
Abstract
Epilepsy is one of the most common neurological disorders, with a prevalence of 1% and lifetime incidence of 3%. There are numerous epilepsy syndromes, most of which are considered to be genetic epilepsies. Despite the discovery of more than 20 genes for epilepsy to date, much of the genetic contribution to epilepsy is not yet known. Copy number variants have been established as an important source of mutation in other complex brain disorders, including intellectual disability, autism and schizophrenia. Recent advances in technology now facilitate genome-wide searches for copy number variants and are beginning to be applied to epilepsy. Here, we discuss what is currently known about the contribution of copy number variants to epilepsy, and how that knowledge is redefining classification of clinical and genetic syndromes.
Similar content being viewed by others


Overview of clinical types and the genetics of epilepsy
The International League Against Epilepsy defines an epileptic seizure as 'a transient occurrence of signs and/or symptoms due to abnormal excessive or synchronous neuronal activity in the brain' [1, 2]. The condition is common, with prevalence around 1% and lifetime incidence around 3% [3]. Most epilepsies can be broadly and easily classified based on their pattern of electroclinical onset as either generalized ('originating at some point from within, and rapidly engaging, bilaterally distributed networks') or focal ('originating within networks limited to one hemisphere') [1]. Within each of these broad classifications are multiple distinct syndromes, more than half of which are considered to be 'genetic epilepsies'. In older terminology, genetic epilepsies were referred to as 'idiopathic epilepsies' [4]. Syndromes, and sometimes subsyndromes, are delineated when the seizures are defined by easily recognizable electroclinical features and similar enough to be regarded as a homogeneous group, distinct from other groups in the same classification level (Table 1). For example, genetic generalized epilepsies are frequently divided into their subsyndromes of childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy and generalized tonic clonic seizures.
There is a subset of epilepsy syndromes that are clearly monogenic, and traditional linkage studies in large families have been useful for identifying causative genes [5, 6]. However, the vast majority of the genetic epilepsies are multifactorial, with an underlying genetic contribution that is polygenic, where few or usually none of the susceptibility genes have been identified. This multifactorial concept dates back to the early works of William Lennox [7] and was well established in the modern era with additional twin data [8]. It is important to note that epilepsy with complex genetics and complex epilepsy are distinct concepts. To the geneticist, complex epilepsy is epilepsy with complex genetics; that is, multifactorial epilepsy that is polygenic and influenced by environmental effects, both internal and external. Complex epilepsy to the epileptologist, on the other hand, refers to the complexity of the seizure pattern. Without an appreciation of the difference, interactions between basic and clinical scientists can be, and have been from personal experience, confused by 'complex epilepsy' meaning different things to different people. In the context of this article, complex epilepsy will mean that which is multifactorial in origin, rather than necessarily having complex seizure patterns.
Monogenic epilepsies
To date, more than 20 genes have been identified for the group of genetic epilepsies that are primarily monogenic [5, 6, 9, 10], prompting a recent update of clinically based classification [1]. While individual syndromes that comprise each of these groups are generally diagnosed through clinical assessment, molecular testing now facilitates more accurate definition of clinically similar disorders that are now known to be caused by mutation of different genes. While gene identity provides an alternative or additional criterion for syndrome classification, it also has clinical efficacy - providing a rapid definitive diagnosis to obviate an otherwise circuitous set of invasive or costly investigative procedures. Furthermore, in some cases, specific therapeutic intervention can be enabled to achieve improved outcomes or more accurate prognosis. Genetic testing for the epilepsies has high clinical utility in cases that may involve SLC2A1 (glucose transporter type 1 deficiency), SCN1A (Dravet syndrome), PCDH19 (familial epilepsy and mental retardation limited to females, 'Dravet-like' PCDH19 syndrome), ARX (X-linked infantile spasms and myoclonic seizures, dystonia, and X-linked lissencephaly with ambiguous genitalia) or STK9 (X-linked infantile spasms) mutations. Testing has high analytical sensitivity (ability to detect the presence of a causative mutation) and high analytical specificity (ability to exclude mutation in a candidate gene) for all of the monogenic epilepsies, but not necessarily high clinical utility apart from some of the syndromes associated with the above genes [9]. It has little or no clinical utility at this time when knowledge of the gene is not needed for accurate syndrome classification, when knowledge of the gene does not direct or affect treatment, or in cases of genetically complex epilepsies triggered by the combined effects of multiple genes spread across the genome, most likely each having only a small effect on phenotype.
Complex epilepsies
Speculation of the genetic architecture for the genetically complex epilepsies centers on the common disease-common variant hypothesis [11] and the common disease-rare variant hypothesis [12]. The general failure of linkage and association studies applied to the complex epilepsies [13–16] argues against the common disease-common variant hypothesis, although the major criticism of such studies is that they are underpowered to detect the magnitude of odds ratios that are likely associated with susceptibility variants in the genetically complex epilepsies [17] and indeed other neuropsychiatric brain disorders.
The common disease-rare variant hypothesis, which suggests a variable subset of multiple rare genetic variants, has greater appeal for complex epilepsy [18, 19], especially given the failure of association studies, which work on the premise of the common disease-common variant hypothesis [16], to deliver consistent findings. A mixture of the two models is also entirely plausible [19] with functional differences in the electrophysiological properties of ion channels demonstrated for both rare and polymorphic genetic variation detected at the GABRD (encoding γ-aminobutyric acid A receptor, δ), CACNA1H (encoding calcium channel, voltage-dependent, T type, α 1 H subunit) and CLCN2 (encoding chloride channel 2) genes [20–23], for example. Computer simulation supports the notion that genetic variations associated with only very small functional changes in ion channel properties are sufficient to make meaningful contributions to increasing susceptibility to epilepsy [24].
