-induced myopathy with SLCO1B1 has been confirmed independently in several studies [50, 51]. It appears likely that additional genetic factors not yet identified may also contribute to susceptibility to statin-induced myopathy, but their effect sizes are likely to be smaller than that for SLCO1B1. Because the overall contribution of SLCO1B1 to hepatic transport varies between different statins, it is likely that the contribution of SCLO1B1*5/*15 to statin-induced myopathy will also vary between the different members of this drug class [49] but more studies on this aspect are needed.
Though the cytochromes P450 represent the best-studied family of genes that contribute to drug disposition and they have been well studied as risk factors for idiosyncratic adverse drug reactions, few positive associations have been reported. One exception to this relates to CYP2B6, which contributes to the metabolism of nevirapine. The non-synonymous CYP2B6 polymorphism 516G>T is associated with decreased catalytic activity with nevirapine and other substrates [52]. It has been demonstrated recently that homozygosity for T516 is associated with an increased risk of nevirapine-related skin rash [35]. Though nevirapine is also associated with DILI in some individuals, there was no evidence that CYP2B6 genotype is a predictor of this adverse reaction.
For DILI, the best example of an association affecting drug disposition is that for NAT2 genotype with isonazid-induced liver injury. There have been a large number of studies on the relationship between polymorphisms in the gene encoding N-acetyltransferase 2 (NAT2), an enzyme important in metabolism of isoniazid, and susceptibility to DILI associated with this drug. Most studies report that individuals who are homozygous for two variant NAT2 alleles (often known as slow acetylators), and therefore predicted to have a complete absence of NAT2 activity, are at increased risk of developing isonziazid-related DILI. Acetylhydrazine, a metabolite of isoniazid that can undergo further metabolism by cytochrome P450 to a toxic metabolite or by NAT2 to the less toxic diacetylhydrazine, is believed to be the cause of toxicity [53]. Individuals with normal levels of NAT2 appear to form diacetylhydrazine efficiently and therefore levels of both acetylhydrazine and the toxic P450 metabolites will be low in these individuals, but high in those with an absence of NAT2 activity [54]. As reviewed recently [55], there are still some unresolved issues on the relevance of NAT2 genotype to isoniazid-related DILI. In particular, not all studies find this association and also many of the patients studied represent cases of mild liver enzyme elevation that often resolves without the drug being withdrawn or does not recur if the drug is withdrawn and reintroduced. There is no evidence that NAT2 genotype is relevant to DILI caused by drugs other than isoniazid.
There are reports of associations between UGT genotype and DILI susceptibility for several different drugs. In a study on tolcapone, which was associated with elevated transaminase levels in some patients during its development, polymorphisms in the UGT1A locus, including several in the main metabolizing enzyme UGT1A6, were significantly associated with elevated transaminase levels [56]. This finding suggested that toxicity might be linked to slow metabolism of the parent drug. In a study on the role of another UDP-glucuronosyltransferase gene, UGT2B7, in susceptibility to diclofenac-related DILI, possession of UGT2B7*2, which is believed to be associated with higher glucuronidating activity, was associated with a significantly increased risk of toxicity [57]. This effect may be due to increased hepatic levels of diclofenac acylglucuronide, which may be involved in the underlying mechanism of toxicity. In a recent GWA study involving DILI cases caused by a variety of different drugs, when polymorphisms relevant to drug disposition only were considered, an apparent association between a polymorphism in UGT1A and susceptibility to DILI associated with fluoroquinolone antimicrobials was detected, though this could not be confirmed in a replication cohort [18].
Drug transporter genes of the ABC transporter superfamily are biologically plausible candidates for a role in DILI susceptibility, especially because some ABC transporter family gene products transport bile acids in addition to drugs [58]. Also, some inherited forms of cholestasis have been demonstrated to result from specific mutations in the ABCB4 (MDR3) and ABCB11 (BSEP) genes [59]. Some evidence for an association between cholestatic liver injury due to a range of drugs and a polymorphism in exon 13 of ABCB11 that had previously been reported to be associated with cholestasis of pregnancy was reported [60]. The association could not be confirmed in a larger cohort of predominantly cholestatic DILI cases [61] or in a GWA study involving DILI caused by a range of drugs [18].
ABCC2 (MRP2) has a major role in the biliary excretion of a variety of glucuronide conjugates. There is evidence that polymorphisms in this gene may be risk factors for some forms of DILI, though effect sizes are unlikely to be very large. In the candidate gene study on diclofenac DILI already discussed above, carriage of an upstream polymorphism in ABCC2 (C-24T) was found to be significantly more common among hepatotoxicity cases [57]. This finding is consistent with increased levels of the reactive diclofenac acyl glucuronide being associated with toxicity since there is evidence that C-24T results in lower production of the MRP2 protein, which would favor cellular accumulation of the glucuronide [62, 63]. In a Korean candidate gene study on DILI caused by a range of drugs, a polymorphism at position -1,549 of ABCC2, which is in linkage disequilibrium with C-24T, was a significant risk factor for the development of hepatocellular toxicity, whereas a second polymorphism at position -1,774 was a risk factor for cholestatic or mixed disease [63]. Further evidence of a modest contribution by ABCC2 to DILI susceptibility is provided from a large GWA study. Though polymorphisms in ABCC2 did not show genome-wide significance, when a subgroup of genes relevant to drug disposition was investigated, a significant association for a number of polymorphisms in ABCC2, including a non-synonymous polymorphism (C1515Y), was seen [18].
Some recent data suggest that genotype for pregnane × receptor (PXR), a transcriptional regulator for various metabolism and transporter genes that are relevant to disposition of both drugs and endogenous factors such as bile acids, may also be a predictor for DILI relating to flucloxacillin [64]. Though the effect size was relatively small, the association involved a polymorphism for which functional significance is well established [65]. Because other drugs are known to act as PXR agonists, the gene encoding PXR has potential as a more general risk factor for DILI.
The relevance of polymorphisms affecting drug disposition to skin reactions has also been assessed, but generally findings are negative. For example, the possible role of microsomal epoxide hydrolase in carbamazepine-induced skin rash has been investigated in considerable detail but with entirely negative results [66, 67]. For sulfamethoxazole-induced skin rash, NAT2 and CYP2C9, which both contribute to metabolism, were found not to be risk factors [68, 69]. There is some borderline significant data for GSTP1 in relation to sulfamethoxazole skin reactions but the biological basis for this association is not clear [69].
Polymorphisms relevant to innate immunity
A number of candidate gene studies have reported that cytokine polymorphisms that may contribute to inflammatory and innate immune responses are predictors for DILI (Table 3). The majority of these reports find relatively small effects that have not been replicated, though the genes involved are biologically plausible risk factors.
