
Abstract
The use of controlled mechanical ventilation (CMV) in patients who experience weaning failure after a spontaneous breathing trial or after extubation is a strategy based on the premise that respiratory muscle fatigue (requiring rest to recover) is the cause of weaning failure. Recent evidence, however, does not support the existence of low frequency fatigue (the type of fatigue that is long-lasting) in patients who fail to wean despite the excessive respiratory muscle load. This is because physicians have adopted criteria for the definition of spontaneous breathing trial failure and thus termination of unassisted breathing, which lead them to put patients back on the ventilator before the development of low frequency respiratory muscle fatigue. Thus, no reason exists to completely unload the respiratory muscles with CMV for low frequency fatigue reversal if weaning is terminated based on widely accepted predefined criteria. This is important, since experimental evidence suggests that CMV can induce dysfunction of the diaphragm, resulting in decreased diaphragmatic force generating capacity, which has been called ventilator-induced diaphragmatic dysfunction (VIDD). The mechanisms of VIDD are not fully elucidated, but include muscle atrophy, oxidative stress and structural injury. Partial modes of ventilatory support should be used whenever possible, since these modes attenuate the deleterious effects of mechanical ventilation on respiratory muscles. When CMV is used, concurrent administration of antioxidants (which decrease oxidative stress and thus attenuate VIDD) seems justified, since antioxidants may be beneficial (and are certainly not harmful) in critical care patients.
Similar content being viewed by others

Introduction
Controlled mechanical ventilation (CMV) is a mode of ventilator support in which each breath is triggered by the ventilator's timer using a respiratory rate set by the clinician. The characteristics of the breath are also set by the clinician, i.e. pressure or flow controlled, volume, flow or time cycled. Because the respiratory muscles are not contracting, the minute ventilation is fully controlled by the ventilator, which takes full responsibility for inflating the respiratory system.
CMV is traditionally used in severely ill patients who cannot tolerate partial ventilatory support (e.g., acute respiratory distress syndrome, septic shock, multiple organ failure), in cases of overt patient-ventilator dysynchrony, and in the immediate postoperative period. CMV is also used when weaning fails (especially T-piece weaning) to rest the respiratory muscle before the next weaning attempt. This review will summarize recent evidence concerning the deleterious effects of CMV on respiratory muscle function and discuss the use of CMV during weaning failure.
Effects of CMV on the respiratory muscles: evidence from animal models
Animal models have been used to unravel the effects of CMV that are beneficial for the respiratory muscles: reversal of respiratory muscle fatigue [1], prevention of muscle fiber injury during a short-term (four hours) model of sepsis [2], and restoration of perfusion to vital organs in shock states when blood flow is 'stolen' by the intensely working respiratory muscles [1, 3].
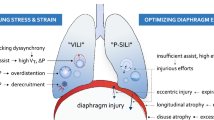
Accumulating experimental evidence suggests, however, that CMV can also induce dysfunction of the diaphragm, resulting in decreased diaphragmatic force generating capacity, diaphragmatic atrophy, and diaphragmatic injury, also called ventilator-induced diaphragmatic dysfunction (VIDD) [4].
Ventilator-induced diaphragmatic dysfunction
In the intact diaphragm of various animal species (including primates) studied in vivo after a period of CMV, transdiaphragmatic pressure generation caused by phrenic nerve stimulation declines at both submaximal and maximal stimulation frequencies (20 to 100 Hz) in a time dependent manner [5–7]. The decline is evident early and worsens as mechanical ventilation is prolonged. Within a few days (3 days in rabbits [7], 5 days in piglets [6], and 11 days in baboons [5]) the pressure-generating capacity of the diaphragm declines by 40% to 50%. The endurance of the diaphragm is also significantly compromised, as suggested by the reduced ability of animals to sustain an inspiratory resistive load [5].
The decreased force-generating capacity is not secondary to changes in lung volume because transpulmonary pressure or dynamic lung compliance do not change. Moreover, it is not caused by changes in abdominal compliance, given the nearly stable abdominal pressure over the observation period and the similar results obtained with abdominal wrapping, which prevents changes in abdominal compliance [5, 6].