Multiple sclerosis is another disorder with complex inheritance where extensive study suggests 'risk variants likely to include hundreds of modest effects and possibly thousands of very small effects' [25]. Similar conclusions with systematic effects of multiple rare variants across the genome have been suggested for schizophrenia and bipolar disorder [26]. We predict the same for epilepsy with complex inheritance, with seizure susceptibility thresholds determined by combinations of many rare to moderately common sequence variants, copy number variants (CNVs) and perhaps non-coding DNA sequences with functional effects. Weak effects will only be detectable by genome-wide association studies using massive sample sizes. Kryukov et al. [27] pre-empted outcomes from deep resequencing by massively parallel sequencing (previously referred to as next-generation sequencing [28]) by promoting an association study approach based on the premise of multiple rare variants present in susceptibility genes in higher numbers for a given disease group (for example, epilepsy) than in their corresponding controls. The statistical tools to support that approach are now surfacing [29].
The heritability of genetic generalized epilepsy suggests a major genetic component [8] but virtually none has yet been identified. This constitutes the 'dark matter' [30]. The task is to find this missing heritability and characterize it in terms of number of loci, effect sizes, allelic frequencies of variants and the nature of the variants [31]. Areas being investigated include cis-acting genome-wide regulatory variants [32], genome-wide copy number variants [33, 34] as discussed below, and, in the future, next-generation sequencing [28].
Copy number variation in epilepsy
CNVs are deletions, duplications or insertions of DNA in the genome that range in size from approximately 1 kb to several megabases. Many CNVs have no apparent clinical significance, and numerous studies have now established that CNVs are dispersed throughout the genomes of healthy individuals and some CNVs are quite common [35–37]. Importantly, CNVs have also been identified as a significant source of mutation. Small CNVs may result in the deletion or duplication of one or more exons of a known disease gene, and there are now many examples in the literature. In patients with intellectual disability (ID) or developmental delay, testing for large CNVs is now commonplace, as large CNVs underlie 15% to 20% of cases of ID [38, 39]. CNVs can be detected by targeted studies directed to specific known CNVs by techniques such as multiplex ligation-dependent probe amplification (MLPA). In the epilepsies, MLPA is generally targeted to exons of known epilepsy genes to detect intragenic deletions or duplications [40–45], some of which are too small to be detected by genome-wide approaches.
Genome-wide methods to detect CNVs include array-comparative genomic hybridization (array-CGH) and SNP genotyping arrays. These technologies can be targeted to specific chromosomal regions [43, 45–49]. However, their real power lies with capability for genome-wide interrogation, where there is no need for a priori knowledge of where a lesion may lie [33, 34, 46, 50]. Using that approach, Depienne et al. [46] discovered a Dravet-like syndrome caused by severe PCDH19 mutations on chromosome X, and McMahon et al. [50] 'rediscovered' the 15q13.3 CNV and found a novel 10q21.2 microduplication. Mefford et al. [33] and Heinzen et al. [34] used genome-wide approaches to establish the extent of rare CNVs in the genetic epilepsies (see below). For CNVs with boundaries extending beyond the target gene, array-CGH is a powerful tool for accurately determining size and gene content. Large epilepsy-associated CNVs detectable by MLPA, but extending well beyond the one gene of special interest (for example, beyond SCN1A), can also be reliably detected by array technologies [40, 43, 45].
The role of CNVs in epilepsy has now been addressed by several groups using both targeted and genome-wide approaches. Helbig and colleagues [51] first directed our attention to the role of the 15q13.3 microdeletion in the etiology of epilepsy. This microdeletion was first described in a series of patients with ID, most of whom also suffered from seizures [52], but is much more common in epilepsy cohorts [51, 53, 54]. This is one of the most prevalent genetic risk factors identified for the genetic generalized epilepsy syndromes. A range of rare mutations within SLC2A1 encoding the GLUT1 glucose transporter are at least as important within the childhood absence epilepsy subsyndrome of genetic generalized epilepsy [55, 56]. Although estimated confidence intervals are broad, the estimated odds risk ratio of 68 (95% confidence interval 29 to 181) for the 15q13.3 deletion [54] greatly exceeds that of most common susceptibility variants detectable by genome-wide association studies in disorders other than epilepsy. Despite its relative 'severity' in relation to risk, its frequency in epilepsy cohorts is relatively high at around 1.3%. Conversely, this variant is difficult to find in the general control population, despite the screening of large numbers of controls, even though family studies following detection of an index case disclose frequent transmissions from non-penetrant carrier parents [54, 57]. Moreover, the position of the original mutation in the pedigree is often not too far back into its living ancestry, suggesting a relatively high recurrent mutation rate. Of the seven genes within the lesion, haploinsufficiency of CHRNA7 (nicotinic acetylcholine receptor, α7) is considered to be the most likely pathogenic element, although it is not the only neuronally expressed gene affected by the deletion. Interestingly, early genome-wide linkage studies implicated the CHRNA7 region in juvenile myoclonic epilepsy [58], but this could not be replicated [59], and screening of CHRNA7 did not detect convincing mutations [60]. Could it be that the families studied by Elmslie et al. [58] contained enough families segregating the 15q13.3 microdeletion to give a linkage signal?