Three recent GWA studies on DILI have generally failed to identify any novel genome-wide significant associations, with only SNPs in HLA genes showing strong effects. However, various additional analyses performed on these data sets have identified some additional interesting genes that may contribute to susceptibility. In the GWA study on flucloxacillin-induced DILI, if data from cases positive for HLA-B*57:01 only were re-analyzed, a novel genome-wide significant association for a SNP adjacent to ST6GAL1, a gene that contributes to B-cell responses, was detected [17, 70]. Since some patients with flucloxacillin DILI show an antibody response [71], this could be of relevance to the toxicity mechanism. For amoxicillin-clavulanate, in addition to performing a conventional GWA study, polymorphisms in genes relevant to drug disposition and to autoimmunity were analyzed separately. No positive associations for drug disposition genes were detected, but for the immune response genes, two SNPs in strong linkage disequilibrium in PTPN22, a gene that contributes to T-cell responses, showed significance after correction for multiple testing [72]. These SNPs had been previously found to be associated with susceptibility to several autoimmune diseases where HLA genotype is also a risk factor, so a contribution to this form of DILI appears biologically plausible. In a similar approach in a larger GWA study involving DILI caused by a range of different drugs, but also including the flucloxacillin and amoxicillin-clavulanate DILI cases in the two previous GWA studies [17, 72], analysis of 256 cases of hepatocellular DILI for autoimmune-related polymorphisms found a significant association for a SNP in STAT4 [18]. STAT4 encodes a transcription factor that transduces IL-12 and IL-23 signals in the T-cell response [73] and the significant SNP has been associated previously with several autoimmune diseases so it represents another biologically plausible association for DILI. The association with hepatocellular DILI was confirmed in a replication cohort and appeared to relate particularly to DILI reactions involving statins.
GWA studies on drug-induced hypersensitivity reactions affecting the skin have generally yielded fewer novel associations of the type seen for liver toxicity up to the present [37, 74], but this may be due in part to the numbers of cases studied being smaller. One exception to this is a GWA concerned with skin rash due to nevirapine [34], which has found that, in addition to confirming a role for B*35:05 in susceptibility, two SNPs in CCHCR1 were significantly associated with the reaction. This gene is a well-established contributor to psoriasis susceptibility and, unlike the autoimmune genes discussed above, does not appear to have a general role in T-cell responses. Instead it has been suggested to be a negative regulator of keratinocyte proliferation [75], which seems relevant to skin rash.
Polymorphisms predicting cardiotoxicity reactions
Cardiotoxicity is currently the most common reason for withdrawal of licensed drugs from the market and a wide range of drugs are known to give rise to idiosyncratic cardiotoxicity (for review, see [76]). As discussed in the Introduction to this article, QT interval prolongation is an imperfect marker for the arrhythmic potential of a drug, but it is currently the only available measure. There are a number of rare congenital syndromes associated with QT prolongation in the absence of any drug treatment and genetic factors associated with some of these syndromes, mainly mutations in ion channel genes, have now been identified [76]. In addition, GWA studies on factors affecting QT length in populations have identified a number of significant SNPs in various genes, including the nitric oxide synthase 1 regulator NOS1AP and a range of sodium and potassium channel genes, including SCN5A and KCNJ2 [77–79]. These factors have also been investigated in candidate gene studies on drug-induced long QT syndrome, as it is considered likely that similar factors contribute to both congenital long QT and drug-induced long QT syndrome [76].
The first genetic study on drug-induced cardiotoxicity sequenced five ion channel genes in 32 patients who had suffered QT prolongation due to a variety of drugs and found previously described rare mutations in four patients, including D85N in KCNE1 in two of these. The general conclusion was that there was a contribution of known ion channel mutations to the reactions but they were not a major risk factor [80]. A larger candidate gene study, involving 317 cases, also found an increased prevalence of D85N in KCNE1 in the cases with an allele frequency increase from 0.8% among controls to 3.9% of cases [81], again suggesting this was a minor risk factor. A further candidate gene study based in Europe and involving 307 patients with drug-induced QT prolongation confirmed a role for KCNE1 D85N with an odds ratio of 9.0, with the variant allele present in 8.6% of cases and 1.8% of population control subjects [82].
Another population genetic risk factor for long QT, a polymorphism in NOS1AP, has also been studied in relation to drug-induced QT prolongation. Verapamil is associated with QT prolongation and in a prospective population study involving over 7,000 individuals, use of this drug was confirmed to be associated with QT prolongation, with individuals positive for the NOS1AP variant (rs10494366) showing the longest QT intervals [83]. More recently, a number of polymorphisms in NOS1AP were genotyped in 87 British drug-induced long QT cases. A single SNP (rs10800397) was significantly associated with increased risk of the adverse drug reaction generally, and this SNP together with two others showed a more pronounced effect when cases due to amiodarone only were considered [84].
The first GWA study on drug-induced QT-prolongation involved 183 patients in a phase III clinical trial of the antipsychotic drug iloperidone who had QT measurements performed 14 days after the start of drug treatment [85]. No genome-wide significant signals were detected but relatively low P values were obtained for several novel loci. No trends towards significance with the SNPs in either ion channels or NOS1AP relevant to QT length in the previous studies were detected. Another recent GWA study examined a cohort of 783 schizophrenia patients taking antipsychotic drugs frequently associated with QT prolongation [86]. Significant effects were seen for SNPs in NOS1AP and NUBPL, a gene concerned with mitochondrial function. In addition, evidence for a role for the transporter gene SLC22A23 in relation to the effect of the drug quetiapine on prolongation was obtained.
Consistent associations with certain genes have started to emerge for drug-induced cardiotoxicity but overall effects are small. The possibility that rare variants are more important contributors requires further investigation. Though genes relevant to drug disposition are plausible candidates for contributors to drug-induced cardiotoxicity and a number of studies have been performed, findings on these have generally been negative.
Concluding remarks and future prospects
As reviewed recently by others [87], considerable progress has been made in understanding the genetics of adverse drug reactions with particular progress made using both candidate gene and GWA approaches in understanding idiosyncratic adverse drug reactions where HLA genotype is a risk factor. Two of these associations that show very high sensitivity and specificity for abacavir hypersensitivity and B*57:01 and carbamazepine toxicity and B*15:02 have been translated to the clinic. There is also potential for translating some of the other associations such as that between A*31:01 and carbamazepine-induced skin rash, and that between B*58:01 and allopurinol-induced skin rash. Most of these advances have been achieved through candidate gene association studies rather than GWA studies.