Neural or neuromuscular transmission remains intact as reflected by the lack of changes in phrenic nerve conduction (latency) and the stable response to repetitive stimulation of the phrenic nerve [6]. In contrast, the decrease in the compound muscle action potential suggests that excitation-contraction coupling or membrane depolarization may be involved in the dysfunction [6]. Thus, the mechanical ventilation induced impairment of force generating capacity appears to reside within the myofibers [4].
In vitro results of isometric (both twitch and tetanic) tension development in isolated diaphragmatic strips confirm the in vivo findings [8–13], and suggest that the decline in contractility is an early (12 hours) [9] and progressive phenomenon [9, 14]. Isometric force development declines by 30% to 50% after 1 to 3 days of CMV in rats and rabbits, though this time course might be prolonged in piglets [6], which might suggest that the bigger the species, the longer it takes for VIDD to develop.
The mechanisms of VIDD have not been fully elucidated. Muscle atrophy, oxidative stress and structural injury have been documented after CMV [4]. The precise contribution of each to the development of VIDD has yet to be defined.
Muscle atrophy results from a combination of decreased protein synthesis and increased proteolysis [15], and both mechanisms have been documented in VIDD [16, 17]. Of the three intracellular proteolytic systems of mammalian cells (lysosomal proteases, calpains and proteasomes), both calpains and proteasomes are activated to induce atrophy secondary to CMV [17]. The proteasome is a multisubunit multicatalytic complex that exists in two major forms: the core 20S proteasome can be free or bound to a pair of 19S regulators to form the 26S proteasome. Although the 26S proteasome is activated with ventilator-induced cachexia [14, 18], Shanely et al. [17] showed that CMV resulted in a five-fold increase in 20S proteasome activity, which is specialized in degrading proteins oxidized by reactive oxygen species [19]. Oxidative damage of a protein results in its partial unfolding, exposing hidden hydrophobic residues; therefore, an oxidized protein does not need to be further modified by ubiquitin conjugation to confer a hydrophobic patch, nor does it require energy from ATP hydrolysis to unfold [20].
This result is in concert with the evidence for oxidative stress-induced modification of proteins obtained from the diaphragms of animals subjected to CMV [17, 21]. Oxidative stress is augmented in the diaphragm after CMV, as indicated by the increased protein oxidation and lipid peroxidation by-products [17, 21]. The onset of oxidative modifications is quite rapid, occurring within the first six hours of the institution of CMV [21]. Oxidative stress can modify many critical proteins involved in energetics, excitation-contraction coupling, and force generation. Accordingly, CMV-induced diaphragmatic protein oxidation was evident in insoluble (but not soluble) proteins with molecular masses of about 200, 128, 85, and 40 kDa [21]. These findings raise the possibility that actin (40 kDa) and/or myosin (200 kDa) undergo oxidative modification during CMV [21]. This intriguing possibility awaits confirmation by more specific identification of the modified proteins.
Structural abnormalities of different subcellular components of diaphragmatic fibers have been found after CMV [7, 22, 23]. The changes consist of disrupted myofibrils, increased numbers of lipid vacuoles in the sarcoplasm, and abnormally small mitochondria containing focal membrane disruptions. Similar alterations were observed in the external intercostal muscles of ventilated animals, but not in the hind limb muscle [22]. The structural alterations in the myofibrils have detrimental effects on diaphragmatic force-generating capacity, the number of abnormal myofibrils being inversely related to the force output of the diaphragm [7].
Clinical relevance of ventilator-induced diaphragmatic dysfunction
Do we have evidence for VIDD in patients? Although conclusive data do not exist, several intriguing observations suggest VIDD may occur in patients. The twitch transdiaphragmatic pressure elicited by magnetic stimulation of the phrenic nerves is reduced in ventilated patients compared with normal subjects [24], and in patients ready to undergo weaning trials [25]. Diaphragmatic atrophy was documented (by ultrasound) in a tetraplegic patient after prolonged CMV [26]. The time course of atrophy, however, was not established. Furthermore, denervation atrophy removes substances originating from the nerve that are trophic for the muscle, which is not the case in VIDD, as neural and neuromuscular functions remain intact. The presence of confounding factors, such as disease state (e.g., sepsis) and drug therapy (e.g., corticosteroids, neuromuscular blocking agents), makes documentation of VIDD difficult in a clinical setting [4]. Nevertheless, retrospective analysis of post-mortem data from neonates who received ventilator assistance for 12 days or more before death revealed diffuse diaphragmatic myofiber atrophy (small myofibers with rounded outlines), which were not present in extradiaphragmatic muscles [27].