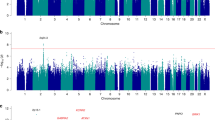
Subsequent studies investigated the role of other large CNVs that had previously been associated with increased risk of ID, autism and schizophrenia [53]. Somewhat surprisingly, significant numbers of the same recurrent CNVs involved in the disorders listed above were implicated as a component of the polygenic pathogenic genetic architecture in the clinically and genetically complex (idiopathic) epilepsies. Two microdeletions commonly associated with epilepsy are at 15q11.2 and 16p13.11 [33, 34, 53]. Together with the 15q13.3 microdeletion, their combined frequency in test populations of genetic generalized epilepsy is approximately 3% [33]. Other large recurrent CNVs associated with ID, autism or schizophrenia that have also been detected in epilepsy are at 1q21.1, 16p12, 22q11 and two regions within 16p11.2 [33, 53]. These CNVs represent clearly defined genetic determinants that overlap with a number of hitherto regarded distinct disorders comprising part or all of their genetic architectures. The three most common recurrent CNVs, which together account for up to 3% of epilepsies, are shown in Figure 1. Notably, the 15q13.3 microdeletion has been consistently present in 0.5% to 1% of all genetic generalized epilepsy cohorts but has not been seen in >3,000 patients who presented with focal epilepsy syndromes [34], and therefore it may be a risk factor specifically for generalized epilepsy syndromes. Deletions at 16p13.11 and 15q11.2 have been found in both generalized and focal epilepsies [33, 34, 53].

Three 'common' recurrent microdeletions in epilepsy. Microdeletion of 15q13.3 (1.5 Mb) in a patient with absence epilepsy. Microdeletion of 16p13.11 (800 kb) in a patient with juvenile myoclonic epilepsy. Microdeletion of 15q11.2 (350 kb) in a patient with infantile seizures. Regions depicted for each panel are as follows: 15q13.3 deletion: chr15, 28.0 to 31.0 Mb; 16p13.11 deletion: chr16, 15.0 to 16.7 Mb; and 15q11.2 deletion: chr15, 20.2 to 20.8 Mb (National Center for Biotechnology Information Build 36/hg18). Red vertical lines represent array-comparative genomic hybridization probes that are deleted. Segmental duplications are represented by orange, yellow and gray blocks. Note that blocks of segmental duplications flank each deleted region. Genes are represented in blue, with key proposed candidate genes in red.
The large, recurrent CNVs described above occur because of specific genomic architecture at each respective chromosome region. CNV is mediated by naturally occurring sets of low copy repeats or segmental duplications [61–63] that facilitate non-allelic homologous recombination [64, 65], resulting in deletion or duplication of the intervening unique sequence. Therefore, each region with such architecture is prone to rearrangement at meiosis, causing recurrence of large CNVs with nearly identical breakpoints in unrelated individuals. Because CNVs at these rearrangement-prone regions of the genome occur with an appreciable frequency, it has been possible to detect a statistically significant difference between cases and controls.
Apart from the recurrent CNVs discussed above, the rare non-recurrent CNVs are also likely to play a significant role in the genetic etiology of epilepsy. Two recent studies applied genome-wide technologies to detect CNVs in affected individuals. Heinzen and colleagues [34] evaluated 3,812 individuals and found an enrichment of large (>1 Mb) deletions in affected individuals, the majority of which were seen in one individual each. Mefford et al. [33] evaluated 517 individuals with various types of epilepsy and found that nearly 10% carried one or more rare CNVs that had not been previously found at an appreciable frequency in controls. Again, the majority of events were seen only once, and represent a subset of the rare non-recurrent CNVs involving genes that have been implicated in ID, autism or schizophrenia.
Syndrome constellations associated with CNVs
Taken literally, a constellation is a number of stars grouped within an outline. Here, we regard the CNV as the 'outline' encompassing a group of its associated syndromes comprising the syndrome constellation. Different combinations of syndromes define the constellations that are packaged within different CNVs. The CNVs can be recurrent in the population, and any recurrent CNV located in a given region is virtually identical from patient to patient. The syndrome constellations include one or more types of ID, dysmorphism, autism, schizophrenia and, more recently, genetic generalized epilepsy. The various syndromes within the constellations are themselves genetically and phenotypically heterogeneous, and in some cases have defined subsyndromes. For example, genetic generalized epilepsy consists of the subsyndromes childhood absence epilepsy, juvenile absence epilepsy, juvenile myoclonic epilepsy and generalized tonic clonic seizures. Recurrent deletions at 15q13.3 (1.5 Mb, seven genes), at 16p13.11 (1.2 Mb, eight genes) and at 15q11.2 (1.3 Mb, four genes) are emerging as the most common genetic determinants for various distinct disorders with complex inheritance. These generally include intellectual disability with or without dysmorphism, autism, schizophrenia or genetic generalized or focal epilepsy. Epilepsy was the latest addition to the constellations of syndromes associated with each of these CNVs, and is now well established [33, 34, 51, 53, 54]. A similar picture is emerging for the rarer recurrent CNVs at 1q21.1, 16p12 and two regions within 16p11.2 [33, 53].
Given the comorbidity of ID and epilepsy, autism and ID, and autism and epilepsy, for example, perhaps it should not be surprising that some CNVs cause overlapping neuropsychiatric features in affected individuals. However, it seems remarkable that the same CNV susceptibility lesion can be a genetic determinant for apparently disparate conditions (for example, only epilepsy in one patient, only schizophrenia in another). One possible explanation might be that odds risk ratios associated with disorders included within a given constellation of syndromes is relatively high in the context of disorders with complex inheritance. For example, genetic generalized epilepsy has an odds risk ratio of 68 (95% confidence interval 29 to 181) for the 15q13.3 deletion [54]; this is far higher than for susceptibility variants generally detected in complex genetic disorders. Certainly another possible explanation is the presence of as yet undetected additional genetic or epigenetic variants that influence the phenotypic outcome. All of the 'common' recurrent CNVs in epilepsy (15q13.3, 16p13.11 and 15q11.2) have probably been identified already, given the extent of the array-CGH genome-wide searches already completed [33, 34]. Some of the less common recurrent microdeletions at 1q21.1, 16p12 and two regions within 16p11.2 may be associated with their own multisyndrome constellations.
