
Abstract
Pharmacogenetic research aims to study how genetic variation may influence drug efficacy and/or toxicity; pharmacogenomics expands this quest to the entire genome. Pharmacogenetic findings may help to uncover new drug targets, illuminate pathophysiology, clarify disease heterogeneity, aid in the fine-mapping of genetic associations, and contribute to personalized treatment. In diabetes, there is precedent for the successful application of pharmacogenetic concepts to monogenic forms of the disease, such as maturity onset diabetes of the young or neonatal diabetes. Whether similar insights will be produced for the common form of type 2 diabetes remains to be seen. With recent advances in genetic approaches, the successive application of candidate gene studies, large-scale genotyping studies and genome-wide association studies has begun to generate suggestive results that may lead to changes in clinical practice. However, many potential barriers to the translation of pharmacogenetic discoveries to the clinical management of diabetes still remain. Here, we offer a contemporary overview of the field in its current state, identify potential obstacles, and highlight future directions.
Similar content being viewed by others

Pharmacogenetics, pharmacogenomics and diabetes
There is great variability in drug response, whether measured in terms of efficacy or toxicity. Part of this variability is due to direct biological factors that influence the bioavailability and effects of a drug on its target, and part is due to indirect factors (psychological or social) related to patient adherence, access to care, and physician prescribing practices. With regard to direct biological factors, the efficacy and toxicity of a medication are determined by the balance between its pharmacokinetics (absorption, distribution, metabolism and elimination of the drug) and its pharmacodynamics (the physiological effect of the active drug at the site of biological action). Inherited genetic variation contributes to this variability, as multiple genes encode proteins directly involved in the pharmacokinetics and pharmacodynamics of drugs. Pharmacogenetic research aims to investigate the association of inherited genetic variants with response to drug therapy, including drug efficacy or adverse effects. Pharmacogenomics brings this search for genetic determinants of drug response to the scope of the entire genome.
As systematic queries of the human genome become more feasible on a large scale, a number of advantages for pharmacogenetic investigation become evident (Table 1). First, by casting an agnostic net in search of genetic determinants of drug response, pharmacogenomics can uncover novel molecular targets for commonly used drugs, which serve to illuminate previously unsuspected mechanisms of action. Second, insofar as such mechanisms point to specific cellular pathways, this approach has the potential to illuminate the pathophysiology of the disease affected by the drug. Third, in so doing, a more nuanced understanding of the processes that give rise to a pathological state can help refine the nosology of disease, clarifying its heterogeneity and establishing particular subtypes. Fourth, pharmacogenetic experiments can help demonstrate that one of several genes within a genomic region previously associated with the trait does indeed cause the disease process: by perturbing a live human with a drug that targets that gene and assessing their response to the perturbation, the investigator may be able to 'close the loop' and demonstrate that the associated gene is indeed involved in producing the phenotype of interest. And fifth, cataloguing the genetic determinants of drug response can guide the personalized choice of therapy based on both safety and efficacy. Examples abound: such personalized therapy is now the standard of care for azathioprine [1, 2] and abacavir [3] to avoid drug-related complications, and for certain forms of cancer to achieve better outcomes [4–7]. Safety and efficacy concerns both suggest that similar precautions may need to be taken for other drugs with a narrow therapeutic index (where the therapeutic dose is close to the dose that produces adverse effects), such as warfarin [8–10] and clopidogrel [11–14].
As far as diabetes is concerned, pharmacogenetic approaches have already had a substantial impact on rare, monogenic forms of diabetes. Maturity onset diabetes of the young (MODY) is characterized by onset at young age, autosomal dominant transmission and a non-ketotic presentation in typically, but not always, non-obese people. It is caused by mutations in the genes that encode glucokinase and several transcription factors relevant to pancreatic β-cell development [15]. Patients with transcription factor MODY respond better to sulfonylureas than to metformin [16], illustrating the pathophysiological mechanisms centered in the β cell (where insulin secretagogues such as sulfonylureas act) for this form of the disease. Similarly, neonatal diabetes, diagnosed by the onset of hyperglycemia within the first 6 months of life, is caused by activating mutations in the islet ATP-sensitive potassium channel Kir6.2 (encoded by KCNJ11) or its associated sulfonylurea receptor SUR1 (encoded by ABCC8), which interfere with the ability of the β cell to respond to a glucose load. These patients can be effectively treated by high-dose sulfonylureas [17–20], allowing children mistakenly diagnosed with type 1 diabetes (based on the early onset of disease and undetectable C peptide) to be safely transitioned from multiple daily insulin injections to an oral agent once the correct genetic diagnosis is made.
Whether pharmacogenetics will have an analogous impact on common type 2 diabetes (T2D), the subject of this review, remains to be seen. T2D is one of the leading causes of cardiovascular disease, microvascular complications and death in the USA and worldwide. Its incidence has been rising steadily over the past few decades, and is predicted to reach epidemic proportions in developing nations. Despite this obvious environmental contribution to the disease, it is now recognized that T2D pathogenesis is driven by multiple genetic factors interacting with a metabolically deleterious environment [21]. In recent years, well-powered candidate gene studies and genome-wide association studies (GWASs) have uncovered over 40 genomic loci that are associated with T2D at genome-wide levels of statistical significance (P < 5 × 10-8, determined empirically to account for the number of independent hypotheses among common variants in the European genome [22]). However, these associations simply point to areas of the genome that are overrepresented in cases of T2D when compared with non-diabetic controls; in most cases, the exact identity of the culprit gene and the causal variant remain unknown [23].