The typical clinical scenario in which to suspect VIDD is a patient who fails to wean after a period of CMV because of respiratory muscle dysfunction [4]. Other known causes of respiratory muscle weakness such as shock, sepsis, major malnutrition, electrolyte disturbances and neuromuscular disorders, have been ruled out. For example, prolonged neuromuscular blockade can be excluded by the lack of an abnormal response to train-of-four stimulation; critical illness polyneuropathy by the absence of neuropathic changes on electrophysiological testing; and acute quadriplegic myopathy by the lack of corticosteroid exposure history (or by muscle biopsy in indeterminate cases) [28].
At the present time, it seems prudent to suggest that the period of time spent in CMV mode be curtailed as much as possible, especially in older individuals. In fact, animal studies suggest that the effects of aging and mechanical ventilation are additive [29]. Although CMV induced similar losses (24%) in diaphragmatic isometric tension in both young and old animals, the combined effects of aging and CMV resulted in 34% decrement in diaphragmatic isometric tension compared to young control animals [29]. Furthermore, partial modes of ventilatory support should be used whenever possible, even in situations where CMV is classically used, such as acute respiratory distress syndrome [30, 31], because assisted modes attenuate the deleterious effects of mechanical ventilation on respiratory muscles [32]. Further studies are needed to determine the amount of activity the respiratory muscles should have to prevent VIDD. Preliminary results (based on the force (Po) data of animals subjected to three days of either assisted mechanical ventilation or CMV and the electromyographic activity of the diaphragm) suggest that partial diaphragm contractions at 25% or more of the spontaneous breathing electromyographic activity can significantly attenuate VIDD (C Sassoon, personal communication). It is also not known whether periods of intermittent activity (i.e., 'exercise' of the respiratory muscles) can prevent or attenuate VIDD. Preliminary results in rats suggest that allowing either 5 or 60 minutes of spontaneous breathing every 6 hours of CMV to 'exercise' the respiratory muscles could not significantly attenuate the decrease in diaphragmatic force production induced by CMV despite being adequate to prevent atrophy [33]. Whether more frequent intervals of spontaneous breathing might be more effective in this regard awaits experimental proof.
The use of CMV for respiratory muscle rest during difficult weaning
The use of CMV in patients who experience weaning failure after a spontaneous breathing trial or after extubation is a strategy based on the premise that respiratory muscle fatigue (requiring rest to recover) is the cause of weaning failure [1, 34]. This is because the load that the respiratory muscles of patients who fail to wean are facing is increased to a range that would predictably produce fatigue of the respiratory muscles [35] if patients were allowed to continue spontaneous breathing without ventilator assistance. Recent evidence, however, does not support the existence of low frequency fatigue (the type of fatigue that is long-lasting, taking more than 24 hours to recover) in patients who fail to wean despite the excessive respiratory muscle load [25]. Twitch transdiaphragmatic pressure elicited by magnetic stimulation of the phrenic nerve was not altered before and after the failing weaning trials [25]. The tension-time index of the diaphragm was 0.17 to 0.22 during failing weaning trials [25]. Bellemare and Grassino [36] reported that the relationship between the tension-time index of the diaphragm (TTdi) and time to task failure in healthy subjects follows an inverse power function: time to task failure = 0.1 (TTdi)-3.6. Based on this formula, the expected times to task failure would be 59 to 28 minutes. The average value of the TTdi during the last minute of the trial was 0.26, and patients undergoing weaning failure would be predicted to sustain this effort for another 13 minutes before development of diaphragmatic fatigue [25]. Thus, the lack of low frequency respiratory muscle fatigue development despite the excessive load is due to the fact that physicians have adopted criteria for the definition of spontaneous breathing trial failure, and thus termination of unassisted breathing, that lead them to put patients back on the ventilator before the development of low frequency respiratory muscle fatigue. Thus, no reason exists to completely unload the respiratory muscles with CMV for low frequency fatigue reversal if weaning is terminated based on widely accepted predefined criteria. Whether high frequency fatigue develops in patients who fail to wean is not known. Even if this were the case, however, animal studies suggest that complete unloading of the respiratory muscles delays high frequency fatigue reversal, and thus CMV should not be used [37, 38].