Rare or unique non-recurrent CNVs are collectively more common than the combined recurrent ones. These lesions provide a wealth of leads to candidate epilepsy genes within or closely adjacent to them. The number, frequency and distribution of each gene-bearing CNV are consistent with the common disease-rare variant model for the genetic architecture for complex epilepsy. Overall genetic profiles of susceptibility genes for each individual are likely to be unique and fit the polygenic heterogeneity concept [18]. Genes within these epilepsy-associated CNVs and genes identified through massively parallel sequencing [66] each represent independent opportunities to break out of the ion channel paradigm that might potentially constrain our thinking when the genetic architecture of epilepsy might extend beyond ion channels. Results of studies performed so far suggest that haploinsufficiency (deletions) or overexpression (duplications) of some of the genes in non-recurrent CNVs may elicit the same syndromes as those in their associated constellations.
There are two common threads in these discussions. First, the constellations of syndromes associated with each recurrent CNV can include a range of diverse phenotypes, including, in most cases, some combination of ID, autism, schizophrenia and epilepsy. Each CNV probably elicits its own specific distribution of phenotypes and frequency of each phenotype, defining the associated constellation. Second, the mechanism for genesis of this extreme clinical heterogeneity observed within virtually identical lesions is not yet known. Several mechanistic possibilities have been outlined [34, 67–69] but none has been proven as a general mechanism, or even a mechanism specific to any given CNV. The clinical heterogeneity is likely to depend upon the nature of the other risk factors or genetic modifiers in the rest of the genome that alone or in combination may specify the phenotype.
Conclusions and future perspectives
The concept of extensive clinical heterogeneity in epilepsy associated with a well-defined genetic lesion is not new. Well known examples are genetic generalized epilepsy with febrile seizures plus [19], caused by mutations in sodium channel genes, and recently, genetic generalized epilepsy caused by the 15q13.3 CNV [70]. These observations have challenged complete reliance on the phenotype-first approach to diagnosis. Investigations will always begin with general clinical evaluation to broadly classify cases into disease categories. Taking genetic generalized epilepsy as an example, is it then necessary to further refine down to subsyndromes using clinical criteria alone, and to even contemplate endophenotyping for deeper clinical refinement? The answer is clearly no in the context of syndromic constellations associated with some CNVs and phenotypic spectrums associated with some familial missense mutations. The aim of that exercise of making phenotypes as clinically homogeneous as possible would be to promote genetic homogenization of study populations so that associations are easier to detect. But for CNVs and missense mutations in some genes, collections of the same CNV or same mutation are already genetically homogeneous, at least for that component of the complex polygenic architecture.
The approach needs to be turned upside down, by adoption of a genotype-first approach where novel genomic disorders such as genetic generalized epilepsy are classified and defined by detection of a common deletion or duplication. The collection of large numbers of patients with the same CNV genotype but wide variety of phenotypes including epilepsy will facilitate genotype-phenotype studies that might provide insight into the mechanisms that influence phenotype diversity in these and other disorders. Conversely, the collection of large numbers of genetic generalized epilepsy patients (not even subtyped into subsyndromes) with significantly more multiple rare DNA sequence changes within the same putative epilepsy susceptibility gene, as compared with unaffected controls, might be an outcome of their pursuit through massively parallel sequencing. That would enable us to work backwards, to endophenotype just those cases with mutations in a defined susceptibility gene to see if they have subtle phenotypic features in common. Thus might emerge a subsyndrome classification that is different to that currently in use, based on more relevant components of the phenotype that better reflect the underlying molecular genetics.
Finally, we agree that careful clinical phenotyping is a vital component of our research, as the constellations associated with each of the CNVs need to be accurately characterized. Consider cohorts comprising 15q13.3 deletions, for example. Some of the cases are regarded as epilepsy only. Others are regarded as having dual phenotypes, of epilepsy and ID, for example. Are these really dual phenotypes? Consider the hypothetical possibility that the haploid content of the 15q13.3 region lowers the seizure threshold and adversely affects intelligence in everyone who carries it. Some carriers will not have epilepsy because their susceptibility profile contains too few susceptibility variants at other loci throughout the genome, in addition to 15q13.3, to take them across the seizure threshold. Some carriers will not have ID because their baseline intelligence quotient will be high enough to begin with that even with some depression of intelligence quotient through the effects of the 15q13.3 deletion they remain within the normal range. Others, toward the lower end of the normal range to begin with, unfortunately drop down into the ID range. We challenge the clinical researchers to prove us wrong or, like us, seriously question the notion of dual phenotypes presenting in only a subset of the 15q13.3 deletion carriers.
Abbreviations
- Array-CGH:
-
array-comparative genomic hybridization
- CNV:
-
copy number variant
- ID:
-
intellectual disability
- MLPA:
-
multiplex ligation-dependent probe amplification
- SNP:
-
single nucleotide polymorphism.
References
Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, Moshe SL, Nordli D, Plouin P, Scheffer IE: Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia. 2010, 51: 676-685. 10.1111/j.1528-1167.2010.02522.x.
Fisher RS, van Emde Boas W, Blume W, Elger C, Genton P, Lee P, Engel J: Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia. 2005, 46: 470-472. 10.1111/j.0013-9580.2005.66104.x.
