
Summary
The presenilins are evolutionarily conserved transmembrane proteins that regulate cleavage of certain other proteins in their transmembrane domains. The clinical significance of this regulation is shown by the contribution of presenilin mutations to 20-50% of early-onset cases of inherited Alzheimer's disease. Although the precise molecular mechanism underlying presenilin function or dysfunction remains elusive, presenilins are thought to be part of a complex of proteins that has 'γ-secretase cleavage' activity, which is clearly central in the pathogenesis of Alzheimer's disease. Mutations in presenilins increase the production of the longer isoforms of amyloid β peptide, which are neurotoxic and prone to self-aggregation. Biochemical studies indicate that the presenilins do not act alone but operate within large heteromeric protein complexes, whose components and enzymatic core are the subject of much study and controversy; one essential component is nicastrin. The presenilin primary sequence is remarkably well conserved in eukaryotes, suggesting some functional conservation; indeed, defects caused by mutations in the nemotode presenilin homolog can be rescued by human presenilin.
Similar content being viewed by others
Gene organization and evolution history
The presenilin 1 (PS1) gene on human chromosome 14 (14q24.3) was initially discovered by genetic analysis of a subset of pedigrees in which the Alzheimer's disease is transmitted as a pure autosomal dominant trait [1]. The closely related PS2 gene on chromosome 1 (1q42.2) was identified subsequently by sequence homology [2,3]. Both PS1 and PS2 genes are organized into ten translated exons that display tissue-specific alternative splicing [2,4,5,6,7]. The functions and biological importance of differentially spliced presenilin variants are poorly understood; differential expression of isoforms may lead to differential regulation of the proteolytic processing of the β-amyloid precursor protein (βAPP; see later). For example, aberrant PS2 transcripts lacking exon 5 increase the rate of production of amyloid β peptide (Aβ, the neurotoxic peptide implicated in Alzheimer's disease) [8], whereas naturally occurring isoforms without exons 3 and 4 and/or without exon 8 do not affect production of Aβ [6,9].
GenBank database searches using the full length PS1 sequence suggest that presenilin-like proteins are phylogenetically ancient and well-conserved across diverse eukaryote species, including plants, molluscs, insects, fish, birds, and mammals [10,11,12,13,14,15,16]. Functional conservation of presenilins in most non-human species is undetermined, except in the nematode Caenorhabditis elegans, in which a deficiency in Sel-12, the PS1 homolog, induces an egg-laying defect that can be rescued by expression of human PS1 [17,18]. Additional presenilin homologs were recently identified in disparate eukaryotes by their homology to the PS1 transmembrane domains, suggesting that the presenilin family may be more common than previously contemplated [19,20].
Characteristic structural features
Mammalian PS1 and PS2 are synthesized as 50 kDa polypeptides, each predicted to traverse the membrane 6-10 times; the ammo and carboxyl termini are both oriented towards the cytoplasm [21]. The current model, with eight transmembrane domains, is shown in Figure 1. More than 100 different missense mutations and two splicing-defect mutations in the PS1 gene have been reported (Table 1) [22,23]. These are dispersed throughout the PS1 sequence, with the majority of mutations clustered near membrane interfaces in the highly conserved transmembrane domains or in hydrophobic residues in either the amino-terminal domain or the putative loop domain between transmembrane domains 6 and 7.

A molecular model of Presenilin-1. The protein is thought to have eight transmembrane domains. Residues associated with mutations found in familial Alzheimer's disease are colored as indicated in the key. 'Endoproteolysis' indicates the approximate site of the imprecise cleavage of the molecule.
Following synthesis, the PS1 and PS2 holoproteins undergo tightly regulated, but imprecise, endoproteolysis in their third cytoplasmic loop domain to generate an approximately 35 kDa amino-terminal fragment and an 18-20 kDa carboxy-terminal fragment, which remain associated with each other [24]. It is clear that cleavage of presenilins following export from the endoplasmic reticulum is governed by additional rate-limiting factors, such as nicastrin (see below), because overexpressed presenilins readily saturate the processing machinery and accumulate as holoproteins [25]. An additional proteolytic pathway is known to involve members of the caspase 3 family of proteases and may be involved in apoptosis [26].
Localization and function
Human PS1 and PS2 have distinct patterns of expression in human tissues. Whereas PS1 is transcribed uniformly throughout the brain and in peripheral tissues, the PS2 transcript is expressed at relatively low levels in the brain, except in the corpus collosum, where it is high; it is highly expressed in some peripheral tissues, such as pancreas, heart, and skeletal muscle [27]. The low PS2 levels in brain and the compensatory activity provided by PS1 may explain why PS2 mutations are infrequent and incompletely penetrant compared with PS1 mutations, which are fully penetrant [28,29].