There has been slower progress on understanding the genetic basis for adverse drug reactions where HLA does not contribute. One success story is the association between SLCO1B1 and statin myopathy. Though the predictive value is probably insufficient for widespread clinical translation and it is likely that the contribution by SLCO1B1 genotype varies between different statins, the finding has increased understanding of the mechanism for this toxicity and genotyping could be beneficial in certain situations. Otherwise, GWA studies have not identified novel genes to any great extent. This may be due to insufficient numbers of cases being studied. Continuing efforts by international consortia to increase cohort sizes for various adverse drug reactions may still enable further progress to be made using a GWA approach. The availability of cohorts of well-phenotyped cases will also be helpful for whole genome sequencing, which is likely to become more routine as processing costs fall and methods for data analysis improve.
Performing genetic association studies on idiosyncratic adverse drug reactions is of particular value because of the lack of animal models for most of the common reactions and also the difficulty in obtaining material from the target organ for most types of reaction. One interesting recent development is the possibility of using induced pluripotent stem (iPS) cells from individuals who have suffered adverse drug reactions to model the reaction. This has been proposed as a means of studying congenital long QT syndrome [88], but should be equally applicable to drug-induced long QT and also to other adverse drug reactions such as DILI since it is now possible to derive hepatocyte-like cells from human iPS cells [89].
In addition to the adverse drug reactions discussed in detail in this article, there are a number of other relatively common reactions, including clozapine-induced agranulocytosis, bisphosphonate-induced osteonecrosis of the jaw (BONJ) and renal toxicity, that are important clinical problems. Clozapine-induced agranulocytosis has recently been shown to be HLA-associated [90] and some genetic risk factors for BONJ [91, 92] have also been described. Genetic aspects of drug-induced renal toxicity are still poorly understood despite being a common form of adverse drug reaction and also a cause of drug attrition, so further studies in this area would be valuable.
Abbreviations
- BONJ:
-
bisphosphonate-induced osteonecrosis of the jaw
- DILI:
-
drug-induced liver injury
- GWA:
-
genome-wide association
- HLA:
-
human leukocyte antigen
- IL:
-
interleukin
- iPS:
-
induced pluripotent stem
- PXR:
-
pregnane × receptor
- SJS:
-
Stevens-Johnson syndrome
- SNP:
-
single nucleotide polymorphism
- TEN:
-
toxic epidermal necrolysis.
References
Pirmohamed M, Breckenridge AM, Kitteringham NR, Park BK: Adverse drug reactions. BMJ. 1998, 316: 1295-1298. 10.1136/bmj.316.7140.1295.
Assis DN, Navarro VJ: Human drug hepatotoxicity: a contemporary clinical perspective. Expert Opin Drug Metab Toxicol. 2009, 5: 463-473. 10.1517/17425250902927386.
Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, Lakey JH, Rahman T, Wang XN, McGovern N, Pagan S, Cookson S, McDonald D, Chua I, Wallis J, Cant A, Wright M, Keavney B, Chinnery PF, Loughlin J, Hambleton S, Santibanez-Koref M, Collin M: Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011, 118: 2656-2658. 10.1182/blood-2011-06-360313.
Zeitz C, Jacobson SG, Hamel CP, Bujakowska K, Neuille M, Orhan E, Zanlonghi X, Lancelot ME, Michiels C, Schwartz SB, Bocquet B, Antonio A, Audier C, Letexier M, Saraiva JP, Luu TD, Sennlaub F, Nguyen H, Poch O, Dollfus H, Lecompte O, Kohl S, Sahel JA, Bhattacharya SS, Audo I: Whole-exome sequencing identifies LRIT3 mutations as a cause of autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet. 2013, 92: 67-75.
Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, Wright FA, Rieder MJ, Tabor HK, Nickerson DA, Barnes KC, Gibson RL, Bamshad MJ: Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012, 44: 886-889. 10.1038/ng.2344.
Albrechtsen A, Grarup N, Li Y, Sparso T, Tian G, Cao H, Jiang T, Kim SY, Korneliussen T, Li Q, Nie C, Wu R, Skotte L, Morris AP, Ladenvall C, Cauchi S, Stancakova A, Andersen G, Astrup A, Banasik K, Bennett AJ, Bolund L, Charpentier G, Chen Y, Dekker JM, Doney AS, Dorkhan M, Forsen T, Frayling TM, Groves CJ, et al: Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia. 2013, 56: 298-310. 10.1007/s00125-012-2756-1.
Bjornsson E: The natural history of drug-induced liver injury. Semin Liver Dis. 2009, 29: 357-363. 10.1055/s-0029-1240004.
Park BK, Kitteringham NR, Maggs JL, Pirmohamed M, Williams DP: The role of metabolic activation in drug-induced hepatotoxicity. Annu Rev Pharmacol Toxicol. 2005, 45: 177-202. 10.1146/annurev.pharmtox.45.120403.100058.
Otsuka S, Yamamoto M, Kasuya S, Ohtomo H, Yamamoto Y, Yoshida TO, Akaza T: HLA antigens in patients with unexplained hepatitis following halothane anesthesia. Acta Anaesthesiol Scand. 1985, 29: 497-501. 10.1111/j.1399-6576.1985.tb02242.x.
Eade OE, Grice D, Krawitt EL, Trowell J, Albertini R, Festenstein H, Wright R: HLA A and B locus antigens in patients with unexplained hepatitis following halothane anaesthesia. Tissue Antigens. 1981, 17: 428-432.
Ranek L, Dalhoff K, Poulsen HE, Brosen K, Flachs H, Loft S, Wantzin P: Drug metabolism and genetic polymorphism in subjects with previous halothane hepatitis. Scand J Gastroenterol. 1993, 28: 677-680. 10.3109/00365529309098271.
Stricker BH, Blok AP, Claas FH, Van Parys GE, Desmet VJ: Hepatic injury associated with the use of nitrofurans: a clinicopathological study of 52 reported cases. Hepatology. 1988, 8: 599-606. 10.1002/hep.1840080327.
Berson A, Freneaux E, Larrey D, Lepage V, Douay C, Mallet C, Fromentry B, Benhamou JP, Pessayre D: Possible role of HLA in hepatotoxicity - an exploratory-study in 71 patients with drug-induced idiosyncratic hepatitis. J Hepatol. 1994, 20: 336-342.
Hautekeete ML, Horsmans Y, van Waeyenberge C, Demanet C, Henrion J, Verbist L, Brenard R, Sempoux C, Michielsen PP, Yap PSH, Rahier J, Geubel AP: HLA association of amoxicillin-clavulanate-induced hepatitis. Gastroenterology. 1999, 117: 1181-1186. 10.1016/S0016-5085(99)70404-X.