Hauser WA, Annegers JF, Rocca WA: Descriptive epidemiology of epilepsy: contributions of population-based studies from Rochester, Minnesota. Mayo Clin Proc. 1996, 71: 576-586. 10.4065/71.6.576.
Annegers JF: The epidemiology of epilepsy. The Treatment of Epilepsy: Principles and Practice. Edited by: Wylie E. 1996, Baltimore: Williams and Wilkins
Helbig I, Scheffer IE, Mulley JC, Berkovic SF: Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol. 2008, 7: 231-245. 10.1016/S1474-4422(08)70039-5.
Heron SE, Scheffer IE, Berkovic SF, Dibbens LM, Mulley JC: Channelopathies in idiopathic epilepsy. Neurotherapeutics. 2007, 4: 295-304. 10.1016/j.nurt.2007.01.009.
Vadlamudi L, Andermann E, Lombroso CT, Schachter SC, Milne RL, Hopper JL, Andermann F, Berkovic SF: Epilepsy in twins: insights from unique historical data of William Lennox. Neurology. 2004, 62: 1127-1133.
Berkovic SF, Howell RA, Hay DA, Hopper JL: Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998, 43: 435-445. 10.1002/ana.410430405.
Ottman R, Annegers JF, Hauser WA, Kurland LT: Seizure risk in offspring of parents with generalized versus partial epilepsy. Epilepsia. 1989, 30: 157-161. 10.1111/j.1528-1157.1989.tb05448.x.
Reid CA, Berkovic SF, Petrou S: Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009, 87: 41-57. 10.1016/j.pneurobio.2008.09.016.
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN: Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet. 2003, 33: 177-182. 10.1038/ng1071.
Pritchard JK, Cox NJ: The allelic architecture of human disease genes: common disease-common variant...or not?. Hum Mol Genet. 2002, 11: 2417-2423. 10.1093/hmg/11.20.2417.
Cavalleri GL, Walley NM, Soranzo N, Mulley J, Doherty CP, Kapoor A, Depondt C, Lynch JM, Scheffer IE, Heils A, Gehrmann A, Kinirons P, Gandhi S, Satishchandra P, Wood NW, Anand A, Sander T, Berkovic SF, Delanty N, Goldstein DB, Sisodiya SM: A multicenter study of BRD2 as a risk factor for juvenile myoclonic epilepsy. Epilepsia. 2007, 48: 706-712. 10.1111/j.1528-1167.2007.00977.x.
Cavalleri GL, Weale ME, Shianna KV, Singh R, Lynch JM, Grinton B, Szoeke C, Murphy K, Kinirons P, O'Rourke D, Ge D, Depondt C, Claeys KG, Pandolfo M, Gumbs C, Walley N, McNamara J, Mulley JC, Linney KN, Sheffield LJ, Radtke RA, Tate SK, Chissoe SL, Gibson RA, Hosford D, Stanton A, Graves TD, Hanna MG, Eriksson K, Kantanen AM, et al: Multicentre search for genetic susceptibility loci in sporadic epilepsy syndrome and seizure types: a case-control study. Lancet Neurol. 2007, 6: 970-980. 10.1016/S1474-4422(07)70247-8.
Hempelmann A, Taylor KP, Heils A, Lorenz S, Prud'homme JF, Nabbout R, Dulac O, Rudolf G, Zara F, Bianchi A, Robinson R, Gardiner RM, Covanis A, Lindhout D, Stephani U, Elger CE, Weber YG, Lerche H, Nurnberg P, Kron KL, Scheffer IE, Mulley JC, Berkovic SF, Sander T: Exploration of the genetic architecture of idiopathic generalized epilepsies. Epilepsia. 2006, 47: 1682-1690. 10.1111/j.1528-1167.2006.00677.x.
Tan NC, Mulley JC, Berkovic SF: Genetic association studies in epilepsy: 'the truth is out there'. Epilepsia. 2004, 45: 1429-1442. 10.1111/j.0013-9580.2004.22904.x.
Tan NC, Berkovic SF: The Epilepsy Genetic Association Database (epiGAD): analysis of 165 genetic association studies, 1996-2008. Epilepsia. 2010, 51: 686-689. 10.1111/j.1528-1167.2009.02423.x.
Dibbens LM, Heron SE, Mulley JC: A polygenic heterogeneity model for common epilepsies with complex genetics. Genes Brain Behav. 2007, 6: 593-597. 10.1111/j.1601-183X.2007.00333.x.
Mulley JC, Scheffer IE, Harkin LA, Berkovic SF, Dibbens LM: Susceptibility genes for complex epilepsy. Hum Mol Genet. 2005, 14 (Spec No 2): R243-249. 10.1093/hmg/ddi355.
Dibbens LM, Feng HJ, Richards MC, Harkin LA, Hodgson BL, Scott D, Jenkins M, Petrou S, Sutherland GR, Scheffer IE, Berkovic SF, Macdonald RL, Mulley JC: GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum Mol Genet. 2004, 13: 1315-1319. 10.1093/hmg/ddh146.
Feng HJ, Kang JQ, Song L, Dibbens L, Mulley J, Macdonald RL: Delta subunit susceptibility variants E177A and R220H associated with complex epilepsy alter channel gating and surface expression of α4β2δ GABAA receptors. J Neurosci. 2006, 26: 1499-1506. 10.1523/JNEUROSCI.2913-05.2006.