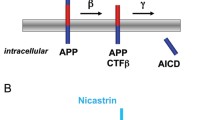
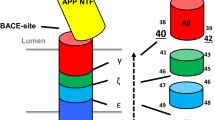
The βAPP protein is cleaved by three different activities, called α-, β- and γ-secretases, to generate Aβ and other fragments. Members of the Notch family, which are involved in developmental signaling in many animals, undergo cleavage at a site (S3) within the transmembrane domain to release an intracellular domain (NICD). It is well established that presenilins are required for the γ-secretase cleavage of βAPP and for the S3 cleavage of Notch-family receptors [30]. For βAPP processing, γ-secretase cleavage is the final step of two distinct proteolytic pathways involving either an α-secretase - which precludes Aβ peptide formation - or a β-secretase, which releases the Aβ peptide, comprising the 40 or 42 carboxy-terminal residues of βAPP. It is uncertain whether the γ-secretase cleavage event occurs at the plasma membrane or during trafficking of βAPP. The usual downstream effect of presenilin mutations in individuals with presenilin-linked familial Alzheimer's disease is the accumulation of Aβ in the brain [31,32] and a shift in the site of the γ-secretase cleavage of βAPP to produce the longer Aβ peptide, spanning residues 1-42 (Aβ42). These main features can be recapitulated in cell culture or in animal models expressing mutant forms of PS1 [33,34,35]. Conversely, PS1-deficient mice are impaired in γ-secretase activity, have reduced Aβ secretion, and accumulate γ-secretase substrates (the carboxy-terminal βAPP fragments derived from α- and β-secretase processing; see Figure 2) [36].

The role of presenilins in the γ-secretase cleavage of Notch and βAPP. Notch is cleaved by tumor necrosis factor α converting enzyme (TACE), and its ligand binds to the part of Notch that remains attached to the membrane. βAPP is cleaved by either the γ-secretase pathway or the γ-secretase pathway to give a membrane-bound carboxy-terminal fragment (APP-CTF). Subsequent γ-secretase cleavage (in the transmembrane domain) of Notch or APP-CTF produces carboxy-terminal intracellular domains, NICD and AICD, respectively, which enter the nucleus and are thought to regulate gene expression. The γ-secretase cleavage of βAPP also produces the neurotoxic Aβ peptide, but only if βAPP has been first cleaved by γ-secretase (not γ-secretase). The γ-secretase complex includes, in addition to PS1, the presenilin-binding protein nicastrin; members of the Armadillo protein family, such as β-catenin, have also been detected in presenilin complexes, although their role is not understood. Aph-1 and Pen-2 may also participate in the γ-secretase complex.
Mutation of two highly conserved aspartate residues in the transmembrane domains of PS1 (Asp257 and Asp385, shown in blue in Figure 1) inactivates γ-secretase activity and reduces Aβ secretion [37]. The sequence motif around Asp385 is somewhat similar to a sequence within prepilins, a family of bacterial peptidases [38]; this has promoted speculation that presenilins are themselves aspartyl proteases responsible for γ-secretase activity and that the critical Asp257 and Asp385 residues form that catalytic center of the γ-secretase. Additional support for the idea that presenilins are the proteases that have γ-secretase activity comes from studies in which photoactivated inhibitors of γ-secretase activity were found to bind to PS1 and PS2 [39,40].
It should be noted that forms of PS1 with the D257A or D385A mutations integrate poorly into the heteromeric complexes that are considered necessary for γ-secretase function, raising the possibility that these transmembrane-domain mutations disable PS1 structurally [41]. Moreover, several lines of evidence show that the regulation of βAPP and Notch cleavage differs, however, and such evidence is difficult to reconcile with a direct enzymatic role for PS1 in γ-secretase cleavage. First, a naturally occurring splice variant of PS1 lacking the region (encoded by exon 8) that contains the critical Asp257 allows Aβ production but not cleavage of Notch [42]. Second, different presenilin mutations differentially affect Aβ production and Notch cleavage [43,44,45]. Third, some recently discovered γ-secretase inhibitors preferentially affect processing βAPP over that of Notch [46]. Together, these findings suggest the presenilins regulate proteolysis indirectly, perhaps by an effect on trafficking of βAPP or Notch or by activation of the γ-secretase.