O'Donohue J, Oien KA, Donaldson P, Underhill J, Clare M, MacSween RM, Mills PR: Co-amoxiclav jaundice: clinical and histological features and HLA class II association. Gut. 2000, 47: 717-720. 10.1136/gut.47.5.717.
Stricker BH, Van den Broek JW, Keuning J, Eberhardt W, Houben HG, Johnson M, Blok AP: Cholestatic hepatitis due to antibacterial combination of amoxicillin and clavulanic acid (augmentin). Dig Dis Sci. 1989, 34: 1576-1580. 10.1007/BF01537113.
Daly AK, Donaldson PT, Bhatnagar P, Shen Y, Pe'er I, Floratos A, Daly MJ, Goldstein DB, John S, Nelson MR, Graham J, Park BK, Dillon JF, Bernal W, Cordell HJ, Pirmohamed M, Aithal GP, Day CP: HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet. 2009, 41: 816-819. 10.1038/ng.379.
Urban TJ, Shen Y, Stolz A, Chalasani N, Fontana RJ, Rochon J, Ge D, Shianna KV, Daly AK, Lucena MI, Nelson MR, Molokhia M, Aithal GP, Floratos A, Pe'er I, Serrano J, Bonkovsky H, Davern TJ, Lee WM, Navarro VJ, Talwalkar JA, Goldstein DB, Watkins PB: Limited contribution of common genetic variants to risk for liver injury due to a variety of drugs. Pharmacogenet Genomics. 2012, 22: 784-795. 10.1097/FPC.0b013e3283589a76.
Svensson CK, Cowen EW, Gaspari AA: Cutaneous drug reactions. Pharmacol Rev. 2001, 53: 357-379.
Roujeau JC, Bracq C, Huyn NT, Chaussalet E, Raffin C, Duedari N: HLA phenotypes and bullous cutaneous reactions to drugs. Tissue Antigens. 1986, 28: 251-254.
Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, Wu JY, Chen YT: Medical genetics: a marker for Stevens-Johnson syndrome. Nature. 2004, 428: 486-10.1038/428486a.
US Food and Drug Administration: Information for Healthcare Professionals: Dangerous or Even Fatal Skin Reactions - Carbamazepine (marketed as Carbatrol, Equetro, Tegretol, and generics). [http://www.fda.gov/Drugs/DrugSafety/PostmarketdrugsafetyinformationforPatientsandProviders/ucm124718.htm]
Ozeki T, Mushiroda T, Yowang A, Takahashi A, Kubo M, Shirakata Y, Ikezawa Z, Iijima M, Shiohara T, Hashimoto K, Kamatani N, Nakamura Y: Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet. 2011, 20: 1034-1041. 10.1093/hmg/ddq537.
McCormack M, Alfirevic A, Bourgeois S, Farrell JJ, Kasperaviciute D, Carrington M, Sills GJ, Marson T, Jia X, de Bakker PI, Chinthapalli K, Molokhia M, Johnson MR, O'Connor GD, Chaila E, Alhusaini S, Shianna KV, Radtke RA, Heinzen EL, Walley N, Pandolfo M, Pichler W, Park BK, Depondt C, Sisodiya SM, Goldstein DB, Deloukas P, Delanty N, Cavalleri GL, Pirmohamed M: HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med. 2011, 364: 1134-1143. 10.1056/NEJMoa1013297.
Mallal S, Nolan D, Witt C, Masel G, Martin AM, Moore C, Sayer D, Castley A, Mamotte C, Maxwell D, James I, Christiansen FT: Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse transcriptase inhibitor abacavir. Lancet. 2002, 359: 727-732. 10.1016/S0140-6736(02)07873-X.
Hetherington S, Hughes AR, Mosteller M, Shortino D, Baker KL, Spreen W, Lai E, Davies K, Handley A, Dow DJ, Fling ME, Stocum M, Bowman C, Thurmond LM, Roses AD: Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002, 359: 1121-1122. 10.1016/S0140-6736(02)08158-8.
Hughes DA, Vilar FJ, Ward CC, Alfirevic A, Park BK, Pirmohamed M: Cost-effectiveness analysis of HLA B*5701 genotyping in preventing abacavir hypersensitivity. Pharmacogenetics. 2004, 14: 335-342. 10.1097/00008571-200406000-00002.
Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, Jagel-Guedes E, Rugina S, Kozyrev O, Cid JF, Hay P, Nolan D, Hughes S, Hughes A, Ryan S, Fitch N, Thorborn D, Benbow A: HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008, 358: 568-579. 10.1056/NEJMoa0706135.
Hung SI, Chung WH, Liou LB, Chu CC, Lin M, Huang HP, Lin YL, Lan JL, Yang LC, Hong HS, Chen MJ, Lai PC, Wu MS, Chu CY, Wang KH, Chen CH, Fann CS, Wu JY, Chen YT: HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci USA. 2005, 102: 4134-4139. 10.1073/pnas.0409500102.
Lonjou C, Borot N, Sekula P, Ledger N, Thomas L, Halevy S, Naldi L, Bouwes-Bavinck JN, Sidoroff A, de Toma C, Schumacher M, Roujeau JC, Hovnanian A, Mockenhaupt M: A European study of HLA-B in Stevens-Johnson syndrome and toxic epidermal necrolysis related to five high-risk drugs. Pharmacogenet Genomics. 2008, 18: 99-107. 10.1097/FPC.0b013e3282f3ef9c.
Littera R, Carcassi C, Masala A, Piano P, Serra P, Ortu F, Corso N, Casula B, La Nasa G, Contu L, Manconi PE: HLA-dependent hypersensitivity to nevirapine in Sardinian HIV patients. Aids. 2006, 20: 1621-1626. 10.1097/01.aids.0000238408.82947.09.
Gatanaga H, Yazaki H, Tanuma J, Honda M, Genka I, Teruya K, Tachikawa N, Kikuchi Y, Oka S: HLA-Cw8 primarily associated with hypersensitivity to nevirapine. Aids. 2007, 21: 264-265. 10.1097/QAD.0b013e32801199d9.
Chantarangsu S, Mushiroda T, Mahasirimongkol S, Kiertiburanakul S, Sungkanuparph S, Manosuthi W, Tantisiriwat W, Charoenyingwattana A, Sura T, Chantratita W, Nakamura Y: HLA-B*3505 allele is a strong predictor for nevirapine-induced skin adverse drug reactions in HIV-infected Thai patients. Pharmacogenet Genomics. 2009, 19: 139-146. 10.1097/FPC.0b013e32831d0faf.