The lack of fatigue, however, does not mean that the loaded breathing associated with weaning failure is not injurious for the respiratory muscles. Both animal models and human data have shown that breathing against such loads (TTdi 0.17 to 0.22) can injure the respiratory muscles [39]. Nevertheless, this injury peaks at about three days after the excessive loading, which coincides with the documented decline in the force-generating capacity of the diaphragm at this later time point [39]. Thus, although weaning failure is not associated with low frequency fatigue of the diaphragm at the time of termination of spontaneous breathing trials, it may lead to the onset of an injurious process in the respiratory muscles, which is expected to peak later.
Whether CMV would be beneficial under these circumstances is not clear. All animal studies of VIDD to date have been performed with previously normal diaphragm muscle. We do not know, therefore, to what extent the response to CMV might be modified by the baseline state of the diaphragm. For instance, oxidative stress is implicated in the loss of diaphragmatic force-generating capacity associated with sepsis, as well as mechanical ventilation. Short-term (four hours) CMV, however, actually improves force-generating capacity of the diaphragm in sepsis and does not appear to alter the level of oxidative stress under these conditions [2]. Along these same lines, the response to CMV could conceivably be quite different in a diaphragm previously loaded to the point of injury, which is also associated with increased oxidative stress. Under these specific circumstances, does CMV favour or prevent the development of further oxidative stress, injury, and contractile dysfunction? Moreover, once diaphragmatic injury has occurred, does CMV facilitate or impair the subsequent muscle repair process, particularly as evidence suggests that CMV alters the expression of myogenic transcription factors involved in muscle regeneration [40]? The answers to these important questions await further study.
Antioxidants attenuate the detrimental effects of CMV
Given the central role of oxidative stress in the development of VIDD, antioxidant supplementation could decrease the oxidative stress and could thus attenuate VIDD. Accordingly, when rats were administered the antioxidant Trolox (an analogue of vitamin E) from the onset of CMV, its detrimental effects on contractility and proteolysis were prevented [41]. Interestingly, a combination of vitamins E and C administered to critically ill surgical (mostly trauma) patients was effective in reducing the duration of mechanical ventilation compared to non-supplemented patients [42]. It is tempting to speculate that part of this beneficial effect was mediated by preventing VIDD. Thus, when CMV is used, concurrent administration of antioxidants seems justified, as a recent metanalysis suggests that they are beneficial (and certainly not harmful) in critical care patients [43].
Abbreviations
- CMV:
-
controlled mechanical ventilation
- TTdi:
-
tension-time index of the diaphragm
- VIDD:
-
ventilator-induced diaphragmatic dysfunction.
References
Vassilakopoulos T, Zakynthinos S, Roussos C: Respiratory muscles and weaning failure. Eur Respir J 1996, 9: 2383-2400. 10.1183/09031936.96.09112383
Ebihara S, Hussain SN, Danialou G, Cho WK, Gottfried SB, Petrof BJ: Mechanical ventilation protects against diaphragm injury in sepsis: interaction of oxidative and mechanical stresses. Am J Respir Crit Care Med 2002, 165: 221-228.
Viires N, Sillye G, Aubier M, Rassidakis A, Roussos C: Regional blood flow distribution in dog during induced hypotension and low cardiac output. Spontaneous breathing versus artificial ventilation. J Clin Invest 1983, 72: 935-947.