Heron SE, Khosravani H, Varela D, Bladen C, Williams TC, Newman MR, Scheffer IE, Berkovic SF, Mulley JC, Zamponi GW: Extended spectrum of idiopathic generalized epilepsies associated with CACNA1H functional variants. Ann Neurol. 2007, 62: 560-568. 10.1002/ana.21169.
Saint-Martin C, Gauvain G, Teodorescu G, Gourfinkel-An I, Fedirko E, Weber YG, Maljevic S, Ernst JP, Garcia-Olivares J, Fahlke C, Nabbout R, LeGuern E, Lerche H, Poncer JC, Depienne C: Two novel CLCN2 mutations accelerating chloride channel deactivation are associated with idiopathic generalized epilepsy. Hum Mutat. 2009, 30: 397-405. 10.1002/humu.20876.
Thomas EA, Reid CA, Berkovic SF, Petrou S: Prediction by modeling that epilepsy may be caused by very small functional changes in ion channels. Arch Neurol. 2009, 66: 1225-1232. 10.1001/archneurol.2009.219.
Bush WS, Sawcer SJ, de Jager PL, Oksenberg JR, McCauley JL, Pericak-Vance MA, Haines JL: Evidence for polygenic susceptibility to multiple sclerosis - the shape of things to come. Am J Hum Genet. 2010, 86: 621-625. 10.1016/j.ajhg.2010.02.027.
Purcell SM, Wray NR, Stone JL, Visscher PM, O'Donovan MC, Sullivan PF, Sklar P: Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009, 460: 748-752.
Kryukov GV, Pennacchio LA, Sunyaev SR: Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007, 80: 727-739. 10.1086/513473.
Mardis ER: The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24: 133-141.
Price AL, Kryukov GV, de Bakker PI, Purcell SM, Staples J, Wei LJ, Sunyaev SR: Pooled association tests for rare variants in exon-resequencing studies. Am J Hum Genet. 2010, 86: 832-838. 10.1016/j.ajhg.2010.04.005.
Maher B: Personal genomes: The case of the missing heritability. Nature. 2008, 456: 18-21. 10.1038/456018a.
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM: Finding the missing heritability of complex diseases. Nature. 2009, 461: 747-753. 10.1038/nature08494.
Sadee W: Measuring cis-acting regulatory variants genome-wide: new insights into expression genetics and disease susceptibility. Genome Med. 2009, 1: 116-10.1186/gm116.
Mefford HC, Muhle H, Ostertag P, von Spiczak S, Buysse K, Baker C, Franke A, Malafosse A, Genton P, Thomas P, Gurnett CA, Schreiber S, Bassuk AG, Guipponi M, Stephani U, Helbig I, Eichler EE: Genome-wide copy number variation in epilepsy: novel susceptibility loci in idiopathic generalized and focal epilepsies. PLoS Genet. 2010, 6: e1000962-10.1371/journal.pgen.1000962.
Heinzen EL, Radtke RA, Urban TJ, Cavalleri GL, Depondt C, Need AC, Walley NM, Nicoletti P, Ge D, Catarino CB, Duncan JS, Kasperaviciute D, Tate SK, Caboclo LO, Sander JW, Clayton L, Linney KN, Shianna KV, Gumbs CE, Smith J, Cronin KD, Maia JM, Doherty CP, Pandolfo M, Leppert D, Middleton LT, Gibson RA, Johnson MR, Matthews PM, Hosford D, et al: Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010, 86: 707-718. 10.1016/j.ajhg.2010.03.018.
Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J, Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME: Origins and functional impact of copy number variation in the human genome. Nature. 2010, 464: 704-712. 10.1038/nature08516.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D, Krauss RM, Myers RM, Ridker PM, Chasman DI, Mefford H, Ying P, Nickerson DA, Eichler EE: Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet. 2009, 84: 148-161. 10.1016/j.ajhg.2008.12.014.
Shaikh TH, Gai X, Perin JC, Glessner JT, Xie H, Murphy K, O'Hara R, Casalunovo T, Conlin LK, D'Arcy M, Frackelton EC, Geiger EA, Haldeman-Englert C, Imielinski M, Kim CE, Medne L, Annaiah K, Bradfield JP, Dabaghyan E, Eckert A, Onyiah CC, Ostapenko S, Otieno FG, Santa E, Shaner JL, Skraban R, Smith RM, Elia J, Goldmuntz E, Spinner NB, et al: High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009, 19: 1682-1690. 10.1101/gr.083501.108.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, et al: Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010, 86: 749-764. 10.1016/j.ajhg.2010.04.006.
Shaffer LG, Kashork CD, Saleki R, Rorem E, Sundin K, Ballif BC, Bejjani BA: Targeted genomic microarray analysis for identification of chromosome abnormalities in 1500 consecutive clinical cases. J Pediatr. 2006, 149: 98-102. 10.1016/j.jpeds.2006.02.006.
Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, Keren B, Abert B, Gautier A, Baulac S, Arzimanoglou A, Cazeneuve C, Nabbout R, LeGuern E: Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009, 46: 183-191. 10.1136/jmg.2008.062323.
Heron SE, Cox K, Grinton BE, Zuberi SM, Kivity S, Afawi Z, Straussberg R, Berkovic SF, Scheffer IE, Mulley JC: Deletions or duplications in KCNQ2 can cause benign familial neonatal seizures. J Med Genet. 2007, 44: 791-796. 10.1136/jmg.2007.051938.
Marini C, Mei D, Temudo T, Ferrari AR, Buti D, Dravet C, Dias AI, Moreira A, Calado E, Seri S, Neville B, Narbona J, Reid E, Michelucci R, Sicca F, Cross HJ, Guerrini R: Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia. 2007, 48: 1678-1685. 10.1111/j.1528-1167.2007.01122.x.