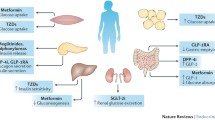
Many oral anti-diabetes medications have been developed and used in clinical practice for years, but some of their biological mechanisms are not completely understood. Figure 1 illustrates the tissues targeted by current anti-diabetes medications. Pharmacogenetics has proven invaluable in guiding therapeutic choices in MODY and neonatal diabetes, and its extension to common T2D is now beginning to take place. In the past 10 years, three major approaches for pharmacogenetic discovery have evolved in parallel with technological development: candidate gene studies opened the way to large-scale genotyping studies, which were followed by GWASs. In the early stages of genetic investigation only common variation in candidate genes could be realistically examined, because of limited efficiency in genotyping and imperfect knowledge of the human genome and its patterns of variation. As large-scale genotyping based on a comprehensive haplotype map of the human genome became available for deployment in larger samples, GWASs became a powerful research tool enabling the transition from pharmacogenetics to pharmacogenomics in T2D. Here, we review the most current pharmacogenetic evidence in T2D based on these different approaches, confining our remarks to the studies and drug classes (sulfonylureas, metformin, and thiazolidinediones) that have gathered the most conclusive evidence in this regard. For a comprehensive list of smaller studies and ongoing clinical trials, see a recent systematic review [24].

Proposed tissue targets of the major classes of anti-diabetes medications. For details, see Table 2. Although the primary tissue targets are shown, many of these drugs have multi-organ effects. The dotted line denotes putative mechanisms that remain to be fully demonstrated in humans. DPP-IV, dipeptidyl peptidase-IV; GI, gastrointestinal tract; GLP-1, glucagon-like peptide-1; TZDs, thiazolidinediones.
Candidate genes
These studies focus on a few plausible candidate genes involved in drug pharmacokinetics/pharmacodynamics, or those associated with T2D as a disease phenotype. Here we focus on sulfonylureas, metformin and thiazolidinediones.
Sulfonylureas
Cytochrome P450 2C9 (CYP2C9) is the rate-limiting enzyme in the metabolism of many sulfonylureas, such as glipizide, glimepiride and tolbutamide. For glyburide, although CYP3A4 contributes more than 50% of the formation of total metabolites, CYP2C9 also contributes 30% of the formation of metabolites. The wild-type allele of the CYP2C9 gene is CYP2C9*1; the two allelic variants CYP2C9*2 (rs1799853) and CYP2C9*3 (rs1057910) encode the loss-of-function missense amino acid polymorphisms *2 (Arg144Cys) and *3 (Ile359Leu), respectively. Pharmacokinetic analysis of glyburide was performed in 21 healthy volunteers with all 6 combinations of the CYP2C9 alleles *1, *2 and *3. Homozygous carriers of the CYP2C9*3/*3 genotypes had reduced clearance of glyburide and increased insulin secretion 12 hours after glyburide ingestion [25], a finding that was confirmed by others [26, 27]. In Japanese T2D patients, those with CYP2C9*1/*3 genotypes had significantly elevated plasma concentrations of glimepiride and a greater reduction in glycated hemoglobin (HbA1C) than those with CYP2C9*1/*1 [28]. Much larger studies have been made possible by the compilation of prescription information and clinical outcomes from electronic medical records, focused on patients with T2D who have also consented to donate a DNA sample. Using this retrospective approach, the GoDARTS investigators in Tayside, Scotland, examined 1,073 incident users of sulfonylureas: patients with two copies of the *2 or *3 alleles were 3.4 times more likely to achieve treatment targets (HbA1C levels under 7%) than patients with two wild-type CYP2C9 alleles [29].
Sulfonylureas bind to the pancreatic β-cell sulfonylurea receptor SUR1 coupled to the ATP-dependent potassium channel Kir6.2, causing channel closure and triggering insulin secretion in a glucose-independent manner. As mentioned above, activating mutations in their respective genes, ABCC8 and KCNJ11 [17, 20], cause neonatal diabetes mellitus. The K (Lys) allele at the common Glu23Lys polymorphism in KCNJ11 has been shown to be associated with increased risk of T2D by several large studies [30–33]. Interestingly, KCNJ11 and ABCC8 lie next to each other on chromosome 11, and are separated by only 5 kb: a large region of linkage disequilibrium imposes strong correlation among several variants across both genes [33]. Another missense polymorphism, Ala1369Ser in ABCC8, is highly correlated with KCNJ11 Glu23Lys in all populations examined, such that any association signal at KCNJ11 Glu23Lys is genetically indistinguishable from ABCC8 Ala1369Ser [33, 34]: in other words, carriers of the risk K allele at Glu23Lys almost always carry the A (Ala) allele at Ala1369Ser. Functional studies have tried to establish which of the two missense variants is causal: in recent elegant work, Hamming and colleagues [35] have demonstrated that the A allele at Ala1369Ser is responsible for the increased responsiveness to gliclazide shown by mutated channels in vitro.