Vassilakopoulos T, Petrof BJ: Ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 2004, 169: 336-341. 10.1164/rccm.200304-489CP
Anzueto A, Peters JI, Tobin MJ, de los SR, Seidenfeld JJ, Moore G, Cox WJ, Coalson JJ: Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med 1997, 25: 1187-1190. 10.1097/00003246-199707000-00021
Radell PJ, Remahl S, Nichols DG, Eriksson LI: Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med 2002, 28: 358-364. 10.1007/s00134-002-1207-8
Sassoon CS, Caiozzo VJ, Manka A, Sieck GC: Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol 2002, 92: 2585-2595.
Le Bourdelles G, Viires N, Boczkowski J, Seta N, Pavlovic D, Aubier M: Effects of mechanical ventilation on diaphragmatic contractile properties in rats. Am J Respir Crit Care Med 1994, 149: 1539-1544.
Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL: Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 2002, 92: 1851-1858.
Yang L, Luo J, Bourdon J, Lin MC, Gottfried SB, Petrof BJ: Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am J Respir Crit Care Med 2002, 166: 1135-1140. 10.1164/rccm.2202020
Gayan-Ramirez G, De Paepe K, Cadot P, Decramer M: Detrimental effects of short-term mechanical ventilation on diaphragm function and IGF-I mRNA in rats. Intensive Care Med 2003, 29: 825-833.
Capdevila X, Lopez S, Bernard N, Rabischong E, Ramonatxo M, Martinazzo G, Prefaut C: Effects of controlled mechanical ventilation on respiratory muscle contractile properties in rabbits. Intensive Care Med 2003, 29: 103-110.
Shanely RA, Coombes JS, Zergeroglu AM, Webb AI, Powers SK: Short-duration mechanical ventilation enhances diaphragmatic fatigue resistance but impairs force production. Chest 2003, 123: 195-201. 10.1378/chest.123.1.195
Zhu E, Sassoon CS, Nelson R, Pham HT, Zhu L, Baker MJ, Caiozzo VJ: Early effects of mechanical ventilation on isotonic contractile properties and MAF-box gene expression in the diaphragm. J Appl Physiol 2005, 99: 747-756. 10.1152/japplphysiol.00126.2005
Hussain SN, Vassilakopoulos T: Ventilator-induced cachexia. Am J Respir Crit Care Med 2002, 166: 1307-1308. 10.1164/rccm.2208004
Shanely RA, Van Gammeren D, Deruisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, Powers SK: Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med 2004, 170: 994-999. 10.1164/rccm.200304-575OC
Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK: Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med 2002, 166: 1369-1374. 10.1164/rccm.200202-088OC
Deruisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, Ordway GA, Powers SK: Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol 2005, 98: 1314-1321. 10.1152/japplphysiol.00993.2004
Davies KJ: Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001, 83: 301-310. 10.1016/S0300-9084(01)01250-0
Shringarpure R, Grune T, Mehlhase J, Davies KJ: Ubiquitin-conjugation is not required for the degradation of oxidized proteins by the proteasome. J Biol Chem 2003, 278: 311-318. 10.1074/jbc.M206279200
Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, Deruisseau KC, Powers SK: Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol 2003, 95: 1116-1124.
Bernard N, Matecki S, Py G, Lopez S, Mercier J, Capdevila X: Effects of prolonged mechanical ventilation on respiratory muscle ultrastructure and mitochondrial respiration in rabbits. Intensive Care Med 2003, 29: 111-118.
Radell P, Edstrom L, Stibler H, Eriksson LI, Ansved T: Changes in diaphragm structure following prolonged mechanical ventilation in piglets. Acta Anaesthesiol Scand 2004, 48: 430-437. 10.1111/j.1399-6576.2004.00352.x
Watson AC, Hughes PD, Louise HM, Hart N, Ware RJ, Wendon J, Green M, Moxham J: Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med 2001, 29: 1325-1331. 10.1097/00003246-200107000-00005
Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ: Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 2003, 167: 120-127. 10.1164/rccm.200210-1246OC
Ayas NT, McCool FD, Gore R, Lieberman SL, Brown R: Prevention of human diaphragm atrophy with short periods of electrical stimulation. Am J Respir Crit Care Med 1999, 159: 2018-2020.