The biological purpose of presenilin-dependent γ-secretase cleavage of βAPP is still unknown. By analogy with the signaling pathway downstream of cleaved Notch and NICD, recent studies have raised the intriguing possibility that the short-lived carboxyl-terminal stub of βAPP, called (βAPP intracellular domain (AICD), is released into the cytoplasm following γ-secretase cleavage and translocates to the nucleus (Figure 2), where it may regulate expression of components involved in mobilizing intracellular calcium stores [47,48,49]. Another proposal implicates βAPP as a regulator of the axonal transport of a subset of vesicles ferrying cargo to nerve terminals. This view is derived from the observations that βAPP interacts directly with the light chain of the transport protein kinesin [50], that the transport of a vesicular compartment containing PS1 and γ-secretase depends on βAPP [51], and that deletion of the Drosophila βAPP-like gene (dAPPL) or overexpression of either dAPPL or human (βAPP in Drosophila disrupts axonal transport [52,53]. In this scheme, γ-secretase cleavage of the βAPP by presenilin-containing complexes releases the carboxy-terminal portion of (βAPP that connects the transport vesicle to the transport machinery through interaction with kinesin, thereby disengaging the vesicle from microtubules upon arrival at its destination. Thus, presenilins may influence diverse cellular processes, such as intracellular signaling and axonal traffic.
In vitro studies of detergent-solubilized membranes show that γ-secretase activity resides within large multisubunit complexes that also contain presenilins. If presenilin molecules are excluded from these complexes, they are rapidly targeted for proteosome-mediated degradation [54]. On density gradients, presenilin holoproteins and the amino-and carboxy-terminal fragments of presenilins co-elute with high-molecular-weight markers (180 kDa for the holoproteins and 250-1000 kDa for the fragments [25,55]), presumably because they are part of larger complexes, and antibodies to PS1 coimmunoprecipitate heteromeric protein complexes that contain γ-secretase activity [56]. Conversely, affinity isolation with γ-secretase inhibitors co-purifies protein complexes containing PS1 [39,40]. Members of the Armadillo protein family (β- and δ-catenin, neural plakophilin-related armadillo protein (NPRAP), and p0071) [55,57,58] interact with presenilins but are not required for γ-secretase activity in vitro [40]. Other interactions whose role in γ-secretase activity is unknown have been reviewed previously [22].
More recently, PS1 and PS2 were found to interact with nicastrin, a novel single-pass transmembrane protein that is essential for processing of βAPP and Notch [59,60,61]. Nicastrin is clearly an important regulator of γ-secretase activity: nicastrin antibodies immunoprecipitate both presenilin and the active γ-secretase complex [40], and missense or deletion mutations within a conserved lumenal domain of nicastrin up- or down-regulate Aβ production in a manner that corresponds with PS1 binding, suggesting that γ-secretase activity is generated only after an obligatory interaction between nicastrin and PS1 [59]. Notch cleavage is affected similarly by nicastrin mutations, albeit to a lesser extent [60]. Moreover, nicastrin is essential for the normal processing of both βAPP and Notch homologs in Drosophila and C. elegans, and human nicastrin can partially rescue mutants of the C. elegans nicastrin homolog Aph-2 [59,61,62,63,64], suggesting that nicastrin function and its interactions with presenilins are conserved widely in non-mammalian species. Only mature glycosylated nicastrin that has passed through the Golgi compartment interacts with PS1 and is included in γ-secretase complexes [65]; overexpressed nicastrin fails to mature normally and accumulates within the endoplasmic reticulum. Moreover, entry of each of nicastrin and PS1 into γ-secretase complexes appears to be regulated by the other protein: the loss of one partner destabilizes the other [61,63,66,67].
Two potential new members of the PS-nicastrin complexes are homologs of Aph-1 and Pen-2, components of the C. elegans Glp-1/Notch signaling cascade that interact genetically with Sel-12/presenilin and Aph-2/nicastrin [68,69]. Primary sequence analysis suggests that Aph-1 and Pen-2 have seven and two membrane spanning domains, respectively, that are conserved in their respective Drosophila and human homologs. Human Aph-1 and Pen-2 can rescue C. elegans mutants lacking their homologs only when both transgenes are present together, implying that they act in concert. Moreover, reduction of Aph-1 and Pen-2 expression in Drosophila cells by RNA inhibition reduces γ-secretase activity [69]. Reduced expression of nematode Aph-1 causes mislocalization of Aph-2/nicastrin [68], and both Aph-1 and Pen-2 are required to maintain presenilin levels [69], suggesting that they regulate, or are components of, the presenilin-nicastrin γ-secretase complexes.
Frontiers
The identification of the additional γ-secretase components within the presenilin complexes is clearly an important task that lies ahead. The complexes purified to date are quite large, partly because of membrane impurities that remain associated following treatment with gentle detergents and partly because of interacting proteins that are not related to γ-secretase activity but are necessary for trafficking and maturation of the complex. The genetic cause of at least half of all cases of early onset familial Alzheimer's disease remain unexplained, and some of the unknown genes may have products that may modulate presenilin activity within γ-secretase complexes.
References
Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K: Cloning of a gene bearing mis-sense mutations in early-onset familial Alzheimer's disease. Nature. 1995, 375: 754-760. 10.1038/375754a0. This study revealed the involvement of presenilin-1 in early onset FAD.