In one early report, diabetic carriers of the risk K allele at KCNJ11 Glu23Lys were found to have a higher risk for secondary sulfonylurea failure, which was defined as fasting plasma glucose greater than 300 mg/dl despite sulfonylurea treatment followed by sulfonylurea-metformin combined therapy [36]. This finding, which could also be interpreted as genetically driven diabetes progression, received some support from a smaller independent study [37]. However, in a much larger prospective study of 1,268 Chinese patients with T2D, carriers of the risk A allele at ABCC8 Ala1369Ser showed improved short-term response to gliclazide, in a manner consistent with the functional work described above and with the results obtained in neonatal diabetes [38]. These two disparate observations might be reconciled if the increased initial responsiveness to sulfonylureas shown by carriers of the risk allele evolves into β-cell exhaustion and earlier sulfonylurea failure, but such hypotheses require longer term follow-up.
Polymorphisms in the transcription factor 7-like 2 gene (TCF7L2) harbor the strongest association with T2D reported to date, which is present in nearly all ethnic groups [39, 40]. Fine-mapping and functional work have shown that the intronic rs7903146 is the likely cause of the association signal [41, 42]. Carriers of the risk allele at this locus show diminished β-cell function [43, 44]. Therefore, it is of great interest to verify whether sulfonylurea therapy works differently depending on genotype at TCF7L2. This hypothesis was also tested in GoDARTS: individuals with the T2D-associated homozygous TT genotype were less likely to respond to sulfonylurea therapy and reach the treatment target of HbA1C under 7% [45]. No such effect was seen for metformin, where genotype at TCF7L2 did not make a difference. Consistent results have been published recently in two independent central European cohorts [46, 47]. The contrast between TCF7L2 (where the T2D risk genotype is associated with a weaker response to sulfonylureas) and KCNJ11/ABCC8 (in which the genotypes that increase risk of T2D are associated with a stronger response) illustrates that disease association does not necessarily predict the direction of pharmacogenetic impact: whether it is beneficial or harmful may depend on the points along the relevant physiological pathway at which the gene and the drug exert their respective effects.
Metformin
Metformin is a safe and effective first-line biguanide agent in T2D therapy [48–51]. It improves insulin sensitivity, reduces hepatic gluconeogenesis and causes modest weight loss [52]. Although it has been shown to activate the cellular fuel sensor AMP-dependent kinase (AMPK) [53, 54], other mechanisms independent of AMPK activity have also been proposed [55]. Its pharmacokinetics involves two key processes in humans: the organic cation transporters OCT1 and OCT2 (encoded by SLC22A1 and SLC22A2, respectively) mediate metformin transmembrane transport into hepatocytes and renal tubular cells, respectively; the multidrug and toxin extrusion protein MATE1 (encoded by SLC47A1) facilitates excretion of unchanged metformin into urine and bile. Nonsynonymous polymorphisms in SLC22A1 have been found to be associated with different plasma concentration of metformin in small European and Asian cohorts [56, 57]; the key results in Europeans were not replicated in the retrospective but much larger GoDARTS cohort [58]. A smaller retrospective study based on clinical records has also been established in Rotterdam: Becker et al. [59] found that the non-coding genetic variant rs622342 in SLC22A1 is associated with changes in HbA1C levels after metformin treatment, a finding that awaits replication. Also in Rotterdam, the rs2289669 non-coding polymorphism in SLC47A1 was associated with metformin response: a 0.30% HbA1C reduction was reported per minor A allele compared with the G allele [60]. This result has been reproduced in the Diabetes Prevention Program (DPP; see below) [61].
Thiazolidinediones
Thiazolidinediones increase glucose uptake by skeletal muscle, enhance lipolysis and suppress hepatic glucose output by enhancing the binding of the peroxisome proliferator-activated receptor γ (PPARγ) to its target DNA response element. The missense mutation in its gene PPARG that causes a proline to alanine change at codon 12 of the protein has been consistently associated with protection from T2D [62]. However, this polymorphism does not predict changes in fasting glucose, HbA1C levels or insulin sensitivity after treatment with a variety of thiazolidinedione agents [63–65]. Putative associations of other variants in this gene with response to troglitazone [66] have not been replicated [65].
Other genes have been investigated for association with thiazolidinedione response. Kang et al. [67] found that two variants in the adiponectin gene (ADIPOQ) were associated with changes in fasting glucose and HbA1C levels after 12 weeks of rosiglitazone treatment. Genetic variation in CYP2C8 was found to be associated with altered clearance rate of rosiglitazone [68]. These results require confirmation.
Large-scale genotyping studies
As genotyping platforms accommodated higher throughput, investigators could focus on hundreds or thousands of variants simultaneously, either selected from the literature or attempting to capture common variation in candidate genes comprehensively. Following this approach, the DPP investigators studied the association of 1,590 single nucleotide polymorphisms (SNPs) with metformin response, defined as its ability to prevent or delay the onset of diabetes, in 2,994 DPP participants who were at high risk of T2D [61]. These SNPs in 40 genes were selected because they are either associated with T2D according to early GWASs, encode drug-metabolizing/transporting enzymes, or were involved in relevant physiological processes. A variant in strong linkage disequilibrium with the polymorphism in the metformin transporter gene SLC47A1 reported by the Rotterdam group [60] (see above) was associated with metformin response. A number of other loci showed suggestive associations, but none of them reached statistical significance after correction for the number of hypotheses tested.