Marini C, Scheffer IE, Nabbout R, Mei D, Cox K, Dibbens LM, McMahon JM, Iona X, Carpintero RS, Elia M, Cilio MR, Specchio N, Giordano L, Striano P, Gennaro E, Cross JH, Kivity S, Neufeld MY, Afawi Z, Andermann E, Keene D, Dulac O, Zara F, Berkovic SF, Guerrini R, Mulley JC: SCN1A duplications and deletions detected in Dravet syndrome: Implications for molecular diagnosis. Epilepsia. 2009, 50: 1670-1678. 10.1111/j.1528-1167.2009.02013.x.
Mulley JC, Nelson P, Guerrero S, Dibbens L, Iona X, McMahon JM, Harkin L, Schouten J, Yu S, Berkovic SF, Scheffer IE: A new molecular mechanism for severe myoclonic epilepsy of infancy: exonic deletions in SCN1A. Neurology. 2006, 67: 1094-1095. 10.1212/01.wnl.0000237322.04338.2b.
Wang JW, Kurahashi H, Ishii A, Kojima T, Ohfu M, Inoue T, Ogawa A, Yasumoto S, Oguni H, Kure S, Fujii T, Ito M, Okuno T, Shirasaka Y, Natsume J, Hasegawa A, Konagaya A, Kaneko S, Hirose S: Microchromosomal deletions involving SCN1A and adjacent genes in severe myoclonic epilepsy in infancy. Epilepsia. 2008, 49: 1528-1534. 10.1111/j.1528-1167.2008.01609.x.
Depienne C, Bouteiller D, Keren B, Cheuret E, Poirier K, Trouillard O, Benyahia B, Quelin C, Carpentier W, Julia S, Afenjar A, Gautier A, Rivier F, Meyer S, Berquin P, Helias M, Py I, Rivera S, Bahi-Buisson N, Gourfinkel-An I, Cazeneuve C, Ruberg M, Brice A, Nabbout R, Leguern E: Sporadic infantile epileptic encephalopathy caused by mutations in PCDH19 resembles Dravet syndrome but mainly affects females. PLoS Genet. 2009, 5: e1000381-10.1371/journal.pgen.1000381.
Heron SE, Scheffer IE, Grinton BE, Eyre H, Oliver KL, Bain S, Berkovic SF, Mulley JC: Familial neonatal seizures with intellectual disability caused by microduplication of chromosome 2q24.3. Epilepsia. 2010, 51: 1865-1869. 10.1111/j.1528-1167.2010.02558.x.
Mei D, Marini C, Novara F, Bernardina BD, Granata T, Fontana E, Parrini E, Ferrari AR, Murgia A, Zuffardi O, Guerrini R: Xp22.3 genomic deletions involving the CDKL5 gene in girls with early onset epileptic encephalopathy. Epilepsia. 2010, 51: 647-654. 10.1111/j.1528-1167.2009.02308.x.
Suls A, Velizarova R, Yordanova I, Deprez L, Van Dyck T, Wauters J, Guergueltcheva V, Claes LR, Kremensky I, Jordanova A, De Jonghe P: Four generations of epilepsy caused by an inherited microdeletion of the SCN1A gene. Neurology. 2010, 75: 72-76. 10.1212/WNL.0b013e3181e62088.
McMahon JM, Scheffer IE, Nicholl JK, Waters W, Eyre H, Hinton L, Nelson P, Yu S, Dibbens LM, Berkovic SF, Mulley JC: Detection of microchromosomal aberrations in refractory epilepsy: a pilot study. Epileptic Disord. 2010, 12: 192-198.
Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Moller RS, et al: 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009, 41: 160-162. 10.1038/ng.292.
Sharp AJ, Mefford HC, Li K, Baker C, Skinner C, Stevenson RE, Schroer RJ, Novara F, De Gregori M, Ciccone R, Broomer A, Casuga I, Wang Y, Xiao C, Barbacioru C, Gimelli G, Bernardina BD, Torniero C, Giorda R, Regan R, Murday V, Mansour S, Fichera M, Castiglia L, Failla P, Ventura M, Jiang Z, Cooper GM, Knight SJ, Romano C, et al: A recurrent 15q13.3 microdeletion syndrome associated with mental retardation and seizures. Nat Genet. 2008, 40: 322-328. 10.1038/ng.93.
de Kovel CG, Trucks H, Helbig I, Mefford HC, Baker C, Leu C, Kluck C, Muhle H, von Spiczak S, Ostertag P, Obermeier T, Kleefuss-Lie AA, Hallmann K, Steffens M, Gaus V, Klein KM, Hamer HM, Rosenow F, Brilstra EH, Kasteleijn-Nolst Trenite D, Swinkels ME, Weber YG, Unterberger I, Zimprich F, Urak L, Feucht M, Fuchs K, Moller RS, Hjalgrim H, De Jonghe P, et al: Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010, 133: 23-32. 10.1093/brain/awp262.
Dibbens LM, Mullen S, Helbig I, Mefford HC, Bayly MA, Bellows S, Leu C, Trucks H, Obermeier T, Wittig M, Franke A, Caglayan H, Yapici Z, Sander T, Eichler EE, Scheffer IE, Mulley JC, Berkovic SF: Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009, 18: 3626-3631. 10.1093/hmg/ddp311.
Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE: Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology. 2010, 75: 432-440. 10.1212/WNL.0b013e3181eb58b4.