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T: Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995, 376: 775-778. 10.1038/376775a0. The authors identify mutations in the PS2 gene as a pathogenic factor in AD.
Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, Bird TD, Schellenberg GD: A familial Alzheimer's disease locus on chromosome 1. Science. 1995, 269: 970-973. Mapping of a familial AD locus to the PS2 gene.
The Alzheimer's disease collaborative group: The structure of the presenilin 1 (S182) gene and the identification of six novel mutations in early onset AD pedigrees. Nat Genet. 1995, 11: 219-222. The first analysis of the intron-exon structure of the PS1 gene.
Hutton M, Busfield F, Wragg M, Crook R, Perez-tur J, Clark RF, Prihar G, Talbot C, Phillips H, Wright K, et al: Complete analysis of the presenilin 1 gene in early onset Alzheimer's disease. Neuroreport. 1996, 7: 801-805. An analysis of the intron-exon structure of the PS1 gene.
Prihar G, Fuldner RA, Perez-tur J, Lincoln S, Duff K, Crook R, Hardy J, Philips CA, Venter C, Talbot C, et al: Structure and alternative splicing of the presenilin-2 gene. Neuroreport. 1996, 7: 1680-1684. A description of intron-exon structure of PS2 gene and the production of alternatively spliced isoforms.
Anwar R, Moynihan TP, Ardley H, Brindle N, Coletta PL, Cairns N, Markham AF, Robinson PA: Molecular analysis of the presenilin 1 (S182) gene in "sporadic" cases of Alzheimer's disease: identification and characterisation of unusual splice variants. J Neurochem. 1996, 66: 1774-1777. The first report detailing the discovery of unexpected splicing of PS1 transcripts.
Sato N, Imaizumi K, Manabe T, Taniguchi M, Hitomi J, Katayama T, Yoneda T, Morihara T, Yasuda Y, Takagi T, et al: Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J Biol Chem. 2001, 276: 2108-2114. 10.1074/jbc.M006886200. Alternative splicing of PS2 is shown to have an affect on the production of the Aβ peptide.
Grunberg J, Walter J, Eckman C, Capell A, Schindzielorz A, Younkin S, Mehta N, Hardy J, Haass C: Truncated presenilin 2 derived from differentially spliced mRNA does not affect the ratio of amyloid beta-peptide 1-42/1-40. Neuroreport. 1998, 9: 3293-3299. Alternatively spliced PS2 variants are expressed as amino-terminally truncated proteins, which support Aβ peptide production.
Levitan D, Greenwald I: Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature. 1995, 377: 351-354. 10.1038/377351a0. Identification of a C. elegans presenilin homolog.
Hong CS, Koo EH: Isolation and characterization of Drosophila presenilin homolog. Neuroreport. 1997, 8: 665-668. Identification of a Drosophila PS1 homolog.
Boulianne GL, Livne-Bar I, Humphreys JM, Liang Y, Lin C, Rogaev E, George-Hyslop P: Cloning and characterization of the Drosophila presenilin homologue. Neuroreport. 1997, 8: 1025-1029. Identification of a Drosophila presenilin homolog.
Theologis A, Ecker JR, Palm CJ, Federspiel NA, Kaul S, White O, Alonso J, Altafi H, Araujo R, Bowman CL, et al: Sequence and analysis of chromosome 1 of the plant Arabidopsis thaliana. Nature. 2000, 408: 816-820. 10.1038/35048500. Chromosome 1 of Arabidopsis thaliana encodes the plant presenilin homolog among its many genes.
Tsujimura A, Yasojima K, Hashimoto-Gotoh T: Cloning of Xenopus presenilin-alpha and -beta cDNAs and their differential expression in oogenesis and embryogenesis. Biochem Biophys Res Commun. 1997, 231: 392-396. 10.1006/bbrc.1996.6043. Identification of a Xenopus presenilin homolog.
Calenda A, Mestre-Frances N, Czech C, Pradier L, Petter A, Perret M, Bons N, Bellis M: Cloning of the presenilin 2 cDNA and its distribution in brain of the primate, Microcebus murinus : coexpression with betaAPP and Tau proteins. Neurobiol Dis. 1998, 5: 323-333. 10.1006/nbdi.1998.0205. Identification of a PS2 homolog in lemurs and immunocytochemical characterization of its distribution in the brain.
Martinez-Mir A, Canestro C, Gonzalez-Duarte R, Albalat R: Characterization of the amphioxus presenilin gene in a high gene-density genomic region illustrates duplication during the vertebrate lineage. Gene. 2001, 279: 157-164. 10.1016/S0378-1119(01)00751-X. Identification of a chordate presenilin homolog.
Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I: Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1996, 93: 14940-14944. 10.1073/pnas.93.25.14940. Expression of human wild-type PS1, and to a lesser extent a mutant form of PS1 implicated in FAD, reversed the phenotype associated with sel-12 deficiency in C. elegans, demonstrating functional conservation between nemotode and human presenilin homologs.
Baumeister R, Leimer U, Zweckbronner I, Jakubek C, Grunberg J, Haass C: Human presenilin-1, but not familial Alzheimer's disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signalling independently of proteolytic processing. Genes Funct. 1997, 1: 149-159. Replacement of defective sel-12 C. elegans mutants with human PS1 reversed the defects in Notch signaling. Only partial reversal was observed with FAD-mutant PS1.
Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, Golde TE: Identification of a novel family of presenilin homologues. Hum Mol Genet. 2002, 11: 1037-1044. 10.1093/hmg/11.9.1037. In this paper and [20], multiple putative presenilin homologs were identified by database analysis based on sequence similarities to the presenilin transmembrane domains.
Grigorenko AP, Moliaka YK, Korovaitseva GI, Rogaev EI: Novel class of polytopic proteins with domains associated with putative protease activity. Biochemistry (Mosc). 2002, 67: 826-835. 10.1023/A:1016365227942. See [19].
Hutton M, Hardy J: The presenilins and Alzheimer's disease. Hum Mol Genet. 1997, 6: 1639-1646. 10.1093/hmg/6.10.1639. A review of presenilin function that presents the eight-transmembrane-domain model for both human presenilin proteins.
Tandon A, Rogaeva EA, Mullan M, St George-Hyslop P: Molecular genetics of Alzheimer's disease: the role of beta-amyloid and the presenilins. Curr Opin Neurol. 2000, 13: 377-384. 10.1097/00019052-200008000-00003. A comprehensive review of presenilin and β-amyloid biology.
Rogaeva E: The solved and unsolved mysteries of the genetics of early-onset Alzheimer's disease. Neuromolecular Med. 2002, 2: 1-10. 10.1385/NMM:2:1:01. A review of the current status of AD genetics.
Thinakaran G, Regard JB, Bouton CM, Harris CL, Price DL, Borchelt DR, Sisodia SS: Stable association of presenilin derivatives and absence of presenilin interactions with APP. Neurobiol Dis. 1998, 4: 438-453. 10.1006/nbdi.1998.0171. The authors report that the amino- and carboxy-terminal fragments of PS1 or PS2 can be co-immunoprecipitated, suggesting that the fragments remain complexed after endoproteolysis.
Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C: The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem. 1998, 273: 3205-3211. 10.1074/jbc.273.6.3205. Amino- and carboxy-terminal fragments of PS1 co-sediment in large protein complexes. Overexpression of PS1 failed to increase the levels of the PS1 proteolytic derivatives, but instead caused accumulation of the holoprotein that was excluded from the larger presenilin complexes.
Kim TW, Pettingell WH, Jung YK, Kovacs DM, Tanzi RE: Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science. 1997, 277: 373-376. 10.1126/science.277.5324.373. Description of an alternative cleavage of PS2 that is mediated by caspase-3 and is upregulated by an FAD mutation.
Rogaev EI, Sherrington R, Wu C, Levesque G, Liang Y, Rogaeva EA, Ikeda M, Holman K, Lin C, Lukiw WJ, et al: Analysis of the 5' sequence, genomic structure, and alternative splicing of the presenilin-1 gene (PSEN1) associated with early onset Alzheimer disease. Genomics. 1997, 40: 415-424. 10.1006/geno.1996.4523. Characterization of regulatory sequences within the 5' untranslated PS1 gene sequence and the tissue distribution of PS1 transcripts.
Sherrington R, Froelich S, Sorbi S, Campion D, Chi H, Rogaeva EA, Levesque G, Rogaev EI, Lin C, Liang Y, et al: Alzheimer's disease associated with mutations in presenilin 2 is rare and variably penetrant. Hum Mol Genet. 1996, 5: 985-988. 10.1093/hmg/5.7.985. Genetical analysis of sixty FAD pedigrees revealed that mutations in PS2 are an uncommon cause of AD and that the penetrance and age of onset associated with PS2 mutations is highly variable.
Bird TD, Levy-Lahad E, Poorkaj P, Sharma V, Nemens E, Lahad A, Lampe TH, Schellenberg GD: Wide range in age of onset for chromosome 1-related familial Alzheimer's disease. Ann Neurol. 1996, 40: 932-936. PS2-linked FAD presents with highly variable age of onset and penetrance.
Kopan R, Goate A: A common enzyme connects notch signaling and Alzheimer's disease. Genes Dev. 2000, 14: 2799-2806. 10.1101/gad.836900. A review linking presenilin to γ-secretase-mediated proteolysis of Notch and βAPP.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, et al: Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996, 2: 864-870. This study was the first to reported elevated plasma Aβ peptide levels in patients with PS1-linked FAD.