The TRoglitazone In the Prevention Of Diabetes (TRIPOD) study was a single-center, randomized, and double-blinded clinical trial aiming to investigate the effect of troglitazone on T2D incidence in Hispanic women with prior gestational diabetes. Out of 93 women who received troglitazone, 63 had improvement in insulin sensitivity and 30 did not. The TRIPOD investigators sequenced 40 kb of PPARG and 133 SNPs were identified in the PPARG region. Eight of 133 SNPs were found to be nominally associated with improvement in insulin sensitivity, but not with change in fasting glucose [66]. However, these associations were not corrected for multiple testing nor substantiated in the DPP [65].
Genome-wide association studies
The first GWAS for glycemic response to any anti-diabetic drug was conducted by a collaboration formed by GoDARTS, the UK Prospective Diabetes Study (UKPDS) clinical trial [50] and the Wellcome Trust Case-Control Consortium [69] and focused on metformin. The discovery GWAS cohort included 1,024 individuals from Tayside, Scotland; follow-up cohorts included an additional 1,783 GoDARTS participants and 1,113 UKPDS participants. The first cohort underwent genome-wide genotyping, and 14 SNPs showing a suggestive association with metformin response as a categorical trait (defined as achieving HbA1C ≤ 7%) at P < 10-6 were taken forward into the other two cohorts. These SNPs concentrated around the Ataxia-telangiectasia mutated (ATM) gene in chromosome 11q22, and showed consistent associations with metformin response as a quantitative trait. During the replication attempt, the minor A allele at rs11212617 was again associated with increased metformin response in both cohorts, and achieved conventional genome-wide statistical significance on meta-analysis (P = 2.9 × 10-9). In functional experiments, the authors further showed that the selective ATM inhibitor KU-55933 inhibited metformin activation of AMPK in rat hepatoma cells as well as phosphorylation of AMPK and a downstream target, implicating ATM itself as the gene responsible for the association signal [69]. Minor limitations of this landmark study include the relatively small sample size for a GWAS, the somewhat arbitrary definition of clinical response, and the unexplained connection between the SNP itself and the effects seen in the in vitro assays.
Translating pharmacogenetic information into clinical practice
Promise
Pharmacogenetic research in T2D has already advanced our understanding of the pathophysiology of hyperglycemia, by highlighting the β cell as a nodal point in its pathogenesis. Furthermore, pharmacogenetic investigation has already begun to deliver on the promise of individualized therapy for some monogenic forms of diabetes. Table 2 summarizes our current understanding of the drug targets, clinical responses, and candidate genes involved in the human response to anti-diabetes medications. In T2D, it seems that slower metabolism due to the CYP2C9*2 and *3 polymorphisms can result in improved glycemic control but more severe hypoglycemia, information that may be useful to the practitioner. In addition, although carriers of the risk genotype at KCNJ11/ABCC8 respond better to gliclazide, carriers of the risk genotype at TCF7L2 are worse candidates for sulfonylurea therapy. Finally, extension of the GWAS approach to larger, better powered meta-analyses and other drug classes may reveal new mechanisms of action.
Barriers
However, several obstacles stand in the way of widespread pharmacogenetic applications. First, T2D is a complex disease (or group of diseases), caused by the higher order interaction of many common (and possibly rare) variants among themselves and with the environment. Thus, deciding which of these genetic factors are clinically actionable requires renewed discovery and experimental testing in appropriately designed and rigorously analyzed pharmacogenetic protocols. These trials should not only demonstrate genetic effects, but also show that acting on genetic information before prescribing specific therapies leads to better outcomes and is cost-effective. Second, drug-drug and gene-environment (lifestyle) interactions may override the genetic determinants of medication response: for example, in the DPP an intensive lifestyle intervention benefits all participants regardless of genetic burden [70], and it is possible that higher pharmacological doses may simply overcome the modest resistance induced by genetics. Third, for pre-prescription genotyping to be practical, all clinically actionable variants (not just for T2D or anti-diabetic drugs, but for all common phenotypes and medications) should be placed on a single array that can be processed efficiently and cheaply in Clinical Laboratory Improvement Amendments (CLIA)-certified laboratories, and only once in the lifetime of each patient. This will require the coordination of investigators involved in multiple areas of human health, constant updating, and the participation of manufacturing companies to produce such arrays on a large scale.
Conclusions
In summary, it appears that genetic variation in the cytochrome P450 system affects response to sulfonylureas. Among T2D-associated loci, carriers of the risk allele at TCF7L2 show a poorer response to sulfonylureas, whereas carriers of the risk alleles at the sulfonylurea receptor complex encoded by ABCC8 and KCNJ11 seem to have a stronger response to gliclazide, a finding supported by in vitro data. Variation in the gene encoding the metformin transporter, SLC47A1, may influence the disposal of metformin, and recent results from the first GWAS for metformin response merit independent confirmation.
Although significant progress has been made in T2D pharmacogenetics, the field is still in its infancy. Much work is needed in performing comprehensive assessments of genetic variation across well phenotyped, sufficiently large sample collections, which can typically be attained only in the setting of international collaborations. For future pharmacogenetic research, the pre-competitive participation of pharmaceutical companies, which could contribute DNA samples and outcomes garnered during multiple clinical trials that compare proprietary compounds with generic drugs, may be crucial. Phenotypes should be harmonized and the definition of drug response should have both clinical value and biological relevance. Once bona fide genetic signals are identified, they should be followed up with targeted pharmacogenetic studies that evaluate whether these associations can be modulated by using different dosing regimens or whether the a priori use of genetic information improves patient outcomes.