Knisely AS, Leal SM, Singer DB: Abnormalities of diaphragmatic muscle in neonates with ventilated lungs. J Pediatr 1988, 113: 1074-1077.
Deem S, Lee CM, Curtis JR: Acquired neuromuscular disorders in the intensive care unit. Am J Respir Crit Care Med 2003, 168: 735-739. 10.1164/rccm.200302-191UP
Criswell DS, Shanely RA, Betters JJ, McKenzie MJ, Sellman JE, Van Gammeren DL, Powers SK: Cumulative effects of aging and mechanical ventilation on in vitro diaphragm function. Chest 2003, 124: 2302-2308. 10.1378/chest.124.6.2302
Zakynthinos SG, Vassilakopoulos T, Daniil Z, Zakynthinos E, Koutsoukos E, Katsouyianni K, Roussos C: Pressure support ventilation in adult respiratory distress syndrome: short-term effects of a servocontrolled mode. J Crit Care 1997, 12: 161-172. 10.1016/S0883-9441(97)90027-7
Putensen C, Zech S, Wrigge H, Zinserling J, Stuber F, Von Spiegel T, Mutz N: Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med 2001, 164: 43-49.
Sassoon CS, Zhu E, Caiozzo VJ: Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 2004, 170: 626-632. 10.1164/rccm.200401-042OC
Gayan-Ramirez G, Testelmans D, Racz G, Maes K, Cadot P, Zador E, Wuytack F, Decramer M: Intermittent spontaneous breathing protects the rat diaphragm from the detrimental effects of mechanical ventilation [abstract]. Am J Respir Crit Care Med 2004, 169: A123.
Vassilakopoulos T, Roussos C, Zakynthinos S: Weaning from mechanical ventilation. J Crit Care 1999, 14: 39-62. 10.1016/S0883-9441(99)90007-2
Vassilakopoulos T, Zakynthinos S, Roussos C: The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med 1998, 158: 378-385.
Bellemare F, Grassino A: Effect of pressure and timing of contraction on human diaphragm fatigue. J Appl Physiol 1982, 53: 1190-1195. 10.1063/1.330569
Uchiyama A, Imanaka H, Nishimura M, Taenaka N, Fujino Y, Yoshiya I: Effects of pressure-support ventilation on recovery from acute diaphragmatic fatigue in rabbits. Crit Care Med 1998, 26: 1225-1230. 10.1097/00003246-199807000-00025
Uchiyama A, Imanaka H, Nishimura M, Taenaka N, Fujino Y, Yoshiya I: Optimal level of pressure support ventilation for recovery from diaphragmatic fatigue in rabbits. Crit Care Med 2000, 28: 473-478. 10.1097/00003246-200002000-00031
Jiang TX, Reid WD, Road JD: Free radical scavengers and diaphragm injury following inspiratory resistive loading. Am J Respir Crit Care Med 2001, 164: 1288-1294.
Racz G, Gayan-Ramirez G, De Paepe K, Zador E, Wuytack F, Decramer M: Early changes in rat diaphragm biology with mechanical ventilation. Am J Respir Crit Care Med 2003, 168: 297-304. 10.1164/rccm.200206-541OC
Betters JL, Criswell DS, Shanely RA, Van Gammeren D, Falk D, Deruisseau KC, Deering M, Yimlamai T, Powers SK: Trolox attenuates mechanical ventilation-induced diaphragmatic dysfunction and proteolysis. Am J Respir Crit Care Med 2004, 170: 1179-1184. 10.1164/rccm.200407-939OC
Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV: Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg 2002, 236: 814-822. 10.1097/00000658-200212000-00014
Heyland D: Antioxidant nutrients:a systematic review of trace elements and vitamins in the critically ill patient. Intensive Care Med 2005, 31: 327-337. 10.1007/s00134-004-2522-z
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Vassilakopoulos, T., Zakynthinos, S. & Roussos, C. Bench-to-bedside review: Weaning failure – should we rest the respiratory muscles with controlled mechanical ventilation?. Crit Care 10, 204 (2005). https://doi.org/10.1186/cc3917
Published:
DOI: https://doi.org/10.1186/cc3917