Tamaoka A, Fraser PE, Ishii K, Sahara N, Ozawa K, Ikeda M, Saunders AM, Komatsuzaki Y, Sherrington R, Levesque G, et al: Amyloid-beta-protein isoforms in brain of subjects with PS1-linked, beta APP-linked and sporadic Alzheimer disease. Brain Res Mol Brain Res. 1998, 56: 178-185. 10.1016/S0169-328X(98)00044-8. Accumulation of the long isoforms of the Aβ peptide is increased in postmortem cerebral cortices of individuals with PS1-linked FAD.
Borchelt DR, Thinakaran G, Eckman CB, Borchelt DR, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, et al: Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996, 17: 1005-1013. Expression of FAD-linked PS1 mutations raised the Aβ42/40 ratio in cell culture and in transgenic mice.
Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, et al: Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996, 383: 710-713. 10.1038/383710a0. Transgenic mice expressing human FAD-linked PS1 mutants have increased Aβ42 production.
Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, et al: Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997, 3: 67-72. Expression of FAD-linked PS1 and PS2 mutations raised significantly the production of Aβ42 in cell culture and in transgenic mice.
De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F: Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998, 391: 387-390. 10.1038/34910. The first report to show that PS1 expression is essential for γ-secretase cleavage of βAPP, and that γ-secretase substrates accumulate in the absence of PS1.
Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ: Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999, 398: 513-517. 10.1038/19077. The authors present data showing that two conserved aspartate residues located in the transmembrane domain of PS1 are essential for γ-secretase activity, and propose that PS1 is itself an aspartyl protease.
Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim ML, Baumeister R, et al: Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat Cell Biol. 2000, 2: 848-851. 10.1038/35041097. The identification of a short stretch of primary sequence homology between the presenilins and bacterial aspartyl protease prompts the suggestion that presenilins belong to a family of aspartyl proteases.
Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al: Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000, 405: 689-694. 10.1038/35015085. Covalent labeling of PS1 with photoactivated crosslinking agents that were designed to behave as transition-state analogs for aspartyl proteases.
Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS: Activity-dependent isolation of the presenilin-gamma-secretase complex reveals nicastrin and a gamma substrate. Proc Natl Acad Sci USA. 2002, 99: 2720-2725. 10.1073/pnas.052436599. Biochemical purification of functional γ-secretase complex reveals the presence of PS1, nicastrin, and surprisingly C83, the βAPP-derived γ-secretase substrate. Other PS1-binding proteins such as the catenins or calsenilin are not required for γ-secretase activity.
Yu G, Chen F, Nishimura M, Steiner H, Tandon A, Kawarai T, Arawaka S, Supala A, Song YQ, Rogaeva E, et al: Mutation of conserved aspartates affects maturation of both aspartate mutant and endogenous presenilin 1 and presenilin 2 complexes. J Biol Chem. 2000, 275: 27348-27353. PS1 with mutations in the conserved aspartate residues fails to enter high-molecular-weight γ-secretase complexes and blocks the maturation of endogenous PS1.
Capell A, Steiner H, Romig H, Keck S, Baader M, Grim MG, Baumeister R, Haass C: Presenilin-1 differentially facilitates endoproteolysis of the beta-amyloid precursor protein and Notch. Nat Cell Biol. 2000, 2: 205-211. 10.1038/35008626. Aβ peptide generation is severely reduced by PS1 mutations at Asp385, but only partially by Asp257 mutations, whereas Notch cleavage is blocked by either mutation, suggesting differential regulation of the γ-secretase and S3-cleavage activities by PS1 mutants.
Kulic L, Walter J, Multhaup G, Teplow DB, Baumeister R, Romig H, Capell A, Steiner H, Haass C: Separation of presenilin function in amyloid beta-peptide generation and endoproteolysis of Notch. Proc Natl Acad Sci USA. 2000, 97: 5913-5918. 10.1073/pnas.100049897. The authors report that γ-secretase and S3-cleavage activities can be distinguished by mutations engineered at Leu286 of PS1, which increase Aβ42 production substantially but inhibit Notch cleavage.
Zhang DM, Levitan D, Yu G, Nishimura M, Chen F, Tandon A, Kawarai T, Arawaka S, Milman P, Holmes E, et al: Mutation of the conserved N-terminal cysteine (Cys92) of human presenilin 1 causes increased Abeta42 secretion in mammalian cells by impaired Notch/Lin12 signalling in C. elegans embryos. Neuroreport. 2000, 11: 3227-3230. Expression of a Cys92Ser PS1 mutant increases Aβ42 production in mammalian cells but causes the loss of Notch signaling in C. elegans, suggesting that the γ-secretase and S3-cleavage activities are distinctly regulated.