In conclusion, we stand at a threshold where the question of whether genetic information will influence prescribing practice can be asked in a definitive manner. Because either an affirmative or a negative answer would be useful, the question must be asked. We do hope the use of individual genetic information can help guide intelligent medication choices in the future: public and private funding bodies should support clinical trials with large sample sizes in an effort to show improved outcomes and cost effectiveness before this promise can be delivered to clinical practice.
Abbreviations
- AMPK:
-
AMP-dependent kinase
- ATM :
-
Ataxia-telangiectasia mutated
- CYP:
-
cytochrome P450
- DPP:
-
Diabetes Prevention Program
- GWAS:
-
genome-wide association study
- HbA1C:
-
glycated hemoglobin
- MODY:
-
maturity onset diabetes of the young
- PPARG :
-
peroxisome proliferator-activated receptor γ
- SNP:
-
single nucleotide polymorphism
- SUR1:
-
sulfonylurea receptor 1
- T2D:
-
type 2 diabetes
- TCF7L2 :
-
transcription factor 7-like 2 gene
- TRIPOD:
-
TRoglitazone In the Prevention Of Diabetes
- UKPDS:
-
United Kingdom Prospective Diabetes Study.
References
Ford LT, Berg JD: Thiopurine S-methyltransferase (TPMT) assessment prior to starting thiopurine drug treatment; a pharmacogenomic test whose time has come. J Clin Pathol. 2010, 63: 288-295. 10.1136/jcp.2009.069252.
Relling MV, Gardner EE, Sandborn WJ, Schmiegelow K, Pui CH, Yee SW, Stein CM, Carrillo M, Evans WE, Klein TE: Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011, 89: 387-391. 10.1038/clpt.2010.320.
Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, Jägel-Guedes E, Rugina S, Kozyrev O, Cid JF, Hay P, Nolan D, Hughes S, Hughes A, Ryan S, Fitch N, Thorborn D, Benbow A: HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008, 358: 568-579. 10.1056/NEJMoa0706135.
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M: EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004, 304: 1497-1500. 10.1126/science.1099314.
Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA: Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004, 350: 2129-2139. 10.1056/NEJMoa040938.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001, 344: 783-792. 10.1056/NEJM200103153441101.
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA: Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011, 364: 2507-2516. 10.1056/NEJMoa1103782.
Ginsburg GS, Voora D: The long and winding road to warfarin pharmacogenetic testing. J Am Coll Cardiol. 2010, 55: 2813-2815. 10.1016/j.jacc.2010.04.006.
Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, Limdi NA, Page D, Roden DM, Wagner MJ, Caldwell MD, Johnson JA: Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009, 360: 753-764.
Manolopoulos VG, Ragia G, Tavridou A: Pharmacogenetics of coumarinic oral anticoagulants. Pharmacogenomics. 2010, 11: 493-496. 10.2217/pgs.10.31.
Mega JL, Close SL, Wiviott SD, Shen L, Hockett RD, Brandt JT, Walker JR, Antman EM, Macias W, Braunwald E, Sabatine MS: Cytochrome p-450 polymorphisms and response to clopidogrel. N Engl J Med. 2009, 360: 354-362. 10.1056/NEJMoa0809171.
Pare G, Mehta SR, Yusuf S, Anand SS, Connolly SJ, Hirsh J, Simonsen K, Bhatt DL, Fox KA, Eikelboom JW: Effects of CYP2C19 genotype on outcomes of clopidogrel treatment. N Engl J Med. 2010, 363: 1704-1714. 10.1056/NEJMoa1008410.
Mega JL, Simon T, Collet JP, Anderson JL, Antman EM, Bliden K, Cannon CP, Danchin N, Giusti B, Gurbel P, Horne BD, Hulot JS, Kastrati A, Montalescot G, Neumann FJ, Shen L, Sibbing D, Steg PG, Trenk D, Wiviott SD, Sabatine MS: Reduced-function CYP2C19 genotype and risk of adverse clinical outcomes among patients treated with clopidogrel predominantly for PCI: a meta-analysis. JAMA. 2010, 304: 1821-1830. 10.1001/jama.2010.1543.
Roden DM, Shuldiner AR: Responding to the clopidogrel warning by the US Food and Drug Administration: real life is complicated. Circulation. 2010, 122: 445-448. 10.1161/CIRCULATIONAHA.110.973362.
Bell G, Polonsky KS: Diabetes mellitus and genetically programmed defects in beta-cell function. Nature. 2001, 414: 788-791. 10.1038/414788a.
Pearson ER, Starkey BJ, Powell RJ, Gribble FM, Clark PM, Hattersley AT: Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet. 2003, 362: 1275-1281. 10.1016/S0140-6736(03)14571-0.
Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njølstad PR, Ashcroft FM, Hattersley AT: Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004, 350: 1838-1849. 10.1056/NEJMoa032922.
Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, Abuelo D, Phornphutkul C, Molnes J, Bell GI, Gloyn AL, Hattersley AT, Molven A, Søvik O, Njølstad PR: Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: Patient characteristics and initial response to sulfonylurea therapy. Diabetes. 2004, 53: 2713-2718. 10.2337/diabetes.53.10.2713.
Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, Slingerland AS, Shield J, Robert JJ, Holst JJ, Clark PM, Ellard S, Søvik O, Polak M, Hattersley AT: Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006, 355: 467-477. 10.1056/NEJMoa061759.
Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P: Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006, 355: 456-466. 10.1056/NEJMoa055068.
McCarthy MI: Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010, 363: 2339-2350. 10.1056/NEJMra0906948.
Pe'er I, Yelensky R, Altshuler D, Daly MJ: Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet Epidemiol. 2008, 32: 381-385. 10.1002/gepi.20303.
Billings LK, Florez JC: The genetics of type 2 diabetes: what have we learned from GWAS?. Ann N Y Acad Sci. 2010, 1212: 59-77. 10.1111/j.1749-6632.2010.05838.x.
Manolopoulos VG, Ragia G, Tavridou A: Pharmacogenomics of oral antidiabetic medications: current data and pharmacoepigenomic perspective. Pharmacogenomics. 2011, 12: 1161-1191. 10.2217/pgs.11.65.
Kirchheiner J, Brockm-Iller J, Meineke I, Bauer S, Rohde W, Meisel C, Roots I: Impact of CYP2C9 amino acid polymorphisms on glyburide kinetics and on the insulin and glucose response in healthy volunteers. Clin Pharmacol Ther. 2002, 71: 286-296. 10.1067/mcp.2002.122476.
Jetter A, Kinzig-Schippers M, Skott A, Lazar A, Tomalik-Scharte D, Kirchheiner J, Walchner-Bonjean M, Hering U, Jakob V, Rodamer M, Jabrane W, Kasel D, Brockmöller J, Fuhr U, Sörgel F: Cytochrome P450 2C9 phenotyping using low-dose tolbutamide. Eur J Clin Pharmacol. 2004, 60: 165-171. 10.1007/s00228-004-0754-z.
Niemi M, Cascorbi I, Timm R, Kroemer HK, Neuvonen PJ, Kivistö KT: Glyburide and glimepiride pharmacokinetics in subjects with different CYP2C9 genotypes. Clin Pharmacol Ther. 2002, 72: 326-332. 10.1067/mcp.2002.127495.
Suzuki K, Yanagawa T, Shibasaki T, Kaniwa N, Hasegawa R, Tohkin M: Effect of CYP2C9 genetic polymorphisms on the efficacy and pharmacokinetics of glimepiride in subjects with type 2 diabetes. Diabetes Res Clin Pract. 2006, 72: 148-154. 10.1016/j.diabres.2005.09.019.
Zhou K, Donnelly L, Burch L, Tavendale R, Doney AS, Leese G, Hattersley AT, McCarthy MI, Morris AD, Lang CC, Palmer CN, Pearson ER: Loss-of-function CYP2C9 variants improve therapeutic response to sulfonylureas in type 2 diabetes: a Go-DARTS study. Clin Pharmacol Ther. 2010, 87: 52-56. 10.1038/clpt.2009.176.
Gloyn AL, Hashim Y, Ashcroft SJ, Ashfield R, Wiltshire S, Turner RC: Association studies of variants in promoter and coding regions of beta-cell ATP-sensitive K-channel genes SUR1 and Kir6.2 with Type 2 diabetes mellitus (UKPDS 53). Diabet Med. 2001, 18: 206-212. 10.1046/j.1464-5491.2001.00449.x.
Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM: Large-scale association studies of variants in genes encoding the pancreatic β-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003, 52: 568-572.
Barroso I, Luan J, Middelberg RPS, Harding A-H, Franks PW, Jakes RW, Clayton D, Schafer AJ, O'Rahilly S, Wareham NJ: Candidate gene association study in type 2 diabetes indicates a role for genes involved in β-cell function as well as insulin action. PLoS Biol. 2003, 1: e20-10.1371/journal.pbio.0000020.
Florez JC, Burtt N, de Bakker PI, Almgren P, Tuomi T, Holmkvist J, Gaudet D, Hudson TJ, Schaffner SF, Daly MJ, Hirschhorn JN, Groop L, Altshuler D: Haplotype structure and genotype-phenotype correlations of the sulfonylurea receptor and the islet ATP-sensitive potassium channel gene region. Diabetes. 2004, 53: 1360-1368. 10.2337/diabetes.53.5.1360.
Inoue H, Ferrer J, Warren-Perry M, Zhang Y, Millns H, Turner RC, Elbein SC, Hampe CL, Suarez BK, Inagaki N, Seino S, Permutt MA: Sequence variants in the pancreatic islet beta-cell inwardly rectifying K+ channel Kir6.2 (Bir) gene: identification and lack of role in Caucasian patients with NIDDM. Diabetes. 1997, 46: 502-507. 10.2337/diabetes.46.3.502.
Hamming KS, Soliman D, Matemisz LC, Niazi O, Lang Y, Gloyn AL, Light PE: Coexpression of the type 2 diabetes susceptibility gene variants KCNJ11 E23K and ABCC8 S1369A alter the ATP and sulfonylurea sensitivities of the ATP-sensitive K(+) channel. Diabetes. 2009, 58: 2419-2424. 10.2337/db09-0143.
Sesti G, Laratta E, Cardellini M, Andreozzi F, Del Guerra S, Irace C, Gnasso A, Grupillo M, Lauro R, Hribal ML, Perticone F, Marchetti P: The E23K variant of KCNJ11 encoding the pancreatic b-cell adenosine 5'-triphosphate-sensitive potassium channel subunit Kir6.2 is associated with an increased risk of secondary failure to sulfonylurea in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006, 91: 2334-2339. 10.1210/jc.2005-2323.