Suls A, Mullen SA, Weber YG, Verhaert K, Ceulemans B, Guerrini R, Wuttke TV, Salvo-Vargas A, Deprez L, Claes LR, Jordanova A, Berkovic SF, Lerche H, De Jonghe P, Scheffer IE: Early-onset absence epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol. 2009, 66: 415-419. 10.1002/ana.21724.
van Bon BW, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, Innis JW, de Ravel TJ, Mercer CL, Fichera M, Stewart H, Connell LE, Ounap K, Lachlan K, Castle B, Van der Aa N, van Ravenswaaij C, Nobrega MA, Serra-Juhe C, Simonic I, de Leeuw N, Pfundt R, Bongers EM, Baker C, Finnemore P, Huang S, Maloney VK, Crolla JA, van Kalmthout M, Elia M, et al: Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009, 46: 511-523. 10.1136/jmg.2008.063412.
Elmslie FV, Rees M, Williamson MP, Kerr M, Kjeldsen MJ, Pang KA, Sundqvist A, Friis ML, Chadwick D, Richens A, Covanis A, Santos M, Arzimanoglou A, Panayiotopoulos CP, Curtis D, Whitehouse WP, Gardiner RM: Genetic mapping of a major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q. Hum Mol Genet. 1997, 6: 1329-1334. 10.1093/hmg/6.8.1329.
Sander T, Schulz H, Vieira-Saeker AM, Bianchi A, Sailer U, Bauer G, Scaramelli A, Wienker TF, Saar K, Reis A, Janz D, Epplen JT, Riess O: Evaluation of a putative major susceptibility locus for juvenile myoclonic epilepsy on chromosome 15q14. Am J Med Genet. 1999, 88: 182-187. 10.1002/(SICI)1096-8628(19990416)88:2<182::AID-AJMG16>3.0.CO;2-Z.
Taske NL, Williamson MP, Makoff A, Bate L, Curtis D, Kerr M, Kjeldsen MJ, Pang KA, Sundqvist A, Friis ML, Chadwick D, Richens A, Covanis A, Santos M, Arzimanoglou A, Panayiotopoulos CP, Whitehouse WP, Rees M, Gardiner RM: Evaluation of the positional candidate gene CHRNA7 at the juvenile myoclonic epilepsy locus (EJM2) on chromosome 15q13-14. Epilepsy Res. 2002, 49: 157-172. 10.1016/S0920-1211(02)00027-X.
Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE: Recent segmental duplications in the human genome. Science. 2002, 297: 1003-1007. 10.1126/science.1072047.
Zody MC, Garber M, Sharpe T, Young SK, Rowen L, O'Neill K, Whittaker CA, Kamal M, Chang JL, Cuomo CA, Dewar K, FitzGerald MG, Kodira CD, Madan A, Qin S, Yang X, Abbasi N, Abouelleil A, Arachchi HM, Baradarani L, Birditt B, Bloom S, Bloom T, Borowsky ML, Burke J, Butler J, Cook A, DeArellano K, DeCaprio D, Dorris L, et al: Analysis of the DNA sequence and duplication history of human chromosome 15. Nature. 2006, 440: 671-675. 10.1038/nature04601.
Makoff AJ, Flomen RH: Detailed analysis of 15q11-q14 sequence corrects errors and gaps in the public access sequence to fully reveal large segmental duplications at breakpoints for Prader-Willi, Angelman, and inv dup(15) syndromes. Genome Biol. 2007, 8: R114-10.1186/gb-2007-8-6-r114.
Lupski JR, Stankiewicz P: Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005, 1: e49-10.1371/journal.pgen.0010049.
Pujana MA, Nadal M, Guitart M, Armengol L, Gratacos M, Estivill X: Human chromosome 15q11-q14 regions of rearrangements contain clusters of LCR15 duplicons. Eur J Hum Genet. 2002, 10: 26-35. 10.1038/sj.ejhg.5200760.
Corbett M, Gecz J: Great Expectations: Using massively parallel sequencing to solve inherited disorders. Expert Rev Mol Diag.
Mefford HC: Genotype to phenotype-discovery and characterization of novel genomic disorders in a 'genotype-first' era. Genet Med. 2009, 11: 836-842. 10.1097/GIM.0b013e3181c175d2.
Mulley JC, Dibbens LM: Chipping away at the common epilepsies with complex genetics: the 15q13.3 microdeletion shows the way. Genome Med. 2009, 1: 33-10.1186/gm33.
Sharp AJ: Emerging themes and new challenges in defining the role of structural variation in human disease. Hum Mutat. 2009, 30: 135-144. 10.1002/humu.20843.
Helbig I, Mefford HC, Sharp AJ, Guipponi M, Fichera M, Franke A, Muhle H, de Kovel C, Baker C, von Spiczak S, Kron KL, Steinich I, Kleefuss-Lie AA, Leu C, Gaus V, Schmitz B, Klein KM, Reif PS, Rosenow F, Weber Y, Lerche H, Zimprich F, Urak L, Fuchs K, Feucht M, Genton P, Thomas P, Visscher F, de Haan GJ, Moller RS, et al: 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009, 41: 160-162. 10.1038/ng.292.
Acknowledgements
JCM is supported by SA Pathology within the South Australian Department of Health. HCM is funded by the NIH (NINDS 1R01NS069605) and is a recipient of the Career Award for Medical Scientists from the Burroughs Wellcome Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
HCM drafted sections of the manuscript, edited the draft and supplied the figure. JCM drafted sections of the manuscript and edited the draft. Both authors have read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Mefford, H.C., Mulley, J.C. Genetically complex epilepsies, copy number variants and syndrome constellations. Genome Med 2, 71 (2010). https://doi.org/10.1186/gm192
Published:
DOI: https://doi.org/10.1186/gm192