Chantarangsu S, Mushiroda T, Mahasirimongkol S, Kiertiburanakul S, Sungkanuparph S, Manosuthi W, Tantisiriwat W, Charoenyingwattana A, Sura T, Takahashi A, Kubo M, Kamatani N, Chantratita W, Nakamura Y: Genome-wide association study identifies variations in 6p21.3 associated with nevirapine-induced rash. Clin Infect Dis. 2011, 53: 341-348. 10.1093/cid/cir403.
Yuan J, Guo S, Hall D, Cammett AM, Jayadev S, Distel M, Storfer S, Huang Z, Mootsikapun P, Ruxrungtham K, Podzamczer D, Haas DW: Toxicogenomics of nevirapine-associated cutaneous and hepatic adverse events among populations of African, Asian, and European descent. Aids. 2011, 25: 1271-1280. 10.1097/QAD.0b013e32834779df.
Pirmohamed M, Arbuckle JB, Bowman CE, Brunner M, Burns DK, Delrieu O, Dix LP, Twomey JA, Stern RS: Investigation into the multidimensional genetic basis of drug-induced Stevens-Johnson syndrome and toxic epidermal necrolysis. Pharmacogenomics. 2007, 8: 1661-1691. 10.2217/14622416.8.12.1661.
Kazeem GR, Cox C, Aponte J, Messenheimer J, Brazell C, Nelsen AC, Nelson MR, Foot E: High-resolution HLA genotyping and severe cutaneous adverse reactions in lamotrigine-treated patients. Pharmacogenet Genomics. 2009, 19: 661-665. 10.1097/FPC.0b013e32832c347d.
Genin E, Schumacher M, Roujeau JC, Naldi L, Liss Y, Kazma R, Sekula P, Hovnanian A, Mockenhaupt M: Genome-wide association study of Stevens-Johnson syndrome and toxic epidermal necrolysis in Europe. Orphanet J Rare Dis. 2011, 6: 52-10.1186/1750-1172-6-52.
Uetrecht J: Immunoallergic drug-induced liver injury in humans. Semin Liver Dis. 2009, 29: 383-392. 10.1055/s-0029-1240007.
Adam J, Pichler WJ, Yerly D: Delayed drug hypersensitivity: models of T-cell stimulation. Br J Clin Pharmacol. 2011, 71: 701-707. 10.1111/j.1365-2125.2010.03764.x.
Illing PT, Vivian JP, Dudek NL, Kostenko L, Chen Z, Bharadwaj M, Miles JJ, Kjer-Nielsen L, Gras S, Williamson NA, Burrows SR, Purcell AW, Rossjohn J, McCluskey J: Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature. 2012, 486: 554-558.
Norcross MA, Luo S, Lu L, Boyne MT, Gomarteli M, Rennels AD, Woodcock J, Margulies DH, McMurtrey C, Vernon S, Hildebrand WH, Buchli R: Abacavir induces loading of novel self-peptides into HLA-B*57: 01: an autoimmune model for HLA-associated drug hypersensitivity. Aids. 2012, 26: F21-29. 10.1097/QAD.0b013e328355fe8f.
Ostrov DA, Grant BJ, Pompeu YA, Sidney J, Harndahl M, Southwood S, Oseroff C, Lu S, Jakoncic J, de Oliveira CA, Yang L, Mei H, Shi L, Shabanowitz J, English AM, Wriston A, Lucas A, Phillips E, Mallal S, Grey HM, Sette A, Hunt DF, Buus S, Peters B: Drug hypersensitivity caused by alteration of the MHC-presented self-peptide repertoire. Proc Natl Acad Sci USA. 2012, 109: 9959-9964. 10.1073/pnas.1207934109.
Monshi M, Faulkner L, Gibson A, Jenkins RE, Farrell J, Earnshaw CJ, Alfirevic A, Cederbrant K, Daly AK, French N, Pirmohamed M, Park BK, Naisbitt DJ: HLA-B*57:01-restricted activation of drug-specific T-cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology. 2012, doi: 10.1002/hep.26077
Wei CY, Chung WH, Huang HW, Chen YT, Hung SI: Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol. 2012, 129: 1562-1569 e1565. 10.1016/j.jaci.2011.12.990.
Ko TM, Chung WH, Wei CY, Shih HY, Chen JK, Lin CH, Chen YT, Hung SI: Shared and restricted T-cell receptor use is crucial for carbamazepine-induced Stevens-Johnson syndrome. J Allergy Clin Immunol. 2011, 128: 1266-1276 e1211. 10.1016/j.jaci.2011.08.013.
Dalakas MC: Toxic and drug-induced myopathies. J Neurol Neurosurg Psychiatry. 2009, 80: 832-838. 10.1136/jnnp.2008.168294.
Link E, Parish S, Armitage J, Bowman L, Heath S, Matsuda F, Gut I, Lathrop M, Collins R: SLCO1B1 variants and statin-induced myopathy - a genomewide study. N Engl J Med. 2008, 359: 789-799.
Niemi M: Transporter pharmacogenetics and statin toxicity. Clin Pharmacol Ther. 2010, 87: 130-133. 10.1038/clpt.2009.197.
Voora D, Shah SH, Spasojevic I, Ali S, Reed CR, Salisbury BA, Ginsburg GS: The SLCO1B1*5 genetic variant is associated with statin-induced side effects. J Am Coll Cardiol. 2009, 54: 1609-1616. 10.1016/j.jacc.2009.04.053.
Donnelly LA, Doney AS, Tavendale R, Lang CC, Pearson ER, Colhoun HM, McCarthy MI, Hattersley AT, Morris AD, Palmer CN: Common nonsynonymous substitutions in SLCO1B1 predispose to statin intolerance in routinely treated individuals with type 2 diabetes: a go-DARTS study. Clin Pharmacol Ther. 2011, 89: 210-216. 10.1038/clpt.2010.255.
Frasco MA, Mack WJ, Van Den Berg D, Aouizerat BE, Anastos K, Cohen M, De Hovitz J, Golub ET, Greenblatt RM, Liu C, Conti DV, Pearce CL: Underlying genetic structure impacts the association between CYP2B6 polymorphisms and response to efavirenz and nevirapine. Aids. 2012, 26: 2097-2106. 10.1097/QAD.0b013e3283593602.
Metushi IG, Cai P, Zhu X, Nakagawa T, Uetrecht JP: A fresh look at the mechanism of isoniazid-induced hepatotoxicity. Clin Pharmacol Ther. 2011, 89: 911-914. 10.1038/clpt.2010.355.
Eichelbaum M, Musch E, Castroparra M, Vonsassen W: Isoniazid hepatotoxicity in relation to acetylator phenotype and isoniazid metabolism. Br J Clin Pharmacol. 1982, 14: P575-P576.
Daly AK, Day CP: Genetic association studies in drug-induced liver injury. Drug Metab Rev. 2012, 44: 116-126. 10.3109/03602532.2011.605790.