Holstein A, Hahn M, Stumvoll M, Kovacs P: The E23K variant of KCNJ11 and the risk for severe sulfonylurea-induced hypoglycemia in patients with type 2 diabetes. Horm Metab Res. 2009, 41: 387-390. 10.1055/s-0029-1192019.
Feng Y, Mao G, Ren X, Xing H, Tang G, Li Q, Li X, Sun L, Yang J, Ma W, Wang X, Xu X: Ser1369Ala variant in sulfonylurea receptor gene ABCC8 is associated with antidiabetic efficacy of gliclazide in Chinese type 2 diabetic patients. Diabetes Care. 2008, 31: 1939-1944. 10.2337/dc07-2248.
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K: Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006, 38: 320-323. 10.1038/ng1732.
Cauchi S, El Achhab Y, Choquet H, Dina C, Krempler F, Weitgasser R, Nejjari C, Patsch W, Chikri M, Meyre D, Froguel P: TCF7L2 is reproducibly associated with type 2 diabetes in various ethnic groups: a global meta-analysis. J Mol Med. 2007, 85: 777-782. 10.1007/s00109-007-0203-4.
Helgason A, Pálsson S, Thorleifsson G, Grant SF, Emilsson V, Gunnarsdottir S, Adeyemo A, Chen Y, Chen G, Reynisdottir I, Benediktsson R, Hinney A, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Schäfer H, Faruque M, Doumatey A, Zhou J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Sigurdsson G, Hebebrand J, Pedersen O, Thorsteinsdottir U, Gulcher JR, et al: Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet. 2007, 39: 218-225. 10.1038/ng1960.
Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, Berney T, Montanya E, Mohlke KL, Lieb JD, Ferrer J: A map of open chromatin in human pancreatic islets. Nat Genet. 2010, 42: 255-259. 10.1038/ng.530.
Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PIW, Shuldiner AR, Knowler WC, Nathan DM, Altshuler D, The diabetes Prevention Program Research Group: TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med. 2006, 355: 241-250. 10.1056/NEJMoa062418.
Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P, Sjögren M, Ling C, Eriksson KF, Lethagen AL, Mancarella R, Berglund G, Tuomi T, Nilsson P, Del Prato S, Groop L: Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007, 117: 2155-2163. 10.1172/JCI30706.
Pearson ER, Donnelly LA, Kimber C, Whitley A, Doney ASF, McCarthy MI, Hattersley AT, Morris AD, Palmer CNA: Variation in TCF7L2 influences therapeutic response to sulfonylureas: A GoDARTs study. Diabetes. 2007, 56: 2178-2182. 10.2337/db07-0440.
Holstein A, Hahn M, Korner A, Stumvoll M, Kovacs P: TCF7L2 and therapeutic response to sulfonylureas in patients with type 2 diabetes. BMC Med Genet. 2011, 12: 30-
Schroner Z, Javorsky M, Tkacova R, Klimcakova L, Dobrikova M, Habalova V, Kozarova M, Zidzik J, Rudikova M, Tkac I: Effect of sulphonylurea treatment on glycaemic control is related to TCF7L2 genotype in patients with type 2 diabetes. Diabetes Obes Metab. 2011, 13: 89-91.
DeFronzo RA, Goodman AM: Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. The Multicenter Metformin Study Group. N Engl J Med. 1995, 333: 541-549. 10.1056/NEJM199508313330902.
Garber AJ, Duncan TG, Goodman AM, Mills DJ, Rohlf JL: Efficacy of metformin in type II diabetes: Results of a double-blind, placebo-controlled, dose-response trial. Am J Med. 1997, 103: 491-497. 10.1016/S0002-9343(97)00254-4.
UKPDS: Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998, 352: 854-865.
Ong CR, Molyneaux LM, Constantino MI, Twigg SM, Yue DK: Long-term efficacy of metformin therapy in nonobese individuals with type 2 diabetes. Diabetes Care. 2006, 29: 2361-2364. 10.2337/dc06-0827.
Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE: Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995, 333: 550-554. 10.1056/NEJM199508313330903.
Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE: Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001, 108: 1167-1174.
Fryer LGD, Parbu-Patel A, Carling D: The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002, 277: 25226-25232. 10.1074/jbc.M202489200.
Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F, Viollet B: Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010, 120: 2355-2369. 10.1172/JCI40671.
Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, Giacomini KM: Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008, 83: 273-280. 10.1038/sj.clpt.6100275.
Chen L, Takizawa M, Chen E, Schlessinger A, Segenthelar J, Choi JH, Sali A, Kubo M, Nakamura S, Iwamoto Y, Iwasaki N, Giacomini KM: Genetic polymorphisms in organic cation transporter 1 (OCT1) in Chinese and Japanese populations exhibit altered function. J Pharmacol Exp Ther. 2010, 335: 42-50. 10.1124/jpet.110.170159.
Zhou K, Donnelly LA, Kimber CH, Donnan PT, Doney AS, Leese G, Hattersley AT, McCarthy MI, Morris AD, Palmer CN, Pearson ER: Reduced function SLC22A1 polymorphisms encoding Organic Cation Transporter 1 (OCT1) and glycaemic response to metformin: A Go-DARTS study. Diabetes. 2009, 58: 1434-1439. 10.2337/db08-0896.
Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH: Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J. 2009, 9: 242-247. 10.1038/tpj.2009.15.