Okochi M, Eimer S, Bottcher A, Baumeister R, Romig H, Walter J, Capell A, Steiner H, Haass C: A loss of function mutant of the presenilin homologue sel-12 undergoes aberrant endoproteolysis in Caenorhabditis elegans and increased A-beta-42 generation in human cells. J Biol Chem. 2000, 275: 40925-40932. 10.1074/jbc.M005254200. Expression of the Cys92Ser PS1 mutation, which corresponds to the loss-of-function sel-12 mutant in C. elegans (Cys60Ser), does not affect Notch cleavage in mammalian cells, but increases Aβ42 production.
Petit A, Bihel F, Alves da Costa C, Pourquie O, Checler F, Kraus JL: New protease inhibitors prevent gamma-secretase-mediated production of Abeta40/42 without affecting Notch cleavage. Nat Cell Biol. 2001, 3: 507-511. 10.1038/35074581. Pharmacological evaluation of non-peptidergic γ-secretase inhibitors that do not affect S3-cleavage of Notch.
Kimberly WT, Zheng JB, Guenette S, Selkoe DJ: The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a Notch-like manner. J Biol Chem. 2001, 276: 40288-40292. The authors show that exogenous expression of the cytoplasmic carboxy-terminal tail of βAPP causes it to translocate into the nucleus; they argue that γ-secretase-derived βAPP intracellular domain (AICD) regulates transcription in the nucleus.
Leissring MA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, Anliker B, Muller U, Saftig P, De Strooper B, et al: A physiologic signaling role for the gamma-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA. 2002, 99: 4697-4702. 10.1073/pnas.072033799. The loss of phosphoinositide-mediated intracellular calcium signaling that is associated with either PS1 deficiency or pharmacological inhibition of γ-secretase activity is reconstituted by overexpression of βAPP intracellular domain.
Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, Kaether C, Zheng H, Ghetti B, Haass C, Steiner H: Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci USA. 2002, 99: 8025-8030. 10.1073/pnas.112686799. Characterization of a PS1 mutation that increases Aβ production but, surprisingly, inhibits the generation of the Notch and βAPP intracellular domains.
Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS: Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000, 28: 449-459. This study characterizes the interaction between βAPP and kinesin light chain and shows that expression of mutant kinesin reduces βAPP axonal transport.
Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS: Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001, 414: 643-648. 10.1038/414643a. The authors report that a vesicular compartment containing γ-secretase activity undergoes βAPP- and kinesin-dependent axonal transport.
Torroja L, Chu H, Kotovsky I, White K: Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol. 1999, 9: 489-492. 10.1016/S0960-9822(99)80215-2. A role for βAPP in axonal trafficking is suggested by the finding that overexpression of the Drosophila βAPP homolog causes an interruption of axonal transport of synaptic proteins, similar to the phenotype induced by kinesin light chain mutants.
Gunawardena S, Goldstein LS: Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001, 32: 389-401. Ablation of the Drosophila βAPP homolog or overexpression of the human βAPP or APPL in Drosophila reproduced the abnormal axonal trafficking phenotypes induced by kinesin light chain mutants.
Fraser PE, Levesque G, Yu G, Mills LR, Thirlwell J, Frantseva M, Gandy SE, Seeger M, Carlen PL, George-Hyslop P: Presenilin 1 is actively degraded by the 26S proteasome. Neurobiol Aging. 1998, 19: S19-S21. 10.1016/S0197-4580(98)00029-3. The authors show that PS1 undergoes proteasome-mediated degradation that is distinct from the normal activity that controls endoproteolysis of the PS1 loop domain.
Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, et al: The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains beta-catenin. J Biol Chem. 1998, 273: 16470-16475. 10.1074/jbc.273.26.16470. PS1 amino- and carboxy-terminal fragments reside in high-molecular-weight proteins located in the endoplasmic reticulum and Golgi compartments, that also contain β-catenin.
Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, Shi XP, Yin KC, Shafer JA, Gardell SJ: Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA. 2000, 97: 6138-6143. 10.1073/pnas.110126897. γ-secretase activity was biochemically isolated in high-molecular-weight protein complexes that contain PS1.
Zhou J, Liyanage U, Medina M, Ho C, Simmons AD, Lovett M, Kosik KS: Presenilin 1 interaction in the brain with a novel member of the Armadillo family. Neuroreport. 1997, 8: 2085-2090. A yeast two-hybrid analysis revealed that δ-catenin is a PS1-interacting protein.
Levesque G, Yu G, Nishimura M, Zhang DM, Levesque L, Yu H, Xu D, Liang Y, Rogaeva E, Ikeda M, et al: Presenilins interact with armadillo proteins including neural-specific plakophilin-related protein and beta-catenin. J Neurochem. 1999, 72: 999-1008. 10.1046/j.1471-4159.1999.0720999.x. A yeast two-hybrid analysis revealed that β-catenin, p0071, and the novel neuronal protein called neural plakophilin-related armadillo protein are PS1-interacting proteins.
Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, et al: Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000, 407: 48-54. 10.1038/35024009. This study identified nicastrin as prominent PS1-binding protein and an essential component of γ-secretase complexes.
Chen F, Yu G, Arawaka S, Nishimura M, Kawarai T, Yu H, Tandon A, Supala A, Song YQ, Rogaeva E, et al: Nicastrin binds to membrane-tethered Notch. Nat Cell Biol. 2001, 3: 751-754. 10.1038/35087069. Nicastrin is shown to interact with Notch and to regulate S3-site cleavage.
Hu Y, Ye Y, Fortini ME: Nicastrin is required for gamma-secretase cleavage of the Drosophila Notch receptor. Dev Cell. 2002, 2: 69-78. Drosophila nicastrin mutants reproduce a Notch-like phenotype, indicating that nicastrin is essential for Notch signaling in the fly, and nicastrin-deficiency destabilizes endogenous presenilin.
Levitan D, Yu G, St George HP, Goutte C: APH-2/nicastrin functions in LIN-12/Notch signaling in the Caenorhabditis elegans somatic gonad. Dev Biol. 2001, 240: 654-661. 10.1006/dbio.2001.0486. Aph-2, the C. elegans nicastrin homolog, is required for Glp-1/Notch signaling.
Chung HM, Struhl G: Nicastrin is required for Presenilin-mediated transmembrane cleavage in Drosophila. Nat Cell Biol. 2001, 3: 1129-1132. 10.1038/ncb1201-1129. Nicastrin deficiency in Drosophila abolishes Notch signaling and γ-secretase-cleavage of βAPP and causes a reduction in PS1 stability.
Lopez-Schier H, St Johnston D: Drosophila nicastrin is essential for the intramembranous cleavage of notch. Dev Cell. 2002, 2: 79-89. Nicastrin is essential for Notch cleavage, and mutations in nicastrin and PS1 disrupt the cytoskeleton, suggesting widespread defects induced by γ-secretase dysfunction.
Yang DS, Tandon A, Chen F, Yu G, Yu H, Arawaka S, Hasegawa H, Duthie M, Schmidt S, Nixon RA, et al: Mature glycosylation and trafficking of nicastrin modulate its binding to presenilins. J Biol Chem. 2002, 277: 28135-28142. 10.1074/jbc.M110871200. This study characterizes the trafficking-dependent maturation of nicastrin and shows that PS1 interacts preferentially with mature nicastrin in the Golgi compartment.
Leem JY, Vijayan S, Han P, Cai D, Machura M, Lopes KO, Veselits ML, Xu H, Thinakaran G: Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J Biol Chem. 2002, 277: 19236-19240. 10.1074/jbc.C200148200. Nicastrin in PS1-deficient cells is mistrafficked and fails to undergo the normal maturation of its oligosaccharide chains.
Edbauer D, Winkler E, Haass C, Steiner H: Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci USA. 2002, 99: 8666-8671. When either PS1 or nicastrin expression is individually reduced by RNA interference, there was a striking reduction in the stability of the other binding partner.
Goutte C, Tsunozaki M, Hale VA, Priess JR: APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA. 2002, 99: 775-779. 10.1073/pnas.022523499. This study identified Aph-1, a potential new member of the γ-secretase complex, and showed that normal trafficking of Aph-2, the nicastrin homolog, requires Aph-1 expression.
Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, et al: aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002, 3: 85-97. This study identified Aph-1 and Pen-2, two potential new members of the γ-secretase complex, using a C. elegans genetic screen to identify proteins that interact with Sel-12/PS1 and Aph-2/nicastrin.
Rogaeva EA, Fafel KC, Song YQ, Medeiros H, Sato C, Liang Y, Richard E, Rogaev EI, Frommelt P, Sadovnick AD, et al: Screening for PS1 mutations in a referral-based series of AD cases: 21 novel mutations. Neurology. 2001, 57: 621-625. Discovery of multiple novel PS1 mutations by sequencing the PS1 gene in 372 individuals with AD and 42 asymptomatic individuals with a family history of AD.
Alzheimer research forum: presenilin mutations directory. [http://www.alzforum.org/res/com/mut/pre/default.asp]
Acknowledgements
We gratefully acknowledge grants from the Alzheimer Society of Ontario, the Canadian Institutes of Health Research, Scottish Rite Charitable Foundation, Ontario Mental Health Foundation, and the Alzheimer Society of Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tandon, A., Fraser, P. The presenilins. Genome Biol 3, reviews3014.1 (2002). https://doi.org/10.1186/gb-2002-3-11-reviews3014
Published:
DOI: https://doi.org/10.1186/gb-2002-3-11-reviews3014