Acuna G, Foernzler D, Leong D, Rabbia M, Smit R, Dorflinger E, Gasser R, Hoh J, Ott J, Borroni E, To Z, Thompson A, Li J, Hashimoto L, Lindpaintner K: Pharmacogenetic analysis of adverse drug effect reveals genetic variant for susceptibility to liver toxicity. Pharmacogenomics J. 2002, 2: 327-334. 10.1038/sj.tpj.6500123.
Daly AK, Aithal GP, Leathart JB, Swainsbury RA, Dang TS, Day CP: Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes. Gastroenterology. 2007, 132: 272-281. 10.1053/j.gastro.2006.11.023.
Geier A, Wagner M, Dietrich CG, Trauner M: Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim Biophys Acta. 2007, 1773: 283-308. 10.1016/j.bbamcr.2006.04.014.
Noe J, Kullak-Ublick GA, Jochum W, Stieger B, Kerb R, Haberl M, Mullhaupt B, Meier PJ, Pauli-Magnus C: Impaired expression and function of the bile salt export pump due to three novel ABCB11 mutations in intrahepatic cholestasis. J Hepatol. 2005, 43: 536-543. 10.1016/j.jhep.2005.05.020.
Lang C, Meier Y, Stieger B, Beuers U, Lang T, Kerb R, Kullak-Ublick GA, Meier PJ, Pauli-Magnus C: Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury. Pharmacogenet Genomics. 2007, 17: 47-60. 10.1097/01.fpc.0000230418.28091.76.
Bhatnagar P, Day CP, Aithal G, Pirmohamed M, Bernal W, Daly AK: Genetic variants of hepatic transporters and susceptibility to drug induced liver injury. Toxicology. 2008, 253: 10-10. 10.1016/j.tox.2008.07.008.
Haenisch S, Zimmermann U, Dazert E, Wruck CJ, Dazert P, Siegmund W, Kroemer HK, Warzok RW, Cascorbi I: Influence of polymorphisms of ABCB1 and ABCC2 on mRNA and protein expression in normal and cancerous kidney cortex. Pharmacogenomics J. 2007, 7: 56-65. 10.1038/sj.tpj.6500403.
Choi JH, Ahn BM, Yi J, Lee JH, Nam SW, Chon CY, Han KH, Ahn SH, Jang IJ, Cho JY, Suh Y, Cho MO, Lee JE, Kim KH, Lee MG: MRP2 haplotypes confer differential susceptibility to toxic liver injury. Pharmacogenet Genomics. 2007, 17: 403-415. 10.1097/01.fpc.0000236337.41799.b3.
Andrews E, Armstrong M, Tugwood J, Swan D, Glaves P, Pirmohamed M, Aithal GP, Wright MC, Day CP, Daly AK: A role for the pregnane × receptor in flucloxacillin-induced liver injury. Hepatology. 2010, 51: 1656-1664. 10.1002/hep.23549.
Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boguski MS, Schuetz E: Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001, 27: 383-391. 10.1038/86882.
Green VJ, Pirmohamed M, Kitteringham NR, Gaedigk A, Grant DM, Boxer M, Burchell B, Park BK: Genetic analysis of microsomal epoxide hydrolase in patients with carbamazepine hypersensitivity. Biochem Pharmacol. 1995, 50: 1353-1359. 10.1016/0006-2952(95)02009-8.
Gaedigk A, Spielberg SP, Grant DM: Characterization of the microsomal epoxide hydrolase gene in patients with anticonvulsant adverse drug reactions. Pharmacogenetics. 1994, 4: 142-153. 10.1097/00008571-199406000-00005.
Alfirevic A, Stalford AC, Vilar FJ, Wilkins EG, Park BK, Pirmohamed M: Slow acetylator phenotype and genotype in HIV-positive patients with sulphamethoxazole hypersensitivity. Br J Clin Pharmacol. 2003, 55: 158-165. 10.1046/j.1365-2125.2003.01754.x.
Wolkenstein P, Loriot MA, Flahault A, Cadilhac M, Caumes E, Eliaszewicz M, Beaune P, Roujeau JC, Chosidow O: Association analysis of drug metabolizing enzyme gene polymorphisms in AIDS patients with cutaneous reactions to sulfonamides. J Invest Dermatol. 2005, 125: 1080-1082. 10.1111/j.0022-202X.2005.23939.x.
Nasirikenari M, Segal BH, Ostberg JR, Urbasic A, Lau JT: Altered granulopoietic profile and exaggerated acute neutrophilic inflammation in mice with targeted deficiency in the sialyltransferase ST6Gal I. Blood. 2006, 108: 3397-3405. 10.1182/blood-2006-04-014779.
Carey MA, van Pelt FN: Immunochemical detection of flucloxacillin adduct formation in livers of treated rats. Toxicology. 2005, 216: 41-48. 10.1016/j.tox.2005.07.015.
Lucena MI, Molokhia M, Shen Y, Urban TJ, Aithal GP, Andrade RJ, Day CP, Ruiz-Cabello F, Donaldson PT, Stephens C, Pirmohamed M, Romero-Gomez M, Navarro JM, Fontana RJ, Miller M, Groome M, Bondon-Guitton E, Conforti A, Stricker BH, Carvajal A, Ibanez L, Yue QY, Eichelbaum M, Floratos A, Pe'er I, Daly MJ, Goldstein DB, Dillon JF, Nelson MR, Watkins PB, Daly AK: Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology. 2011, 141: 338-347. 10.1053/j.gastro.2011.04.001.
Korman BD, Kastner DL, Gregersen PK, Remmers EF: STAT4: genetics, mechanisms, and implications for autoimmunity. Curr Allergy Asthma Rep. 2008, 8: 398-403. 10.1007/s11882-008-0077-8.
Shen Y, Nicoletti P, Floratos A, Pirmohamed M, Molokhia M, Geppetti P, Benemei S, Giomi B, Schena D, Vultaggio A, Stern R, Daly MJ, John S, Nelson MR, Pe'er I: Genome-wide association study of serious blistering skin rash caused by drugs. Pharmacogenomics J. 2012, 12: 96-104. 10.1038/tpj.2010.84.
Tiala I, Wakkinen J, Suomela S, Puolakkainen P, Tammi R, Forsberg S, Rollman O, Kainu K, Rozell B, Kere J, Saarialho-Kere U, Elomaa O: The PSORS1 locus gene CCHCR1 affects keratinocyte proliferation in transgenic mice. Hum Mol Genet. 2008, 17: 1043-1051.
Kannankeril P, Roden DM, Darbar D: Drug-induced long QT syndrome. Pharmacol Rev. 2010, 62: 760-781. 10.1124/pr.110.003723.
Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY, Larson MG, Wichmann HE, Marban E, O'Donnell CJ, Hirschhorn JN, Kaab S, Spooner PM, Meitinger T, Chakravarti A: A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006, 38: 644-651. 10.1038/ng1790.
Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, Bis JC, Marciante K, Rivadeneira F, Noseworthy PA, Sotoodehnia N, Smith NL, Rotter JI, Kors JA, Witteman JC, Hofman A, Heckbert SR, O'Donnell CJ, Uitterlinden AG, Psaty BM, Lumley T, Larson MG, Stricker BH: Common variants at ten loci influence QT interval duration in the QTGEN Study. Nat Genet. 2009, 41: 399-406. 10.1038/ng.364.
Nolte IM, Wallace C, Newhouse SJ, Waggott D, Fu J, Soranzo N, Gwilliam R, Deloukas P, Savelieva I, Zheng D, Dalageorgou C, Farrall M, Samani NJ, Connell J, Brown M, Dominiczak A, Lathrop M, Zeggini E, Wain LV, Newton-Cheh C, Eijgelsheim M, Rice K, de Bakker PI, Pfeufer A, Sanna S, Arking DE, Asselbergs FW, Spector TD, Carter ND, Jeffery S, et al: Common genetic variation near the phospholamban gene is associated with cardiac repolarisation: meta-analysis of three genome-wide association studies. PLoS One. 2009, 4: e6138-10.1371/journal.pone.0006138.
Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, Schulze-Bahr E, Haverkamp W, Breithardt G, Cohen N, Aerssens J: Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug-induced long QT syndrome patients. J Mol Med. 2004, 82: 182-188. 10.1007/s00109-003-0522-z.
Nishio Y, Makiyama T, Itoh H, Sakaguchi T, Ohno S, Gong YZ, Yamamoto S, Ozawa T, Ding WG, Toyoda F, Kawamura M, Akao M, Matsuura H, Kimura T, Kita T, Horie M: D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J Am Coll Cardiol. 2009, 54: 812-819. 10.1016/j.jacc.2009.06.005.
Kaab S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze-Bahr E, Guicheney P, Bishopric NH, Myerburg RJ, Schott JJ, Pfeufer A, Beckmann BM, Martens E, Zhang T, Stallmeyer B, Zumhagen S, Denjoy I, Bardai A, Van Gelder IC, Jamshidi Y, Dalageorgou C, Marshall V, Jeffery S, Shakir S, Camm AJ, Steinbeck G, Perz S, Lichtner P, et al: A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug-induced torsades de pointes. Circ Cardiovasc Genet. 2012, 5: 91-99. 10.1161/CIRCGENETICS.111.960930.
van Noord C, Aarnoudse AJ, Eijgelsheim M, Sturkenboom MC, Straus SM, Hofman A, Kors JA, Newton-Cheh C, Witteman JC, Stricker BH: Calcium channel blockers, NOS1AP, and heart-rate-corrected QT prolongation. Pharmacogenet Genomics. 2009, 19: 260-266. 10.1097/FPC.0b013e328324e556.
Jamshidi Y, Nolte IM, Dalageorgou C, Zheng D, Johnson T, Bastiaenen R, Ruddy S, Talbott D, Norris KJ, Snieder H, George AL, Marshall V, Shakir S, Kannankeril PJ, Munroe PB, Camm AJ, Jeffery S, Roden DM, Behr ER: Common variation in the NOS1AP gene is associated with drug-induced QT prolongation and ventricular arrhythmia. J Am Coll Cardiol. 2012, 60: 841-850. 10.1016/j.jacc.2012.03.031.
Volpi S, Heaton C, Mack K, Hamilton JB, Lannan R, Wolfgang CD, Licamele L, Polymeropoulos MH, Lavedan C: Whole genome association study identifies polymorphisms associated with QT prolongation during iloperidone treatment of schizophrenia. Mol Psychiatry. 2009, 14: 1024-1031. 10.1038/mp.2008.52.
Aberg K, Adkins DE, Liu Y, McClay JL, Bukszar J, Jia P, Zhao Z, Perkins D, Stroup TS, Lieberman JA, Sullivan PF, van den Oord EJ: Genome-wide association study of antipsychotic-induced QTc interval prolongation. Pharmacogenomics J. 2012, 12: 165-172. 10.1038/tpj.2010.76.
Pavlos R, Mallal S, Phillips E: HLA and pharmacogenetics of drug hypersensitivity. Pharmacogenomics. 2012, 13: 1285-1306. 10.2217/pgs.12.108.
Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, Feldman O, Gepstein A, Arbel G, Hammerman H, Boulos M, Gepstein L: Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011, 471: 225-229. 10.1038/nature09747.
Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-Doran I, Griffin J, Ahrlund-Richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L: Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010, 120: 3127-3136. 10.1172/JCI43122.
Athanasiou MC, Dettling M, Cascorbi I, Mosyagin I, Salisbury BA, Pierz KA, Zou W, Whalen H, Malhotra AK, Lencz T, Gerson SL, Kane JM, Reed CR: Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011, 72: 458-463. 10.4088/JCP.09m05527yel.
Katz J, Gong Y, Salmasinia D, Hou W, Burkley B, Ferreira P, Casanova O, Langaee TY, Moreb JS: Genetic polymorphisms and other risk factors associated with bisphosphonate induced osteonecrosis of the jaw. Int J Oral Maxillofac Surg. 2011, 40: 605-611. 10.1016/j.ijom.2011.02.002.
Nicoletti P, Cartsos VM, Palaska PK, Shen Y, Floratos A, Zavras AI: Genomewide pharmacogenetics of bisphosphonate-induced osteonecrosis of the jaw: the role of RBMS3. Oncologist. 2012, 17: 279-287. 10.1634/theoncologist.2011-0202.
Andrade RJ, Lucena MI, Alonso A, Garcia-Cortes M, Garcia-Ruiz E, Benitez R, Fernandez MC, Pelaez G, Romero M, Corpas R, Duran JA, Jimenez M, Rodrigo L, Nogueras F, Martin-Vivaldi R, Navarro JM, Salmeron J, de la Cuesta FS, Hidalgo R: HLA class II genotype influences the type of liver injury in drug-induced idiosyncratic liver disease. Hepatology. 2004, 39: 1603-1612. 10.1002/hep.20215.
Donaldson PT, Daly AK, Henderson J, Graham J, Pirmohamed M, Bernal W, Day CP, Aithal GP: Human leucocyte antigen class II genotype in susceptibility and resistance to co-amoxiclav-induced liver injury. J Hepatol. 2010, 53: 1049-1053. 10.1016/j.jhep.2010.05.033.