Becker ML, Visser LE, van Schaik RHN, Hofman A, Uitterlinden AG, Stricker BHC: Genetic variation in the multidrug and toxin extrusion 1 transporter protein influences the glucose-lowering effect of metformin in patients with diabetes: A preliminary study. Diabetes. 2009, 58: 745-749.
Jablonski KA, McAteer JB, de Bakker PI, Franks PW, Pollin TI, Hanson RL, Saxena R, Fowler S, Shuldiner AR, Knowler WC, Altshuler D, Florez JC: Common variants in 40 genes assessed for diabetes incidence and response to metformin and lifestyle intervention in the diabetes prevention program. Diabetes. 2010, 59: 2672-2681. 10.2337/db10-0543.
Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES: The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000, 26: 76-80. 10.1038/79216.
Bluher M, Lubben G, Paschke R: Analysis of the relationship between the Pro12Ala variant in the PPAR-γ2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care. 2003, 26: 825-831. 10.2337/diacare.26.3.825.
Snitker S, Watanabe RM, Ani I, Xiang AH, Marroquin A, Ochoa C, Goico J, Shuldiner AR, Buchanan TA: Changes in insulin sensitivity in response to troglitazone do not differ between subjects with and without the common, functional Pro12Ala peroxisome proliferator-activated receptor-γ2 gene variant: results from the Troglitazone in Prevention of Diabetes (TRIPOD) study. Diabetes Care. 2004, 27: 1365-1368. 10.2337/diacare.27.6.1365.
Florez JC, Jablonski KA, Sun MW, Bayley N, Kahn SE, Shamoon H, Hamman RF, Knowler WC, Nathan DM, Altshuler D, The Diabetes Prevention Program Research Group: Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab. 2007, 92: 1502-1509. 10.1210/jc.2006-2275.
Wolford JK, Yeatts KA, Dhanjal SK, Black MH, Xiang AH, Buchanan TA, Watanabe RM: Sequence variation in PPARG may underlie differential response to troglitazone. Diabetes. 2005, 54: 3319-3325. 10.2337/diabetes.54.11.3319.
Kang ES, Park SY, Kim HJ, Kim CS, Ahn CW, Cha BS, Lim SK, Nam CM, Lee HC: Effects of Pro12Ala polymorphism of peroxisome proliferator-activated receptor γ2 gene on rosiglitazone response in type 2 diabetes. Clin Pharmacol Ther. 2005, 78: 202-208. 10.1016/j.clpt.2005.04.013.
Kirchheiner J, Thomas S, Bauer S, Tomalik-Scharte D, Hering U, Doroshyenko O, Jetter A, Stehle S, Tsahuridu M, Meineke I, Brockmöller J, Fuhr U: Pharmacokinetics and pharmacodynamics of rosiglitazone in relation to CYP2C8 genotype. Clin Pharmacol Ther. 2006, 80: 657-667. 10.1016/j.clpt.2006.09.008.
Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, Donnelly LA, Schofield C, Groves CJ, Burch L, Carr F, Strange A, Freeman C, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Craddock N, Deloukas P, Dronov S, Duncanson A, Edkins S, Gray E, Hunt S, Jankowski J, Langford C, Markus HS, Mathew CG, et al: Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011, 43: 117-120. 10.1038/ng.735.
Hivert MF, Jablonski KA, Perreault L, Saxena R, McAteer JB, Franks PW, Hamman RF, Kahn SE, Haffner S, Meigs JB, Altshuler D, Knowler WC, Florez JC: Updated genetic score based on 34 confirmed type 2 diabetes loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes. 2011, 60: 1340-1348. 10.2337/db10-1119.
Bailey CJ, Turner RC: Metformin. N Engl J Med. 1996, 334: 574-579. 10.1056/NEJM199602293340906.
Inzucchi SE: Oral antihyperglycemic therapy for type 2 diabetes: scientific review. JAMA. 2002, 287: 360-372. 10.1001/jama.287.3.360.
Yki-Jarvinen H: Thiazolidinediones. N Engl J Med. 2004, 351: 1106-1118. 10.1056/NEJMra041001.
Drucker DJ, Nauck MA: The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006, 368: 1696-1705. 10.1016/S0140-6736(06)69705-5.
Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, Zinman B: Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2009, 32: 193-203. 10.2337/dc08-9025.
Nathan DM, Buse JB, Davidson MB, Ferrannini E, Holman RR, Sherwin R, Zinman B: Medical management of hyperglycaemia in type 2 diabetes mellitus: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2009, 52: 17-30. 10.1007/s00125-008-1157-y.
Rodbard HW, Jellinger PS, Davidson JA, Einhorn D, Garber AJ, Grunberger G, Handelsman Y, Horton ES, Lebovitz H, Levy P, Moghissi ES, Schwartz SS: Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract. 2009, 15: 540-559.
Acknowledgements
JCF is supported by NIH/NIDDK R01 DK072041 and R01 DK088214 and a Clinician Scientist Development Award granted by the Doris Duke Charitable Foundation. CH is supported by NIH training grant 5 T32 DK007540-25.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JCF has received consulting honoraria from Daiichi-Sankyo and AstraZeneca.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Huang, C., Florez, J.C. Pharmacogenetics in type 2 diabetes: potential implications for clinical practice. Genome Med 3, 76 (2011). https://doi.org/10.1186/gm292
Published:
DOI: https://doi.org/10.1186/gm292