Kindmark A, Jawaid A, Harbron CG, Barratt BJ, Bengtsson OF, Andersson TB, Carlsson S, Cederbrant KE, Gibson NJ, Armstrong M, Lagerstrom-Fermer ME, Dellsen A, Brown EM, Thornton M, Dukes C, Jenkins SC, Firth MA, Harrod GO, Pinel TH, Billing-Clason SM, Cardon LR, March RE: Genome-wide pharmacogenetic investigation of a hepatic adverse event without clinical signs of immunopathology suggests an underlying immune pathogenesis. Pharmacogenomics J. 2008, 8: 186-195. 10.1038/sj.tpj.6500458.
Hirata K, Takagi H, Yamamoto M, Matsumoto T, Nishiya T, Mori K, Shimizu S, Masumoto H, Okutani Y: Ticlopidine-induced hepatotoxicity is associated with specific human leukocyte antigen genomic subtypes in Japanese patients: a preliminary case-control study. Pharmacogenomics J. 2008, 8: 29-33. 10.1038/sj.tpj.6500442.
Singer JB, Lewitzky S, Leroy E, Yang F, Zhao X, Klickstein L, Wright TM, Meyer J, Paulding CA: A genome-wide study identifies HLA alleles associated with lumiracoxib-related liver injury. Nat Genet. 2010, 42: 711-714. 10.1038/ng.632.
Spraggs CF, Budde LR, Briley LP, Bing N, Cox CJ, King KS, Whittaker JC, Mooser VE, Preston AJ, Stein SH, Cardon LR: HLA-DQA1*02:01 is a major risk factor for lapatinib-induced hepatotoxicity in women with advanced breast cancer. J Clin Oncol. 2011, 29: 667-673. 10.1200/JCO.2010.31.3197.
Martin AM, Nolan D, James I, Cameron P, Keller J, Moore C, Phillips E, Christiansen FT, Mallal S: Predisposition to nevirapine hypersensitivity associated with HLA-DRB1*0101 and abrogated by low CD4 T-cell counts. Aids. 2005, 19: 97-99. 10.1097/00002030-200501030-00014.
Huang YS, Chern HD, Su WJ, Wu JC, Lai SL, Yang SY, Chang FY, Lee SD: Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002, 35: 883-889. 10.1053/jhep.2002.32102.
Ohno M, Yamaguchi I, Yamamoto I, Fukuda T, Yokota S, Maekura R, Ito M, Yamamoto Y, Ogura T, Maeda K, Komuta K, Igarashi T, Azuma J: Slow N-acetyltransferase 2 genotype affects the incidence of isoniazid and rifampicin-induced hepatotoxicity. Int J Tuberc Lung Dis. 2000, 4: 256-261.
Cho HJ, Koh WJ, Ryu YJ, Ki CS, Nam MH, Kim JW, Lee SY: Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis (Edinb). 2007, 87: 551-556. 10.1016/j.tube.2007.05.012.
Possuelo LG, Castelan JA, de Brito TC, Ribeiro AW, Cafrune PI, Picon PD, Santos AR, Teixeira RL, Gregianini TS, Hutz MH, Rossetti ML, Zaha A: Association of slow N-acetyltransferase 2 profile and anti-TB drug-induced hepatotoxicity in patients from Southern Brazil. Eur J Clin Pharmacol. 2008, 64: 673-681. 10.1007/s00228-008-0484-8.
Bozok Cetintas V, Erer OF, Kosova B, Ozdemir I, Topcuoglu N, Aktogu S, Eroglu Z: Determining the relation between N-acetyltransferase-2 acetylator phenotype and antituberculosis drug induced hepatitis by molecular biologic tests. Tuberk Toraks. 2008, 56: 81-86.
Kim SH, Kim SH, Bahn JW, Kim YK, Chang YS, Shin ES, Kim YS, Park JS, Kim BH, Jang IJ, Song J, Kim SH, Park HS, Min KU, Jee YK: Genetic polymorphisms of drug-metabolizing enzymes and anti-TB drug-induced hepatitis. Pharmacogenomics. 2009, 10: 1767-1779. 10.2217/pgs.09.100.
Yamada S, Tang M, Richardson K, Halaschek-Wiener J, Chan M, Cook VJ, Fitzgerald JM, Elwood RK, Brooks-Wilson A, Marra F: Genetic variations of NAT2 and CYP2E1 and isoniazid hepatotoxicity in a diverse population. Pharmacogenomics. 2009, 10: 1433-1445. 10.2217/pgs.09.66.
Lee SW, Chung LS, Huang HH, Chuang TY, Liou YH, Wu LS: NAT2 and CYP2E1 polymorphisms and susceptibility to first-line anti-tuberculosis drug-induced hepatitis. Int J Tuberc Lung Dis. 2010, 14: 622-626.
Ben Mahmoud L, Ghozzi H, Kamoun A, Hakim A, Hachicha H, Hammami S, Sahnoun Z, Zalila N, Makni H, Zeghal K: Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatotoxicity in Tunisian patients with tuberculosis. Pathol Biol (Paris). 2012, 60: 324-330. 10.1016/j.patbio.2011.07.001.
Bose PD, Sarma MP, Medhi S, Das BC, Husain SA, Kar P: Role of polymorphic N-acetyl transferase2 and cytochrome P4502E1 gene in antituberculosis treatment-induced hepatitis. J Gastroenterol Hepatol. 2011, 26: 312-318. 10.1111/j.1440-1746.2010.06355.x.
Sotsuka T, Sasaki Y, Hirai S, Yamagishi F, Ueno K: Association of isoniazid-metabolizing enzyme genotypes and isoniazid-induced hepatotoxicity in tuberculosis patients. Vivo. 2011, 25: 803-812.
Aithal GP, Ramsay L, Daly AK, Sonchit N, Leathart JB, Alexander G, Kenna JG, Caldwell J, Day CP: Hepatic adducts, circulating antibodies, and cytokine polymorphisms in patients with diclofenac hepatotoxicity. Hepatology. 2004, 39: 1430-1440. 10.1002/hep.20205.
Carr DF, Alfirevic A, Tugwood JD, Barratt BJ, Sherwood J, Smith J, Pirmohamed M, Park BK: Molecular and genetic association of interleukin-6 in tacrine-induced hepatotoxicity. Pharmacogenet Genomics. 2007, 17: 961-972. 10.1097/FPC.0b013e3282f00919.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that they have no competing interests.
Rights and permissions
About this article
Cite this article
Daly, A.K. Pharmacogenomics of adverse drug reactions. Genome Med 5, 5 (2013). https://doi.org/10.1186/gm409
Published:
DOI: https://doi.org/10.1186/gm409